
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical determination of trace pesticide residues based on multiwalled carbon nanotube grafted acryloyloxy ferrocene carboxylates with different spacers†
Feng Xu *,
Zhuo-Miao Cui,
He Li and
Yan-Ling Luo*
*,
Zhuo-Miao Cui,
He Li and
Yan-Ling Luo*
Key Laboratory of Macromolecular Science of Shaanxi Province, School of Chemistry and Chemical Engineering, Shaanxi Normal University, Xi'an 710062, P. R. China. E-mail: luoyanl@snnu.edu.cn; fengxu@snnu.edu.cn; Fax: +86 29 81530727
First published on 24th January 2017
Abstract
Food safety has always been a topic that attracts attention. The residues of pesticides will directly or potentially affect and threaten human health when they are used to protect crops. Therefore, it is important to rapidly, promptly and accurately monitor pesticide residues in food. In this study, a novel nanohybrid composite with good electrochemical responses was developed, and it was prepared by the esterification reaction of hydroxyl-terminated polybutadiene (HTPB) with MWCNTs-COOH, followed by atom transfer radical polymerization of 4-acryloyloxybutyl(ethyl) ferrocene carboxylates with different spacers. The nanohybrid composites were characterized by FTIR, TGA, Raman, XRD, XPS, SEM and TEM techniques. Cyclic voltammetry (CV) determination showed that a longer spacer between the side ferrocene groups and main chains endowed the electrochemical modified electrodes with active electroresponse, obvious redox current and reversible electrochemical properties because of the faster electron transfer rates. The modified electrode sensor with a longer spacer was used to detect melamine and trichlorfon residues by CV and differential pulse voltammetry (DPV) techniques. The sensor had a good linear relationship over a wide concentration range, a maximal recovery of ca. 112.4% and a low detection limit of about 1.5 × 10−7 and 3.5 × 10−8 mol L−1, respectively.
1. Introduction
In the recent decades, the need to develop rapid, sensitive and cost-effective pesticide assay techniques to solve the worldwide eco-environmental and food-safety problems has become urgent because food ingredient fraud and organophosphorus pesticide (OP) residues are very serious.1 Melamine and trichlorfon are typical examples that are used in a range of products and are being abused for economic interests. These substances cause damage to the ecosystem and human health. For example, melamine, an illegal food adulterant, may cause kidney stones, kidney failure or even death.2 High concentrations of melamine were reported in contaminated Chinese infant formula in September, 2008. More than 51![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 infants and young children in China were hospitalized for urinary problems, possible renal tube blockages, and kidney stones due to the consumption of the melamine-contaminated infant formula and related dairy products.3 OPs, for example trichlorfon, are used widely for agriculture, vector control and domestic purposes as insecticides because of their high activity, low bioaccumulation and moderately rapid degradation in the environment. Nevertheless, a certain amount of pesticides migrates from agriculture land and can cause a spectrum of toxic effects on the eco-environment, aquatic organisms and human beings.4
900 infants and young children in China were hospitalized for urinary problems, possible renal tube blockages, and kidney stones due to the consumption of the melamine-contaminated infant formula and related dairy products.3 OPs, for example trichlorfon, are used widely for agriculture, vector control and domestic purposes as insecticides because of their high activity, low bioaccumulation and moderately rapid degradation in the environment. Nevertheless, a certain amount of pesticides migrates from agriculture land and can cause a spectrum of toxic effects on the eco-environment, aquatic organisms and human beings.4
Conventional methods to detect melamine in milk and trichlorfon in food and fruits mainly include liquid chromatography (LC),5 gas chromatography (GC),6 gas chromatography-mass spectrometry (GC-MS),7 high performance liquid chromatography-diode array detector (HPLC-DAD),8 liquid chromatography-mass spectrometry (LC-MS),9 etc. Although these methods can give precise analytical data, they require large and expensive instruments, inconvenient utilization, time-consuming analysis, complicated sample treatments, etc. Consequently, it is imperative to explore a new detection method to realize the fast and sensitive determination of melamine and trichlorfon. Electrochemical methods have attracted attention because of their simple design and preparation, low cost, high sensitivity and fast response for determinations.10 Various new functional materials are employed to modify electrochemical glass carbon electrodes, and this offers excellent prospects for designing novel detecting systems and enhancing the performance of electrochemical sensors.11 Liang et al.12 reported a potentiometric sensor based on a molecularly imprinted polymer for melamine detection with a detection limit of 1.6 × 10−6 mol L−1 that exhibits a short response time and good stability. Ai et al.13 took advantage of inter-particle crosslinking of gold nanoparticles to realize the simple and visual detection of melamine, which enables the detection of melamine in raw milk and infant formula at a concentration as low as 2.5 ppb without the aid of any advanced instruments. However, this colorimetric sensor requires a complicated pretreatment to get rid of the interferences in the real samples. Sundarmurugasan et al.14 prepared a acetylcholinesterase–zinc oxide modified platinum electrode that was used to simultaneously detect monocrotophos and dichlorvos in orange samples with a detection limit of 1.2 × 10−5 mol L−1.
Ferrocene-based polymers are a new class of functional polymers that have attracted significant attention because of their unique redox, electrochemical, mechanical, semiconductive, photophysical, optoelectronic, electronic and magnetic properties.15,16 They can be constructed into various systems and devices, and they show important values in academic research and extensive application prospects in the fields of semiconductor materials, phase separation materials, catalysts, redox-responsive drug carriers, biosensors, magnetic ceramic precursors, and smart surfaces,17–19 which is a very active field in functional materials in recent years. Especially, ferrocene groups (Fc) can enhance the electron transfer process between electrode surfaces and immobilized (bio)catalysts, leading to more sensitive and stable (bio)sensors. Modified electrodes comprised of Fc-based polymer films, for instance polyvinylferrocene, have thus gained considerable attention.20 The diversity of the side-chain structure and elemental valence states of the ferrocene-based polymers are convenient for manipulating their structure and properties. Li et al.21 found that with the increasing length of the side chains, the graft copolymers were difficult to oxidize, and the reversibility of the process was confined. Nevertheless, the chain length influence, especially the length of the spacers between the ferrocene groups in the side chains and main chains, on the electrochemical properties has not been discussed. Multi-walled carbon nanotubes (MWCNTs) show excellent physical, mechanical, electrochemical and optical properties, fast electron transportation, large specific surface areas and strong adsorption.22,23 Sensors based on carbon nanotubes have found applications in electrochemical detection of metal ions, pesticides and other pollutants, and these applications have been summarized in detail in a review paper.24 These findings motivated us to construct novel nanohybrid composites with increased electrochemical activities combining MWCNTs with ferrocene-based polymers and to fabricate novel electrochemical sensors for the detection of trace pesticide residues.
