
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Probing the binding mechanism of photoresponsive azobenzene polyamine derivatives with human serum albumin†
M. Deianaa,
Z. Pokladekb,
M. Ziemianekb,
N. Tarnowicza,
P. Mlynarzb,
M. Samoca and
K. Matczyszyn *a
*a
aAdvanced Materials Engineering and Modelling Group, Faculty of Chemistry, Wroclaw University of Science and Technology, Wyb. Wyspianskiego 27, 50-370 Wroclaw, Poland. E-mail: katarzyna.matczyszyn@pwr.edu.pl
bDepartment of Bioorganic Chemistry, Faculty of Chemistry, Wroclaw University of Science and Technology, Wyb. Wyspianskiego 27, 50-370 Wroclaw, Poland
First published on 17th January 2017
Abstract
Control over chemical and biochemical processes by agents sensitive either to internal or external stimuli has attracted much attention in recent years. In particular photosensitive polyamines have been recently used to photo-trigger the hybridization/melting of DNA as well as to modify its intrinsic morphology. These results prompted us to synthesize azobenzene-based polyamine derivatives and study their impact towards human serum albumin (HSA), the principal extracellular protein in plasma, which is highly responsible for the proper biological activity exerted by exogenous compounds. It turns out that to assess and understand the binding mechanism of relevant compounds towards the HSA active sites is a critical step for the design of biomolecules-targeted probes. Herein, we show that both the mono-substituted Azo-4N and bi-substituted bis-Azo-4N azobenzene derivatives bind the protein template with moderate affinity and the number of positive charges along the polyamine moiety plays a pivotal role in stabilizing the photochrome–HSA adduct. Changes taking place in the fluorescence intensity of the tryptophan residue enabled us to determine the Ksv and kq parameters which provide evidence for a quenching driven by a static mechanism. Both ΔH and ΔS of the binding process being negative indicates that the HSA–photochrome association is mainly stabilized by a combination of long-range interactions of ionic nature. The overlap between the donor (Trp-214) and acceptor (photochrome) spectra allowed to calculate the distance (r) and the rate (kET) of energy transfer. Investigation of the HSA structural components reveals that the azobenzene derivatives, in both their conformations, slightly affect the overall protein secondary structure and they do not change its native state. Direct comparison of our results achieved by using photosensitive polyamines with those previously reported for biogenic and analogous polyamines bound to HSA reveals that the azo motif does not enhance to a large degree the overall binding affinity of the photoswitches towards the globular protein and contributes only a little to affect its intrinsic morphology.
Introduction
Control over biological processes through the use of non-covalently bound photochromic units sensitive to internal or external stimuli represents an alternative strategy over the counter covalent approaches.1 Among the various types of stimuli used, such as electrical-field strength,2 temperature,3 molecular recognition,4 and pH value,5 photoregulation is particularly advantageous for modulating movement and conformation of the host material in which the photochrome is placed since the timing, location and strength of the light can be easily adjusted and controlled without loss of efficiency.6 In a recent series of papers the control over DNA compaction and decompaction through the use of photoresponsive azobenzene-based cationic molecules has been discussed.7–10 Zhou and coworkers showed that the formation and dissociation of G-quadruplexes of DNA could be induced by interconversion of the trans and cis forms of the azo compounds.6,11 Recently, Bergen and coworkers reported on the effective manipulation of DNA melting and hybridization by azobenzene-based guanidinium units.12 Even more recently, our group demonstrated that the intrinsic DNA chirality could be reversibly triggered by changing either the shape or the concentration of the non-covalently bound photosensitive polyamines (B-to-A and B-to-A-to-Z intermediate DNA transitions).13,14 Along this direction, our group investigated the isomerization pathway of light-responsive azobenzenes through nonlinear optics (NLO) and quantum mechanics approaches in both solutions and liquid crystalline phases.15–18 This overall framework prompted us to investigate the interaction mechanism of azobenzene-based polyamines derivatives with human serum albumin (HSA). This study is highly relevant since HSA, the most abundant protein in the circulatory system, is well known to interact with a wide range of exogenous compounds modifying their pharmacodynamic and pharmacokinetic properties.19–21 Hence, understanding of the association process between the relevant compounds and serum proteins is a critical step for designing more specific and efficient biomolecules-targeted probes. In this paper, we report on the synthesis and bio-physical characterization of the binding interaction between the mono-substituted “Azo-4N” and bi-substituted “bis-Azo-4N” photoresponsive polyamines and HSA. We show that both the number of positive charges distributed along the substituent moiety and the changes in shape occurring on the photo-isomerization play a key role in stabilizing the HSA–photochrome adduct. Moreover, we demonstrate that the interconversion of the planar trans and bent cis forms do not lead to severe changes of the protein secondary structure in contrast with the results reported for the interaction of BSA with an azobenzene-based photoresponsive surfactant (azoTAB).22 A direct comparison of our results with those previously reported for analogue23 and biogenic polyamines24 categorically establish that the azo moiety contributes only weakly to the overall adduct stabilization. This study represent the first attempt to shed light onto the complexation process between this emerging new class of photosensitive binders and human serum albumin.Experimental
Materials
Common reagent-grade chemicals were used without further purification. Human serum albumin (HSA) and sodium cacodylate trihydrate were purchased from Sigma Aldrich Chem. Co. Deionized water was used throughout all experiments. HSA, Azo-4N and bis-Azo-4N were dissolved in deionized water and kept under dark, except during UV irradiation. HSA concentration was determined by absorption spectroscopy.23 Sodium cacodylate trihydrate (1 × 10−2 M) was used to control pH of the solutions (pH 7.2).Sample irradiation
The photo-induced isomerization reactions of Azo-4N and bis-Azo-4N were performed by using a high pressure Hg Oriel lamp equipped with interference filters at 313 and 436 nm. The resulting light intensity was 0.41 mW cm−2 at 313 nm (trans–cis) and 1.04 mW cm−2 at 436 nm (cis–trans).The Azo-4N and bis-Azo-4N trans-to-cis and cis-to-trans ratio was assessed by 1H NMR spectroscopy according to the intensity ratios of the corresponding signals (data not shown). The trans![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :cis and cis:trans composition of the photostationary state was found to be 80:20 and 70:30 for Azo-4N and bis-Azo-4N, respectively. The effect of the UV irradiation on the protein structure was assessed by irradiating the HSA sample for 90 minutes and comparing the resulting emission and CD spectra with those of the non-irradiated biomolecule.
:cis and cis:trans composition of the photostationary state was found to be 80:20 and 70:30 for Azo-4N and bis-Azo-4N, respectively. The effect of the UV irradiation on the protein structure was assessed by irradiating the HSA sample for 90 minutes and comparing the resulting emission and CD spectra with those of the non-irradiated biomolecule.
