
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mn(OAc)3 catalyzed intermolecular oxidative peroxycyclization of naphthoquinone†
Alex Meye Biyogoa,
Christophe Curtia,
Hussein El-Kashefb,
Omar Khoumeria,
Thierry Termea and
Patrice Vanelle*a
aAix-Marseille Université, CNRS, Institut de Chimie Radicalaire ICR, UMR 7273, Laboratoire de Pharmaco-Chimie Radicalaire, Faculté de Pharmacie, 27 boulevard Jean Moulin CS 30064, Marseille Cedex 05, 13385, France. E-mail: patrice.vanelle@univ-amu.fr
bChemistry Department, Faculty of Science, Assiut University, Egypt
First published on 22nd December 2016
Abstract
Manganese(III) acetate-mediated peroxycyclization between 2-hydroxy-3-methylnaphthoquinone and various alkenes was performed to obtain dihydronaphtho[2,3-c][1,2]dioxine-5,10(3H,10aH)-diones. The reactivity of symmetrical or unsymmetrical 1,1-disubstituted alkenes and monosubstituted alkenes allowed the synthesis of more than 50 original molecules. Focusing on the excellent reactivity of 2-hydroxy-3-methylnaphthoquinone, we describe the first example of Mn(OAc)3 reactivity with nitro-substituted alkenes. The scope, limitations and stereochemistry of the products synthesized are discussed. Starting from monosubstituted alkenes, the instability of a pair of diastereoisomers was observed, leading to ring opening.
Introduction
Manganese(III) acetate is a useful tool in organic chemistry, but remains underestimated in medicinal chemistry. Over the past four decades, the literature has contained accounts of its applications in C–C bond-forming reactions starting from various substrates.1–3 Among these substrates, the reactivity of quinones was studied under anaerobic conditions with stoichiometric quantities of Mn(OAc)3.4Mn(OAc)3 under aerobic conditions was also described as an efficient catalytic system, leading to C–C bond formation and oxygen insertion via a radical pathway, yielding 1,2-dioxane derivatives.5–7
Other synthetic pathways to heterocyclic peroxides were described in a recent review.8 They are based on three key reagents: oxygen, ozone and hydrogen peroxide. In particular, synthesis of 1,2-dioxanes can be realized with oxygen. Such reactions were described with singlet oxygen,9 with thiols by oxidation of 1,5-dienes10,11 or by the Isayama–Mukaiyama method.12 Ozonolysis of double bonds also afforded hydroperoxides, which can be then cyclized.13 The Kobayashi method involving an urea–hydrogen peroxide complex allowed the peroxidation of carbonyl group of insaturated ketones followed by a cyclization.14,15
Saturated heterocyclic endoperoxides, such as 1,2-dioxane, were described as powerful antiinfectious compounds, with applications against trypanosomiasis,16,17 leishmaniasis,16 helminthiasis,18 and trichomoniasis.19 Despite these applications, heterocyclic endoperoxides are mainly known for being potent antimalarial compounds. In addition to the well-known commercial drug artemisinin,20 trioxolanes arterolane and artefenomel,21 a wide variety of 1,2-dioxanes showed potential antimalarial activity.22–26 Quinone derivatives have important biological applications, mainly as anticancer27 and antiparasitic28 compounds. The 2-hydroxy-3-alkylnaphthoquinone lapachol derivatives antimalarial activity29 encouraged us to seek an easily reproducible procedure to obtain chimeric 2-hydroxy-3-alkylnaphthoquinone/endoperoxide molecules.
Expanding on our previous work on the synthesis of antiparasitic compound30–33 and Mn(OAc)3 reactivity,34–37 we present herein the first example of this original reactivity on 2-hydroxy-3-methylnaphthoquinone with various alkenes, under Mn(OAc)3 catalytic conditions.
Results and discussion
First, best reaction conditions were studied with 1,1-diphenylethylene (Scheme 1). According to previous Mn(OAc)3 reactivity studies, good yields can be obtained with this alkene, as the intermediate benzylic tertiary radical is highly stabilized.38,39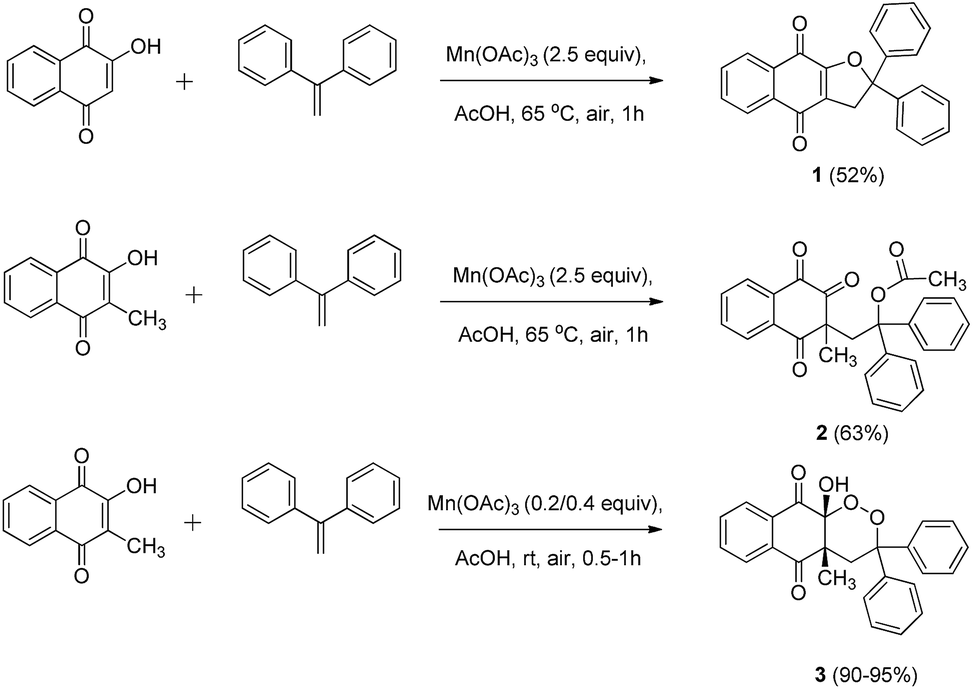 | ||
| Scheme 1 Mn(OAc)3 mediated cyclization of 2-hydroxynaphthoquinones with 1,1-diphenylethylene under aerobic conditions. | ||
2-Hydroxynaphthoquinones were chosen both without and with a methyl in position 3. Results are summarized in Table 1.