In this context, our aim is to design and prepare novel MWCNTs and graft polybutadiene-block-ferrocene derivative copolymer nanohybrids by an ATRP approach and fabricate electrochemical sensors for the detection of melamine and trichlorfon residues. The influence of the different lengths of spacers between the ferrocene groups in the side chains and main chains on the electrochemical performance is investigated to more efficiently enhance the charge transfer rate and to acquire excellent electrocatalytic activities. Cyclic voltammetry (CV) and differential pulse voltammetry (DPV) were employed to determine trace melamine and trichlorfon so as to attain a highly sensitive, simple and quick electrochemical method for the detection of melamine in milk and trichlorfon in fruit and vegetables.
2. Materials and experiments
2.1 Materials and reagents
Carboxylated multi-walled carbon nanotubes (MWCNTs-COOH, 95%), with particle diameters of 10–30 nm, –COOH content of 3.86% (ca. 0.86 mmol g−1), and length of 10–30 μm, were provided by the Chengdu Institute of Organic Chemistry, CAS, and dewatered in an oven at 120 °C before use. Ferrocenecarboxylic acid (FCA, 98%) and hydroxyl-terminated polybutadiene (HTPB, MW of 2300 and –OH value of 1.1 mmol g−1) were purchased from J & K Reagent Company, China. 4-Hydroxybutyl acrylate (HBA, 98%) and 2-hydroxyethyl acrylate (HEA, 96%) were provided by Zahn Chemical Technology Co., Ltd., Shanghai and were used without further purification. 2-Bromoisobutyryl bromide (BIB, 98%), 4-dimethylaminopyridine (DMAP, a highly effective nucleophilic acylation catalyst, 99%), tetrabutylammonium hexafluorophosphate (TABP, 98%), and dicyclohexylcarbodiimide (DCC, 99%) came from the Sigma-Aldrich (Shanghai) Trading Co., Ltd., China. CuBr (98%, Aldrich) was purified by stirring overnight in CH3COOH at room temperature, followed by washing the solid residues with ethanol, acetone and diethyl ether in sequence, and drying at 40 °C in vacuo for 24 h. N,N,N′,N′′,N′′-Pentamethyldiethylenetriamine (PMDETA, 98%) was purchased from the Aladdin (Shanghai) Reagent Company, China and was used without further purification. Triethylamine (TEA, 98%) was provided by the Tianjin Fuchen Chem Reagent Factory, China and was refluxed over CaH2 for 2 h and distilled before use. Tetrahydrofuran (THF, 99.5%), dichloromethane (CH2Cl2, 99.5%), N,N-dimethylformamide (DMF, 99.5%) were obtained from the Sinopharm Chemical Reagent Co., Ltd., China and were used without further purification. Melamine (99.5%) and trichlorfon (90%) were purchased from Tianjin Kermel Chemical Industry Co., Ltd., China. Perfluorinated ion-exchange resin (5% Nafion solution in aliphatic alcohols/water (85–80/15–20, w/w) mixture) was obtained from the Aladdin (Shanghai) Reagent Company, China. Samples including fresh liquid milk, pear, apple and cucumber were purchased from a local supermarket (Xi'an, China).2.2 Synthesis processes
The synthesis of MWCNTs-g-HTPB-b-PAEFC and MWCNTs-g-HTPB-b-PABFC polymers was carried out through a three-step reaction procedure, as shown in Scheme 1. Preparation of the monomers is described in ESI,† and the 1H NMR spectra are shown in Fig. S1.† All experiments for the polymerization reaction were conducted under purified nitrogen with the use of standard Schlenk tubes. | ||
| Scheme 1 Synthetic strategies of MWCNTs grafted block copolymer nanohybrid composites. | ||
:[monomer]:[ligand]:[catalyst] of 1:50:6:5. In a typical procedure, 0.02 g (0.008 mmol) of MWCNTs-g-HTPB-Br were added to a 25 mL dried Schlenk flask and dispersed in 4 mL DMF for 2 h under ultrasonication, and then 0.25 g (0.4 mmol) of AEFC was put into the flask. The reaction system was degassed by vacuum and purging with N2, and 10.2 μL (0.048 mmol) PMDETA and 2.0 mL DMF were then introduced into the mixed solution via a gastight syringe. After a similar degassing operation, 0.0058 g (0.04 mmol) CuBr was put into the Schlenk flask under the protection of N2. The reaction flask was sealed with a rubber plug, and it was further degassed thrice by means of a freezing–pumping–thawing process. The flask was immersed in an oil bath at 90 °C to start the polymerization. After 48 h, the polymerization was terminated by putting the flask into liquid N2. The reaction mixture solution was vacuum filtered, and the solid was rinsed with DMF thrice. The purified sample was diluted with CHCl3 to replace the residual DMF that remains in the resultant product. After filtering and rinsing with CHCl3 twice, the solid sample was dried in a vacuum oven at 45 °C overnight, giving MWCNTs-g-HTPB-b-PAEFC (mean yield: 88.4%). The MWCNTs-g-HTPB-b-PABFC were synthesized in the same process and feed ratios as the MWCNTs-g-HTPB-b-PABFC with a yield of 85.6%. It should be noted that further ultrasonic treatment was conducted to disperse the MWCNTs-g-HTPB-Br after ABFC was put into the flask.2.3 Preparation of modified electrode sensors
The 0.01 g of the resulting nanohybrid materials was dispersed in 1 mL chloroform via an ultrasonic technique for 2 h. Homogeneously dispersed conducting composite dispersions (6 μL) were uniformly deposited on the clean surface of the pre-treated glass carbon electrodes (GCEs, see also ESI†) using a micro-syringe, and then they were dried with an infrared lamp to obtain finished GCEs coated with the nanohybrid materials. To ensure the proper adherence of the nanostructures on the electrode surface, 5 μL of a 1 wt% Nafion solution was cast on the modified layers and dried before use. The modified electrodes with MWCNTs-g-HTPB-b-PAEFC and MWCNTs-g-HTPB-b-PABFC were named MHPEC and MHPBC sensors, respectively.2.4 Characterization and measurements
Fourier transform infrared (FI-IR) spectra were recorded on an EQUINX55 FTIR (Brucker Corp., Germany) using KBr pellets. Raman spectra were recorded on an AlMEGA dispersive Raman spectrometer (AlMEGA-TM, Therm Nicolet Corp., USA) using an Ar+ laser with an excitation wavelength of 532 nm. Thermal gravimetric analysis (TGA) was carried out on a thermoanalyzer system (Q1000DSC + lNCS + FACS Q600SDT, TA Corp., USA) at a heating rate of 10 °C min−1 under a N2 atmosphere (flow rate: 40 mL min−1). X-ray diffraction (XRD) patterns were recorded on a D/Max-2550 VB+/PC automatic X-ray diffractometer (Rigaku Corp., Japan) employing Cu radiation at a voltage of 40 kV, a current of 30 mA, and a scanning rate of 10° min−1. X-ray photoelectron spectroscopy (XPS) profiles were determined at room temperature on an AXIS ULTRA spectrometer (Kratos Analytical Ltd., Shimadzu Corp., Japan) using a monochromatic Al-Kα X-ray source (1486.6 eV) at a voltage of 15 kV. A high-resolution spectrum was obtained in the peak region of the photoemission for C 1s, O 1s and Fe 2p electrons, and the curve fitting of the XPS spectra was performed using the commercially available CasaXPS software.The morphologies were observed on a transmission electron microscope (TEM, JEM-2100, Electron Corp., Japan) with an accelerating potential of 200 kV. Before observation, the samples were ground into powders and sufficiently dispersed in ethyl alcohol by ultrasonication for about 20 min. After that, a drop of the dispersion was dropped on the copper grid with carbon films and dried for 6 h. Scanning electron microscopy (SEM) images were obtained using a SU-8020 field emission electron microscope (FESEM, Hitachi limited Corp., Japan) at an operating voltage of 15 kV. All measurements were carried at room temperature.