UV/Vis spectroscopy
The UV-Vis absorption spectra were recorded on a Perkin Elmer Lambda 20 UV-Vis spectrometer at 298 K.Fluorescent measurements
Steady-state fluorescence studies were carried out with a Hitachi F-4500 spectrofluorometer. An electronic thermostated water bath (PTP-1 Peltier system DBS) was used for controlling the temperature. The HSA concentration was kept constant (1 × 10−5 M) and samples were titrated with incremental addition of Azo-4N or bis-Azo-4N (1–7 × 10−5 M dark solution) until no appreciable changes of the emission intensity were seen, indicating saturation of the interaction. Under those circumstances (excess of ligand and reaching of the stoichiometry) the quantitative binding data analysis were carried out taking into account the composition of the photostationary state and the inner filter effect (IFE). It is worth noting that, in order to avoid dilution issues, the titration experiments were done by keeping the volume of the samples constant throughout the whole investigation.Circular dichroism
CD measurements were performed at 298 K in the wavelength range of 180–300 nm at different photochrome/HSA ratios and keeping constant the HSA concentration. Before use, the optical chamber of the CD spectrometer was deoxygenated with dry nitrogen and was held under nitrogen atmosphere during the measurements. Each spectrum was averaged from five successive accumulations. The protein secondary structure was computed by using the K2D3 software.25Results and discussion
Synthesis
The compounds Azo-4N and bis-Azo-4N (Fig. 1) were synthesized following the procedure recently reported by us for the analogous mono-substituted Azo-2N and Azo-3N.13 Detailed information on the synthesis, characterization and computation are given in the (ESI) section p.S2-17.† The pKa values of the amine groups along the substituent chain were determined by using the MarvinSketch software.9,14 These values enabled to calculate the net positive charge of the azobenzene derivatives being the object of the present study at physiological pH (see ESI for details concerning the equations used and the derived values, p.S17†). It turned out that the insertion of various amine groups along the azo moiety allowed us to obtain polyamines with a different overall charge distribution.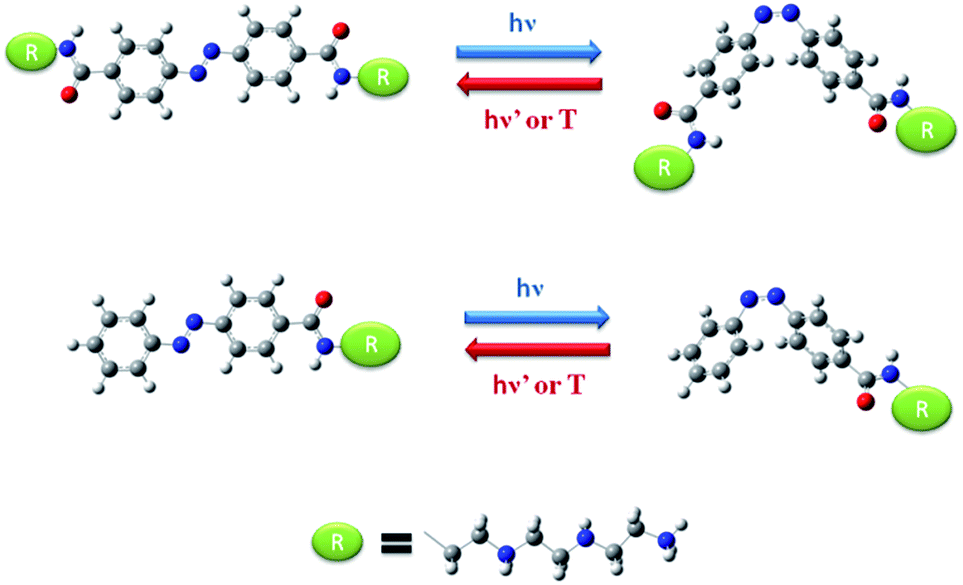 | ||
| Fig. 1 Molecular structure and interconversion pathway of bis-Azo-4N (top panel) and Azo-4N (lower panel). The red, blue and white colors indicate oxygen, nitrogen and hydrogen atoms, respectively. | ||
Assessing the photophysical properties of Azo-4N, bis-Azo-4N and HSA
The UV-Vis absorption spectra of both Azo-4N and bis-Azo-4N in dark condition featured mainly two bands located at ∼330 and ∼430 nm corresponding to π–π* and n–π* electronic transitions, respectively. The π–π* absorption band was characterized by a molar extinction coefficient ε ≈ 3 × 104 M−1 cm−1, whereas the weak n–π* band had a ε ≈ 2 × 103 M−1 cm−1. Upon UV light irradiation a monotonic hypochromism along with a shift of the emission maximum towards shorter wavelength was observed for the band located in the near UV region, whereas the band in the visible region underwent remarkable hyperchromism. Fig. 2 shows the interconversion pathway between the relevant forms of the azobenzene photoswitches.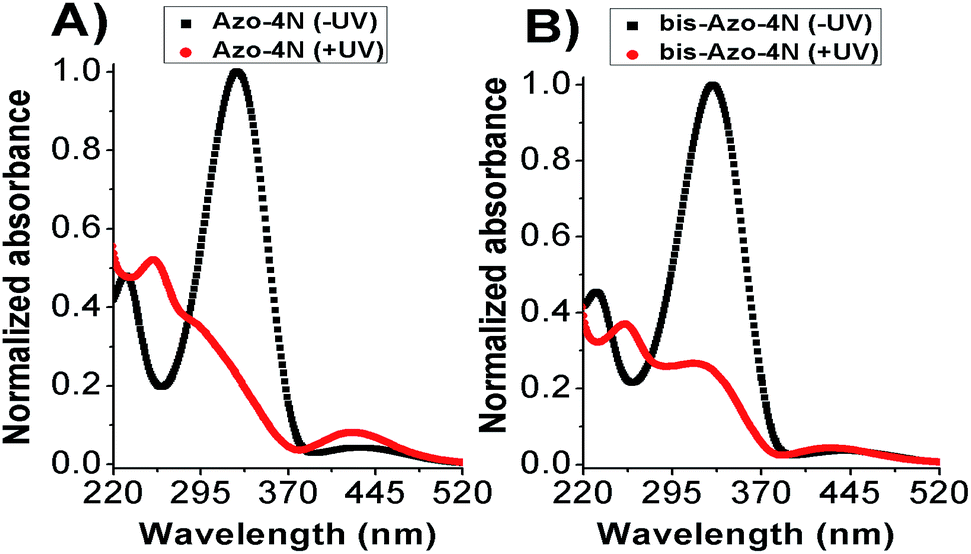 | ||
| Fig. 2 Photoisomerization process of (A) Azo-4N and (B) bis-Azo-4N. Black and red lines depict the photochromes in trans (−UV) and cis (+UV) forms, respectively. [Azo-4N (dark)] = 2 × 10−5 M. | ||
Before assessing the azobenzene derivatives response towards HSA, the inability of the photochromes to induce aggregates was verified. As reported in detail in the ESI section (Fig. S3 and S4†), a linear dependence of absorbance vs. photochrome concentration was observed ruling out any formation of higher order molecular structure within the concentration range used throughout the whole experiment.13
The intrinsic fluorescent intensity of HSA has been usually ascribed to the tryptophan residue (Trp-214) located within the cavity of subdomain IIA and to the tyrosine residues distributed along the whole polypeptide chain.21,26 When excited with a 280 nm wavelength the free protein displayed an emission maximum located at 340 nm (Fig. 3).
