
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA rapid ion chromatography column-switching method for online sample pretreatment and determination of L-carnitine, choline and mineral ions in milk and powdered infant formula†
Dan Wei a,
Xu Wangb,
Nani Wangc and
Yan Zhu*a
a,
Xu Wangb,
Nani Wangc and
Yan Zhu*a
aDepartment of Chemistry, Zhejiang University, Xixi Campus, Hangzhou, Zhejiang, China. E-mail: zhuyan@zju.edu.cn
bDepartment of Medical Laboratory, Hangzhou Medical College, Hangzhou, Zhejiang, China
cDepartment of Medicine, Zhejiang Academy of Traditional Chinese Medicine, Hangzhou, Zhejiang, China
First published on 17th January 2017
Abstract
A simple and rapid column-switching ion chromatography with non-suppressed conductivity detection method was developed for simultaneous determination of L-carnitine, choline and mineral ions (free sodium, potassium, magnesium and calcium ions) in milk and powdered infant formula samples. On-line sample clean-up was achieved on the pretreatment column Dionex IonPac NG1 (50 mm × 4 mm, i.d.), with pretreatment elution (3 mmol L−1 MSA) and washing solution (100% acetonitrile) at a flow rate of 1.0 mL min−1. After column switching, the separation and determination of analytes were performed using guard column Dionex IonPac SGS (50 mm × 4 mm, i.d.) and analytical column Dionex IonPac SCS (250 mm × 4 mm, i.d.) with an isocratic mobile phase consisting of 3 mmol L−1 MSA and 10% (v/v) acetonitrile, at flow-rate of 1 mL min−1, followed by non-suppressed conductivity detection. The organic matrices clean-up performance of the proposed method was studied using an UV detector working at 200–400 nm. Under the optimal conditions, detection limits (S/N = 3) ranging from 0.05–0.30 mg L−1, good linearities in the range of 1–100 mg L−1 with a correlation coefficient over 0.9973, and satisfactory recoveries in the range between 94% and 105% with relative standard deviation less than 2.1% for real samples were attainted. The proposed method was successfully applied to rapid, simultaneous, and labour-saving detection of L-carnitine, choline, and mineral ions in milk and powdered infant formula samples.
1. Introduction
Infant formula has been recognized as a very important dairy product regarding the wide consumption for children under 1 year. The composition of infant formula, especially L-carnitine, choline, and mineral ions (free sodium, potassium, magnesium and calcium ions), play essential roles in both optimal growth (e.g. bone, brain and visual development) and the development of a newborn's immune system against an array of diseases and infections.1–3 In detail, L-carnitine, a critical substance needed for the maintenance of health, is used as a carrier to transport long-chain fatty acids into the mitochondria of a cell for beta-oxidation to produce energy. Carnitine deficiency may lead to insufficient transport of fatty acids through the mitochondrial membrane therefore impairing the mitochondrial energy metabolism. Choline is essential for the normal function of all cells. Adequate choline intake is very important for fetal development, memory function, bone formation, normal liver and kidney functions and the prevention of various diseases.4–7 The mineral ions are the main cations in milk and their fraction contributes to the nutritional quality of milk and largely determines its physicochemical state, thereby determining the conformation and stability of milk proteins.8 More specifically, sodium is the principal cation in extracellular fluid, which is involved in nerve and muscle function, regulation of plasma volume and acid–base balance.9 Potassium, as the major cation in intracellular fluid, plays an important role in the regulation of osmotic pressure, blood pressure and acid–base balance.10 Magnesium is primarily an intracellular nutrient and plays roles in protein and carbohydrate metabolism, synthesis of DNA, and muscle relaxation. Furthermore, magnesium forms a complex with ATP, which serves as the true substrate for biochemical reactions involving energy utilization. Calcium is important in the development and maintenance of strong bones and teeth and also involved in nerve function, muscle contraction and blood clotting.11 The excess or absence of these nutritional elements can also cause several health problems. Thus, it is necessary to develop a simple and rapid method for these nutritional elements determination in milk and powered infant formulas to ensure rigorous control of their composition, including elements added routinely in order to satisfy such nutrients content requirements.Numerous methods for analysis of L-carnitine, choline, and mineral ions (free sodium, potassium, magnesium and calcium ions) either alone or in various combinations have been developed, including gas chromatographic (GC)12 and conventional HPLC and ion chromatography (IC). The limitation of GC analysis is the need to conversion of choline into a volatile derivative. The conventional HPLC with different detectors, such as evaporative light scattering and mass spectrometry detection,13 electrochemical detection and,14 suppression conductivity detection15 has restrictions in terms of weak chromophore, derivatization and redissolution of the extract in organic solvent. Also, IC with conductivity detection has been evaluated for the separation and determination of choline,16 L-carnitine17 sodium, potassium, magnesium and calcium ions18,19 in food samples20,21 and is an attractive alternative. It is well known that IC is a powerful analytical technique for the separation of cations, anions and polar and hydrophilic compounds. However, the direct determination of the target analytes in milk samples is difficult. Complex matrices, including proteins, lipids, carbohydrates, vitamins, minerals and enzymes, would damage the IC columns, decrease sensitivity, selectivity, and reproducibility. Solid-phase extraction (SPE) is the most commonly used methods for clean-up and extraction,22,23 and usually carried out in off-line mode. Using these sample pretreatment methods could be accompanied with risk of analytes losses during sample manipulation and greater organic hazard. Recent application of column-switching technique for on-line extraction is the growing trend in sample preparation procedures.24 Column-switching technique using special sorbents for complex matrix samples can work without the laborious and time-consuming clean-up protocols. From this perspective, column-switching technique for direct injection, on-line preparation and simultaneously analysis of samples is modern approach in implementation of all of analytical processes to one step. The main advantage of this technique is strong reduction of time and sample manipulation during sample preparation steps. Moreover, its application has expanded to the analysis of honeydew and nectar, Colla corii asini.25,26 However, there are few applications in quantification of nutrients in milk products.