Electrochemical measurements, including cyclic voltammetry (CV) and differential pulse voltammetry (DPV), were conducted on a CHI 660E electrochemical workstation (Shanghai Chenhua Co., China) with a conventional three-electrode cell, which is composed of bare or modified glassy carbon electrodes (3.0 mm in diameter) as working electrodes, platinum electrodes as counter electrodes, and saturated calomel electrodes (SCE, 0.2415 V vs. SHE (standard hydrogen electrode)) as reference electrodes. For the detection of melamine, the CV experiment was performed in a 1 mol L−1 H2SO4 solution at a rate of 50 mV s−1. For the detection of trichlorfon, the DPV measurements were performed in PBS at pH = 7 with a modulation amplitude of 50 mV, a pulse width of 50 ms and a step potential of 5 mV.
2.5 Real sample analysis
For the detection of residual melamine, the liquid milk was directly spiked with appropriate amounts of a melamine standard solution. Specifically, 500 mL of 20 mg L−1 melamine in a 1 mol L−1 H2SO4 solution was prepared, and 6.25 mL of the solution was mixed with 250 mL of fresh milk to get a 0.50 mg L−1 (3.9 × 10−6 mol L−1) melamine solution. The melamine concentration was detected by our electrochemical modified electrodes using cyclic voltammetry.For the detection of residual trichlorfon, 10 grams of each sample (pear, apple and cucumber) were cleaned and cut into small pieces and added into a 50 mL beaker. The samples were spiked with PBS standard buffer solutions containing 1 × 10−6, 4 × 10−9 and 4 × 10−12 mol L−1 trichlorfon and immersed for 6 h. The spiked samples were ultrasonicated in 30 mL of PBS at pH 7.0 for 1–2 h and filtered to obtain the extracts. The extracts were transferred into a 50 mL calibrated flask and diluted to the mark with PBS solution. The prepared sample solutions were stored in a refrigerator at 4 °C and determined by DPV. The recovery percentage and relative standard deviations were determined for 3 different concentrations. Before the measurements, the resulting solutions were filtered through a 0.45 μm membrane.
3. Results and discussion
3.1 Structural characterization
FTIR spectroscopy was used to qualitatively confirm the chemical structure of the MWCNTs grafted block copolymer nanohybrid composites, as shown in Fig. 1(A). For the MWCNTs-COOH, a wide and strong peak at 3460 cm−1 is attributed to the stretching vibration mode of the –OH groups; the peak at 1635 cm−1 is ascribed to the stretching vibration of the –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in carboxylic acid, and the vibration band at 1573 cm−1 is assigned to the stretching mode of the benzenoid (νCC) in the MWCNTs backbone, indicating the graphite structure of MWCNTs-COOH.26,27 After HTPB is grafted from MWCNTs-COOH, new and more prominent vibration bands at 1574, 1720 and 3465 cm−1 emerge, which are attributable to the –CC, –CO and –OH stretching modes in HTPB, respectively. It should be noted that the –OH stretching modes do not completely disappear upon the addition of excess HTPB, which guarantees the existence of end hydroxyl groups, restraint of the possible crosslinking reaction and the preparation of the subsequent ATRP initiators. The structural diagram of the MWCNTs-g-HTPB-OH is shown in Scheme 1. The two resulting graft block copolymer nanohybrid materials give identical FTIR spectra because of their almost identical chemical structure. The vibration bands at 3100 cm−1 are attributable to the –CH stretching mode of the ferrocenyl rings; the bands at 2858–2963 cm−1 are ascribed to the –C–H stretching; the peaks at 1715 cm−1 are assigned to the ester carbonyl stretching modes; the peaks at 1555 cm−1 are attributed to the –CC stretching modes of the ferrocenyl rings or/and HTPB; the bands at 1134–1275 cm−1 are related to the –C–O stretching; and the bands at 773–822 and 485–510 cm−1 reflect the C–H in the ferrocene rings and Fe–C or Cp–Fe stretching modes, respectively.28 These results suggest that AEFC and ABFC were chemically attached onto MWCNTs-COOH after ATRP via successful grafting.