| Entrya | R | Mn(OAc)3 (equiv.) | Temperature (time) | Product (yield) |
|---|---|---|---|---|
| a Reaction conditions: 2-hydroxynaphthoquinone (1 equiv.), 1,1-diphenylethylene (0.5 equiv.).b Isolated yields based on number of alkenes equiv.c Reaction conditions: 2-hydroxynaphthoquinone (1 equiv.), 1,1-diphenylethylene (1.1 equiv.).d Isolated yields based on number of naphthoquinone equiv.e Reaction under an oxygen atmosphere. | ||||
| 1 | –H | 2.5 | 65 °C (1 h) | 1 (52%)b |
| 2 | –H | 2.5 | RT (3 h) | — |
| 3 | –H | 0.4 | RT (24 h) | — |
| 4 | –H | 0.4 | 65 °C (1 h) | 1 (30%)b |
| 5 | –CH3 | 2.5 | 65 °C | 2 (63%)b |
| 6 | –CH3 | 0.2 | RT (1 h) | 3 (95%)b |
| 7 | –CH3 | 0.4 | RT (0.5 h) | 3 (90%)b |
| 8c | –CH3 | 0.2 | RT (1 h) | 3 (90%)d |
| 9e | –CH3 | 0.2 | RT (1 h) | 3 (82%)b |
Starting from 2-hydroxynaphthoquinone in the presence of Mn(OAc)3 at 65 °C, dihydrofuronaphthoquinone 1 was obtained in moderate yields (entry 1) according to a previously reported mechanism.4 We obtained lower yields (52%) than those reported with similar 1,1-diaromatic alkenes, perhaps due to the presence of oxygen under our conditions. Catalytic quantities of Mn(OAc)3 only decreased yields without any oxygen insertion (entry 4), and lower temperature (RT) left the unchanged starting material (entries 2 and 3).
With 2-hydroxy-3-methylnaphthoquinone in the presence of 2.5 equiv. of Mn(OAc)3 at 65 °C, product 2 was formed, without traces of 1,2-dioxane ring formation (entry 5). This structure was obtained via the same mechanism observed for product 1, but the presence of a methyl group in position 3 did not allow cyclization. Instead, acetate reacted with the intermediate carbocation to form product 2. Reactivity allowing the purification of product 3 in very good yields was finally observed with catalytic quantities of Mn(OAc)3 at room temperature in only 1 h (entry 6). When Mn(OAc)3 quantities were doubled to 0.4 equiv., time reaction was decreased to 30 min with a moderate yield decrease (entry 7). When alkene quantities were increased to 1.1 equiv., yields were also slightly decreased (entry 8). Moreover, lower yields were observed when reaction was launched under an oxygen atmosphere (entry 9), as previously reported in similar Mn(OAc)3 catalyzed reactivity.40,41
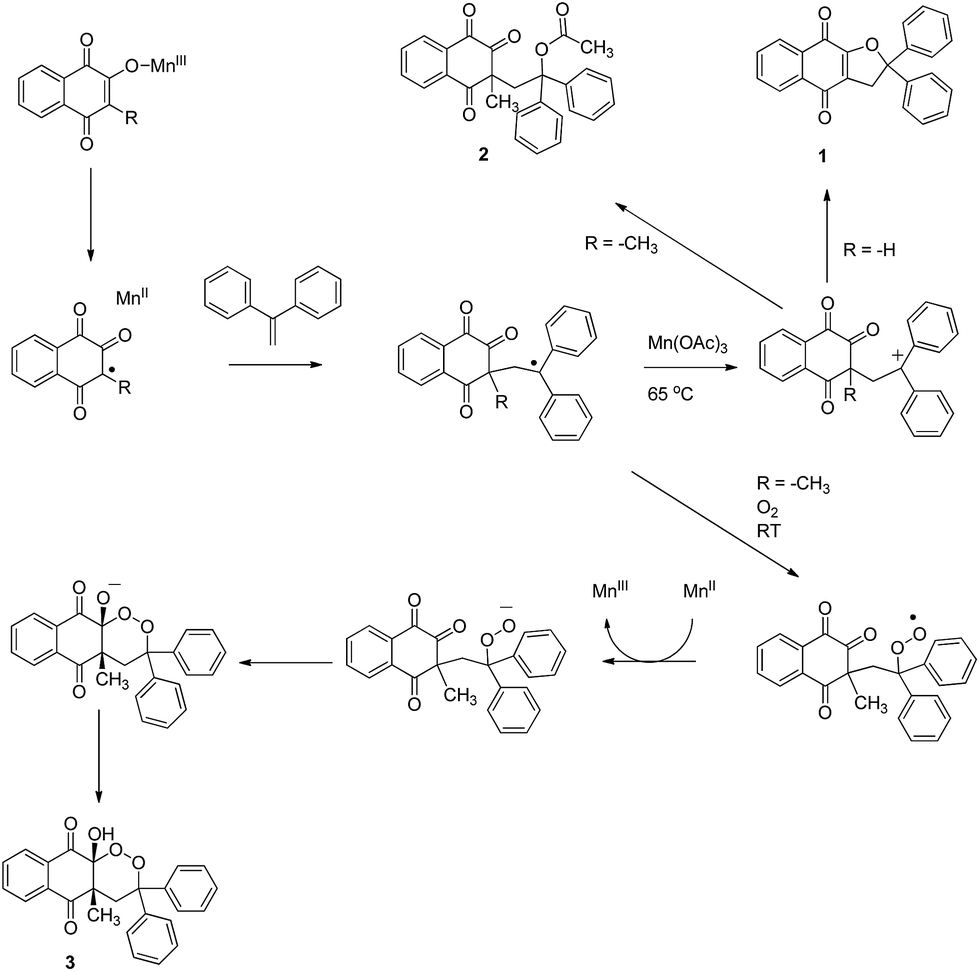
Temperature was previously reported to influence the Mn(OAc)3 reactivity.5,38,39 At high temperature, under anaerobic conditions, a dihydrofuran ring is obtained, and at room temperature under aerobic conditions, oxygen trapping led to a 1,2-dioxane ring. Catalytic quantities of Mn(OAc)3 are sufficient for this mechanism, summarized in Scheme 2.
