
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe oxygen reduction reaction mechanism on Sn doped graphene as an electrocatalyst in fuel cells: a DFT study†
Xiaoxu Sunab,
Kai Lia,
Cong Yinc,
Ying Wanga,
Feng Heab,
Xiaowan Baia,
Hao Tang*c and
Zhijian Wu*a
aState Key Laboratory of Rare Earth Resource Utilization, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, P. R. China. E-mail: zjwu@ciac.ac.cn
bUniversity of Science and Technology of China, Hefei, Anhui 230026, P. R. China
cEnergy Conversion R&D Center, Central Academy of Dongfang Electric Corporation, Chengdu 611731, P. R. China. E-mail: tanghao@dongfang.com
First published on 3rd January 2017
Abstract
Heteroatom doped graphene has caused particular interest in recent years due to its promising ORR (oxygen reduction reaction) activity in fuel cells. Sn doped divacancy graphene (Sn–Gra) was predicted to be a good candidate as a cathode catalyst in the previous study. In this work, the detailed ORR mechanism has been studied for Sn–Gra. The calculated charge transfer indicates that Sn and its adjacent four C atoms are the catalytic reaction sites. The unstable intermediate HOOH suggests that Sn–Gra experiences a four-electron ORR process. The most favorite pathway is the hydrogenation of the O2 molecule. The rate determining step is the hydrogenation of OOH to form H2O + O with the energy barrier of 0.75 eV. This value is slightly smaller than 0.80 eV for Pt, implying that Sn–Gra is a potential cathode catalyst for ORR. The predicted working potential is 0.16 V for the most favorite pathway. We expect that this study could provide new insights for the design of low-cost and highly efficient electrocatalysts in fuel cells.
1. Introduction
A global energy shortage and environmental pollution have stimulated massive research on next-generation energy conversion and storage systems, which should be highly efficient, low cost and environmentally friendly. In this aspect, fuel cells and metal–air batteries are the best alternatives.1–3 For fuel cells and metal–air batteries, however, the current bottleneck lies in the slow kinetics of the cathodic oxygen reduction reaction (ORR).4,5 As a result, catalysts with fast ORR rate are at the heart of key renewable-energy technologies. Although the Pt/C catalyst has been the most active catalyst for ORR to date, the high price, instability and poor CO tolerance severely hindered the commercialization of these state-of-the-art devices.6–8 Therefore, it is quite challenging but desirable to develop efficient alternative catalysts, such as non-precious metals and metal-free materials. By far, the state-of-the-art metal-free catalysts are sp2-hybridized carbon and its derivatives.9 By introducing heteroatom into carbon materials, their electron properties would be tailored and consequently their catalytic activities would be enhanced. Diverse heteroatom-doped carbon materials, such as carbon nanoparticles,10 carbon nanotubes11 and graphene,12 have attracted extensive interests because of their lower price, outstanding mechanical flexibility and superior structural durability.Among these carbon materials, graphene as the ultrathin two-dimensional network has drawn a lot of attention for ORR activity due to its large surface area, high carrier mobility and superior chemical stability.13–15 Continuous efforts have been made to synthesize the heteroatom doped graphene that exhibit excellent ORR performance, such as nitrogen (N),12 boron (B),16 phosphorus (P),17 sulfur (S),18 antimony (Sb),19 and their mixtures.20,21 The catalytic current at the N doped graphene electrode was about three times higher than that at the Pt/C electrode, which opens a way to fabricate other new heteroatom doped carbon materials as ORR catalysts.12 Boron atom possesses strong electron-withdrawing capability and due to its unique electronic structure and properties, the boron doped graphene exhibits pronounced catalytic activity towards ORR and better CO tolerance than Pt/C catalyst in alkaline electrolytes.16 P doped graphene exhibits enhanced cycle and rate capabilities as well as outstanding ORR activity compared with the undoped graphene.17 In addition, S-doped graphene shows a higher electrocatalytic activity than Pt/C catalyst.18
Although great progress has been achieved in experimental studies, the ORR mechanism is only partially understood. In this regard, density functional theory (DFT) is an effective theoretical method to identify the catalytic sites and explore the ORR pathways from the thermodynamic and dynamic perspectives. For N doped graphene in acidic environment, theoretical study indicates that the ORR process is a direct four-electron pathway,22 which is consistent with the experimental observations. For P doped graphene, the OOH formation and dissociation pathway is the most favorable.23 S doped graphene could exhibit ORR catalytic properties comparable to Pt/C catalyst based on DFT calculations,24 which is in agreement with the experimental result.
