
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural stability and electronic property in K2S under pressure†
Ying Li ,
Xilian Jin*,
Tian Cui*,
Quan Zhuang,
Qianqian Lv,
Gang Wu,
Xing Meng,
Kuo Bao,
Bingbing Liu and
Qiang Zhou
,
Xilian Jin*,
Tian Cui*,
Quan Zhuang,
Qianqian Lv,
Gang Wu,
Xing Meng,
Kuo Bao,
Bingbing Liu and
Qiang Zhou
State Key Laboratory of Superhard Materials, College of Physics, Jilin University, Changchun 130012, China. E-mail: jinxilian@jlu.edu.cn; cuitian@jlu.edu.cn; Fax: +86-431-85168825; Tel: +86-431-85168825
First published on 23rd January 2017
Abstract
In this work, the structures, phase sequence, and metallic properties of K2S have been systematically explored. We confirm that the P63/mmc phase is the best possible candidate for the stable structure of K2S at low pressure range. Although the phases of P63/mmc and Cmcm K2S are semiconductors, two new structures of P6/mmm and P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m1 emerge with metallic characters at high pressures. The analyses of electronic localization functions reveal that the conductivity mainly comes from the electrons surrounding S atom chains, which supplies a potential way to improve the conductivity of sulfur to enhance the electrode recharge ability and rate capability in alkali sulfide battery under pressure.
m1 emerge with metallic characters at high pressures. The analyses of electronic localization functions reveal that the conductivity mainly comes from the electrons surrounding S atom chains, which supplies a potential way to improve the conductivity of sulfur to enhance the electrode recharge ability and rate capability in alkali sulfide battery under pressure.
Introduction
The storage and conversion of energy continue to be important to society. Batteries, which interconvert chemical and electrical energy, are widely used in industry and for consumer applications, e.g. appliances and laptop computers. The battery energy storage system, e.g. lead-acid battery, solid oxide fuel battery, solar battery, lithium ion battery and so on, are focused and studied for a long time. The lead-acid battery is the most-used battery system with advantages including large current discharge, rechargeable, and low-cost. On the other hand, the disadvantage of lead-acid battery is obvious, for examples, the heavy weight and pollution.1,2 Solid oxide fuel battery is an energy conversion device that can convert chemical energy directly into electricity without pollution, nevertheless, the high operating temperature limits its widely application in our daily life.3,4 Solar battery is excepted to have large-scale applications if the problem of low efficiency is solved.5,6 Lithium ion battery is an attractive battery for the property of portable type, high energy density, and non-pollution, and is widely used for digital products, electric vehicles, household appliances, etc.7–9 The principle of operation lithium ion battery is that Li is removed from an intercalation cathode, for example LiCoO2, on charging and is inserted between sheets of carbon atoms in the graphite negative electrode; discharge reverses this process.10 Though the compound of Li2S and Na2S with high specific energy (energy per unit weight) and energy density (energy per unit volume) are candidates for applications in solid state batteries,11,12 it is confronted with major challenge, i.e., poor electrode recharge ability and limited rate capability owing to the insulating nature of sulfur and the solid reduction products.13The ground-state properties of alkali metal sulfides of X2S (X = Li, Na, K, Rb), e.g., elastic properties, electronic structure, and optical properties are widely studied.14–17 The calculated band structures show the Li2S, K2S, and Rb2S are indirect bandgap materials, whereas Na2S is a direct gap material.14 At the same pressure, bulk modulus of the alkali metal sulfides show a clear decrease with heavier mass of alkali atoms.15 The similar tendency of decrease with atomic mass of X in X2S has been also reported on the elastic constants under the same pressure. When the pressure is enhanced, except for the linear decrease of the elastic constant of C44, the bulk modulus and the elastic constant of C11, C12 increase in the crystals of Rb2S, K2S and Na2S. Similar trends are also reported in the Li2S except for the elastic constants C44. It is reported that C44 increases linearly with pressure which is related to the difference in the bonding nature of the bottom of the conduction band.14
K2S has been extensively investigated by theoretical and experimental methods since it was first synthesized by Zintl et al.18 It is reported that K2S can crystallize in the antifluorite structure, i.e. Fmm18 at ambient pressure, then transforms to an orthorhombic crystal with the space group of Pnma at about 2.7 GPa.16 Theoretical prediction points out that Pnma crystal can transform to a distorted P63/mmc at 4.4 GPa.16,19 Nonetheless, the Pnma phase is not observed by experiment to date.19 In this work, four stable phases of P63/mmc, Cmcm, P6/mmm and Pm1 have been found at different pressures through ab initio ELocR code.20 The low-pressure phase of Pmma proposed by an experimental analysis19 is found unstable by our theoretical calculations. Based on the reported X-ray diffraction and our theoretical analysis, we update the low-pressure phase with a hexagonal crystal of P63/mmc. Our calculations reveal the insulating character of the two phases of P63/mmc and Cmcm, and the Raman spectra are subsequently analyzed for the experimental convenience. Electronic band structure and the partial density of states show the metallic properties of P6/mmm and Pm1 at pressures. As shown in the chart of electronic localization functions (ELF), the delocalization of electrons around S atoms in the phases of P6/mmm and Pm1 denotes the improvement of conductivity of sulfur in Li2S battery under pressure.