 | ||
| Scheme 2 Naphthoquinone/Mn(OAc)3 proposed reactivity mechanism. | ||
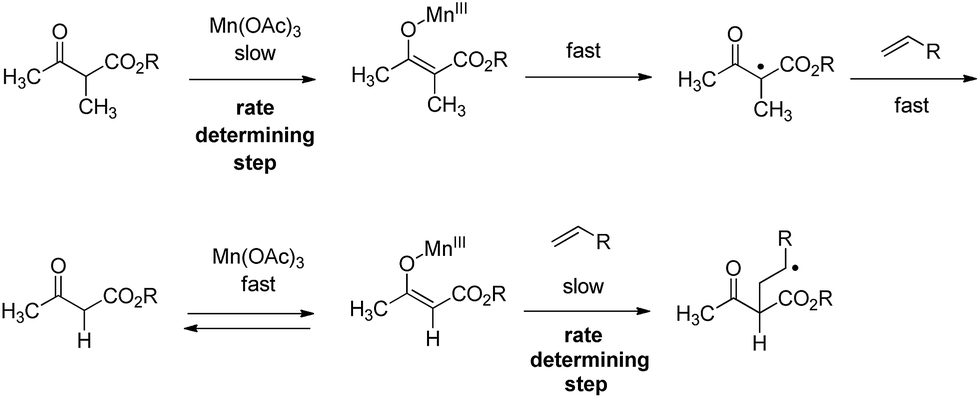
Methyl group substitution in position 3 of naphthoquinone highly influenced Mn(OAc)3 reactivity, appearing to be an important parameter leading to the formation of the 1,2-dioxane ring of product 3. The influence of the alkyl groups on Mn(OAc)3 reactivity was previously studied, for example with α-alkyl-β-keto esters versus α-unsubstituted-β-ketoesters as summarized in Scheme 3.42
 | ||
| Scheme 3 Mn(OAc)3 reactivity with β-ketoesters.42 | ||
2-Hydroxynaphthoquinones are already enolized and the Mn(III) enolate should form readily. Moreover, a methyl group in position 3 should facilitate oxidation, forming a stabilized radical. Without this methyl group, 2-hydroxynaphthoquinone reactivity would probably not be sufficient to initiate the mechanism with catalytic quantities of Mn(OAc)3 and/or room temperature.
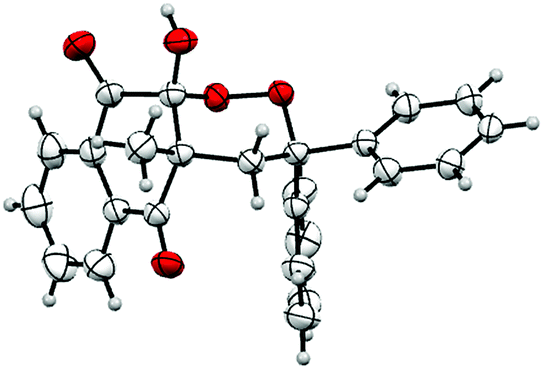
The structure of product 3 was confirmed by X-ray crystallography and an ORTEP view is reported to Fig. 1. This crystallographic analysis confirmed that oxidative cyclization, theoretically possible on carbon 2 or on carbon 4 of naphthoquinones, only took place on carbon 2, probably because of its greater electrophilic nature.
 | ||
| Fig. 1 ORTEP view of product 3. | ||
Crystallographic analysis also showed that 10-hydroxyl and 4-methyl groups of product 3 were cis-substituted. A hydroxyl group occupies the axial position, due to the anomeric effect.43,44 During the intramolecular cyclization (Scheme 2), nucleophilic addition on carbonyl only occurs on the opposite side of the methyl group, yielding to a mixture of two cis-enantiomers.
The reaction was extended to various substrates, starting from symmetrical alkenes (Scheme 4). Results are summarized in Table 2.