In this paper, we presents a rapid on-line column-switching IC method for simultaneous determination of L-carnitine, choline, sodium, potassium, magnesium and calcium ions in milk and powdered infant formula samples. IonPac NG1, a kind of polymer-based reversed-phase C-18 column, was used as precolumn. This column could enable direct injection of the samples into the flow analytical systems, because it would absorb organic interferences to the hydrophobic part of the stationary phase and, at the same time, exclude target ionic molecules. The proposed method was developed and validated and may have useful applications in milk products quality control.
2. Materials and methods
2.1. Instrumentation
Chromatographic analysis was performed on ICS-3000 (Thermo-Fisher Scientific Waltham, MA, USA) consisting of two quaternary pumps with automated membrane eluent degassing, a column heater, two six port valves. On-line sample pretreatments were carried out using Dionex IonPac NG1 guard column (50 mm × 4 mm, i.d.). Separations were accomplished using a Dionex IonPac SCS (250 mm × 4 mm, i.d.) analytical column and IonPac SGS (50 mm × 4 mm, i.d.) guard column. The separated cations were determined by non-suppressed conductivity detection; an UV detector (UVD 340U, Thermo-Fisher Scientific Waltham, MA, USA) was applied in validation experiment. 25 μL of prepared sample was injected into the loop of the chromatograph and a 1 mL polyether ether ketone tube used as a collection loop. Polyether ether ketone (PEEK) tubes with the lengths as short as possible were used to connect all chromatographic hardware. The eluent flow rate was 1.0 mL min−1. Dionex Chromeleon 6.8 software (Thermo-Fisher Scientific Waltham, MA, USA) was used for instrument control and data handling.A SBL-5DTS thermostatic sonic water bath (Scientz biotechnology Co., LTD, Ningbo, China) was used to control the temperature and sonicate the samples. A centrifuge model LG16-B (Lab centrifuge Co., LTD, Beijing, China) was utilized for the centrifugation.
2.2. Reagents and samples
Choline chloride and L-carnitine was purchased from Sigma-Aldrich (St. Louis, MO, USA). 1000 mg mL−1 of sodium, potassium, magnesium and calcium were obtained from National Research Center for Certified Reference Materials (Beijing, China). Deionized water employed in all experiments was obtained from a Genpure water system (TKA, Niederelbert, Germany), with resistivity not lower than 18.2 MΩ cm. Extra pure methanesulfonic acid (MSA) and acetonitrile of HPLC grade was obtained from ThermoFisher Scientific (Waltham, MA, USA). All the reagents used in this study were of analytical-reagent grade. Six real samples from six different manufactures were purchased from local supermarket in Hangzhou, China, and stored at 4 °C until used in this study.2.3. Preparation of the standard solutions and spiked milk samples
Stock solutions containing 1000 mg L−1 of L-carnitine and chloride were prepared by dissolving of substances using deionized water. The stock solutions were kept at 0 to 4 °C for no longer than one month. The stock solution was further diluted with deionized water to obtain working standard solutions used for method development and validation. The calibration standard solutions were prepared at seven concentration levels (1, 2, 5, 10, 20, 30, 50, 75 and 100 mg L−1) for the establishment of the calibration curve. All solutions were sealed and kept at 0 to 4 °C for no longer than one week before analysis.Prior to analysis, 1 g of milk powder was accurately weighed and transferred to a 250 mL flask, to which 8 mL of deionized water was added, and the solution was stirred to dissolve the powder, ultrasonicated for 10 min at 70 °C, 40 Hz and cooled down to the room temperature. Then, 24 mL of acetonitrile was added, centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min. The supernatant solutions were diluted 5 times with deionized water and filtered through a 0.45 μm nylon filter before IC analysis. In the case of liquid milk, 1 mL was transferred to 10 mL centrifugal tube, to which 3 mL of acetonitrile was added, and then centrifuged at 10000 rpm for 10 min. The supernatant solutions were diluted 10 times with deionized water and filtered through a 0.45 μm nylon filter before IC analysis.
000 rpm for 10 min. The supernatant solutions were diluted 5 times with deionized water and filtered through a 0.45 μm nylon filter before IC analysis. In the case of liquid milk, 1 mL was transferred to 10 mL centrifugal tube, to which 3 mL of acetonitrile was added, and then centrifuged at 10000 rpm for 10 min. The supernatant solutions were diluted 10 times with deionized water and filtered through a 0.45 μm nylon filter before IC analysis.