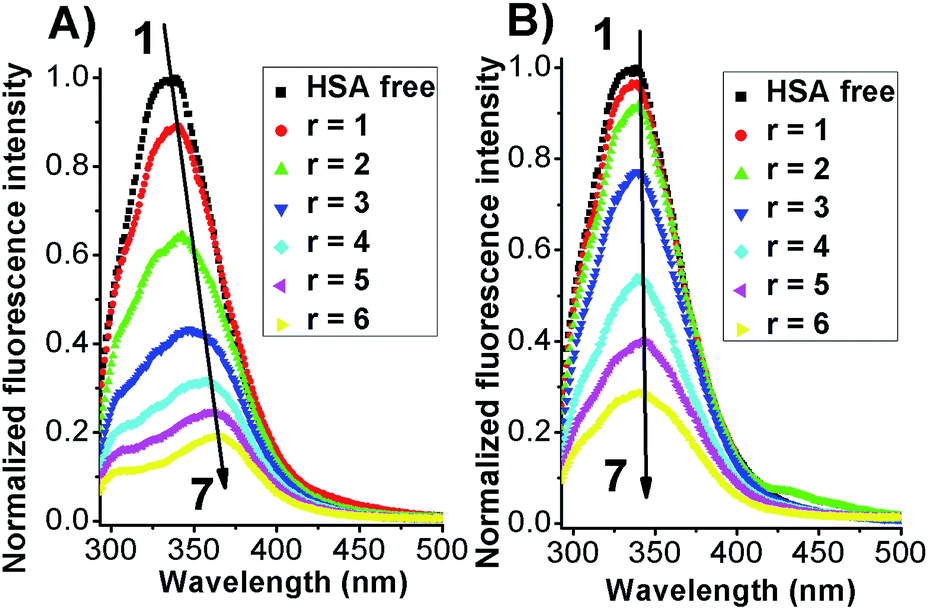 | ||
| Fig. 3 Fluorescence emission spectra of HSA in the absence (black line) and presence of Azo-4N in (A) trans form (−UV) and (B) cis form (+UV). [HSA] = 1 × 10−5 M and [Azo-4N (dark)] = 0–6 × 10−5 M. r is the molar ratio defined as [Azo-4N (dark)]/[HSA]. | ||
Before testing the HSA fluorescence response in the presence of the photochromes the dependence of the protein as a function of UV irradiation was assessed through fluorescence and circular dichroism methods. As reported in the ESI section (Fig. S5 and S6†) both emission and CD spectra were not affected by the presence of the UV light even after an hour and half of irradiation pointing out that the changes that will be discussed further on are not an artifact of the experimental conditions used but the direct outcome of the effective interaction between HSA and the photochromes.
When both Azo-4N (Fig. 3) and bis-Azo-4N (Fig. S7†) were added to the free protein solution a monotonic steady-state quenching of the intrinsic HSA fluorescence intensity was observed. These changes can be ascribed to the effective interaction between the photochromes and the Trp-214 residue located in the hydrophobic cavity of the subdomain IIA of the serum protein.21 It is worth noting that, even if the extent of quenching found was similar for both the isomers (>70%), the elongated planar trans forms gave rise to a significant bathochromic effect (ΔλAzo-4N–HSA = 25 nm; Δλbis-Azo-4N–HSA = 20 nm) whereas the bent cis form did not. These changes can be ascribed to the effect that the isomerization had on the dipole moment of the molecules. The trans form is planar and presents a more hydrophobic character, whereas the non-planar cis form is more hydrophilic and thus more easily exposed to a water-like environment.27 It turns out that the planar conformation allowed the azobenzene motif to fit easily the hydrophobic cavity of the protein where the tryptophan residue is located, whereas the bent form decreased to some extent its binding ability. It also interesting to note that the red shift appeared larger for Azo-4N compared to bis-Azo-4N. The difference may be due to the bigger free volume that the bis-derivatives take up along the protein binding site that of the analogous mono-substituted which could hamper the proper overlapping of the π orbitals of the ligand with those of the amino acid residues.
Quantitative evidence of the HSA–photochrome interactions
At this stage we wanted to analyse quantitatively the binding strength of the azobenzene derivatives toward the protein template. In order to achieve exclusively the contribution of the pure isomeric forms the complex, uncomplexed and free concentrations of the photosensitive polyamines were systematically corrected based on both the trans:cis ratio and the IFE effect.28
It is known that the Stern–Volmer analysis enables one to describe properly the quenching mechanisms encountered in the study of biological systems.29 The nature of quenching is usually classified as either dynamic or static. Dynamic quenching results from the diffusive collision of the excited fluorophore and the quencher and has a marked entropic character, whereas static quenching is usually observed when the fluorophore and the quencher form a stable non-fluorescent ground-state adduct.29–31 In order to distinguish between these two different types of mechanism we processed our data using the following equation:29–31
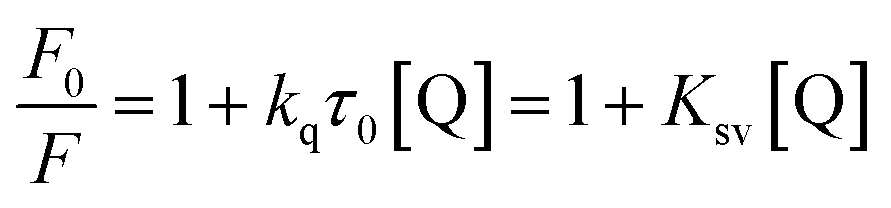 | (1) |
Fig. 4 shows the dependence of the ratio of the emission intensities and the corrected photochrome concentrations.