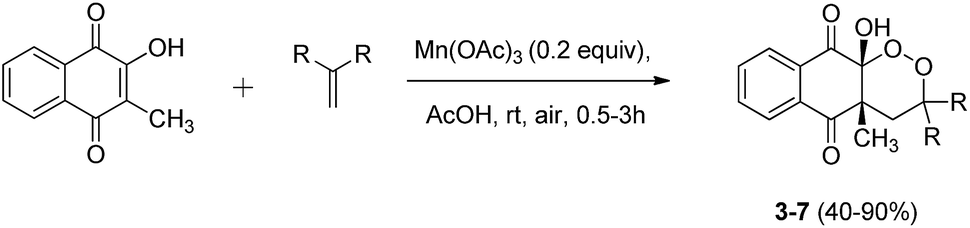 | ||
| Scheme 4 Mn(OAc)3 catalyzed cyclization of 2-hydroxy-3-methylnaphthoquinone and symmetrical alkenes under aerobic conditions. | ||
| Entry | R | Product (yield)a |
|---|---|---|
| a Isolated yields based on number of alkenes equiv.b Reaction conditions: 2-hydroxy-3-methylnaphthoquinone (1 equiv.), alkene (0.5 equiv.).c Reaction conditions: 2-hydroxy-3-methylnaphthoquinone (1 equiv.), alkene (1 equiv.). | ||
| 1b | –Ph | 3 (90%) |
| 2b | –4-Cl-Ph | 4 (86%) |
| 3b | –4-F-Ph | 5 (79%) |
| 4c | –CH2–CH3 | 6 (54%) |
| 5c | –(CH2)5– | 7 (40%) |
cis-Substitution was observed for every product obtained from aromatic and aliphatic symmetrical alkenes. Yields obtained ranged from excellent with aromatic alkenes (entries 1–3), even when they carried electron-withdrawing substituents such as –Cl or –F, to moderate (entries 4 and 5) with aliphatic alkenes. This can be explained by the stability of intermediate radical formed during the reaction just after the C–C bond formation. For each type of alkene, a tertiary radical was formed, sufficiently stabilized to react with oxygen, but for aromatic alkenes the stability of this tertiary benzylic radical was increased by resonance.
From these promising results, we extended this reactivity to 1,1-disubstituted unsymmetrical alkenes, in order to generate a new asymmetric center (Scheme 5). Results are summarized in Table 3.
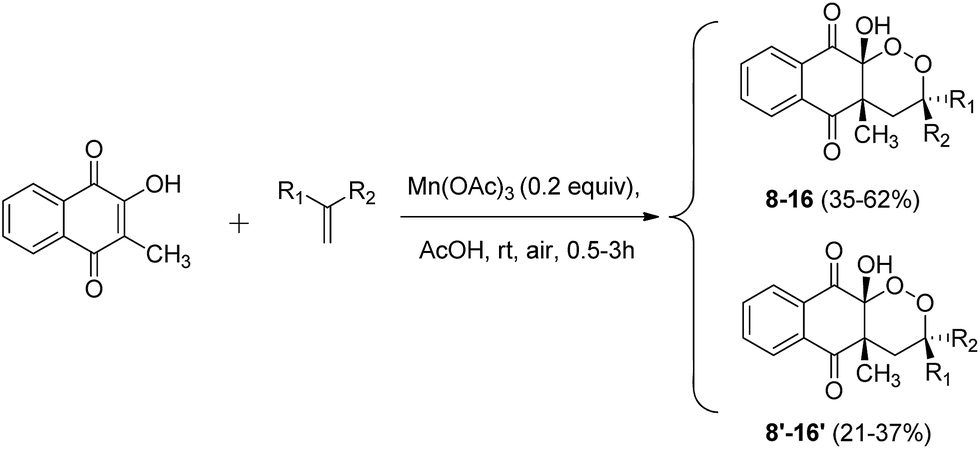 | ||
| Scheme 5 Mn(OAc)3 catalyzed cyclization of 2-hydroxy-3-methylnaphthoquinone and unsymmetrical alkenes under aerobic conditions. | ||
| Entrya | R1 | R2 | Products (yields; diastereoisomeric ratio)b |
|---|---|---|---|
| a Reaction conditions: 2-hydroxy-3-methylnaphthoquinone (1 equiv.), alkene (0.5 equiv.).b Isolated yields based on number of alkenes equiv. | |||
| 1 | –Ph | –4-NO2-Ph | 8/8′ (83%; 75/25) |
| 2 | –CH3 | –Ph | 9/9′ (83%; 55/45) |
| 3 | –CH3 | –4-CH3-Ph | 10/10′ (82%; 60/40) |
| 4 | –CH3 | –3-OCH3-Ph | 11/11′ (59%; 60/40) |
| 5 | –CH3 | –4-Cl-Ph | 12/12′ (80%; 60/40) |
| 6 | –CH3 | –2,5-diCl-Ph | 13/13′ (63%; 60/40) |
| 7 | –CH3 | –4-CN-Ph | 14/14′ (70%; 60/40) |
| 8 | –CH3 | –4-NO2-Ph | 15/15′ (75%; 60/40) |
| 9 | –CH3 | –3-NO2-Ph | 16/16′ (58%; 60/40) |
| 10 | –CH3 | –2-NO2-Ph | — |
As expected, the reactivity of 2-hydroxy-3-methylnaphthoquinone and unsymmetrical alkenes led to a mixture of diastereoisomers. Two fractions were easily separable by column chromatography. The first fraction corresponded to products with the R2 and 4-methyl group cis-substituted (8–16), and the second fraction to products with the R2 and 4-methyl group trans-substituted (8′–16′).