The spiked samples were prepared by spiking the proper amounts of each working solution into 1.0 mL milk or milk powder as prepared above. Samples with higher concentration were diluted with deionized water to obtain concentration inside calibration range. All solutions were stored at 4 °C before analysis.
2.4. IC column switching analysis
The simultaneous on-line sample clean-up and determination was performed using a two columns system. The sample pretreatment column was IonPac NG1 guard column (50 mm × 4 mm, i.d.), with pretreatment elution (3 mmol L−1 MSA) and washing solution (100% acetonitrile) at a flow rate of 1.0 mL min−1. The chromatographic separation was performed on an analytical column Dionex IonPac SCS (250 mm × 4 mm, i.d.) and guard column Dionex IonPac SGS (50 mm × 4 mm, i.d.), using isocratic mobile phase consisting of 3 mmol L−1 MSA and 10% (v/v) acetonitrile and flow rate of 1 mL min−1. Column temperature was kept at 30 °C for both columns.Fig. 1 shows the IC column switching procedures: (1) loading sample (Fig. 1A), (2) on-line sample clean-up pretreatment (Fig. 1B), (3) analyzing and rinsing the pretreatment column (Fig. 1C), and (4) analyzing and the pretreatment column achieving the equilibrium (Fig. 1C). First, 25 μL volume of sample was loaded into the sample loop (Fig. 1A), and then injected into the IonPac NG1 guard column with washing mobile phase of 3 mmol L−1 MSA at the flow-rate of 1 mL min−1 generated by one of the quaternary pumps (Fig. 1B). This condition aided six analytes rapidly eluted and removal of most organic components from samples in the IonPac NG1 guard column. After being completely collected, the analytes in the 1 mL collection loop were sent to the analytical columns for separation with washing mobile phase of 3 mmol L−1 MSA containing 10% acetonitrile at the flow-rate of 1 mL min−1 generated by another the quaternary pump, while