Computational methods
The structural searching of K2S has been carried out by ab initio ELocR code,20 and the further confirmation with high accuracy of structural relaxations and electronic localization functions (ELF) are completed by the Vienna ab initio simulation package (VASP code)21 with a plan wave cutoff energy of 1000 eV and Brillouin zone sampling grid with a spacing of 2π × 0.04 Å−1. Total energy calculations are performed using the VASP codes with the Perdew–Burke–Ernzerhof (PBE) parameterization of generalized gradient approximation (GGA)22 used to treat the exchange–correlation energy. The partial augmented wave (PAW)23 method is adopted with the PAW potentials where 2s2p and 3s3p4s are treated as valence electrons. The electronic self-consistent calculation is stopped until the energy convergence less than 1 × 10−5 eV.The electronic projected density of states and the electronic band structure are calculated by CASTEP24 code with the cutoff energy of 720 eV, GGA-PBE exchange–correlation functional, Brillouin zone sampling grid with a spacing of 2π × 0.03 Å−1, and norm-conserving pseudopotentials. The Raman spectra are obtained within the density functional theory (DFT) formalism, and PBE exchange–correlation functional with norm-conserving pseudopotentials in the CASTEP code.25 The dynamical stability properties phonon calculations are carried out by a finite displacement approach26 through the PHONOPY code.27 The supercell are: 2 × 2 × 2, 2 × 2 × 3, 2 × 2 × 3, 3 × 3 × 3 for P63/mmc, Cmcm, P6/mmm, and Pm1, respectively. The force constants are calculated from forces on atoms with atomic finite displacements, and the finite displacement size is 0.01 Å in this work.
Results and discussion
We have performed extensive structural searches on K2S compounds at selected pressures of 0, 50, 100, 150, and 200 GPa. Five phases with lowest enthalpy, i.e. Fmm, P63/mmc, Cmcm, P6/mmm, and Pm1, are predicted as presented in Fig. 1. According to our enthalpy curves, the K2S can crystallize in a face-centered cubic lattice with the space group of Fmm at ambient pressure, and keep the symmetry up to 3 GPa. The Fmm phase has been synthesized experimentally at ambient pressure and reported by Zintl et al.18 as shown in Fig. 2(a). The unstable phase Pmma19 is also rebuilt in Fig. 2(b) for the convenience of comparison. Elevating pressure above 3 GPa, a hexagonal phase with P63/mmc symmetry emerges and is stable till 88 GPa, it has been reported by experiment.28 The layer stacking sequence in the c-axis can be denoted by the repeated ABAC stacking. K atoms occupy the crystallographic 2a position with m symmetry along a axis in A layers, the other nonequivalent K atoms occupy the crystallographic 2d position with ![[6 with combining macron]](https://www.rsc.org/images/entities/char_0036_0304.gif) m2 symmetry, locating in B and C layers. S atoms occupy the crystallographic 2c position with m2 symmetry and locate at C and B positions in B and C layers respectively, as shown in Fig. 2(c). The base-centered orthorhombic phase of Cmcm is found with the lowest enthalpy in the pressure range of 88 GPa to 136 GPa, which is about 0.03 eV lower than another body-centered orthorhombic structure of Immm per K2S at 100 GPa. There are two nonequivalent atoms in the crystal lattice, and thus consists of one K2S unit as shown in Fig. 2(d). K atoms occupy the crystallographic 8g position (site symmetry is ..m) while S atoms occupy the crystallographic 4c position (site symmetry is m2m). Each K is surrounding by four S atoms to form a triangular pyramid geometry, and the distance between S and nearest K is 2.492 Å. After 136 GPa, another hexagonal crystal with P6/mmm symmetry takes over Cmcm phase and becomes most competitive on enthalpy up to 163 GPa. K atoms occupy the crystallographic 2d position with m2 symmetry, S atoms occupy the crystallographic 1a position with 6/mmm symmetry. There, K atoms form an interesting graphene-like layered structure in the ab plane, and the distance between S and nearest K is 2.590 Å as shown in Fig. 2(e). At high pressure range from 163 GPa to 200 GPa, a trigonal phase of Pm1 obtains the minimum enthalpy, which includes two nonequivalent atoms of K and S consisting of layered K–S geometry. The layer stacking sequence in the c-axis can be denoted by the repeated ABA stacking. K atoms occupy the crystallographic 2d position with 3m. symmetry along b axis in A layers. S atoms occupy the crystallographic 1b position with m symmetry, locating in layers B, as shown in Fig. 2(f). The calculated structural parameters, volume, and Wyckoff positions for each phase are summarized in Table 1.