O in carboxylic acid, and the vibration band at 1573 cm−1 is assigned to the stretching mode of the benzenoid (νCC) in the MWCNTs backbone, indicating the graphite structure of MWCNTs-COOH.26,27 After HTPB is grafted from MWCNTs-COOH, new and more prominent vibration bands at 1574, 1720 and 3465 cm−1 emerge, which are attributable to the –CC, –CO and –OH stretching modes in HTPB, respectively. It should be noted that the –OH stretching modes do not completely disappear upon the addition of excess HTPB, which guarantees the existence of end hydroxyl groups, restraint of the possible crosslinking reaction and the preparation of the subsequent ATRP initiators. The structural diagram of the MWCNTs-g-HTPB-OH is shown in Scheme 1. The two resulting graft block copolymer nanohybrid materials give identical FTIR spectra because of their almost identical chemical structure. The vibration bands at 3100 cm−1 are attributable to the –CH stretching mode of the ferrocenyl rings; the bands at 2858–2963 cm−1 are ascribed to the –C–H stretching; the peaks at 1715 cm−1 are assigned to the ester carbonyl stretching modes; the peaks at 1555 cm−1 are attributed to the –CC stretching modes of the ferrocenyl rings or/and HTPB; the bands at 1134–1275 cm−1 are related to the –C–O stretching; and the bands at 773–822 and 485–510 cm−1 reflect the C–H in the ferrocene rings and Fe–C or Cp–Fe stretching modes, respectively.28 These results suggest that AEFC and ABFC were chemically attached onto MWCNTs-COOH after ATRP via successful grafting.
 | ||
| Fig. 1 FTIR (A) and Raman spectra (B) of (a) MWCNTs-COOH, (b) MWCNTs-g-HTPB, (c) MHPEC and (d) MHPBC. | ||
Raman spectroscopy was further used to confirm the preparation of the resulting nanohybrid composites, as depicted in Fig. 1(B). The original pristine MWCNTs-COOH exhibit two typical vibration bands at about 1339 and 1574 cm−1, which are assigned to the D- and G-band, respectively. The D band reflects defects and/or disorder-induced sp3-hybridized carbon atoms, whereas the G band is ascribed to the vibration of the sp2-bonded carbon atoms in a graphitic layer. The relative peak intensity ratio of the D to G peak (ID:IG) was calculated to be about 0.95, signifying that some defects exist in the MWCNTs-COOH.29 After esterification and the follow-up ATRP, the two main Raman bands in the MWCNTs-COOH remained the same. The ID:IG values, however, increased to 1.03, 1.08 and 1.12, respectively, presumably because HTPB, HTPB-b-PAEFC or HTPB-b-PABFC was covalently linked with the MWCNTs-COOH. Consequently, the HTPB-b-PABFC or HTPB-b-PAEFC copolymer was chemically grafted onto the surface or/and at the defect dots of the MWCNTs.
The relative amount of AEFC and ABFC grafted onto the MWCNT-COOH was estimated from TGA, as shown in Fig. 2(A). The original pristine MWCNTs-COOH exhibit high thermostability and produce only about 4.6% mass loss up to 800 °C, which is ascribed to absorbed moisture and ashes.29 In the case of the MWCNTs-g-HTPB, the mass loss of ca. 3.3% at 360 °C is ascribed to some solvent, DMAP and DCC or DCU residues, and that of 7.4% from 360 to 500 °C is related to the decomposition of HTPB on the surface of the MWCNTs-COOH.30 For the MWCNTs-g-HTPB-b-PAEFC, the mass loss of 11.2% up to 330 °C results from residual solvents and monomer molecules, and the follow-up mass loss of about 16.3% from 330 to 800 °C is attributable to the gradual decomposition of the HTPB and PAEFC backbones covalently attached onto the MWCNTs-COOH. The residual amount is about 72.5%, which is ascribed to the MWCNTs and Fe atoms. In contrast, the HTPB-b-PAEFC copolymer generates about a 39.3% mass loss from 330 to 800 °C, and the residues amount for 40.7%. The difference in the residues is about 31.8%. From these data, it was inferred that the relative amount of the HTPB-b-PAEFC grafted onto the MWCNTs-COOH was about 57.9 wt%, and the relative amount of the MWCNTs-COOH was ca. 42.1 wt%. Likewise, the relative content of HTPB-b-PABFC in the MWCNTs-g-HTPB-b-PABFC graft copolymer nanohybrids was inferred to be approximately 88.4 wt%, and the relative amount of the MWCNTs-COOH was ca. 11.6 wt%.
 | ||
| Fig. 2 TGA curves (A) and XRD spectra (B) of (a) MWCNTs-COOH, (b) MWCNTs-g-HTPB, (c) MHPEC, (c′) HTPB-b-PAEFC, (d) MHPBC, and (d′) HTPB-b-PABFC. | ||
Fig. 2(B) shows the XRD patterns of the CNTs graft polymer nanohybrid composites. The original pristine MWCNTs-COOH produce a sharp characteristic diffraction pattern of the graphite structure at ca. 26.0° and a weak and wide diffraction at 42.7° ascribed to the graphite (002) and (100) reflections, respectively.29,31,32 After it was associated with HTPB, HTPB-b-PABFC and HTPB-b-PAEFC through ATRP, the nanohybrid composites possess almost exactly the same characteristic diffraction patterns as the MWCNTs-COOH. Consequently, the covering of the polymers does not affect the graphite structure of the MWCNTs-COOH. The diffraction peaks, however, were found to widen due to the entangling effect of the amorphous polymer molecular chains, which generally produce a broad scattering pattern. In particular, lower diffraction intensities for the MWCNTs-g-HTPB-b-PABFC emerged, presumably due to the grafting of more amorphous HTPB-b-PABFC onto the surface of the MWCNTs-COOH, as described in the TGA analysis. We therefore inferred that the MWCNTs-COOH still existed in their unique graphite structure, and the nanohybrid composites were successfully prepared.
To better confirm the chemical structure in Scheme 1, XPS was used to identify the surface elemental compositions and the chemical valent states of the as-prepared nanohybrid composites, as shown in Fig. 3 and S2 (ESI†). In the XPS survey spectra of the MWCNTs-COOH in Fig. 3(A), two photoemission peaks at binding energies (BEs) of 285.1 and 532.2 eV were detected, and they are related to the C 1s and O 1s signals.30 The atomic ratios were 96.0 and 4.0%, respectively. The high-resolution photoemission spectra of the C 1s signals in Fig. S2(a) (ESI†) show a dominant peak at 284.5 eV ascribed to sp2-hybridized CC in π-conjugated systems; a peak at 285.2 eV was assigned to the sp3-hybridized C–C bonds at defective locations and the tubular structure asymmetry; a peak at 290.2 eV was ascribed to the π–π* electronic transition due to a high degree of electron delocalization in the graphitic structure. In addition, three peaks appeared at about 286.2, 287.1 and 288.7 eV, which were attributed to the characteristic carbon atoms related to the C–O, carbonyl groups (CO) and carboxylic groups (–COOH) or O–CO, respectively. These findings are consistent with those reported in the literature.33,34 The O 1s core-level spectrum in Fig. S2(b) of the ESI† shows the existence of the oxidation peaks associated with the C–O and CO bonds at 532.3 and 533.5 eV, respectively. These types of oxygen-containing functional moieties on MWCNTs offers possibilities for surface chemical modifications.