the pretreatment column is simultaneously washed by 100% acetonitrile to remove matrices, and the matrices were sent to waste (Fig. 1C). After the analytes analysis was finished, the IC system kept at the initial state (Fig. 1A) to achieve the equilibrium for the next sample. The detailed procedures were described in the Table 1.
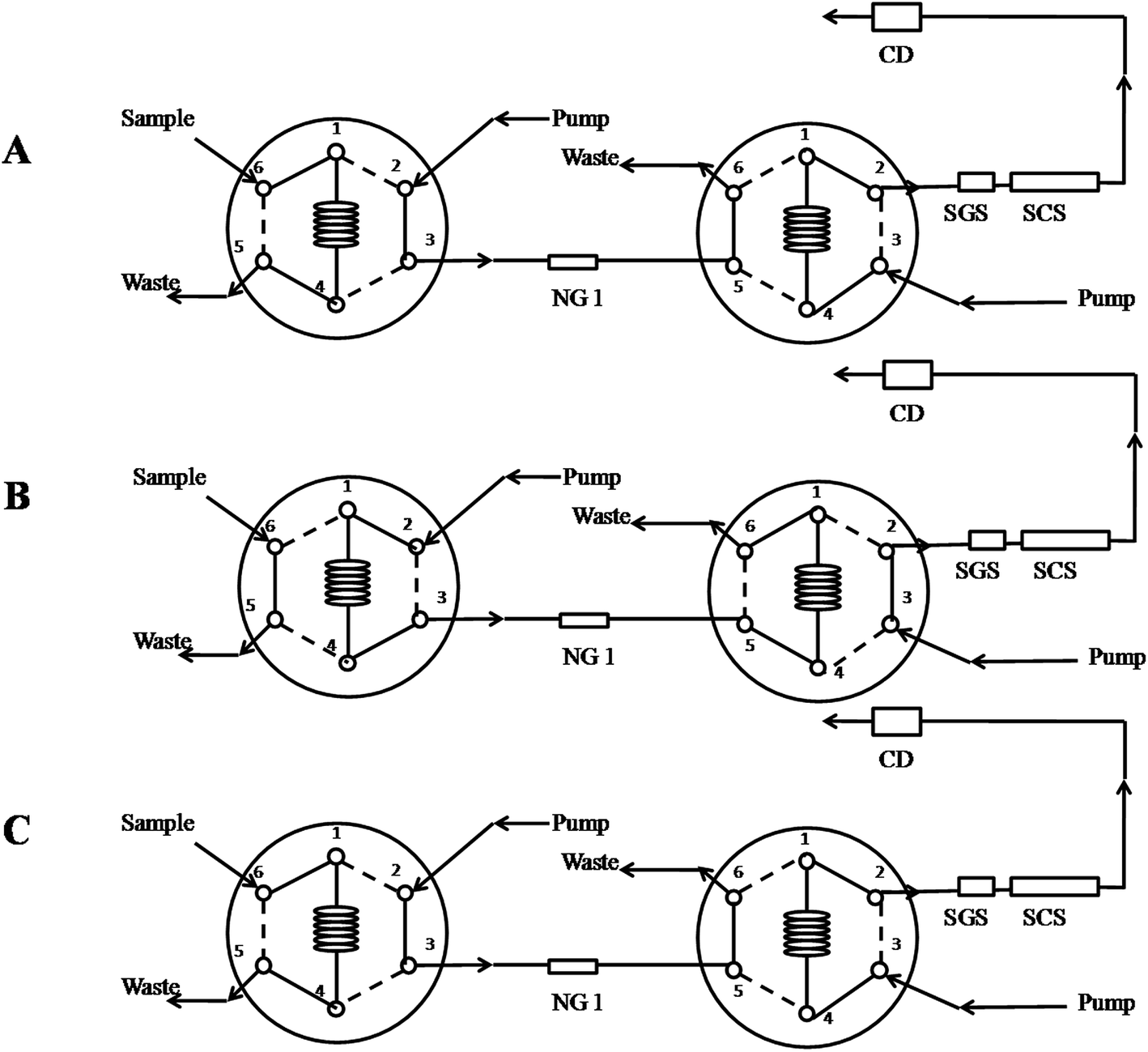 | ||
| Fig. 1 Chromatographic instrument configuration for the column-switching system. (A) Filling the sample loop; (B) on-line sample pretreatment; (C) analyzing six analytes and washing the pretreatment column. | ||
| Procedure | Time (min) | Elution of the pretreatment column | Elution of the analytical column | Valve 1 | Valve 2 |
|---|---|---|---|---|---|
| (1) Sampling | 3 mmol L−1 MSA | 3 mmol L−1 MSA + 10% acetonitrile | Load | Inject | |
| (2) Sample pretreatment | 0–0.5 | 3 mmol L−1 MSA | 3 mmol L−1 MSA + 10% acetonitrile | Inject | Load |
| (3) Analyzing and rinsing of NG1 | 0.5–10.5 | Acetonitrile | 3 mmol L−1 MSA + 10% acetonitrile | Load | Inject |
| (4) Analyzing and NG1 achieving the equilibrium | 10.5–20 | 3 mmol L−1 MSA | 3 mmol L−1 MSA + 10% acetonitrile | Load | Inject |
3. Results and discussion
3.1. Optimization of chromatographic conditions
Prior to the column-switching IC analysis, minimal sample-preparation steps, such as centrifugation or filtration through 0.45 μm filters, are recommended to prolong the lives of the pretreatment column and the analytical column. Centrifuged or filtered samples have micro particles removed as much as possible to avoid plugging the system. Therefore, minimal procedures (proteins precipitation, centrifugation and filtration) are needed prior to column-switching IC system for simultaneous determination of L-carnitine, choline and mineral ions in milk products.Sodium, potassium, calcium and magnesium are the main cations in milk. L-Carnitine and