m2 symmetry, locating in B and C layers. S atoms occupy the crystallographic 2c position with m2 symmetry and locate at C and B positions in B and C layers respectively, as shown in Fig. 2(c). The base-centered orthorhombic phase of Cmcm is found with the lowest enthalpy in the pressure range of 88 GPa to 136 GPa, which is about 0.03 eV lower than another body-centered orthorhombic structure of Immm per K2S at 100 GPa. There are two nonequivalent atoms in the crystal lattice, and thus consists of one K2S unit as shown in Fig. 2(d). K atoms occupy the crystallographic 8g position (site symmetry is ..m) while S atoms occupy the crystallographic 4c position (site symmetry is m2m). Each K is surrounding by four S atoms to form a triangular pyramid geometry, and the distance between S and nearest K is 2.492 Å. After 136 GPa, another hexagonal crystal with P6/mmm symmetry takes over Cmcm phase and becomes most competitive on enthalpy up to 163 GPa. K atoms occupy the crystallographic 2d position with m2 symmetry, S atoms occupy the crystallographic 1a position with 6/mmm symmetry. There, K atoms form an interesting graphene-like layered structure in the ab plane, and the distance between S and nearest K is 2.590 Å as shown in Fig. 2(e). At high pressure range from 163 GPa to 200 GPa, a trigonal phase of Pm1 obtains the minimum enthalpy, which includes two nonequivalent atoms of K and S consisting of layered K–S geometry. The layer stacking sequence in the c-axis can be denoted by the repeated ABA stacking. K atoms occupy the crystallographic 2d position with 3m. symmetry along b axis in A layers. S atoms occupy the crystallographic 1b position with m symmetry, locating in layers B, as shown in Fig. 2(f). The calculated structural parameters, volume, and Wyckoff positions for each phase are summarized in Table 1.
 | ||
| Fig. 1 The calculated enthalpy values per K2S for various structures relative to previously reported Pmma19 structure as a function of pressure. | ||
 | ||
| Fig. 2 The structures of K2S. (a) The Fmm phase at ambient pressure, (b) the unstable Pmma19 phase at 4.4 GPa, (c) the P63/mmc phase at 10 GPa, (d) the Cmcm phase at 100 GPa, (e) the P6/mmm phase at 150 GPa, (f) the Pm1 phase at 200 GPa. Purple and yellow atoms are K and S, respectively. | ||
| Space group | Pressure (GPa) | Volume (Å3/K2S) | Lattice parameter (Å) | Atom (Wyckoff) | Atomic coordinates | ||
| X | Y | Z | |||||
| P63/mmc | 10 | 66.86821 | a = b = 4.9652 | S(2c) | 0.33333 | 0.66667 | 0.25000 |
| c = 6.2634 | K(2a) | 1.00000 | 0.00000 | 1.00000 | |||
| K(2d) | 0.33333 | 0.66667 | 0.75000 | ||||
| Cmcm | 100 | 37.7420125 | a = 7.7793 | S(4c) | 0.00000 | −0.70000 | −0.25000 |
| b = 4.4456 | K(8g) | −0.16510 | −0.19323 | −0.25000 | |||
| c = 4.3653 | |||||||
| P6/mmm | 150 | 32.81625 | a = b = 3.9740 | S(1a) | 0.00000 | 0.00000 | 0.00000 |
| c = 2.3994 | K(2d) | 0.33333 | 0.66667 | 0.50000 | |||
| Pm1 |
200 | 29.64067 | a = b = 3.9022 | S(1b) | 1.00000 | 1.00000 | −0.50000 |
| c = 2.2477 | K(2d) | 2.33333 | 1.66666 | −0.90932 | |||
In order to describe the response of a material to an externally applied stress, the single-crystal elastic constants of proposal structures are calculated and analyzed, as shown in Table 2. The matrix of elastic constants Cij must be positive definite,29 it is worth noting that negative values are not prohibited for Cij.30 For hexagonal structure P63/mmc and P6/mmm, there are five independent elastic constants (C11, C12, C13, C33, and C44), they all satisfy the inequality C44 > 0, C11 > |C12|, (C11 + 2C12)C33 > 2C132, it confirms that P63/mmc and P6/mmm are mechanically stable. As to orthorhombic structure Cmcm, independent elastic constants C22, C33, C44, C55, C66, C12, C13, and C23 meet the inequality C11 > 0, C22 > 0, C33 > 0,C11 + C22 + C33 + [2(C12 + C13 + C23)] > 0, (C11 + C22 − 2C13) > 0, C44 > 0, C55 > 0, C66 > 0, (C22 + C33 − 2C23) > 0, which conforms mechanical stability of Cmcm at corresponding pressure range. Pm1 belongs to trigonal system, the independent elastic constants also satisfy the inequality C44 > 0, C11 > |C12|, (C11 + 2C12)C33 > 2C132, indicating the mechanical stability of Pm1. So, the proposal structures all satisfy Born–Huang criterion,31 representing the stability of the mechanical property.