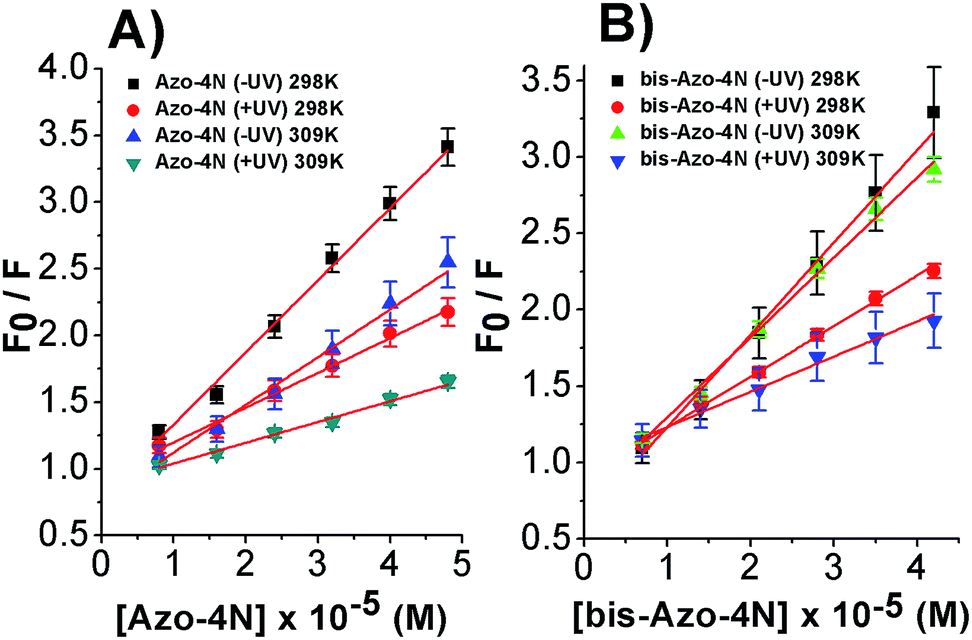 | ||
| Fig. 4 Stern–Volmer plots of the fluorescence quenching of the (A) Azo-4N (±UV)–HSA and (B) bis-Azo-4N (±UV)–HSA systems at 298 and 309 K. [HSA] = 1 × 10−5 M, [Azo-4N (±UV)] = 0 to 4.8 × 10−5 M and [bis-Azo-4N (±UV)] = 0 to 4.2 × 10−5 M. | ||
The linear behaviour of the Stern–Volmer plots suggests that a single class of fluorophore-quencher takes part in the association process. As reported in Table 1, the magnitudes of the Ksv values decreased on increasing the temperature. It is known that higher temperatures lead to faster diffusion and thus larger amount of collisional quenching, whereas in the case of static mechanism the increase in temperature can lead to dissociating the non-fluorescent complex held together by weak ionic and non-ionic forces.29 The Ksv-temperature trend observed here is a clear indication that the photosensitive polyamines form a non-fluorescent complex with the serum protein through a static mechanism of interaction. Further evidence was also provided by calculating the bimolecular quenching rate constants of the complexed protein. As shown in Table 1, the kq values were found to range between 1012 and 1013 M−1 s−1. These values are much higher than the maximum diffusion collisional quenching rate of various quenchers with biopolymers ≈2.0 × 1010 M−1 s−1, unambiguously emphasizing the static nature of the quenching encountered.21,30,31
| Ksv (M−1) | kq (M−1 s−1) | |
|---|---|---|
| Azo-4N (−UV) 298 K | (5.5 ± 0.2) × 104 | (1.6 ± 0.2) × 1013 |
| Azo-4N (+UV) 298 K | (2.6 ± 0.1) × 104 | (7.4 ± 0.1) × 1012 |
| Azo-4N (−UV) 309 K | (3.7 ± 0.3) × 104 | (1.0 ± 0.3) × 1013 |
| Azo-4N (+UV) 309 K | (1.6 ± 0.1) × 104 | (4.5 ± 0.1) × 1012 |
| Bis-Azo-4N (−UV) 298 K | (6.3 ± 0.5) × 104 | (1.8 ± 0.5) × 1013 |
| Bis-Azo-4N (+UV) 298 K | (3.2 ± 0.2) × 104 | (9.1 ± 0.2) × 1012 |
| Bis-Azo-4N (−UV) 309 K | (4.2 ± 0.3) × 104 | (1.2 ± 0.3) × 1013 |
| Bis-Azo-4N (+UV) 309 K | (2.2 ± 0.2) × 104 | (6.2 ± 0.2) × 1012 |
The changes occurring in the intrinsic fluorescence intensity of the protein upon complexation gave the opportunity to calculate the association constants and the number of binding sites (Table 2). In order to accomplish this task both the modified Stern–Volmer equation (eqn (2)) and the Scatchard analysis (eqn (3)) were used:33
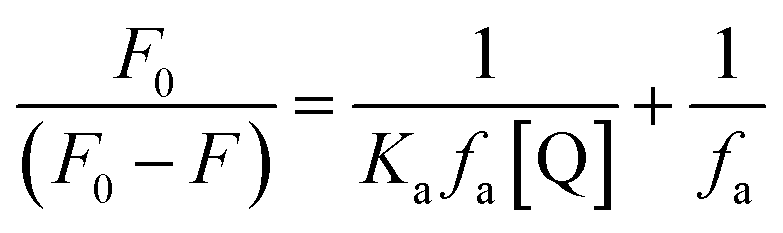 | (2) |
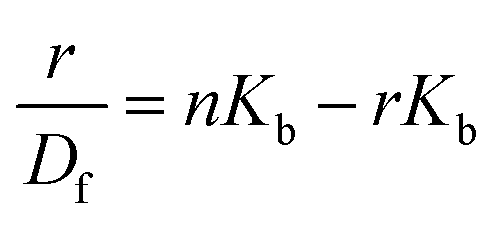 | (3) |
| Ka (M−1) | Kb (M−1) | fa | n | |
|---|---|---|---|---|
| Azo-4N (−UV) 298 K | (5.7 ± 0.3) × 104 | (5.4 ± 0.3) × 104 | 1.1 | 1.0 |
| Azo-4N (+UV) 298 K | (2.1 ± 0.2) × 104 | (2.2 ± 0.2) × 104 | 1.0 | 1.2 |
| Azo-4N (−UV) 309 K | (3.5 ± 0.4) × 104 | (3.3 ± 0.4) × 104 | 1.0 | 1.1 |
| Azo-4N (+UV) 309 K | (1.4 ± 0.2) × 104 | (1.3 ± 0.2) × 104 | 0.9 | 1.2 |
| Bis-Azo-4N (−UV) 298 K | (6.5 ± 0.5) × 104 | (6.1 ± 0.4) × 104 | 1.3 | 1.0 |
| Bis-Azo-4N (+UV) 298 K | (2.8 ± 0.2) × 104 | (2.7 ± 0.2) × 104 | 1.1 | 1.1 |
| Bis-Azo-4N (−UV) 309 K | (4.2 ± 0.3) × 104 | (4.5 ± 0.4) × 104 | 1.2 | 1.0 |
| Bis-Azo-4N (+UV) 309 K | (1.7 ± 0.3) × 104 | (1.6 ± 0.2) × 104 | 0.9 | 1.2 |
As depicted in Fig. 5 and S8–S11,† the binding isotherms were found to be linear either using eqn (2) or (3) indicating that a single class of sites binding through a non-cooperative process take part in the association mechanism between the protein and the azo compounds. This statement was also further confirmed by the values found for fa and n being close to unity.