Motivated by the above studies, in this work, we have studied Sn doped divacancy graphene (Sn–Gra). Previous study shows that single Sn doped divacancy graphene is a good candidate for ORR at the cathode of fuel cells.25 To get further insights, in present study, the specific adsorption sites for various ORR species are identified by calculating the adsorption energies. In addition, the energy barriers and free energy changes are calculated to identify the optimal reaction pathway.
2. Computational details
The catalyst was modeled as a 5 × 5 hexagonal graphene super cell containing 49 atoms with lattice parameters a = b = 12.3 Å, and with periodic boundary conditions in three dimensions. A vacuum layer of 15 Å perpendicular to the graphene sheets was adopted to avoid the artificial interactions between graphene layers.All the geometry optimization and energy calculations were performed within density functional theory framework26,27 using the Vienna ab initio Simulation Package (VASP) code.28,29 Projector augmented wave (PAW)30 potentials were adopted to describe the electronic exchange and correlation within the generalized gradient approximation (GGA) using the Perdew–Burke–Ernzerhof (PBE).31 The Kohn–Sham one-electron wave functions at each k-point were expanded with a kinetic energy cutoff of 400 eV. The Brillouin zone was sampled using a 4 × 4 × 1 k-point grid in the Monkhorst–Pack scheme.32
The atomic positions were optimized until the forces were below 0.01 eV Å−1. The transition state (TS) structures of different reaction pathways were identified through the climbing image nudged elastic band (CI-NEB) method.33 The minimum energy path was optimized with the force-based conjugate-gradient method until the maximum force was below 0.05 eV Å−1. In addition, zero point energy corrections were taken into consideration in all the involved energies. To describe the van-der-Waals (vdW) interactions between the reactants and the substrate, a semiempirical dispersion-corrected density functional theory (DFT-D2)34,35 force-field approach was adopted.
The formation energy (ΔEf) was calculated as:
| ΔEf = ESn–Gra + xμc − (EGra + ESn) | (1) |
The adsorption energy (ΔEads) was calculated as:
| ΔEads = ESn–Gra+adsorbate − (ESn–Gra + Eadsorbate) | (2) |
To explore the effect of the electrode potentials on the ORR activity, we studied the free energy changes of the ORR on Sn doped graphene under various electrode potentials based on the approach developed by Norskov and coworkers.5 The change in free energy (ΔG) of every elementary reaction step was given by the expression:5,37