m1 at 10, 100, 150, and 200 GPa, respectively
| Space group | C11 | C12 | C13 | C33 | C44 |
|---|---|---|---|---|---|
| C22 | C23 | C55 | C66 | ||
| P63/mmc | 101.19881 | 43.82810 | 20.79056 | 114.65024 | 22.52524 |
| 101.19881 | 20.79056 | 22.52524 | 28.68536 | ||
| Cmcm | 568.50040 | 245.86790 | 170.10752 | 465.26990 | 27.42380 |
| 545.48660 | 134.70090 | 108.92205 | 107.29615 | ||
| P6/mmm | 786.32800 | 412.58015 | 276.12338 | 585.58230 | 149.94920 |
| 786.32800 | 276.12338 | 149.94920 | 186.87393 | ||
| Pm1 |
905.68978 | 124.02977 | 100.68456 | 966.62216 | 307.39376 |
| 124.02977 | 100.68456 | 307.39376 | 390.83000 |
The mechanically and thermodynamically stability of proposal phases have been discussed, and they are all stable at relevant pressure range. Furthermore, the phonon band structure and partial phonon density of states (PHDOS) for atoms with nonequivalent Wyckoff positions are calculated to judge its dynamical stability as shown in Fig. 3, and the absence of imaginary frequency modes in the entire Brillouin zone indicates dynamic stability of the structures. From the PHDOS on nonequivalent atoms in P63/mmc crystal at 50 GPa as plotted in Fig. 3(a), we can see clearly that the low frequency modes mainly dominated by the K atoms occupying the crystallographic 2a position, but the other nonequivalent K atoms occupying the crystallographic 2d position contribute more than K atoms with 2a position at high frequency modes. In the Cmcm phase, S atoms mainly contribute to the frequency upon 14 THz while the low frequency below 14 THz mainly comes from the vibrations of K atoms at 100 GPa, see the Fig. 3(b). At higher pressure range from 150 GPa to 200 GPa, there are no distinguishing characteristics in P6/mmm and Pm1 K2S except K atoms contribute more than S atoms as a whole because the number of K atoms is twice the number of S atoms in K2S crystals as shown in Fig. 3 (c) and (d).