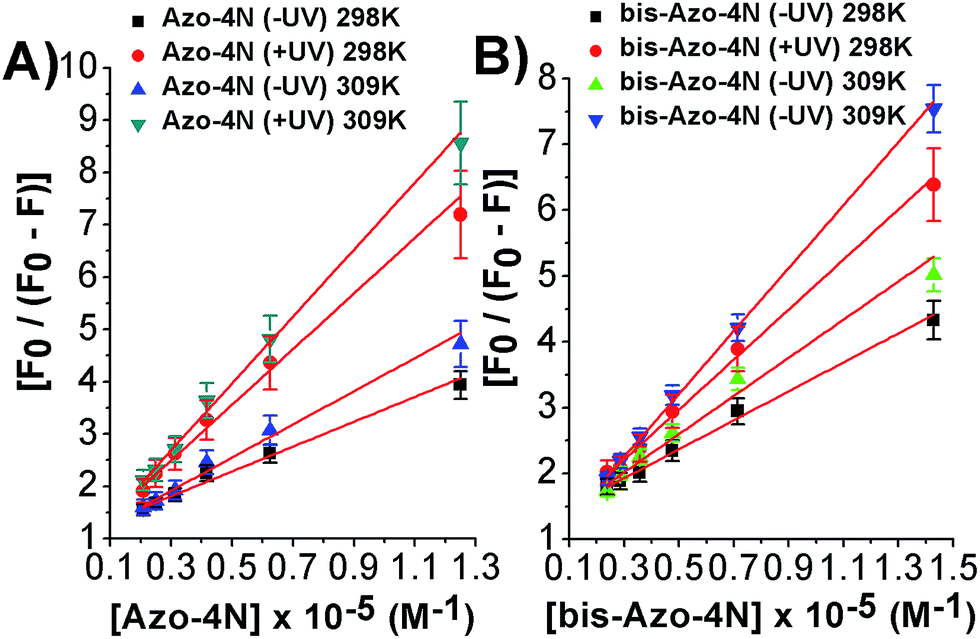 | ||
| Fig. 5 Modified Stern–Volmer plots for the: (A) Azo-4N (±UV) and (B) bis-Azo-4N (±UV)–HSA systems at 298 and 309 K. | ||
It is important to note that the calculated binding constants followed the order: bis-Azo-4N (−UV) 298 K > Azo-4N (−UV) 298 K > bis-Azo-4N (−UV) 309 K > Azo-4N (−UV) 309 K > bis-Azo-4N (+UV) 298 K > Azo-4N (+UV) 298 K > bis-Azo-4N 309 K > Azo-4N (−UV) 309 K. At this stage some conclusions can be drawn:
(i) The planar conformer (reduced free-volume) showed a larger affinity to HSA than the bent cis form indicating that the photoisomerization of the compounds could affect the binding strength of the photoswitches.
(ii) As a direct consequence, the orientation and magnitude of the dipole moment of the azobenzene derivatives played a pivotal role in stabilizing the HSA–photochrome adducts.
(iii) The number of the positive charges distributed along the polyamine moieties could unambiguously direct and orient the probes toward the protein active sites.
(iv) The temperature effect, as shown previously, can reduce long and short-range interactions between the studied ligands and the peptide chains.
The calculated binding constants are in good agreement with those found for a wide variety of HSA-binders and their relatively moderate strength might not compromise the ability of the compounds to exert functions towards other biomolecules.34
In order to better assess the parameters calculated for the newly synthesized photosensitive polyamines a direct comparison with the data reported for biogenic24 and analogue23 polyamines bound to HSA was undertaken. Tajmir-Riahi et al. have shown that polyamines displayed association constants ranging from 103 to 104 M−1 when bound to HSA and that the biogenic polyamines–HSA affinity was strictly a function of the number of positive charges at the nitrogen atoms. It turns out that the photochromes being the object of the present study behave in a similar way to the polyamines previously described and that the azobenzene motif only moderately contributes to enhancing the overall binding constant of the photoswitches toward the serum protein.
The thermodynamic parameters
The effect of temperature on the equilibrium binding constant provided the possibility to elucidate the thermodynamic parameters (ΔH, ΔS and ΔG) involved in the interaction processes. To accomplish this goal we used the van't Hoff equation:31,33
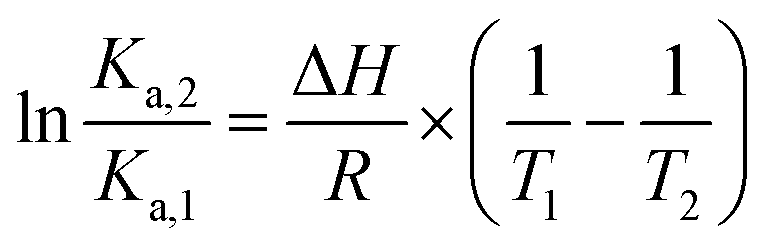 | (4) |
The values are summarized in Table S2† and shown in Fig. 6.
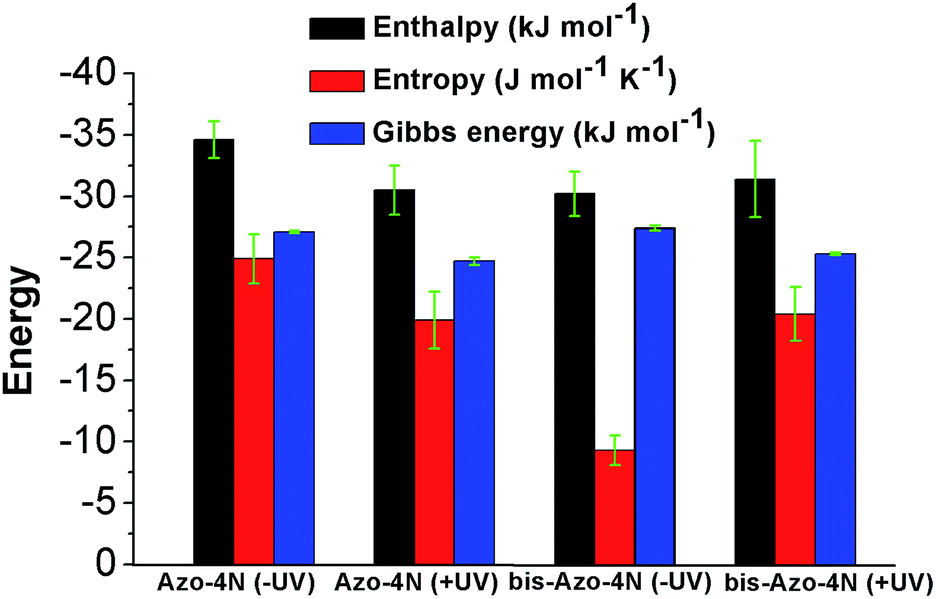 | ||
| Fig. 6 Thermodynamic forces involved in the azo-derivative–HSA systems. | ||