 | ||
| Fig. 3 (A) Wide-survey XPS spectra of (a) MWCNTs-COOH, (b) MWCNTs-g-HTPB, (c) MWCNTs-g-HTPB-b-PABFC (MHPBC) and (d) MWCNTs-g-HTPB-b-PAEFC (MHPEC) nanohybrid composites; and (B) high-resolution XPS spectra of Fe 2p in typical MHPBC and MHPEC nanohybrid composites. | ||
After the HTPB oligomers were grafted onto the surface of the MWCNTs, no new elemental peaks appeared except the C 1s and O 1s signals, but the peak intensities and atomic ratios changed. The atomic ratios of C and O elements were 93.2 and 6.8%, respectively (Fig. 3A(b), and Table S1 in ESI†). Furthermore, in the C 1s and O 1s core-level spectra in Fig. S2(c) and (d) in the ESI,† new signals emerged at the BEs of about 287.6 and 531.9 eV, revealing the presence of chemically different C and O species attributed to the –C–O–H bonds of the terminal hydroxyl groups in the HTPB moieties. This differs from the O 1s and C 1s signals in the MWCNTs-COOH, implying the grafting of HTPB on the surface of the MWCNTs. For the MHPBC and MHPEC nanohybrid composites in Fig. 3A(c) and (d), new element signals assigned to the electrons emitted from the Fe 2p and Fe 3s levels at BEs of about 708.4–720.7 and 56.1 eV in addition to the C 1s and O 1s levels were observed. The emergence of the Fe signals gives evidence for the presence of ferrocene-containing moieties such as PABFC or/and PAEFC on the MWCNT surfaces. MHPEC possesses increased Fe and O atomic ratios but a decreased C amount in comparison with MHPBC, as summarized in Table S1 of the ESI,† which is consistent with their respective molecular structures. The Fe 2p core-level XPS spectra in Fig. 3(B) clearly show that the Fe 2p peaks are deconvoluted into a spin–orbit-coupled doublet with the BEs of ca. 705.3 and 717.9 eV, which is ascribed to the electrons from the Fe 2p3/2 and Fe 2p1/2 electronic levels, respectively, in agreement with those of the ferrocene derivatives.35,36 The C 1s core-level spectra in Fig. S2(e) and (g) of the ESI† display several different C species, and their BEs are approximately 284.5–284.6, 284.8, 285.2–285.3, 286.3, 287.0, 288.6–288.7 and 289.6–289.8 eV assigned to the CC, ferrocenyl carbons (C–Fe), C–C, C–O, (R–)CO, O–CO and π–π* electronic transition, respectively.36–38 The larger peak areas of the C–C signals than the CC signals are consistent with the incorporation of more C–C bonds in the ferrocene-containing blocks. The O 1s core-level spectra disclose the presence of two types of chemically different O species at the BEs of about 532.6–532.8 and 533.9–534.0 eV, which are attributed to the C–O and CO bonds from the ester groups, as shown in Fig. S2(f) and (h) in the ESI.† These results suggest that the ferrocene-containing copolymer moieties were grafted onto the surface of the MWCNTs-COOH, forming the MHPBC and MHPEC nanohybrid composites.
To investigate the morphologies of the prepared nanohybrid composites, SEM observations were made, as shown in Fig. 4. The pure MWCNTs-COOH showed a smooth and randomly entangled surface morphology in clusters. After the surface graft reaction with HTPB and HTPB-b-PABFC or HTPB-b-PABFC, the polymer layers were found to closely cover the surface of the MWCNTs or around the MWCNTs, forming an organic whole connected by the graft polymers. The tubes thickened at different levels, ranging from 3 to 15 nm, because the polymer layers with various chain lengths covered the MWCNTs surface. The encapsulation of these polymers on the surface of the MWCNTs was further observed by TEM, as displayed in Fig. 5. The crude MWCNTs-COOH had outer diameters between 10 and 30 nm, and the wall surface was smooth, without any detectable amorphous layers except for several blobs of amorphous carbon. The TEM images of the MWCNTs-g-HTPB-b-PAEFC and MWCNTs-g-HTPB-b-PABFC clearly show that the surface of the MWCNTs-COOH is covered with amorphous polymer layers with a slightly shallower color. The MWCNTs-COOH layers were thicker by 3–8 nm, and the tubular diameters increased up to 30–38 nm. Considering that the HTPB-b-PAEFC and HTPB-b-PABFC polymer layers are covalently linked to the MWCNTs-COOH, the polymers will be left on the surface of the MWCNTs after the washing operation is completed. Therefore, the HTPB-b-PAEFC and HTPB-b-PABFC copolymers are indeed chemically grafted onto the surface of the MWCNTs-COOH.
 | ||
| Fig. 4 FESEM morphologies of (a) MWCNTs-COOH, (b) MWCNTs-g-HTPB, (c) MHPBC and (d) MHPEC nanohybrids. | ||
 | ||
| Fig. 5 TEM images of (a) MWCNTs-COOH, (b) MWCNTs-g-HTPB, (c) MHPBC, and (d) MHPEC nanohybrids. | ||
3.2 Electrochemical characteristics
CV is a popular and important technique for the study of electrochemistry and has proven to be very useful in obtaining thermodynamic and kinetic information of electrochemical reactions such as the peak potentials and reaction rates.39 The electrochemical properties of the MWCNTs grafted polymer modified electrodes with different chain length spacers (ethyl and butyl groups) between the main chains and ferrocene groups were investigated, as depicted in Fig. 6(A). Clearly, the bare GCE and the modified GCE electrode coated with the MWCNTs-COOH failed to generate any obvious redox response. In contrast, the HTPB-b-PAEFC/GCE and HTPB-b-PABFC/GCE modified electrodes produced well-defined and quasi-reversible redox peak currents and small peak potential differences or peak separation ΔE (ΔEp = Ep,anodic − Ep,cathodic) lower than 115 mV, which was ascribed to the reversible Fe(III)/Fe(II) redox process.40 Particularly, the HTPB-b-PABFC/GCE sensor produced stronger redox peak currents than the HTPB-b-PAEFC/GCE