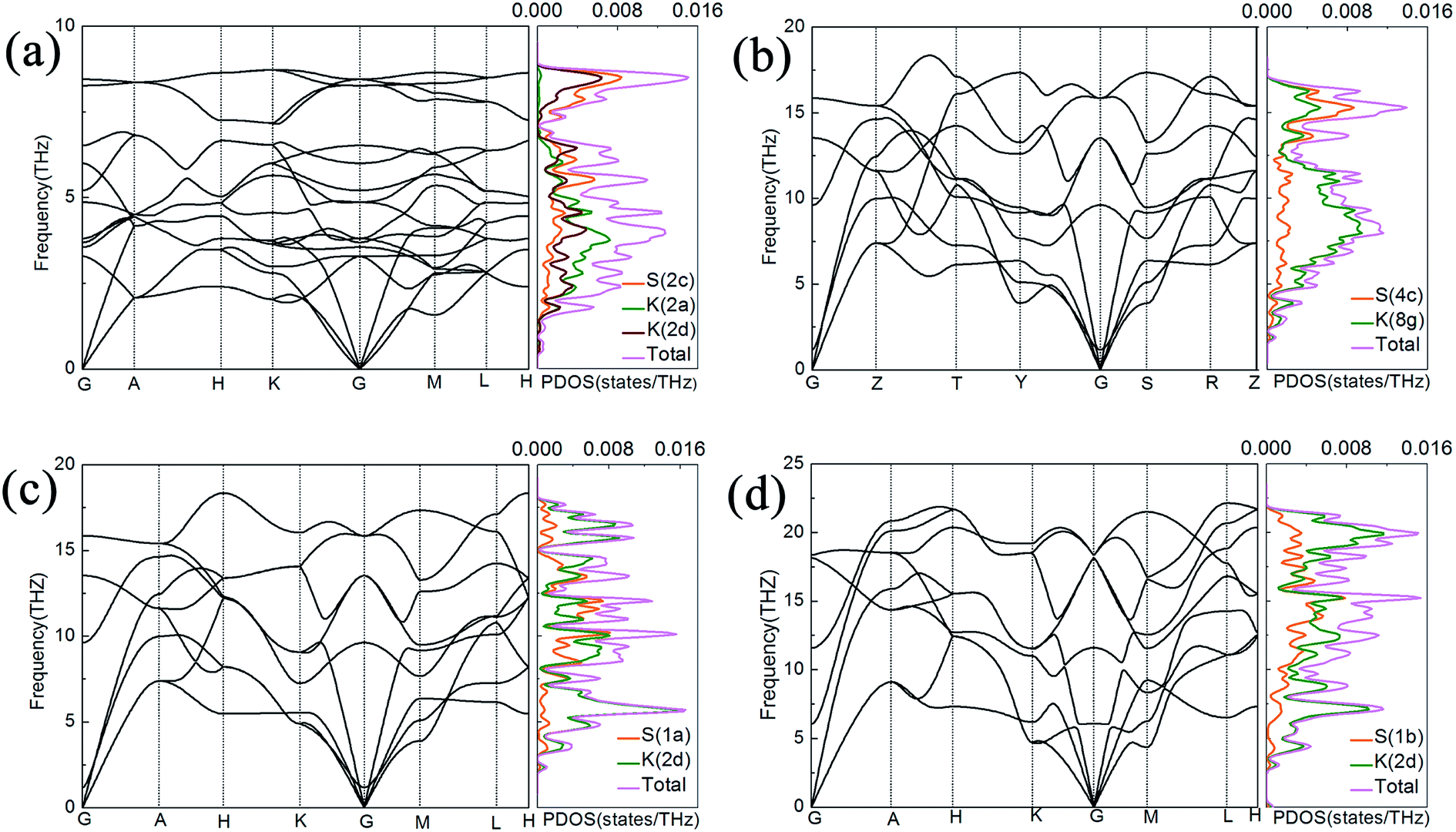 | ||
| Fig. 3 The phonon band structure and partial phonon density of states for atoms with nonequivalent Wyckoff positions are plotted for the proposal phases. (a) P63/mmc phase at 50 GPa, (b) Cmcm phase at 100 GPa, (c) P6/mmm phase at 150 GPa, (d) Pm1 phase at 200 GPa, respectively. | ||
There is a discussion needed with the proposal phase in low pressure range. Experiment proposes a crystal with the space group of Pmma19 at 4.4 GPa, which is not consistent with the structure of P63/mmc proposed in this work. We rebuild Pmma phase according to the space group and atomic coordinates given by the experiment report, and calculate the phonon dispersion and elastic constants. The matrix of elastic constants Cij are as shown in ESI,† which contains C11 < 0, C55 < 0, C66 < 0, C11 + C22 − 2C12 < 0, C11 + C33 − 2C13 < 0. For the orthorhombic structure, elastic constants do not satisfy Born–Huang criterion,31 representing the instability of the mechanical property. Phonon band structure is added in ESI.† There are imaginary frequency modes in the entire Brillouin zone indicates dynamic instability of the structures. Comparing the experimental X-ray powder diffractograms as shown in Fig. 4(a) and (c), the peak position and intensity of P63/mmc K2S coincide well with reported experiment observation. So, the reported Pmma structure is unstable theoretically, and the P63/mmc phase is the best possible candidate for the stable structure of K2S at low pressure range.