choline are highly polar and hydrophilic compounds, and can be converted into cations on acidic conditions. Thus, it is easy to exclude these ionic molecules on the ion excluding column of Dionex IonPac NG1, while water-solvable organic matrixes still remaining in samples can be retained in IonPac NG1 column. Due to the distinct physicochemical characteristics of the substances, ion excluding column (Dionex IonPac NG1) for pretreatment and ion exchange columns (Dionex IonPac SCS and IonPac SGS column) for separation were used in method development.
The column-switching IC method was optimized by changing the analytical conditions in order to obtain the optimal chromatographic performances. The conditions evaluated include the mobile phase of pretreatment column and analytical column, column-switching time and effects of possible matrix ions. All optimization studies were carried out in triplicates.
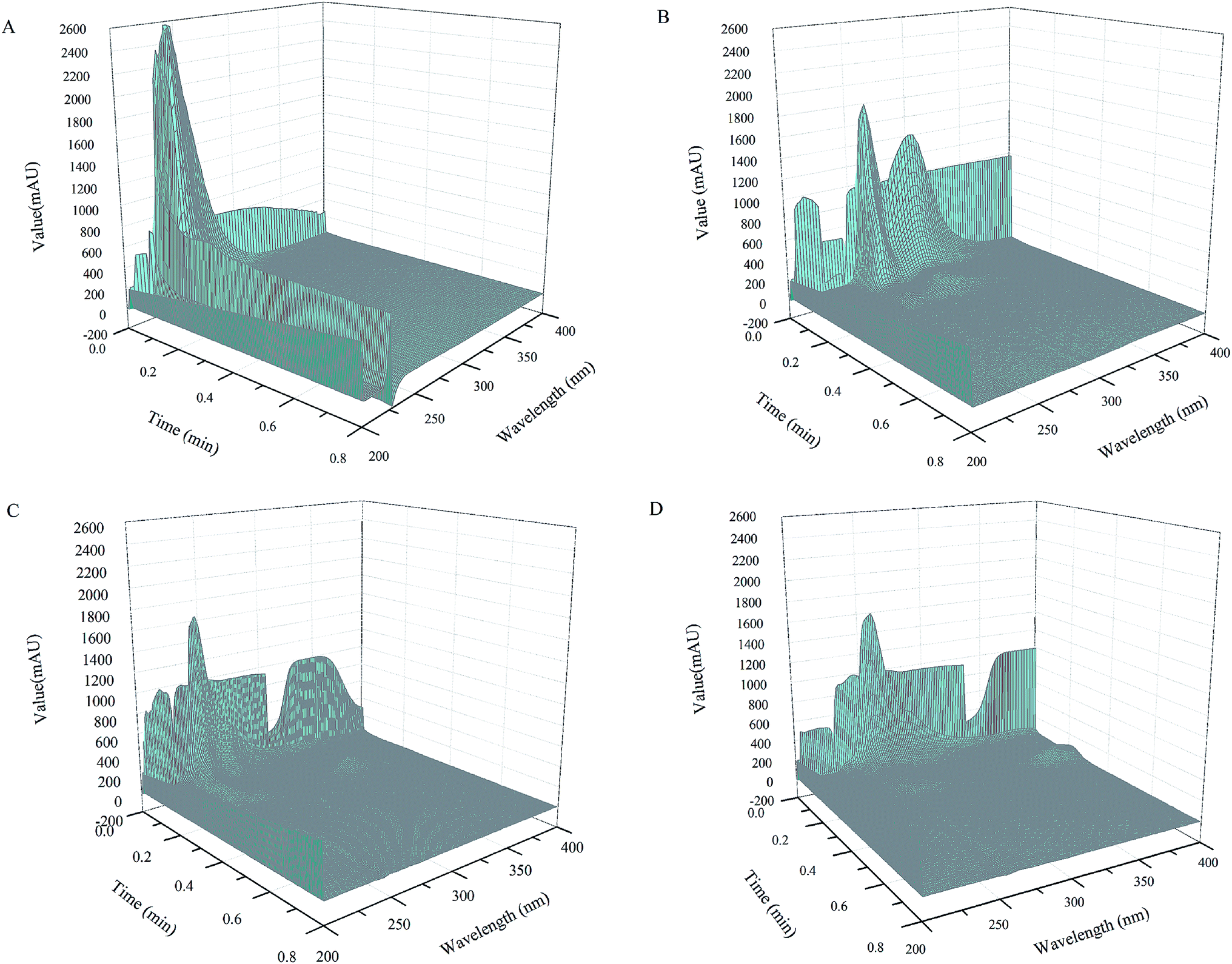 | ||
| Fig. 2 Three dimensional diagram of the real sample, analysis of milk sample by direct injection at the flow rate of 1 mL min−1 (A), analysis of milk sample by on-line pretreatment method at the flow rate of 1.5 mL min−1 (B), 1 mL min−1 (C) and 0.5 mL min−1 (D). Injection volume: 25 μL; UV detection at 200–400 nm, mobile phase: 3 mmol L−1 MSA. | ||
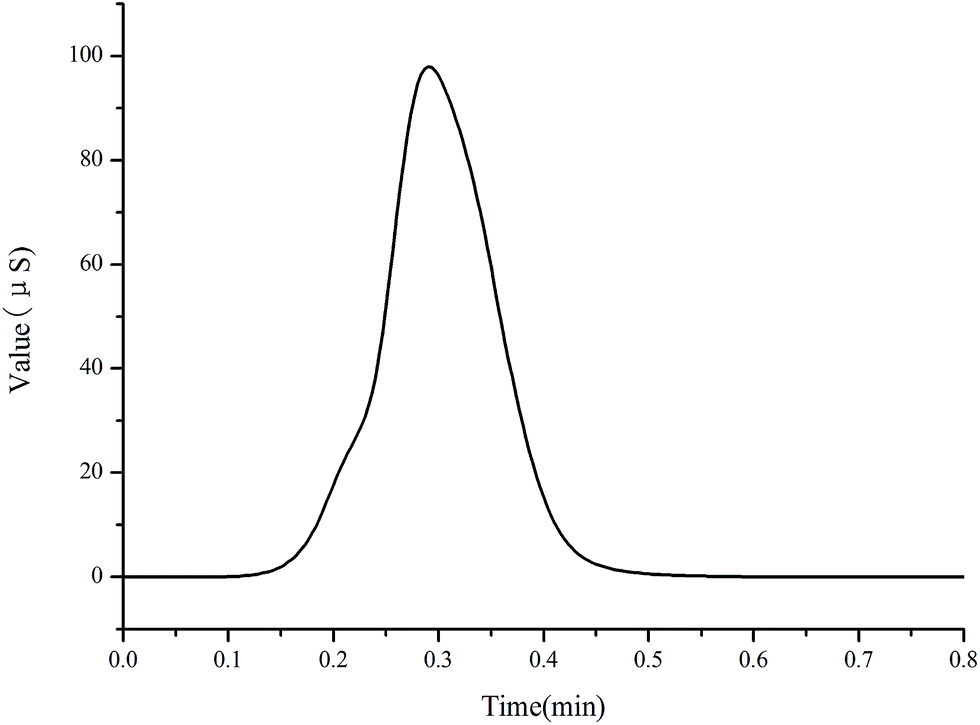 | ||
| Fig. 3 Chromatogram of standard solution (15 mg L−1 sodium, potassium, magnesium, calcium; 10 mg L−1 L-carnitine, choline) by the NG1 column. Injection volume: 25 μL; flow rate: 1.0 mL min−1; non-suppressed conductivity detector, mobile phase: 3 mmol L−1 MSA. | ||
3.2. Method validation
To evaluate the proposed method, experiments with regard to the linearity, limit of detection (LOD), limit of quantification (LOQ), precision selectivity and robustness of the method were performed.:1 and 10:1 respectively. The values were shown in Table 2.