These results are evidence of the excellent reactivity of 2-hydroxy-3-methylnaphthoquinone for Mn(OAc)3 C–C bond forming reactions, even with deactivated alkenes such as nitrile- (entry 7) or nitro- (entries 1, 8 and 9) substituted alkenes. Mn(OAc)3 reactivity of the double bond decreases with aromatic substitution of electron-withdrawing substituents. To our knowledge, this is the first example of a C–C bond forming reaction mediated with Mn(OAc)3 and nitro-substituted alkenes. We did not observe reactivity for 1-nitro-2-(prop-1-en-2-yl)benzene (entry 10), probably because of steric hindrance due to the proximity of nitro and methyl groups with a double bond. Methoxy-substituted alkenes allowed the synthesis of products 12/12′ (entry 4) in lower yields than other alkenes. This phenomenon was already observed with methoxy-substituted alkenes, and an interaction between oxygen and Mn(OAc)3 was hypothesized.37 Under conditions involving stoichiometric quantities of manganese, this limitation dealt by increasing manganese equivalents, but it remains problematic with catalytic quantities.
The structure of products 15/15′ was confirmed by X-ray crystallography and an ORTEP view is reported in Fig. 2.
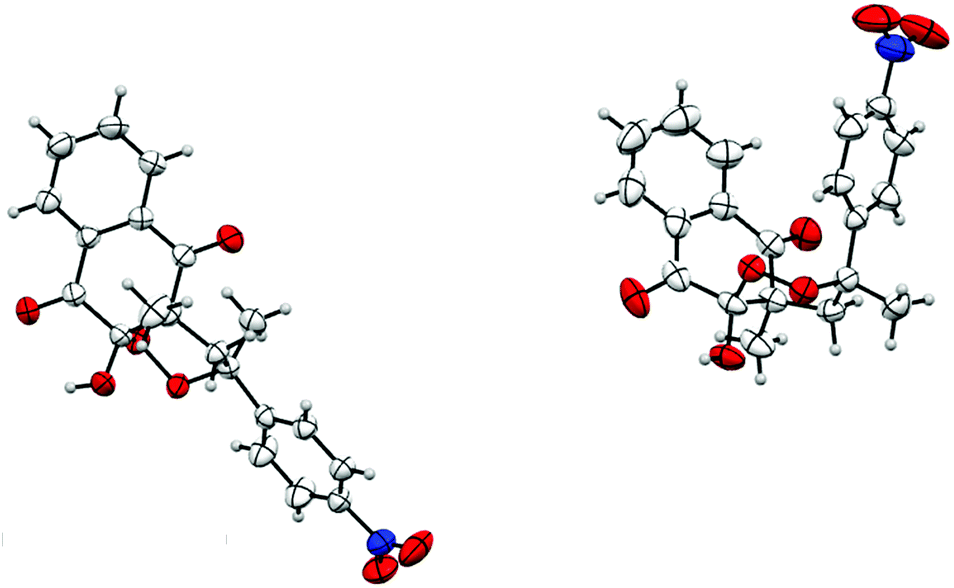 | ||
| Fig. 2 ORTEP view of products 15/15′. | ||
As previously described for 1,1-disubstituted symmetrical alkenes, 10-hydroxyl and 4-methyl groups were cis-substituted. Moreover, for product 15, the R2 substituent and 4-methyl group were also cis-substituted, while they were trans-substituted for product 15′. We noticed a slight stereoselectivity, with a diastereoisomeric excess for products cis-substituted with 10-hydroxyl and R2 substituents. Similar selectivity was previously described and discussed for radical induced peroxycyclization.45,46
Next, given the excellent reactivity of 2-hydroxy-3-methylnaphthoquinone, we extended reactivity to monosubstituted terminal alkenes (Scheme 6). Several vinyl derivatives reacted with naphthoquinone in moderate to excellent yields. Results are summarized in Table 4.
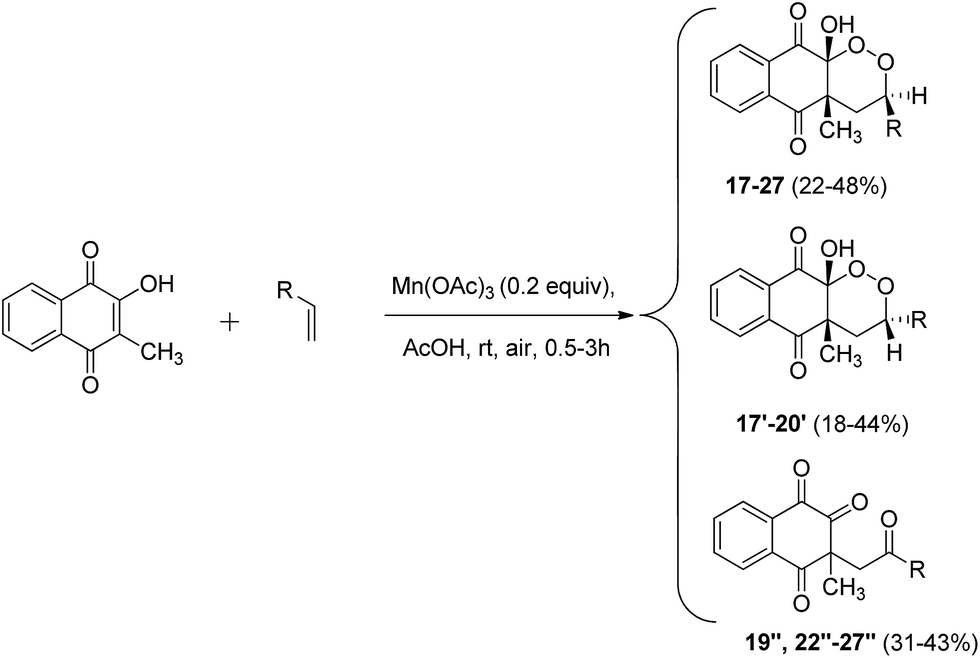 | ||
| Scheme 6 Mn(OAc)3 catalyzed cyclization of 2-hydroxy-3-methylnaphthoquinone and monosubstituted alkenes under aerobic conditions. | ||
| Entrya | R | Diastereoisomer products (yield; diastereoisomeric ratio)b | Ring opening products (yield)b |
|---|---|---|---|
| a Reaction conditions: 2-hydroxy-3-methylnaphthoquinone (1 equiv.), alkene (0.5 equiv.).b Isolated yields based on number of alkenes equiv.c Reaction was left under stirring for 2 hours after starting material consumption. | |||
| 1 | –Ph | 17/17′ (88%; 50/50) | |
| 2 | –4-CH3-Ph | 18/18′ (80%; 60/40) | |
| 3 | –3-CH3-Ph | 19/19′ (58%; 60/40) | |