 | ||
| Fig. 4 (a) Calculated difference X-ray power diffraction for P63/mmc and Pmma which is rebuilt though data from experiment at 4.4 GPa, (b) Raman active modes of P63/mmc phase, (c) experiment X-ray power diffraction for Pmma at 4.4 GPa,19 (d) Raman active modes of Cmcm phase. | ||
Moreover, Raman spectroscopies of P63/mmc and Cmcm are simulated and display in Fig. 4(b) and (d). The Raman spectroscopy of P63/mmc can be classified by the irreducible representation of the point group D6h, a weak characteristic peak is observed at 96 cm−1 correspond to E2g, the another peak appear at 196 cm−1 also belong to E2g irreducible representation. Cmcm vibrational modes belong to the D3h point group, the peaks at 82 cm−1, 442 cm−1 and 519 cm−1 are related to Ag irreducible representation, the peaks at 253 cm−1, 341 cm−1 and 484 cm−1 are correlated with B1g. The following peaks at 255 cm−1 and 551 cm−1 are connected with B3g, the positions of the peak at 337 cm−1 is corresponding to B2g. The two close peaks at 253 cm−1 and 255 cm−1 produce an intense peak, and the other two close peaks at 337 cm−1 and 341 cm−1 combine the most intense peak.
In order to analyze the electronic properties of phases P63/mmc, Cmcm, P6/mmm, and Pm1, the electronic band structure and the partial density of states are further explored in Fig. 5. Band gap is discovered in the P63/mmc phase at 50 GPa, revealing the nonmetallic character which coincides with previously report.14 The electronic band structure of Cmcm phase shows the semiconductor character, as displayed in Fig. 5(c). The band gap decreases with pressure from P63/mmc to Cmcm by contrast to the values of gaps at 50 GPa and 100 GPa in Fig. 5, respectively. Elevating the pressure to 150 GPa, three bands marked with magenta, green, and blue crossing over the Fermi level contribute large total electronic density distribution, and revel the strong metallic character of P6/mmm, as displayed in Fig. 5(b). Three bands intersecting the Fermi level, the Pm1 phase also behaves as a metal, and the bands colored by blue and magenta closing to Fermi level are found to contribute more to the density of states (DOS) of Fermi level, as displayed in Fig. 5(d). From the partial density of states (PDOS) in Fig. 5(b) and (d), the metallic character of P6/mmm and Pm1 is confirmed by the high level of total electronic density distribution at Fermi level. The majority of occupied states come from K(p) state, whereas the contribution from K(s), S(s) and S(p) to the states around the Fermi level is quite small.
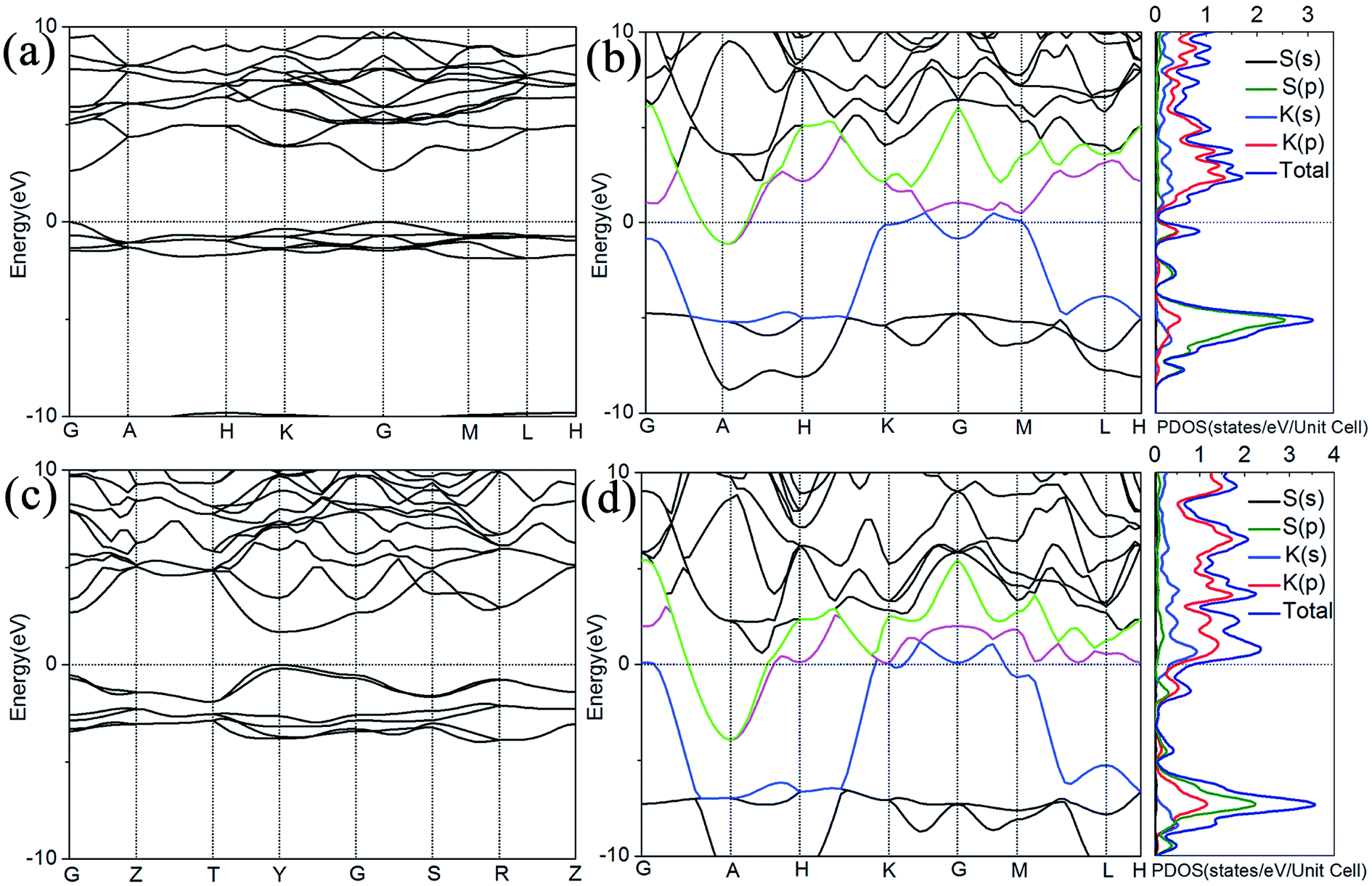 | ||
| Fig. 5 The electronic band structure and PDOS of K2S. (a) The electronic band structure of P63/mmc, (b) the electronic band structure and PDOS of P6/mmm, (c) the electronic band structure of Cmcm, (d) the electronic band structure and PDOS of Pm1. | ||