| Analyte | Linear equation | r2 | Linear (mg L−1) | LOD (mg L−1) | LOQ (mg L−1) | RSD (n = 5)% | |
|---|---|---|---|---|---|---|---|
| Peak area | Retention time (min) | ||||||
| Na | y = 0.0153x − 0.0043 | 0.9977 | 1–100 | 0.05 | 0.18 | 1.5 | 1.1 |
| K | y = 0.0030x + 0.0027 | 0.9976 | 1–100 | 0.15 | 0.50 | 0.9 | 0.4 |
| L-Carnitine | y = 0.0120x + 0.0003 | 0.9962 | 2–200 | 0.25 | 0.82 | 0.9 | 0.7 |
| Choline | y = 0.0124x + 0.0373 | 0.9954 | 1–100 | 0.10 | 0.35 | 1.2 | 0.6 |
| Mg | y = 0.0214x − 0.0496 | 0.9974 | 1–100 | 0.30 | 0.99 | 1.0 | 0.9 |
| Ca | y = 0.0119x − 0.0073 | 0.9996 | 2–200 | 0.25 | 0.85 | 0.7 | 0.2 |
Accuracy and precision were determined by recovery test using spiked purchased milk samples. The non-spiked samples and fortified samples with three concentration levels were subjected to the entire sample pretreatment, column-switching procedures and analytical sequences. All determinations were performed in five times and the means of the results were used. The results are summarized in the Table 3. The average recoveries ranged from 94% to 105% for the proposed method, with the RSD below 3% (n = 5). The chromatograms of spiked real samples by the column-switching IC procedure are shown in Fig. 4. The accurate determination of L-carnitine, choline and sodium, potassium, magnesium and calcium ions in milk and powdered infant formula can be attained by this method.
| Analyte | Sample concentration (mg kg−1) | Spiked concentration (mg kg−1) | Found concentration (mg kg−1) | Average recovery (%) | RSD (%) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Infant formula | Milk powder | Milk | Infant formula | Milk powder | Milk | Infant formula | Milk powder | Milk | Infant formula | Milk powder | Milk | Infant formula | Milk powder | Milk | |
| Na | 1602 | 1694 | 594 | 960 | 1360 | 480 | 2513 | 3182 | 1064 | 98.1 | 104.2 | 99.1 | 1.8 | 1.1 | 0.9 |
| 1200 | 1690 | 600 | 2869 | 3248 | 1223 | 102.4 | 96.2 | 102.4 | 0.9 | 0.9 | 0.9 | ||||
| 1440 | 2030 | 710 | 2926 | 3575 | 1363 | 96.2 | 96.1 | 104.5 | 1.1 | 0.9 | 1.1 | ||||
| K | 4055 | 4302 | 1500 | 3000 | 3400 | 1200 | 6992 | 7871 | 2784 | 99.1 | 102.2 | 103.1 | 0.9 | 1.1 | 0.9 |
| 4000 | 4000 | 1500 | 7354 | 8642 | 3123 | 91.3 | 104.1 | 104.1 | 0.9 | 0.9 | 0.9 | ||||
| 4560 | 5100 | 1800 | 8787 | 8904 | 3188 | 102 | 94.7 | 96.6 | 0.9 | 1.3 | 1.3 | ||||
| L-Carnitine | 76 | 65 | ND | 69 | 52 | 20 | 145 | 121 | 21 | 103.7 | 98.3 | 103.2 | 1.2 | 0.9 | 0.9 |
| 86 | 60 | 40 | 162 | 130 | 42 | 104.1 | 95.6 | 104.0 | 1.3 | 0.5 | 0.9 | ||||
| 90 | 80 | 60 | 166 | 140 | 58 | 96.5 | 98.2 | 96.8 | 1.1 | 0.9 | 0.6 | ||||
| Choline | 1227 | 922 | ND | 960 | 740 | 480 | 2163 | 1559 | 471 | 98.9 | 93.8 | 98.2 | 0.6 | 0.9 | 0.9 |
| 1200 | 900 | 600 | 2502 | 1718 | 580 | 103.1 | 94.3 | 96.6 | 0.9 | 1.2 | 1.8 | ||||
| 1440 | 1100 | 720 | 2712 | 1992 | 693 | 101.7 | 98.5 | 96.3 | 1.8 | 1.3 | 0.7 | ||||
| Mg | 612 | 520 | 132 | 490 | 420 | 100 | 1143 | 907 | 201 | 97.4 | 96.9 | 230 | 0.7 | 0.9 | 1.6 |
| 620 | 500 | 130 | 1126 | 984 | 204 | 94.9 | 90.8 | 239 | 2.1 | 0.9 | 0.7 | ||||
| 734 | 620 | 160 | 1360 | 1100 | 251 | 102 | 94.0 | 295 | 1.1 | 1.2 | 1.6 | ||||
| Ca | 3600 | 3780 | 1010 | 2870 | 2900 | 800 | 6735 | 6660 | 1828 | 104.1 | 99.7 | 101.0 | 1.2 | 1.3 | 1.1 |
| 3600 | 3800 | 1000 | 6826 | 7716 | 1978 | 94.8 | 101.8 | 98.4 | 1.2 | 1.1 | 1.2 | ||||