one. This signifies that the HTPB-b-PABFC/GCE electrode accelerates the electron transfer rates more than HTPB-b-PAEFC/GCE, which is beneficial for the redox reaction between active substances. The MHPBC modified electrode was found to produce more obvious current responses than the HTPB-b-PABFC/GCE counterpart, presumably due to a synergetic effect induced by the two constituents, the ferrocene moieties and MWCNTs. Although the ΔE remains almost the same, ranging from 220 to 218 mV as the length of the spacers increases, the redox peak current significantly increased from the MHPBC to MHPEC electrodes, and the symmetry of the CV curves was enhanced. Therefore, the modified electrode with a longer spacer exhibited more reversible electrochemical properties. The possibility is that the longer spacer for the MHPBC modified electrode decreases the disturbance against the ferrocene moieties and facilitates electron transfer at the electrode interface.21,41,42 In fact, the electrical resistance measurements in Table S2 of the ESI† show that the long chain MHPBC modified electrode sensor has a higher electric conductivity than the short chain MHPEC one, greatly increasing the conduction of the electrons. Therefore, it was concluded that the MHPBC modified electrode with a longer spacer is anticipated to be able to fabricate an electrochemical sensor. | ||
| Fig. 6 (A) CVs of (a) bare GCE, and GCE coated with (b) MWCNTs, (c) HTPB-b-PAEFC, (d) MHPEC, (e) HTPB-b-PABFC and (f) MHPBC in acetonitrile with 0.1 M tetrabutylammoniumperchlorate (TBAP) at a scan rate of 50 mV s−1; and (B) CVs of the MHPBC modified electrodes in a 1 M H2SO4 solution at various scan rates. The scan rate is 20, 30, 40, 50, 60, 70, 80, 90, 100 and 120 mV s−1 in order of the arrows. The inset shows the dependence of the peak currents on the scan rates. | ||
Considering that the scan rate may affect the electroactivity of the nanohybrid materials or the conducting rate of the electrons,43 the effect of the scan rate on the CV profiles was examined, as shown in Fig. 6(B). It can be seen that with the increasing scan rates, the oxidation peak currents strengthened, and the oxidation peak potentials moved slightly in the positive direction. The peak currents were directly proportional to the scan rate in the range from 20 to 120 mV s−1, exhibiting a good linear relationship between the scan rates and peak currents and giving a well-defined linear equation Ip,a (10−5 A) = 0.0720ν (mV s−1) + 4.1587 (R2 = 0.9924), as illustrated in the inset of Fig. 6(B). The cathodic peak current (Ip,c) dependence on the scan rates had similar patterns, offering a linear equation of Ip,c (10−5 A) = −0.12027ν (mV s−1) − 4.0874 (R2 = 0.9910). These findings suggested that the oxidation and reduction were a diffusion-controlled electrode reaction process for the MHPBC sensor, and the electrode processes are quasi-reversible.44
3.3 Electrochemical detection of melamine and trichlorfon
In consideration of the excellent electrochemical responses of the MHPBC modified electrode sensor, it was expected to detect melamine and trichlorfon residues.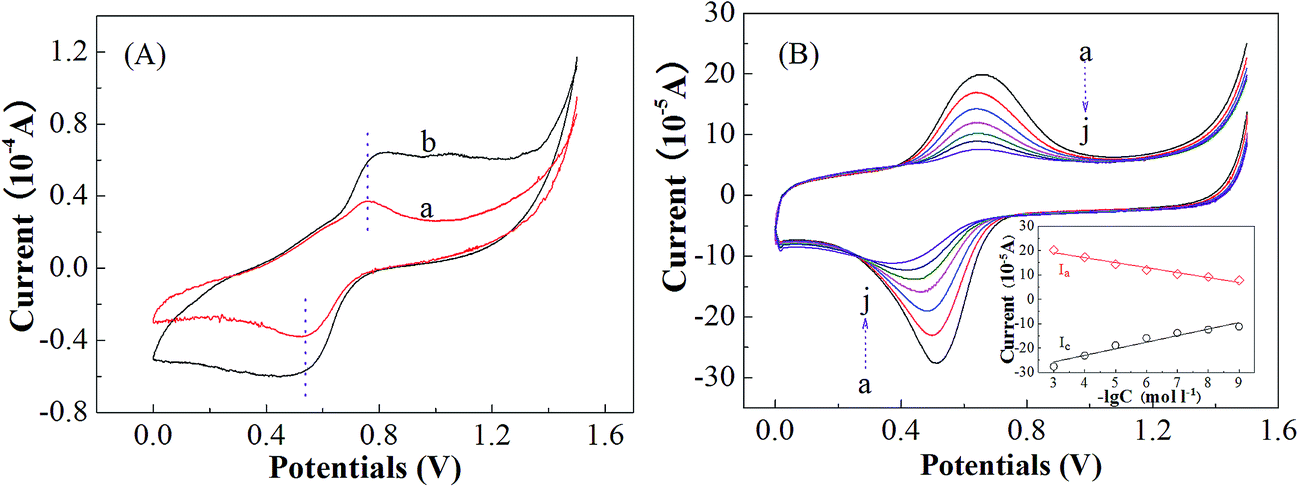 | ||
| Fig. 7 (A) CVs of the MHPBC modified electrodes: (a) in the absence of and (b) in the presence of 1 × 10−3 melamine in a 1 M H2SO4 solution at a scan rate of 50 mV s−1; (B) CVs of the MHPBC modified electrodes in a 1 M H2SO4 solution for various melamine concentrations expressed as mol L−1: 1 × 10−3, 1 × 10−4, 1 × 10−5, 1 × 10−6, 1 × 10−7, 1 × 10−8 and 1 × 10−9 at a scan rate of 50 mV s−1. The inset shows calibration curves for the determination of melamine. | ||
To detect trace melamine, calibration curves were prepared using CV, and the results are shown in Fig. 7(B). The redox peak currents were linearly dependent on the concentrations of melamine over a wide range of 1 × 10−3 to 1 × 10−9 M. The oxidation peak current (Ip,a) was proportional to the logarithm of the melamine concentration (CMel). The linear equation can be expressed as Ip,a (10−5 A) = 2.04lgCMel + 25.25, with the correlation coefficient 0.9632, whilst the reduction current (Ipc) and the logarithm of the melamine concentration show a good linear relationship in a concentration range from 1 × 10−3 to 1 × 10−9 M with the linear calibration equation Ip,c (10−5 A) = −2.69lgCMel − 33.82 (R2 = 0.9319). The limit of detection (LOD) is estimated using three times the noise, viz., signal/noise = 3, together with the following equation as reported elsewhere:45
| LOD = KSb/r | (1) |
To determine the feasibility and reproducibility of the proposed technique, the MHPBC sensor was independently examined under the same conditions by analyzing the residues of melamine in real milk. The Ip,a was determined to be 13.51 × 10−5 A (Fig. S4 of ESI†), and the concentration of melamine was calculated to be about 1.72 × 10−6 mol L−1 using the equation Ip,a (10−5 A) = 2.04lgCMel + 25.25, with a relative standard deviation (RSD) of 2.3% for 9 independent determinations, indicating that the method is feasible and repeatable. The proposed method is also simple and environmentally friendly because it needs no complex subsequent procedures like centrifugation and filtration. The durability or lifetime of the method was examined using the change in the peak currents with time, as shown in Fig. S5 of the ESI.† It was found that the recovery rate of the peak currents was 91.8 and 85.8% with an RSD of 3.98 and 9.98% after 7 and 30 days, respectively. In particular, the peak currents after the second day remained almost unchanged, indicating good stability.