The electronic localization functions (ELF) is derived from an earlier idea of Lennard-Jones32 and first reported by A. D. Becke and K. E. Edgecombe.33 It has been calculated to characterize the degree of electron localization in the proposal K2S crystals. ELF as defined runs from 0 to 1, the values of ELF equaling to 1.0 and 0.5 reflect the extremely strong localization and homogeneous electrons distribution, respectively.33,34 Three-dimensional electron location function (3D ELF) of P6/mmm and Pm1 show free-electron channels with isosurface value of 0.5 (ELF = 0.5) around S atoms, which reveals that the conductivity comes from the contributions of the electrons around the S atoms as presented in Fig. 6(a) and (c). Furthermore, the two-dimensional electron location function (2D ELF) confirm the character of conductivity in two crystals, where connected regions with the ELF equaling to 0.5 are surrounding S atoms as shown in Fig. 6(b) and (d). Potassium is an alkali element and always described as a typical free-electron-like metal at ambient pressure; on the contrary, sulfur is often described as a good insulator at the same condition. Nevertheless, free electrons assemble around only S atom chains demonstrating the conductivity, and not K atoms under high pressure. At ambient pressure, K2S is crystallized in face-centered cubic antifluorite structure with Fmm symmetry which is an ionic crystal14 and performed insulator characters. Nevertheless, the two phases of P6/mmm and Pm1 exhibit metallic features as discussed above, which implies the available improvement of the conductivity of sulfur to enhance the electrode recharge ability and rate capability in alkali sulfide battery under pressure.
 | ||
| Fig. 6 Electron localization function of P6/mmm and Pm1 phase. (a) P6/mmm 3D electron localization function (3D ELF) map with the ELF values of 0.5, (b) P6/mmm 2D electron location function (2D ELF) slice along the (110) plane, (c) Pm1 3D electron localization function (3D ELF) map with the ELF values of 0.5, (d) Pm1 2D electron location function (2D ELF) slice along the (010) plane. | ||
Conclusion
In summary, we have systematically explored phase sequence of K2S at pressures from ambient pressure to 200 GPa using the ab initio ELocR, and stable structures of Fmm, P63/mmc, Cmcm, P6/mmm, and Pm1 have been explored thoroughly. Based on enthalpies, phonon band structure, elastic constants, and X-ray power diffraction, we confirm that the reported Pmma structure is unstable and can distort into P63/mmc by our calculations. Moreover, we also simulate the Raman spectroscopies of P63/mmc and Cmcm phases for the convenience of the further studies of experiments. The two phases of P63/mmc and Cmcm K2S are semiconductors under pressure, P6/mmm and Pm1 emerge with the nature of metallic characters at higher pressures. The analyses of ELF show that the conductivity comes from the electrons surrounding S atom chains, which implies the potential improvement of the conductivity of sulfur to enhance the electrode recharge ability and rate capability in alkali sulfide battery under pressure.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 51632002, 51572108, 11634004, 11174102), Program for Changjiang Scholars and Innovative Research Team in University (No. IRT_15R23), National Found for Fostering Talents of basic Science (No. J1103202), the Development Program of Science and Technology of Jilin Province, China (No. 20150312002ZG). Parts of calculations were performed in the High Performance Computing Center (HPCC) of Jilin University.References
- H. Bode, Lead-acid batteries, John Wiley, New York, 1977 Search PubMed.