| 4296 | 4500 | 1200 | 7785 | 8023 | 2279 | 98.6 | 96.9 | 103.1 | 1.1 | 1.2 | 1.7 | ||||
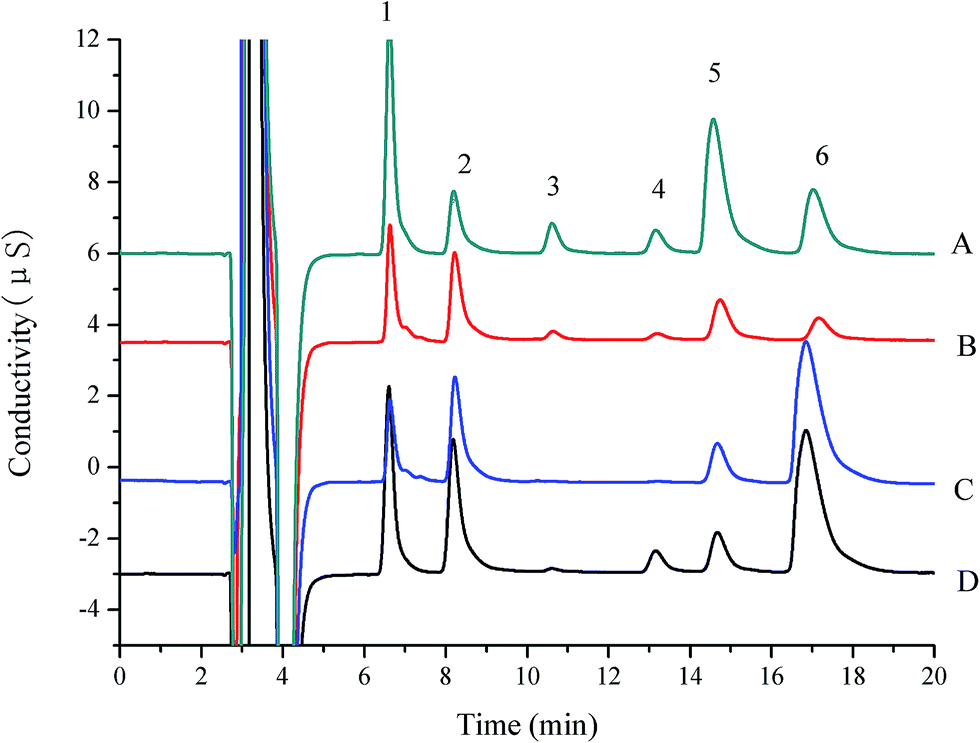 | ||
| Fig. 4 Chromatograms of standard solution (15 mg L−1 sodium, potassium, magnesium, calcium; 10 mg L−1 L-carnitine, choline) (A), infant formula (B), milk (C) and milk powder (D); peaks: (1) sodium; (2) potassium; (3) L-carnitine; (4) choline; (5) magnesium; (6) calcium. | ||
3.3. Application of the method
To verify the applicability of the proposed method, determination of these nutrients in six milk products from different local supermarkets was conducted. The chromatogram of target analytes obtained from milk samples was shown in Fig. 3, and the compositional data of six milk samples was tabulated in Table 4. Peak purity in sample run showed that no interference was observed in the quantitative analysis of six analytes. The suggested procedure of sample preparation was simple, fast and reliable. Much of the nutrients contents measured by the proposed method had values close to label declared. It was clear that the predominant components were potassium and calcium, which up to 60% of all the quantitative analytes. It has been showed that concentrations of the minerals have no statistically significant differences from the label claims. Surprisingly, without mentioned in the label claim, L-carnitine was detected in milk powder 2 and choline was detected in milk 1. It may be that natural milk contains L-carnitine and choline.| Analyte | Declared concentration (mg kg−1) | Found concentration in samples (mg kg−1) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Infant formula 1 | Infant formula 2 | Milk powder 1 | Milk powder 2 | Milk 1 | Milk 2 | Infant formula 1 | Infant formula 2 | Milk powder 1 | Milk powder 2 | Milk 1 | Milk 2 | |
| Na | 1260 | 2000 | 1940 | 2000 | 620 | 720 | 1602 | 2010 | 1694 | 1880 | 594 | 700 |
| K | 3790 | 7910 | 3800 | — | — | — | 4055 | 8000 | 4302 | 6450 | 1500 | 990 |
| L-Carnitine | 80 | 93 | — | — | — | — | 76 | ND | 65 | 20 | ND | ND |
| Choline | 1240 | 880 | 850 | — | — | — | 1227 | ND | 922 | 1050 | ND | 600 |
| Mg | 620 | 520 | 490 | 380 | — | — | 612 | 480 | 520 | 300 | 132 | 100 |
| Ca | 3580 | 6600 | 3640 | 7000 | 1000 | 1250 | 3600 | 6480 | 3780 | 6910 | 1010 | 1200 |
4. Conclusions
In this paper, a simple and automated column-switching IC method with non-suppressed conductivity detection has been developed for the simultaneous determination of L-carnitine, choline, mineral ions (free sodium, potassium, magnesium, calcium ions) in infant formula, milk powder and milk. The developed method proved to be simple, efficient and reliable and may have the large potential applications in measurement and assessment of the nutritional quality of milk products.Acknowledgements
This research was financially supported by Key Laboratory of Health Risk Appraisal for Trace Toxic Chemicals of Zhejiang Province, Science and Technique Plan of Zhejiang TCM (No. 2016ZQ003), Zhejiang Provincial Health and Medicinal Science Foundation (2016RCB003) and Zhejiang Provincial Natural Science Foundation of China (No. Y4110532, LY12B05003, LY13B020001, LQ13B050001).References
- F. A. R. Martino, M. L. F. Sanchez and A. S. Medel, J. Anal. At. Spectrom., 2000, 15, 163–168 RSC.