| ΔG = ΔE + ΔZPE − TΔS + ΔGU + ΔGpH + ΔGfield | (3) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10 × pH, where kB is the Boltzmann constant and T = 300 K. In acid medium, pH is defined as 0.38,39 ΔGfield is the free energy correction owing to the electrochemical double layer, which is negligible. The free energy of O2 was obtained from the reaction O2 + 2H2 → 2H2O, in which the free energy change is 4.92 eV at the temperature of 298.15 K and pressure of 0.035 bar.5,40 According to the computational hydrogen electrode model proposed by Nørskov,5 the free energy of H+ in solution under standard conditions of pH = 0 and U = 0 was defined as that of half H2. Gas phase H2O at 0.035 bar was used as the reference, since the gas phase H2O is in equilibrium with liquid phase at this pressure. For the molecules (such as O2, H2, H2O, etc.) in the gas phase, the entropies and vibrational frequencies were obtained from the NIST database,41 while the zero point energy and the entropies of the ORR intermediates were calculated from the vibrational frequencies.5
10 × pH, where kB is the Boltzmann constant and T = 300 K. In acid medium, pH is defined as 0.38,39 ΔGfield is the free energy correction owing to the electrochemical double layer, which is negligible. The free energy of O2 was obtained from the reaction O2 + 2H2 → 2H2O, in which the free energy change is 4.92 eV at the temperature of 298.15 K and pressure of 0.035 bar.5,40 According to the computational hydrogen electrode model proposed by Nørskov,5 the free energy of H+ in solution under standard conditions of pH = 0 and U = 0 was defined as that of half H2. Gas phase H2O at 0.035 bar was used as the reference, since the gas phase H2O is in equilibrium with liquid phase at this pressure. For the molecules (such as O2, H2, H2O, etc.) in the gas phase, the entropies and vibrational frequencies were obtained from the NIST database,41 while the zero point energy and the entropies of the ORR intermediates were calculated from the vibrational frequencies.5
3. Results and discussion
3.1. Adsorption of the intermediates
First of all, we have studied the adsorption of Sn on relaxed divacancy graphene by putting Sn atom on the different surface sites (Fig. 1). Our results indicate that the adsorption of Sn on the divacancy site (Fig. 1a) is the strongest (−3.70 eV), implying that Sn prefers to be doped on the divacancy graphene. Since the atomic radius of Sn is 1.40 Å which is larger than 0.91 Å of C, Sn lies 1.53 Å above the graphene surface.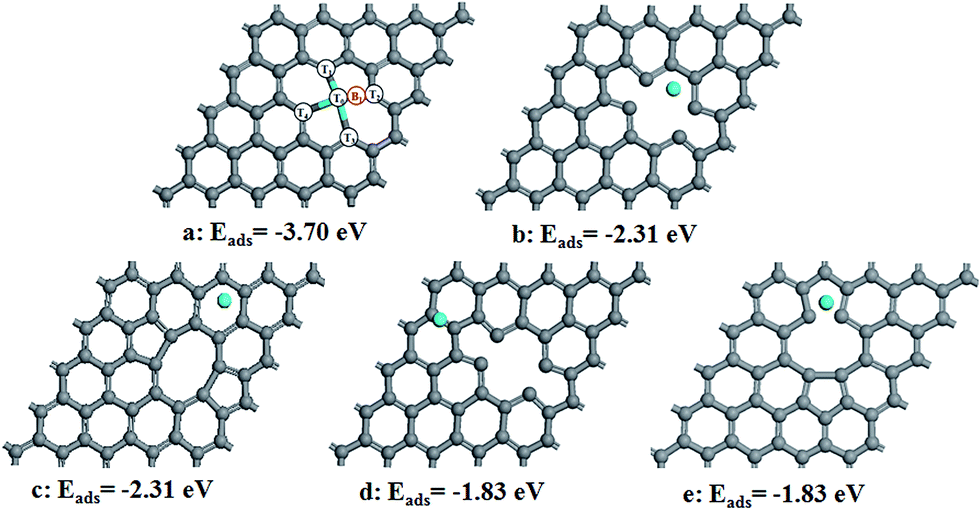 | ||
| Fig. 1 (a–e) Configurations refer to possible adsorption sites for Sn atom at a divacancy in graphene. Eads refers to the corresponding adsorption energy. T0, T1, T2, T3 and T4 in a configuration refer to the top sites, while B1 refers to the bridge site between Sn and C atoms. The gray and blue balls represent C and Sn atoms, respectively. | ||
To indentify the possible reaction sites, we then calculated the Bader charge transfer on Sn–Gra (Table 1). For Sn–Gra without intermediates, Sn has a positive charge of 0.91e, while the total charge of its adjacent four C atoms is −1.02e. This is in agreement with the large electronegativity of C. The NC of Sn and its adjacent four carbon atoms is about −0.11e. This indicates that the Sn and its adjacent four carbon atoms are catalytic active, i.e., the Sn top site (T0 in Fig. 1a), the four adjacent C top atoms (T1, T2, T3, T4 in Fig. 1a) and the bridge site between Sn and C (B1 in Fig. 1a).