- Z. M. Salameh, M. A. Casacca and W. A. Lynch, IEEE Transactions on Energy Conversion, 1992, 7, 93–98 CrossRef.
- S. C. Singhal, Solid State Ionics, 2000, 135, 305–313 CrossRef CAS.
- N. Q. Minh, Solid State Ionics, 2004, 174, 271–277 CrossRef CAS.
- W. Shockley and H. J. Queisser, J. Appl. Phys., 1961, 32, 510 CrossRef CAS.
- M. A. Green, K. Emery and Y. Hishikawa, Prog. Photovoltaics, 2015, 23, 1–9 Search PubMed.
- L. Gao, S. Liu and R. A. Dougal, IEEE Trans. Compon. Packag. Technol., 2002, 25, 495–505 CrossRef CAS.
- Y. Idota, T. Kubota and A. Matsufuji, Science, 1997, 276, 1395–1397 CrossRef CAS.
- J. O. Besenhard, J. Yang and M. Winter, J. Power Sources, 1997, 68, 87–90 CrossRef CAS.
- P. G. Bruce, L. J. Hardwick and K. M. Abraham, MRS Bull., 2011, 36, 506–512 CrossRef CAS.
- P. G. Bruce, S. A. Freunberger and L. J. Hardwick, Nat. Mater., 2012, 11, 19–29 CrossRef CAS PubMed.
- H. Momida, T. Yamashita and T. Oguchi, J. Phys. Soc. Jpn., 2014, 83, 124713 CrossRef.
- Y. J. Choi, K. W. Kim and H. J. Ahn, J. Alloys Compd., 2008, 449, 313–316 CrossRef CAS.
- H. Khachai, R. Khenata and A. Bouhemadou, Solid State Commun., 2008, 147, 178–182 CrossRef CAS.
- R. D. Eithiraj, G. Jaiganesh and G. Kalpana, Phys. Status Solidi, 2007, 244, 1337–1346 CrossRef CAS.
- J. C. Schön, Ž. Čančarević and M. Jansen, J. Chem. Phys., 2004, 121, 2289 CrossRef PubMed.
- H. Khachai, R. Khenata and A. Bouhemadou, J. Phys., 2009, 21, 095404 CAS.
- E. Zintl, A. Harder and B. Dauth, Z. Elektrochem. Angew. Phys. Chem., 1934, 40, 588–593 CAS.
- A. Vegas, A. Grzechnik and M. Hanfland, Solid State Sci., 2002, 4, 1077–1081 CrossRef CAS.
- A code for crystal structural prediction and analysis is based on evolutional local random computational method, and is developed by our group. Some details have been provided in ESI.†.
- G. Kresse and J. Fürthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 77, 3865–3868 CrossRef PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- S. J. Clark, M. D. Segall and C. J. Pickard, Crystalline Materials, 2005, 220, 567–570 CAS.
- M. D. Segall, P. J. D. Lindan and M. J. Probert, J. Phys.: Condens. Matter, 2002, 14, 2717–2744 CrossRef CAS.
- K. Parlinski, Z. Q. Li and Y. Kawazoe, Phys. Rev. Lett., 1997, 78, 4063–4066 CrossRef CAS.
- A. Togo, F. Oba and I. Tanaka, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 134106 CrossRef.
- H. F. Fischmeister, Monatsh. Chem., 1962, 93, 420–434 CrossRef CAS.
- J. F. Nye, Physical Properties of Crystal, Oxford Univ. Press, Oxford, 1985, pp. 142–143 Search PubMed.
- J. P. Perdew and K. Burke, Phys. Rev. Lett., 1997, 77, 3865–3868 CrossRef PubMed.
- Z. Wu, E. Zhao and H. Xiang, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 054115 CrossRef.
- J. E. J. Lennard-Jones, Chem. Phys., 1952, 20, 1024 CAS.
- A. D. Becke and K. E. Edgecombe, J. Chem. Phys., 1990, 92, 5397–5403 CrossRef CAS.
- J. K. Burdett and T. A. McCormick, J. Phys. Chem. A, 1998, 102, 6366–6372 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra25409h |
| This journal is © The Royal Society of Chemistry 2017 |