| 4c | –3-CH3-Ph | 19 (23%) | 19′′ (31%) |
| 5 | –2-CH3-Ph | 20/20′ (52%; 65/35) | |
| 6 | –4-Cl-Ph | 21 (28%) | 21′′ (40%) |
| 7 | –4-F-Ph | 22 (23%) | 22′′ (34%) |
| 8 | –4-CN-Ph | 23 (22%) | 23′′ (33%) |
| 9 | –3-Br-Ph | 24 (25%) | 24′′ (37%) |
| 10 | –4-NO2-Ph | 25 (29%) | 25′′ (43%) |
| 11 | –3-NO2-Ph | 26 (24%) | 26′′ (35%) |
| 12 | –2-NO2-Ph | 27 (22%) | 27′′ (32%) |
Starting from styrene or monosubstituted alkenes bearing electron-donating substituents, diastereoisomer products were synthesized in good yields in a similar way to that described previously. Surprisingly, stereoselectivity was not observed for styrene (entry 1). para-Substitution for alkenes led to better yields than meta- or ortho-substitution (entries 2, 3 and 5). When alkenes bearing electron-withdrawing substituents were used as starting material, only one diastereoisomeric product was purified and a major product identified as a ring opening structure (entries 6–12). To confirm whether this new product came from a secondary reaction or from the degradation of peroxycyclized products, a reaction that only yielded to diastereoisomeric products was left under stirring for 3 hours (entry 4). Similar formation of ring opening product 19′′ was observed, clearly indicating that diastereoisomer 19′ was totally cleaved.
Decomposition of a 1,2-dioxane ring to afford γ-diketones was previously described.47,48 Thus, the formation of the ring opening structure could be explained by the acid-catalyzed rearrangement of the less stable diastereoisomer (Scheme 7).
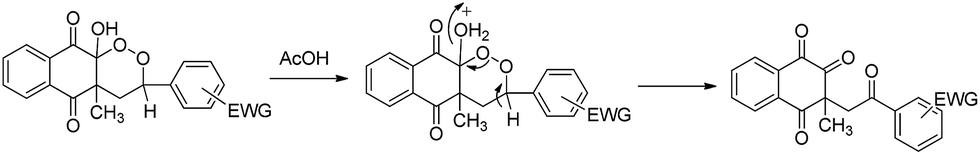 | ||
| Scheme 7 Acid-catalyzed decomposition of peroxycyclized diastereoisomer. | ||
Electron-withdrawing substituents on the benzene ring decrease the stability of the 1,2-dioxane ring by their influence on O–O bond stability and on benzylic hydrogen acidity. Therefore, spontaneous decomposition of peroxycyclized products could occur.
The structure of products 27/27′′ was confirmed by X-ray crystallography and an ORTEP view is shown in Fig. 3.
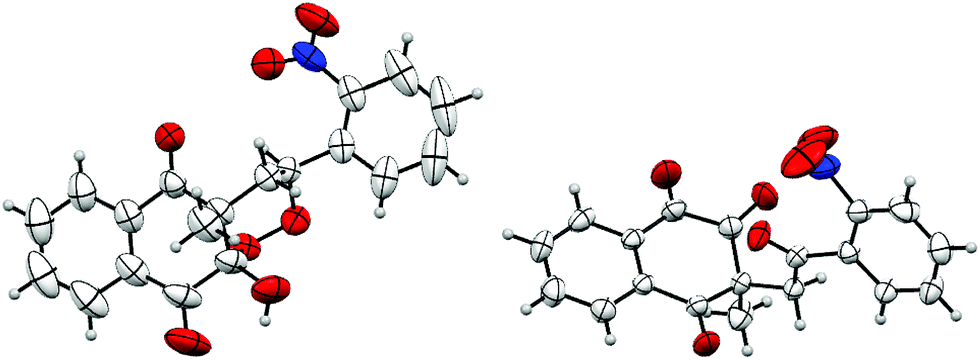 | ||
| Fig. 3 ORTEP view of products 27/27′′. | ||
Peroxycyclized products trans-substituted with 10-hydroxyl and R substituent appeared to be less stable than cis-substituted ones. This difference in stability may also explain the stereoselectivity observed during cyclization. Moreover, starting from monosubstituted alkenes, when R bears an electron-withdrawing substituent, trans-substituted diastereoisomers were probably spontaneously cleaved very close to their formation. On the other hand, if R is substituted by an electron-donating group, the increased reaction time could lead to the same decomposition on trans-substituted diastereoisomers.