In this study, all the experiments were carried out in a conventional electrochemical cell containing 10 mL of a 0.2 mol L−1 PBS buffer solution (pH = 7.0) with various concentrations of trichlorfon from 1.0 × 10−5 to 1.0 × 10−10 mol L−1 and from 5.0 × 10−12 to 3 × 10−13 mol L−1 at room temperature, as shown in Fig. 8. Under the chosen experimental conditions, the linear regression equations can be expressed as Ip,a (μA) = 0.26(−lgC) − 4.41 (R2 = 0.9899) and Ip,c (μA) = 0.096(−lgC) − 2.76 (R2 = 0.9941), respectively. The LOD was estimated to be approximately 3.5 × 10−8 mol L−1 (9.0 × 10−3 mg L−1), a significantly lower LOD in comparison with other techniques.14,47–49 It was therefore concluded that the prepared electrochemical sensor exhibits remarkable advantages with a high sensitivity, wide linear range and low LOD.
 | ||
| Fig. 8 DPV curves of MHPBC modified electrodes for the detection of trichlorfon with a concentration range (mol L−1): (A) 1 × 10−5, 1 × 10−7, 1 × 10−8, 1 × 10−9, and 1 × 10−10 corresponding to (a) to (e), and (B) 5 × 10−12, 3 × 10−12, 2 × 10−12, 1 × 10−12, 5 × 10−13, and 3 × 10−13 from (f) to (k), in PBS at pH 7.0 at a scan rate of 50 mV s−1. The insets show the linear relationship between the peak currents (Ip) and trichlorfon concentrations. | ||
To evaluate the applicability of the MHPBC sensor, pear, apple and cucumber samples immersed in trichlorfon were prepared, and the trichlorfon residues were detected by DPV under the chosen conditions. Each sample was measured five times and averaged, and the results are given in Table 1. It was seen that the MHPBC sensors gave similar data between the determined amount and the added amount of trichlorfon in pear, apple and cucumber samples with low RSDs of 0.7–3.6%, 0.8–8.1% and 0.6–6.3% and high recoveries of 82.0–101.5%, 93.5–112.4% and 90.0–96.2% for pear, apple and cucumber, respectively. Consequently, the developed MHPBC modified electrode sensor proved to be a promising and reliable tool for the rapid and simple determination of trichlorfon residues in real samples.
| Sample | Added (10−7 mol L−1) | Founda (10−7 mol L−1) | RSD (%) | Recovery (%) |
|---|---|---|---|---|
| a Represents the mean value of five measurements in this experiment. | ||||
| Pear | 0.4 | 0.41 | 3.6 | 101.5 |
| 2.0 | 1.68 | 2.3 | 82.0 | |
| 3.5 | 3.26 | 0.7 | 93.1 | |
| Apple | 0.4 | 0.37 | 6.1 | 93.5 |
| 2.0 | 1.77 | 8.1 | 88.5 | |
| 3.5 | 3.93 | 0.8 | 112.4 | |
| Cucumber | 0.4 | 0.38 | 6.3 | 95.0 |
| 2.0 | 1.81 | 0.9 | 90.5 | |
| 3.5 | 3.37 | 0.6 | 96.2 | |
4. Conclusion
In conclusion, we have demonstrated an efficient approach to prepare novel carbon nanotube grafted block copolymer nanohybrid composites, MHPBC and MHPEC, as confirmed by FTIR, Raman, XRD, XPS and TGA measurements. The MHPBC modified electrodes with longer side chains or spacers between the side ferrocene groups and main chains exhibited good electrochemical responses and quasi-reversibility because of the easier electron transfer at the electrode interface. The MHPBC electrochemical modified electrodes had good reproducibility, stability and a linear relationship over a wide concentration range. A significantly low detection limit of 3.5 × 10−8 mol L−1 allows us to detect trace melamine and trichlorfon residues in food or fruits. Therefore, the newly developed nanohybrid composites can potentially be fabricated into novel electrochemical sensors for applications in the inspections of food safety.Acknowledgements
This work is supported by the National Natural Science Foundation of China (grant 21572123, 21273142 and 21172138) and the Fundamental Research Funds for the Central Universities (GK201601003).References
- L. F. M. C. Aquino, A. C. O. Silva, M. Q. Freitas, T. L. Felicio, A. G. Cruz and C. A. Conte-Junior, Food Res. Int., 2014, 62, 233–237 CrossRef CAS
.