| Adsorbate | Q(Sn) | Q(4C) | Q(ads) | NC |
|---|---|---|---|---|
| Sn–Gra | 0.91 | −1.02 | — | −0.11 |
| O2 | 1.59 | −0.77 | −0.53 | 0.29 |
| O | 1.08 | 0.11 | −1.10 | 0.09 |
| H | 1.43 | −0.90 | −0.26 | 0.27 |
| OH | 1.65 | −1.08 | −0.53 | 0.04 |
| OOH | 1.70 | −0.79 | −0.60 | 0.31 |
| H2O | 1.55 | −1.78 | 0.04 | −0.19 |
| 2O | 1.78 | 0.29 | −1.93 | 0.14 |
| O + OH | 1.73 | −0.32 | −1.37 | 0.04 |
After the optimization of all the ORR intermediates (O2, O, H, OH, OOH, H2O, O + O and O + OH) at possible sites, the obtained stable adsorption configurations and corresponding adsorption energies are given in Fig. S1, ESI.† The most stable adsorption configurations and corresponding adsorption energies for each intermediate (except for H atom) are displayed in Fig. 2. After the intermediates are adsorbed on the surface, Sn loses more electrons while the intermediates gain electrons except for H2O molecule. The net charge (NC) of the intermediates is around 0.04–0.31|e|. This confirms again that all the possible ORR elementary reactions could take place around the Sn atom and its adjacent four carbon atoms, which is similar to that of S doped and N doped graphene.24,42 Among the intermediates shown in Table 1, single atomic O gains the most electrons, even making the four carbon atoms positively charged. For two separated O atoms adsorbed on the surface, the total electron gain is −1.93e, i.e., −0.965e for each atom, smaller than −1.10e for single atomic O. This suggests that the interaction between two separated O atoms and Sn–Gra is weaker, which would facilitate the following ORR process. The calculated adsorption energies also support this conclusion, which is −4.64 eV for single atomic O (Fig. 2B), −2.54 eV for each O atom in two separated O atoms (Fig. 2G).
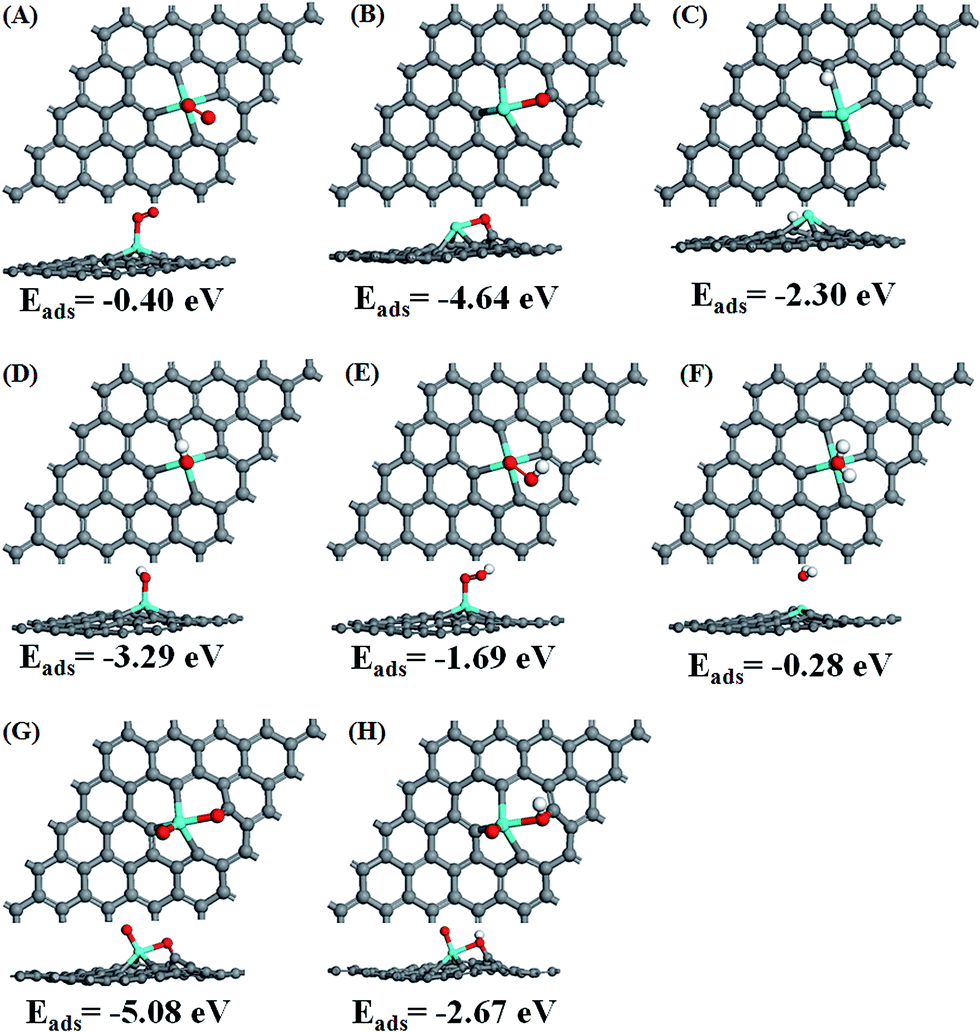 | ||
| Fig. 2 Atomic structures with the lowest adsorption energy for the ORR species (the second lowest adsorption energy for H atom) adsorbed on Sn–Gra. (A) O2, (B) O, (C) H, (D) OH, (E) OOH, (F) H2O, (G) two separated O atoms; (H) O + OH. Eads represents the absorption energy. | ||
The prerequisite for initializing the ORR is that the O2 molecule must be able to be adsorbed on the catalyst surface. There are two configurations for O2 molecule adsorption (Fig. S1†), i.e., side-on configuration in which two O atoms bind to Sn atom with the same bond distance, and end-on configuration in which one O atom binds to Sn atom, while the other O atom is pointing away from the surface. Our calculations show that the end-on configuration