- F. Masotti, D. Erba, I. De Noni and L. Pellegrino, J. Dairy Sci., 2012, 95, 2872–2881 CrossRef CAS PubMed.
- I. Nadaoka, E. Hatakeyama, C. Tanada, T. Sakamoto, S. Fukaya, T. Akiba, K. Inoue, Y. Yamano and T. Toyo'oka, Anal. Sci., 2014, 30, 839–844 CrossRef CAS PubMed.
- Y. P. Xiong, Y. Y. Zhao and S. Goruk, J. Chrom. B, 2012, 911, 170–179 CrossRef CAS PubMed.
- S. H. Zeisel, Nutrition, 2000, 16, 669–671 CrossRef CAS PubMed.
- J. R. Hartwell, M. J. Cecava and S. S. Donkin, J. Dairy Sci., 2000, 83, 2907–2917 CrossRef CAS PubMed.
- J. Sales, P. Homolka and V. Koukolová, J. Dairy Sci., 2010, 93, 3746–3754 CrossRef CAS PubMed.
- R. Gao, E. J. M. Temminghoff, H. P. van Leeuwen, H. J. F. van Valenberg, M. D. Eisner and M. A. J. van Boekel, Int. Dairy J., 2009, 19, 431–436 CrossRef CAS.
- L. Noël, M. Carl, C. Vastel and T. Guérin, Int. Dairy J., 2008, 18, 899–904 CrossRef.
- G. Yellen, Nature, 2002, 419, 35–42 CrossRef CAS PubMed.
- M. C. Latham, Human nutrition in the developing world, Popul. Dev. Rev., 1998, 24, 177 Search PubMed.
- S. H. Zeisel and K. A. Dacosta, J. Nutr. Biochem., 1990, 1, 55–59 CrossRef CAS PubMed.
- P. Donato, F. Cacciola, F. Cichello, M. Russo, P. Dugo and L. Mondello, J. Chromatogr. A, 2011, 1218, 6476–6482 CrossRef CAS PubMed.
- N. Tyrefors and A. Carlsson, J. Chromatogr. A, 1990, 502, 337–349 CrossRef CAS.
- S. H. Chen, V. Soneji and J. Webster, J. Chromatogr. A, 1996, 739, 351–357 CrossRef CAS PubMed.
- K. Oates, L. Chen, B. De Borba, D. Mohindra, J. Rohrer and D. Dowell, J. AOAC Int., 2013, 96, 1400–1406 CrossRef CAS PubMed.
- D. C. Suo, L. Li, S. Zhang and X. O. Su, Anal. Methods, 2013, 5, 59–63 RSC.
- L. B. de Caland, E. L. C. Silveira and M. Tubino, Anal. Chim. Acta, 2012, 718, 116–120 CrossRef PubMed.
- J. M. Liu, C. C. Liu, G. Z. Fang and S. Wang, RSC Adv., 2015, 5, 58713–58726 RSC.
- M. Laikhtman and J. S. Rohrer, J. AOAC Int., 1999, 82, 1156–1162 CAS.
- J. J. Zhang and Y. Zhu, J. Chromatogr. A, 2007, 1170, 114–117 CrossRef CAS PubMed.
- A. Azzouz, A. J. Rascón and E. Ballesteros, J. Pharm. Biomed. Anal., 2016, 119, 16–26 CrossRef CAS PubMed.
- L. Campone, A. L. Piccinelli, R. Celano, I. Pagano, M. Russo and L. Rastrelli, J. Chromatogr. A, 2016, 1428, 212–219 CrossRef CAS PubMed.
- B. Ivana, H. Markéta, Š. Dalibor and S. Petr, Food Chem., 2013, 141, 1433–1437 CrossRef PubMed.
- C. Z. Ni, B. H. Zhu, N. N. Wang, M. H. Wang, S. Q. Chen, J. J. Zhang and Y. Zhu, Food Chem., 2016, 194, 555–560 CrossRef CAS PubMed.
- Z. Zhu, L. Xi, Q. Subhani, Z. Huang and Y. Zhu, Talanta, 2013, 113, 113–117 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra25711a |
| This journal is © The Royal Society of Chemistry 2017 |