Both enthalpy (ΔH) and entropy (ΔS) changes were found to be negative for all the azobenzenes studied. It turned out that the forming of complex between this class of light-activated probes and the protein template is enthalpic driven. We can deduce that long-range interactions such as: van der Waals forces, hydrogen bonding, dipole–dipole interactions and dehydration effects play a pivotal role in the stabilization of the adducts. The negative free energy (ΔG) highlights the spontaneity of the processes. Such results could be expected, regarding the intrinsic polarization of the HSA structure at physiological pH, since its isoelectric point pI ∼ 5 confers to the protein a net overall negative charge which might direct and orientate the positive charged ligands toward its active sites.33
Förster resonance energy transfer
As shown in Fig. 7 and S12,† the fluorescence emission spectrum of the protein overlaps with the absorption spectrum of the azo-compounds in both their relevant isomeric forms. Thus we speculate that energy transfer between the donor (Trp-214) and acceptor (photosensitive polyamines) molecules can occur with a high probability.21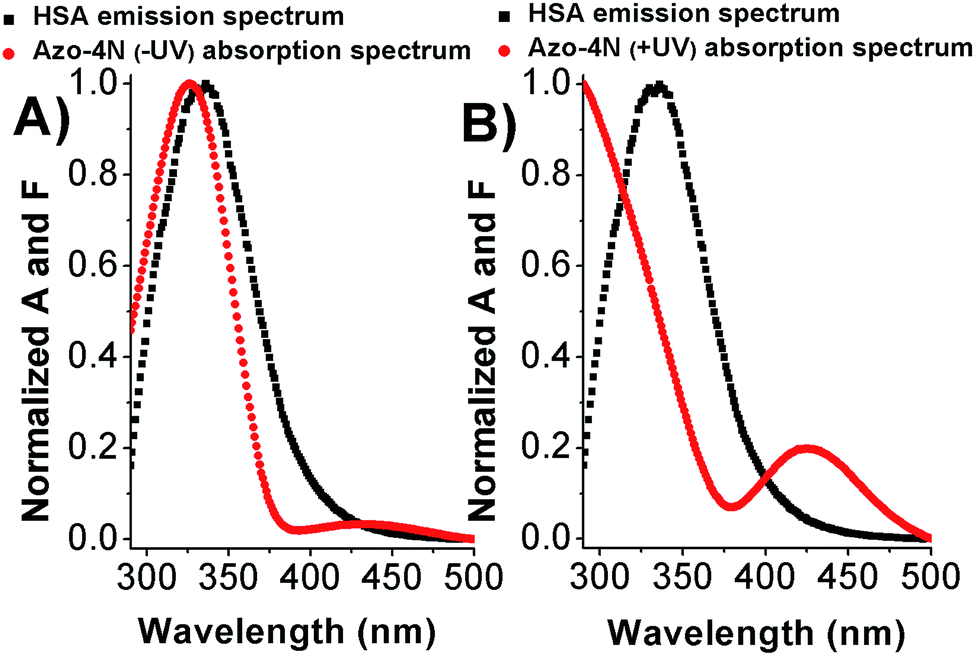 | ||
| Fig. 7 Overlap between the HSA emission spectrum and (A) Azo-4N (−UV) and (B) Azo-4N (+UV) absorption spectrum. [HSA] = [Azo-4N] (±UV) = 1 × 10−5 M. | ||
To gain insight in the proximity between the excited (donor) and neighbour (acceptor) molecules the Förster theory for non-radiative energy transfer (FRET) was used. First, we calculated the overlap integral for each photochrome(±UV)–HSA system by using the following equation:35
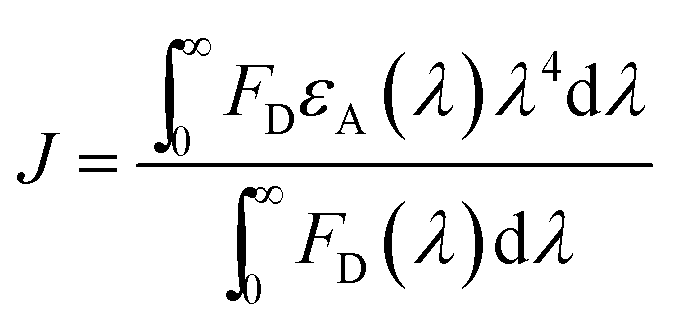 | (5) |
The relative values were than processed to calculate the critical energy transfer distance defined as:
| R06 = 8.79 × 10−25K2n−4φJ | (6) |
Finally by using eqn (7) we determined the Förster radius (r) and the relative efficiency of transfer (E):
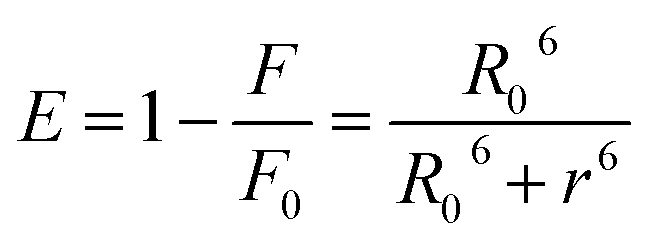 | (7) |
The calculated parameters are reported in Table S3.† The Förster radius (r) was found to be ≈3 nm for each studied system while the efficiency of transfer was between ≈0.3 and 0.5. The r values fell within the 2-to-8 nm and 0.5R0 < r < 1.5R0 range pointing to a high probability of energy transfer between the acceptor and donor molecules (Trp-214) and confirming the origin of the static quenching discussed previously.21 These results constitute a clear evidence that the photochromic probes are located in the proximity of the tryptophan residue within the subdomain IIA. This statement has been also further confirmed by displacement assay in which the well-known site II-binder warfarin was replaced by the addition of increasing concentration of the azobenzene derivatives (see ESI p.S22†).