of O2 molecule exhibits much stronger adsorption (Eads = −0.40 eV in Fig. S1†) than the side-on configuration (Eads = −0.16 eV in Fig. S1†). Thus we chose the O2 molecule configuration with stronger adsorption energy of −0.40 eV (Fig. 2A) as the initial state for the ORR study. The adsorption energy of −0.40 eV is quite close to the value in ref. 25. The end-on configuration of O2 and the corresponding adsorption energy are similar to P doped divacancy graphene.43
For single O atom, the most stable site is the bridge site between Sn and C with adsorption energy of −4.64 eV (Fig. 2B), similar to the P doped43 and silicon doped divacancy graphene.44 Different from single O atom, the most stable adsorption site for H atom is the Sn top site (Fig. S1†). The second stable site is the C top site with the adsorption energy of −2.30 eV (Fig. 2C). Since during the ORR process, Sn top site is always occupied by other species containing O atom, the second most stable site is more favorable for the adsorption of H atom. For OH and OOH species, they prefer to adsorb at Sn top site rather than at C top site with adsorption energies of −3.29 eV and −1.69 eV, respectively (Fig. 2D and E). For H2O molecule, its weak adsorption energy of −0.28 eV (Fig. 2F) is in agreement with the previous study.25 The weak adsorption is also in accordance with the small amount of charge transfer (0.04e). Thus, H2O molecule could be easily desorbed from the Sn–Gra surface once it is formed. In addition, HOOH is found to be unstable during the geometry optimization, suggesting that the ORR mechanism on Sn–Gra is a four-electron process, not a two-electron process.
3.2. ORR mechanism on Sn–Gra
The chemisorption of O2 on Sn–Gra is the first step for the ORR process. Following the adsorption, there are two possible pathways for O2 molecule: dissociation into two O atoms or hydrogenation to OOH species. Once the O2 molecule dissociated into two O atoms, it would undergo sequential hydrogenations to form two H2O molecules. For O2 hydrogenation, the OOH species could either be decomposed to generate O + OH or hydrogenated to form HOOH, O + H2O or two OH species. Since HOOH species could not exist on the surface of Sn–Gra (it will decompose into O + H2O after the hydrogenation), there are only three remaining pathways for following the formed OOH species.The summarized reaction pathways on Sn–Gra are shown in Fig. 3, while the geometric structures of the initial state, transition state and final state for all the possible reaction steps are shown in Fig. 4.