- J. L. Dorne, D. R. Doerge and M. Vandenbroeck, Toxicol. Appl. Pharmacol., 2013, 270, 218–229 CrossRef CAS PubMed
- M. S. Filigenzi, B. Puschner, L. S. Aston and R. H. Poppenga, J. Agric. Food Chem., 2008, 56, 7593–7599 CrossRef CAS PubMed
- R. Basanta, A. Nuñez, E. Lopez, M. Fernandez and F. Diaz-Fierros, Int. J. Environ. Stud., 2007, 48, 211–219 CrossRef
- J.-D. Yang, X. W. Sun, Z. J. Feng, D. F. Hao, M. Q. Wang, X. J. Zhao and C. D. Sun, Toxicol. Lett., 2011, 206, 306–313 CrossRef CAS PubMed
- L. Amendolaa, F. Botrèb, A. S. Carolloa, D. Longoa and L. Zoccolilloa, J. Anal. Chim. Acta., 2002, 461, 97–108 CrossRef
- R. C. Wei, R. Wang, Q. F. Zeng, M. Chen and T. Z. Liu, J. Chromatogr. Sci., 2009, 47, 581–584 CAS
- U. T. Sireli, H. Ekici, H. Y. Can and A. Karagoz, J. Dairy Sci., 2012, 95, 602–608 CrossRef PubMed
- J. Draher, V. Pound and T. M. Reddy, J. Chromatogr. A., 2014, 1373, 106–113 CrossRef CAS PubMed
- Q. Cao, H. Zhao, Y.-J. He, N. Ding and J. Wang, Anal. Chim. Acta, 2010, 675, 24–28 CrossRef CAS PubMed
- X. L. Luo, A. J. Killard and M. R. Smyth, Electroanalysis, 2006, 18, 1131–1134 CrossRef CAS
- R. N. Liang, R. M. Zhang and W. Qin, Sens. Actuators, B, 2009, 141, 544–550 CrossRef CAS
- K. L. Ai, Y. L. Liu and L. H. Lu, J. Am. Chem. Soc., 2009, 131, 9496–9497 CrossRef CAS PubMed
- R. Sundarmurugasan, M. B. Gumpu, B. L. Ramachandra, N. Nesakumar, S. Sethuraman, U. M. Krishnan and J. B. B. Rayappana, Sens. Actuators, B, 2016, 230, 306–313 CrossRef CAS
- V. Bellas and M. Rehahn, Angew. Chem., Int. Ed., 2007, 46, 5082–5104 CrossRef CAS PubMed
- G. R. Whittell and I. Manners, Adv. Mater., 2007, 19, 3439–3468 CrossRef CAS
- Y. J. Ma, W. F. Dong, M. A. Hempenius, H. Möhwald and G. J. Vancso, Nat. Mater., 2006, 5, 724–729 CrossRef CAS PubMed
- B. Y. Kim, E. L. Ratcliff, N. R. Armstrong, T. Kowalewski and J. Pyun, Langmuir, 2010, 26, 2083–2092 CrossRef CAS PubMed
- J. Elbert, M. Gallei, C. Ruettiger, A. Brunsen, H. Didzoleit, B. Stuehn and M. Rehahn, Organometallics, 2013, 32, 5873–5878 CrossRef CAS
- J. Isaksson, C. Tengstedt, M. Fahlman, N. Robinson and M. Berggren, Adv. Mater., 2004, 16, 316–320 CrossRef CAS
- P. Li, H. Kang and N. Che, Polym. Int., 2015, 64, 1015–1022 CrossRef CAS
- A. Q. Shi, J. Wang, X. W. Han, X. A. Fang and Y. Z. Zhang, Sens. Actuators, B, 2014, 200, 206–212 CrossRef CAS
- C. Y. Neo, N. K. Gopalan and J. Y. Ouyang, J. Mater. Chem. A, 2014, 2, 9226–9235 CAS
- A. T. Lawal, Mater. Res. Bull., 2016, 73, 308–350 CrossRef CAS
- H. Kong, C. Gao and D. Yan, J. Am. Chem. Soc., 2004, 126, 412–413 CrossRef CAS PubMed
- B. Scheibe, E. Borowiak-Palen and R. J. Kalenczuk, Mater. Charact., 2010, 61, 185–191 CrossRef CAS
- L. Stobinski, B. lesiak, L. K. Övér, J. Tóth, S. Biniak, G. Trykowski and J. Judek, J. Alloys Compd., 2010, 501, 77–84 CrossRef CAS
- F. J. Xiao and Y. J. Luo, Adv. Mater. Res., 2011, 197, 1225–1230 CrossRef
- H. X. Wu, R. Tong, X. Q. Qiu, H. F. Yang, Y. H. Lin, R. F. Cai and S. X. Qian, Carbon, 2007, 45, 152–159 CrossRef CAS
- A. Tanver, M. H. Huang and Y. J. Luo, RSC Adv., 2016, 6, 49101–49112 RSC
- T. A. Saleh, S. Agarwal and V. K. Gupta, Appl. Catal., B, 2011, 106, 46–53 CAS
- T. A. Saleh and V. K. Gupta, J. Colloid Interface Sci., 2012, 371, 101–106 CrossRef CAS PubMed
- T. I. T. Okpalugo, P. Papakonstantinou, H. Murphy, J. McLaughlin and N. M. D. Brown, Carbon, 2005, 43, 153–161 CrossRef CAS
- W. M. da Silva, H. Ribeiro, J. C. Neves, A. R. Sousa and G. G. Silva, J. Appl. Polym. Sci., 2015, 132, 42587 Search PubMed
- E. J. Lawrence, G. G. Wildgoose, L. Aldous, Y. A. Wu, J. H. Warner, R. G. Compton and P. D. McNaughter, Chem. Mater., 2011, 23, 3740–3751 CrossRef CAS
- Y. Peng, J. E. Lu, C. P. Deming, L. Chen, N. Wang, E. Y. Hirata and S. Chen, Electrochim. Acta, 2016, 211, 704–710 CrossRef CAS
- L. P. M. De Leo, E. de la Llave, D. Scherlis and F. J. Williams, J. Chem. Phys., 2013, 138, 114707 CrossRef PubMed
- C. Nietzold, P. M. Dietrich, A. Lippitz, U. Panne and W. E. S. Unger, Surf. Interface Anal., 2016, 48, 606–610 CrossRef CAS
- M. X. Zhang and J. B. Hu, Electroanalysis, 2013, 25, 780–786 CrossRef CAS
- M. Senel, E. Cevik and M. F. Abasiyanik, Sens. Actuators, B, 2010, 145, 444–450 CrossRef CAS
- D. Nkosi, J. Pillay, K. I. Ozoemena, K. Nounehcd and M. Oyamac, Phys. Chem. Chem. Phys., 2010, 12, 604–613 RSC
- Y. W. Pei, J. Travas-Sejdic and D. E. Williams, Langmuir, 2012, 28, 8072–8083 CrossRef CAS PubMed
- J. L. Gong, F. C. Gong, G. M. Zeng, G. L. Shen and R. Q. Yu, Talanta, 2003, 61, 447–453 CrossRef CAS PubMed
- H. R. Zare, M. Namazian and M. L. Coote, Electrochim. Acta, 2009, 54, 5353–5357 CrossRef CAS
- C. M. Yu, W. Y. Ji, Y. D. Wang, N. Bao and H. Y. Gu, Nanotechnology, 2013, 24, 115502–115510 CrossRef PubMed
- S. Palanisamy, S. Sakthinathan, S. M. Chen and X. H. Liu, Carbohydr. Polym., 2016, 135, 267–273 CrossRef CAS PubMed
- L. Hem, S. Khay, J. H. Choi, E. D. Morgan, A. M. A. El-Aty and J. H. Shim, Toxicol. Res., 2010, 26, 149–155 CrossRef CAS PubMed
- K. Reybier, S. Zairi, N. Jaffrezic-Renault and B. Fahys, Talanta, 2002, 56, 1015–1020 CrossRef CAS PubMed
- M. Ye, Y. Yu, X. He, L. Jiang and C. Wu, J. Shanghai Ocean Univ., 2010, 19, 189–194 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra26436k |
| This journal is © The Royal Society of Chemistry 2017 |