According to the FRET theory the rate of energy transfer could be evaluated by using the following equation:32
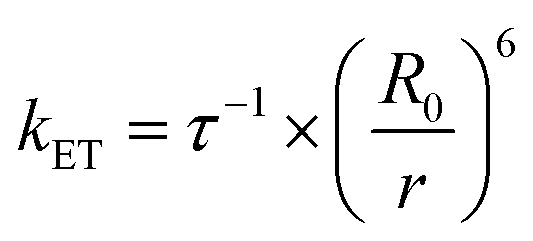 | (8) |
Probing the conformational regulation of the HSA structure by the photochromic units
To determine if the azobenzene derivatives were able to perturb the HSA secondary structure circular dichroism measurement were carried out at fixed protein concentration. As shown in Fig. 8 and S14† the free-HSA spectrum is comprised of two negative dichroic maxima located at 208 and 222 nm. Changes in shape or shifting in λmax of the negative ellipticities are thus indicative of modification occurring to the native state of the biomolecule.38 Upon addition of the photochromes, in both their conformations, a slight decrease of the dichroic band without shifting of the maxima were observed indicating weak changes in the HSA α-helix percentage.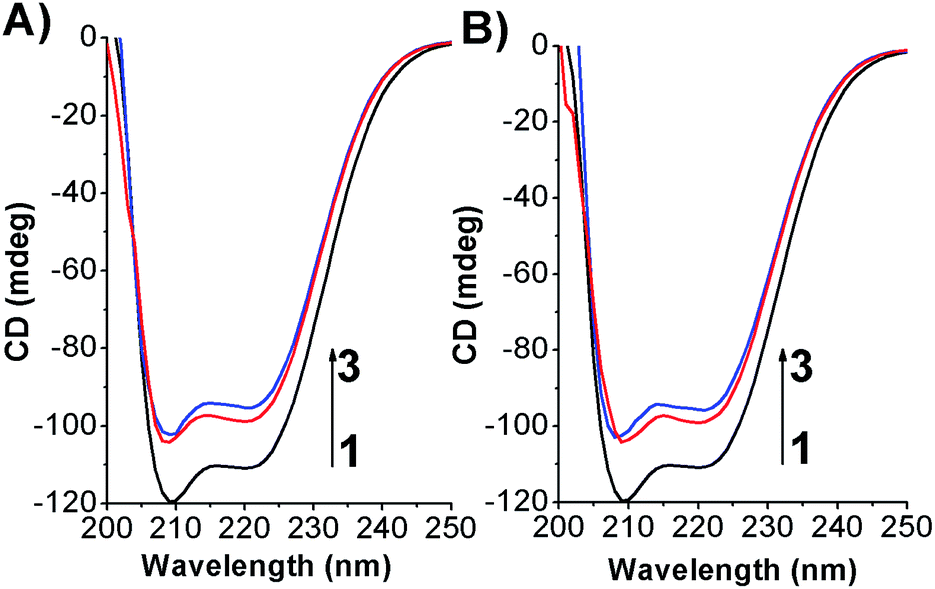 | ||
| Fig. 8 CD spectra of HSA in its free state and complexed form by (A) Azo-4N (−UV) and (B) Azo-4N (+UV) at 298 K. [HSA] 1 × 10−6 M and [Azo-4N (dark)] = 3 and 5 × 10−5 M. | ||
In order to assess quantitatively the structural variation caused by the photochrome on the HSA structure the data were processed by using the K2D3 software25 and the results are depicted in Fig. 9 and reported in Table S4.†
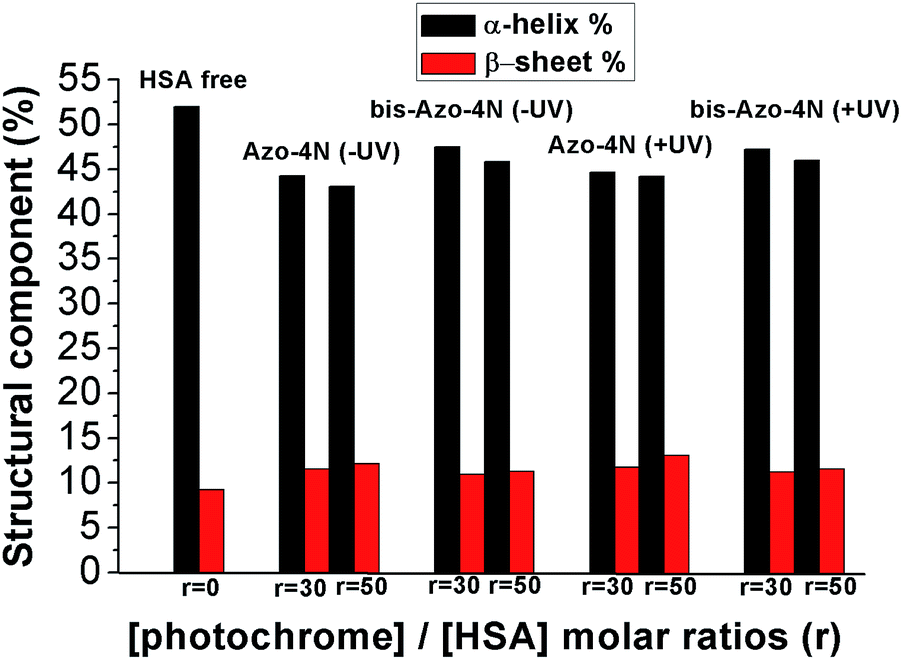 | ||
| Fig. 9 Variation of the HSA secondary structure by ligand addition. r is the molar ratio defined as: [photochrome]/[HSA]. | ||
Interestingly, both the mono and the bis-substituted compounds decreased to similar extent the α-helix component <10%, whereas they increased in a comparable way the β-sheet percentage ≈3%. These changes are indicative of a weak destabilization of the protein moiety occurring without any denaturation process and unambiguously highlight the inability of the photosensitive molecules to fold and unfold the protein structure reversibly. These findings are in stark contrast with those reported for the interaction of BSA with an azobenzene-based photoresponsive surfactant (azoTAB).22 We speculate that the nature of this inconsistency can be ascribed to both the different physicochemical properties of the photosensitive molecules and the experimental conditions used. AzoTAB consists of a surfactant motif which can undergo aggregation in solution, whereas the photosensitive polyamines, the object of the present contribution, did not show formation of higher order molecular structure within the concentration range used. A further discrepancy can be attributed to the fact that we performed our secondary structure investigation by CD spectroscopy while the impact of azo-TAB on the BSA template was monitored by FT-IR measurements. The latter technique required a much higher concentration of ligand (≈10−2 M) compared to the former (≈10−5 M) which could influence the interaction mechanism of the photochromes with the serum proteins. Furthermore, the sequence homology between HSA and BSA is reported to be around 70%, a factor that can affect the specificity of binding.39 On the other hand, it is worth noting that the secondary structure changes occurring in the HSA moiety upon complexation with our photoswitches are in good agreement with those reported for biogenic24 and analogues23 polyamines bringing clear evidence that the unsubstituted azobenzene motif affects the overall tertiary structure of the protein in its native state only weakly (vide supra).
Conclusions
In conclusion, we have discussed the synthesis and binding characteristics of two azobenzene-based polyamines with various distributed positive charges toward human serum albumin. We have unambiguously determined that the nature of the substituent groups along the azobenzene moiety plays a pivotal role in stabilization of the HSA–photochrome adducts. The bio-physical parameters calculated by exploiting the emission quenching of the intrinsic tryptophan residue occurring upon complexation point to the static nature of the quenching observed. These results were also supported by the calculation of the Förster radius and rate of energy transfer for each photochrome(±UV)–HSA system. The magnitude of the binding constants found for the photosensitive polyamines as well as the perturbations on the protein secondary structure are comparable with those found for biogenic polyamines bound to HSA. Moreover, the slight changes occurring in the native state of the protein upon switching the photochrome conformation revealed that the azobenzene motif contributes weakly to either effect or stabilize the structural components of the serum protein. These results represent a step forward for understanding the biological effects that this new emerging class of photochromes exert towards biomacromolecules and sheds light onto this important in vitro complexation.Acknowledgements
The authors gratefully acknowledge the support of the Wroclaw Centre for Networking and Supercomputing. The funding from NCN Opus grant DEC-2013/09/B/ST5/03417 is highly acknowledged. The Leading National Research Centre (KNOW), Wroclaw Centre of Biotechnology programme provided funding for an open access of the paper.Notes and references
- J. Andersson, S. Li, P. Lincoln and J. Andréasson, J. Am. Chem. Soc., 2008, 130, 11836–11837 CrossRef CAS PubMed.
- Y. Osada, H. Okuzaki and H. Hori, Nature, 1992, 355, 242–244 CrossRef CAS.
- M. Yamato, Y. Akiyama, J. Kobayashi, J. Yang, A. Kikuchi and T. Okano, Prog. Polym. Sci., 2007, 32, 1123–1133 CrossRef CAS.