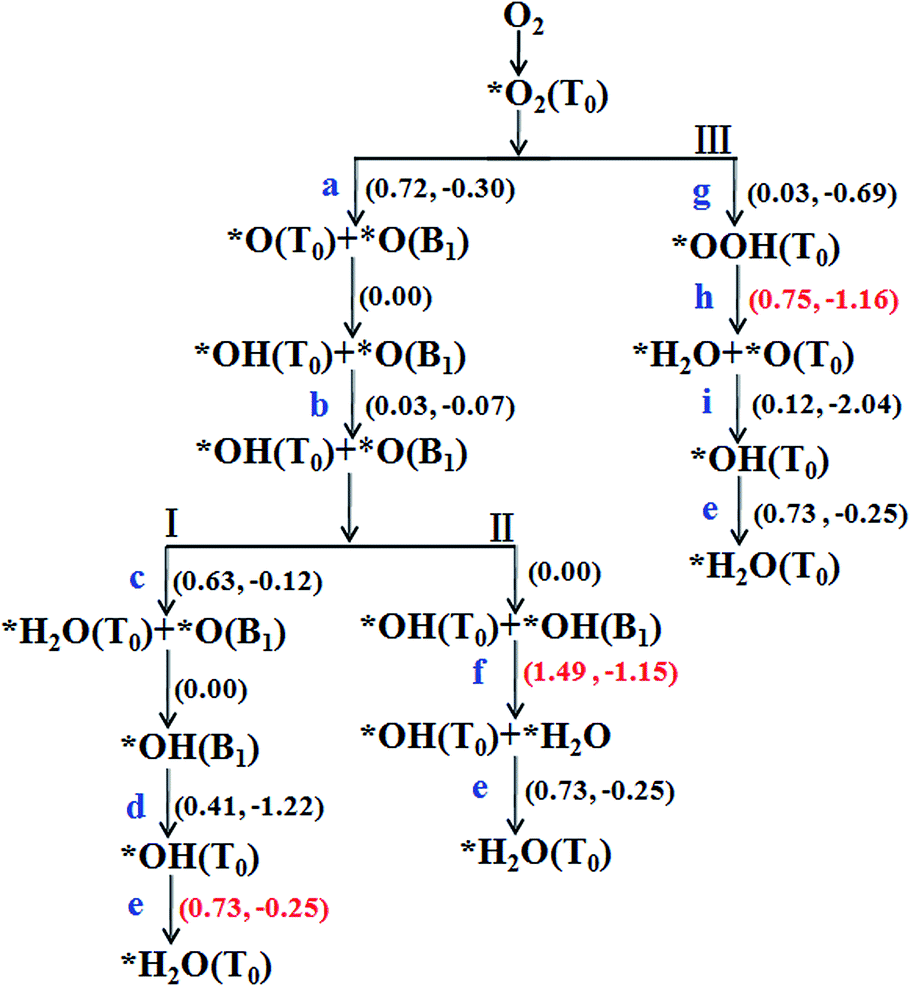 | ||
| Fig. 3 Possible reaction pathways for ORR on Sn–Gra. The numbers in parenthesis are the energy barriers (left) and reaction energies (right) in units of eV. For the details of labels (a–i) see Fig. 4. * denotes that the ORR species is adsorbed on the catalyst surface. | ||
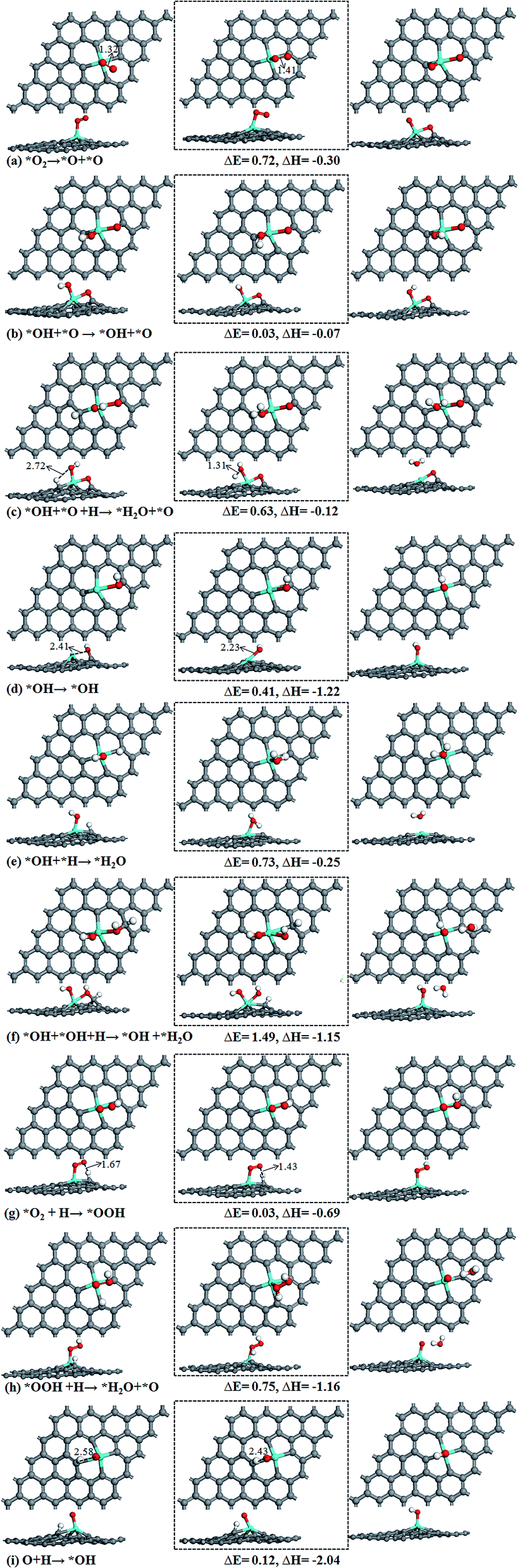 | ||
| Fig. 4 Atomic structures of the initial state (left panel), transition state (middle panel), and final state (right panel) for reaction pathways on Sn–Gra. ΔE is the energy barrier and ΔH is the reaction energy (in units of eV). | ||
Pathway I, formation of H2O + O (Fig. 3). In the initial state, the H atom at the C top site has a bond distance of 2.72 Å with O atom. It decreases to 1.31 Å in the transition state (Fig. 4c). The energy barrier of this process is 0.63 eV with small exothermic energy of −0.12 eV. After the removal of the first H2O molecule from the catalyst surface, the remaining O atom at bridge site will be hydrogenated. This is a spontaneous process without energy barrier. Then, the OH species on the bridge site moves to the most stable Sn top site. This process requires to overcome an energy barrier of 0.41 eV and large exothermic energy of −1.22 eV is released (Fig. 4d). The hydrogenation of OH on Sn top site forms the second H2O with an energy barrier of 0.73 eV and exothermic