Conclusions
A new protocol of manganese(III) acetate catalyzed peroxycyclization of 2-hydroxy-3-methylnaphthoquinone was established and extended to a wide variety of alkenes. The high reactivity of quinones allowed deactivated alkenes to react with good yields. Thus, we report herein the first example of nitro-substituted alkene Mn(OAc)3 radical reactivity. Symmetrical or unsymmetrical 1,1-disubstituted alkenes and monosubstituted alkenes allowed the synthesis of 26 original molecules as a mixture of diastereoisomers or enantiomers. Differences in stability between diastereoisomers could explain stereoselectivity. Moreover, reactivity of monosubstituted alkenes bearing an electron-withdrawing group led to ring opening of the less stable diastereoisomers.Experimental section
Typical procedure for the synthesis of 3
A solution of 2-hydroxy-3-methyl-1,4-naphthoquinone (1.06 mmol, 200 mg, 1 equiv.) in 25 mL of glacial acetic acid was stirred at room temperature in an open vessel. 1,1-Diphenylethylene (0.53 mmol, 96 mg, 0.5 equiv.) and manganese(III) acetate dihydrate (0.21 mmol, 57 mg, 0.2 equiv.) were added and the reaction was controlled by TLC until consumption of alkene. After 1 h, the reaction mixture was poured into 60 mL of cold water and extracted with dichloromethane (3 × 20 mL). The organic extracts were collected and dried (Na2SO4). Solvent was evaporated under reduced pressure and crude product was purified by column chromatography (CH2Cl2/petroleum ether, 70/30), and the product obtained was recrystallized to afford product 3 (202 mg, 95%) as a white solid, mp = 167–168 °C (petroleum ether/CH2Cl2, 9/1). 1H NMR (250 MHz, CDCl3): δ = 7.96–7.99 (m, 1H, Ar-H), 7.53–7.77 (m, 3H, Ar-H), 7.23–7.40 (m, 7H, Ar-H), 7.11–7.14 (m, 3H, Ar-H), 5.04 (s, 1H, OH), 3.68 (d, J = 13.6 Hz, 1H, CH), 2.88 (d, J = 13.6 Hz, 1H, CH), 1.25 (s, 3H, CH3). 13C NMR (62.5 MHz, CDCl3): δ = 195.5 (C), 190.5 (C), 142.8 (C), 140.4 (C), 135.6 (CH), 134.0 (CH), 133.9 (C), 131.1 (C), 128.4 (2CH), 128.3 (CH), 127.7 (2CH), 127.5 (2CH), 127.4 (CH), 126.8 (CH), 126.7 (CH), 126.4 (2CH), 98.8 (C), 86.2 (C), 49.0 (C), 37.7 (CH2), 23.8 (CH3). HRMS (ESI): m/z [M + Na]+ calcd for C25H20O5: 423.1203; found: 423.1204.Acknowledgements
This work was supported by the CNRS and Aix-Marseille University. The authors thank V. Remusat for the NMR spectra recording and V. Monnier for the mass spectra recording.Notes and references
- A. S. Demir and M. Emrullahoglu, Curr. Org. Synth., 2007, 4, 223 CrossRef , and references therein.
- B. B. Snider, Chem. Rev., 1996, 96, 339 CrossRef CAS PubMed , and references therein.
- G. G. Melikyan, Synthesis, 1993, 9, 833 CrossRef , and references therein.
- O. Alagöz, M. Yilmaz, A. T. Pekel, C. Graiff and R. Maggi, RSC Adv., 2014, 4, 14644 RSC.
- S. Tategami, T. Yamada, H. Nishino, J. D. Korp and K. Kurosawa, Tetrahedron Lett., 1990, 31, 6371 CrossRef CAS.
- R. Kumabe, H. Nishino, M. Yasutake, V. H. Nguyen and K. Kurosawa, Tetrahedron Lett., 2001, 42, 69 CrossRef CAS.
- R. Kumabe and H. Nishino, Tetrahedron Lett., 2004, 45, 703 CrossRef CAS.
- A. O. Terent'ev, D. A. Borisov, V. V. Vil' and V. M. Dembitsky, Beilstein J. Org. Chem., 2014, 10, 34 CrossRef PubMed.
- J. J. La Clair, Angew. Chem., Int. Ed., 2006, 45, 2769 CrossRef CAS PubMed.
- A. L. J. Beckwith and R. D. Wagner, J. Am. Chem. Soc., 1979, 101, 7099 CrossRef CAS.
- J. Kim, H. B. Li, A. S. Rosenthal, D. Sang, T. A. Shapiro, M. D. Bachi and G. H. Posner, Tetrahedron, 2006, 62, 4120 CrossRef CAS.
- T. Tokuyasu, S. Kunikawa, M. Abe, A. Masuyama, M. Nojima, H.-S. Kim and Y. Wataya, J. Org. Chem., 2003, 68, 7361 CrossRef CAS PubMed.
- P. Dai and P. H. Dussault, Org. Lett., 2005, 7, 4333 CrossRef CAS PubMed.
- N. Murakami, M. Kawanishi, S. Itagaki, T. Horii and M. Kobayashi, Tetrahedron Lett., 2001, 42, 7281 CrossRef CAS.
- H.-X. Jin, H.-H. Liu, Q. Zhang and Y. Wu, J. Org. Chem., 2005, 70, 4240 CrossRef CAS PubMed.
- J. Howarth and D. Wilson, Bioorg. Med. Chem. Lett., 2003, 13, 2013 CrossRef CAS PubMed.
- Y.-J. Feng, R. A. Davis, M. Sykes, V. M. Avery, D. Camp and R. J. Quinn, J. Nat. Prod., 2010, 73, 716 CrossRef CAS PubMed.
- K. Ingram, C. E. Schiaffo, W. Sittiwong, E. Benner, P. H. Dussault and J. Keiser, J. Antimicrob. Chemother., 2012, 67, 1979 CrossRef CAS PubMed.