- T. Miyata, N. Asami and T. Uragami, Nature, 1999, 399, 766–769 CrossRef CAS PubMed.
- Y. Bae, A. Fukushima, A. Harada and K. Kataoka, Angew. Chem., Int. Ed., 2003, 42, 4640–4643 CrossRef CAS PubMed.
- X. Wang, J. Huang, Y. Zhou, S. Yan, X. Weng, X. Wu, M. Deng and X. Zhou, Angew. Chem., Int. Ed., 2010, 49, 5305–5309 CrossRef CAS PubMed.
- A. Diguet, N. K. Mani, M. Geoffroy, M. Sollogoub and D. Baigl, Chem.–Eur. J., 2010, 16, 11890–11896 CrossRef CAS PubMed.
- A. L. M. Le Ny and C. T. Lee Jr, J. Am. Chem. Soc., 2006, 128, 6400–6408 CrossRef CAS PubMed.
- A. Venancio-Marques, A. Bergen, C. Rossi-Gendron, S. Rudiuk and D. Baigl, ACS Nano, 2014, 8, 3654–3663 CrossRef CAS PubMed.
- S. Rudiuk, K. Yoshikawa and D. Baigl, Soft Matter, 2011, 7, 5854–5860 RSC.
- X. Xing, X. Wang, L. Xu, Y. Tai, L. Dai, X. Zheng, W. Mao, X. Xu and X. Zhou, Org. Biomol. Chem., 2011, 9, 6639–6645 CAS.
- A. Bergen, S. Rudiuk, M. Morel, T. Le Saux, H. Ihmels and D. Baigl, Nano Lett., 2016, 16, 773–780 CrossRef CAS PubMed.
- M. Deiana, Z. Pokladek, J. Olesiak-Banska, P. Młynarz, M. Samoc and K. Matczyszyn, Sci. Rep., 2016, 6, 28605 CrossRef CAS PubMed.
- M. Deiana, Z. Pokladek, K. Matczyszyn, P. Młynarz, M. Buckle and M. Samoc, J. Mater. Chem. B, 2016 10.1039/c6tb02732f.
- P. Krawczyk, A. Kaczmarek, R. Zalesny, K. Matczyszyn, W. Bartkowiak, M. Ziolkowski and P. Cysewski, J. Mol. Model., 2009, 15, 581–590 CrossRef CAS PubMed.
- K. Matczyszyn, A. Chwialkowska and J. Sworakowski, Thin Solid Films, 2008, 516, 8899–8904 CrossRef CAS.
- R. Zalesny, K. Matczyszyn, A. Kaczmarek, W. Bartkowiak and P. Cysewski, J. Mol. Model., 2007, 13, 785–791 CrossRef CAS PubMed.
- K. Matczyszyn, W. Bartkowiak and J. Leszczynski, J. Mol. Struct., 2001, 565–566, 53–57 CrossRef CAS.
- E. Lazaro, P. J. Lowe, X. Briand and B. Faller, J. Med. Chem., 2008, 51, 2009–2017 CrossRef CAS PubMed.
- F. P. Nicoletti, B. D. Howes, M. Fittipaldi, G. Fanali, M. Fasano, P. Ascenzi and G. Smulevich, J. Am. Chem. Soc., 2008, 130, 11677–11688 CrossRef CAS PubMed.
- O. K. Abou-Zied and O. I. K. Al-Shihi, J. Am. Chem. Soc., 2008, 130, 10793–10801 CrossRef CAS PubMed.
- S.-C. Wang and T. L. Lee Jr, J. Phys. Chem. B, 2006, 110, 16117–16123 CrossRef CAS PubMed.
- R. Beauchemin, C. N. N'soukpoé-Kossi, T. J. Thomas, T. Thomas, R. Carpentier and H. A. Tajmir-Riahi, Biomacromolecules, 2007, 8, 3177–3183 CrossRef CAS PubMed.
- A. Ahmed-Ouameur, E. Mangier, S. Diamantoglou, S. Rouillon, R. Carpentier and H. A. Tajmir-Riahi, Biopolymers, 2004, 73, 503–509 CrossRef PubMed.
- C. Louis-Jeune, M. A. Andrade-Navarro and C. Perez-Iratxeta, Proteins, 2012, 80, 374–381 CrossRef CAS PubMed.
- N. Ibrahim, H. Ibrahim, S. Kim, J.-P. Nallet and F. Nepveu, Biomacromolecules, 2010, 11, 3341–3351 CrossRef CAS PubMed.
- E. Merino and M. Ribagorda, J. Org. Chem., 2012, 8, 1071–1090 CAS.
- Z. Chi and R. Liu, Biomacromolecules, 2011, 12, 203–209 CrossRef CAS PubMed.
- J. K. Lakowicz, Principles of Fluorescence Spectroscopy, Plenum Press, New York, 2nd edn, 1999 Search PubMed.
- M. Deiana, K. Matczyszyn, J. Massin, J. Olesiak-Banska, C. Andraud and M. Samoc, PLoS One, 2015, 10, e0129817 Search PubMed.
- M. Deiana, B. Mettra, K. Matczyszyn, K. Piela, D. Pitrat, J. Olesiak-Banska, C. Monnereau, C. Andraud and M. Samoc, Phys. Chem. Chem. Phys., 2015, 17, 30318–30327 RSC.
- M. El-Kemary, M. Gil and A. Douhal, J. Med. Chem., 2007, 50, 2896–2902 CrossRef CAS PubMed.
- Y.-J. Hu, Y. Liu and X.-H. Xiao, Biomacromolecules, 2009, 10, 517–521 CrossRef CAS PubMed.
- L. M. Berezhkovskiy, J. Pharm. Sci., 2007, 96, 249–257 CrossRef CAS PubMed.
- Modern Quantum Chemistry, ed. T. Förster and O. Sinanoglu, Academic Press, New York, 1996 Search PubMed.
- T. Förster, Discuss. Faraday Soc., 1959, 27, 7–17 RSC.
- A. Sytnik and I. Litvinyuk, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 12959–12963 CrossRef CAS.
- M. Deiana, B. Mettra, K. Matczyszyn, D. Pitrat, J. Olesiak-Banska, C. Monnereau, C. Andraud and M. Samoc, Biomacromolecules, 2016, 17, 3609–3618 CrossRef CAS PubMed.
- J. Fan, W. Sun, Z. Wang, X. Peng, Y. Li and J. Cao, Chem. Commun., 2014, 50, 9573–9576 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthesis, characterization and computation of the photoresponsive polyamines, calculation of the positive net charges of the ligands, absence of aggregation, influence of the UV lamp on the HSA template, quenching of the emission intensity of HSA by bis-Azo-4N, Scatchard plots, plots of the effective overlap between the HSA emission spectra and the absorption spectra of bis-Azo-4N (±UV), site-marker experiments, CD spectra of HSA and bis-Azo-4N (±UV), tables reporting the values of the thermodynamic parameters, energy transfer and secondary structure components. See DOI: 10.1039/c6ra26033k |
| This journal is © The Royal Society of Chemistry 2017 |