by −0.25 eV (Fig. 4e). The highest energy barrier (0.73 eV) in pathway I (Fig. 3) suggests that the second hydrogenation step to form H2O molecule is the rate determining step. The weak adsorption of H2O molecule on Sn–Gra implies that the H2O molecule can easily be desorbed from the surface and leaving the Sn–Gra refreshed.
Pathway II, formation of OH + OH (Fig. 3). The hydrogenation of O atom at bridge site needs no energy barrier. After this step, the hydrogenation of OH at bridge site to form OH + H2O needs to overcome a very high energy barrier of 1.49 eV with exothermic energy of −1.15 eV (Fig. 4f). After the release of the first H2O molecule, the hydrogenation of the remaining OH at the Sn top site needs an energy barrier of 0.73 eV (Fig. 4e), the same as in pathway I.
In a word, from Fig. 3, we can conclude that pathway I and III are competitive with similar energy barriers of 0.73 eV and 0.75 eV, respectively. These values are smaller than 0.88 eV for P doped single vacancy graphene,23 0.85 eV for P doped divacancy graphene,43 0.80 eV for Pt (100)45 and 0.79 eV for Pt (111).46 In addition, compared with O2 dissociation with an energy barrier of 0.72 eV, the hydrogenation of O2 to form OOH with an energy barrier of 0.03 eV is much easier. Once the formed OOH species occupies the Sn top site, it could hinder the adsorption of O or OH species.