- B. Camuzat-Dedenis, O. Provot, L. Cointreaux, V. Perroux, J.-F. Berrien, C. Bories, P. M. Loiseau and J. Mayrargue, Eur. J. Med. Chem., 2001, 36, 837 CrossRef CAS PubMed.
- E. Fernandez-Alvaro, W. D. Hong, G. L. Nixon, P. M. O'Neill and F. Calderon, J. Med. Chem., 2016, 59, 5587 CrossRef CAS PubMed.
- P. J. Rosenthal, Lancet Infect. Dis., 2016, 16, 6 CrossRef PubMed.
- M. Brindisi, S. Gemma, S. Kunjir, L. Di Cerbo, S. Brogi, S. Parapini, S. D'Alessandro, D. Taramelli, A. Habluetzel, S. Tapanelli, S. Lamponi, E. Novellino, G. Campiani and S. Butini, MedChemComm, 2015, 6, 357 RSC.
- K. A. El Sayed, D. C. Dunbar, D. K. Goins, C. R. Cordova, T. L. Perry, K. J. Wesson, S. C. Sanders, S. A. Janus and M. T. Hamann, J. Nat. Toxins, 1996, 5, 261 CAS.
- H.-X. Jin, Q. Zhang, H.-S. Kim, Y. Wataya, H.-H. Liu and Y. Wu, Tetrahedron, 2006, 62, 7699 CrossRef CAS.
- J. Howarth and D. Wilson, Bioorg. Med. Chem. Lett., 2003, 13, 2013 CrossRef CAS PubMed.
- A. M. Szpilman, E. E. Korshin, H. Rozenberg and M. D. Bachi, J. Org. Chem., 2005, 70, 3618 CrossRef CAS PubMed.
- P. L. Gutierrez, Free Radical Biol. Med., 2000, 29, 263 CrossRef CAS PubMed.
- O. Kayser, A. F. Kiderlen and S. L. Croft, Parasitol. Res., 2003, 90, S55 CrossRef PubMed.
- L. H. Carvalho, E. M. Rocha, D. S. Raslan, A. B. Oliveira and A. U. Krettli, Braz. J. Med. Biol. Res., 1988, 21, 485 CAS.
- P. Vanelle, M. P. De Meo, J. Maldonado, R. Nouguier, M. P. Crozet, M. Laget and G. Dumenil, Eur. J. Med. Chem., 1990, 25, 241 CrossRef CAS.
- P. Vanelle, M. P. Crozet, J. Maldonado and M. Barreau, Eur. J. Med. Chem., 1991, 26, 167 CrossRef CAS.
- L. A. Dunn, A. G. Burgess, K. G. Krauer, L. Eckmann, P. Vanelle, M. D. Crozet, F. D. Gillin, P. Upcroft and J. A. Upcroft, Int. J. Antimicrob. Agents, 2010, 36, 37 CrossRef CAS PubMed.
- L. Paloque, P. Verhaeghe, M. Casanova, C. Castera-Ducros, A. Dumètre, L. Mbatchi, S. Hutter, M. Kraiem-M'Rabet, M. Laget, V. Remusat, S. Rault, P. Rathelot, N. Azas and P. Vanelle, Eur. J. Med. Chem., 2012, 54, 75 CrossRef CAS PubMed.
- A. Bouhlel, C. Curti, M. P. Bertrand and P. Vanelle, Tetrahedron, 2012, 68, 3596 CrossRef CAS.
- A. Bouhlel, C. Curti, C. Tabele and P. Vanelle, Molecules, 2013, 18, 4296 CrossRef PubMed.
- A. Bouhlel, C. Curti and P. Vanelle, Molecules, 2012, 17, 4313 CrossRef CAS PubMed.
- C. Tabele, A. Cohen, C. Curti, A. Bouhlel, S. Hutter, V. Remusat, N. Primas, T. Terme, N. Azas and P. Vanelle, Eur. J. Med. Chem., 2014, 87, 440 CrossRef CAS PubMed.
- K. Asahi and H. Nishino, Tetrahedron, 2008, 64, 1620 CrossRef CAS.
- K. Asahi and H. Nishino, Eur. J. Org. Chem., 2008, 14, 2404 CrossRef.
- H. Nishino, S.-I. Tategami, T. Yamada, J. D. Korp and K. Kurosawa, Bull. Chem. Soc. Jpn., 1991, 64, 1800 CrossRef CAS.
- C.-Y. Qian, H. Nishino and K. Kurosawa, Bull. Chem. Soc. Jpn., 1991, 64, 3557 CrossRef CAS.
- B. B. Snider, J. J. Patricia and S. A. Kates, J. Org. Chem., 1988, 53, 2137 CrossRef CAS.
- C.-J. Qian, J.-I. Hirose, H. Nishino and K. Karusawa, J. Heterocycl. Chem., 2009, 31, 1219 CrossRef.
- J. T. Edward, Chem. Ind., 1955, 1102 CAS.
- J.-I. Yoshida, K. Sakagushi, S. Isoe and K. Hirotsu, Tetrahedron Lett., 1987, 28, 667 CrossRef CAS.
- C. Daeppen, M. Kaiser, M. Neuburger and K. Gademann, Org. Lett., 2015, 17, 5420 CrossRef CAS PubMed.
- C.-J. Qian, T. Yamada, H. Nishino and K. Kurosawa, Bull. Chem. Soc. Jpn., 1992, 65, 1371 CrossRef CAS.
- V.-H. Nguyen, H. Nishino and K. Kurosawa, Synthesis, 1997, 8, 899 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental part, NMR spectra. CCDC 1495135, 1495137, 1495138, 1495140 and 1495141. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra25138b |
| This journal is © The Royal Society of Chemistry 2017 |