3.3. Effect of electrode potentials on ORR
All the above calculations are performed under zero electrode potential, while in reality, the cathode electrocatalysts for ORR work under positive electrode potentials. Therefore, the influence of different external potentials (U) on the ORR pathways have been studied. The free energy diagrams of the ORR on Sn–Gra have been explored by applying the method developed by Norskov and coworkers.5 The results are shown in Fig. 5 for O2 hydrogenation, Fig. S2 and S3 (ESI†) for O2 dissociation. It is seen from Fig. 5 (pathway III) that at U = 0.00 V, all the elementary steps are downhill. With the increase of the electrode potentials, the formation of the second H2O molecule starts to be uphill at U = 0.16 V. For the O2 dissociation, however, even at U = 0.00 V, the formation of first H2O molecule becomes uphill for pathway I (Fig. S2†), while the formation of OH becomes uphill for pathway II (Fig. S3†). This indicates that pathway III is more favorable for the ORR on Sn–Gra.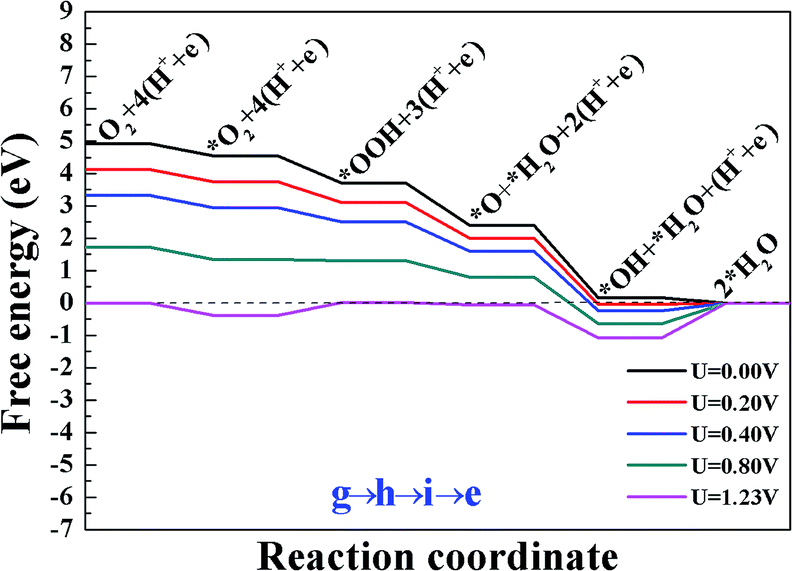 | ||
| Fig. 5 The free energy diagram for the most favorite reaction pathway III (g → h → i → e) on Sn–Gra at different electrode potentials. | ||
4. Conclusions
Sn doped divacancy graphene (Sn–Gra) has been studied by using the density functional method. Our results suggest that Sn and its adjacent four C atoms are the catalytic active sites. For each intermediate, the most stable adsorption site is determined. Four-electron mechanism is predicted for ORR on Sn–Gra. O2 hydrogenation is the most favorable pathway with the energy barrier of 0.75 eV, smaller than ∼0.80 eV for Pt. The calculated working potential is 0.16 V. Thus, we expect that Sn–Gra could be a promising electrocatalyst for ORR.Acknowledgements
The acknowledgements come at the end of an article after the conclusions and before the notes and references. This work is supported by the National Natural Science Foundation of China (21503210, 21521092, 21673220), Jilin Province Youth Fund (20130522141JH), Jilin Province Natural Science Foundation (20150101012JC) and Special Program for Applied Research on Super Computation of the NSFC-Guangdong Joint Fund (the second phase). The authors also thank the financial support from Department of Science and Technology of Sichuan Province (2011GZX0077, 2012JZ0007, 2014HH0049). Part of the computational time is supported by the High Performance Computing Center of Jilin University, Changchun Normal University, and Jilin Province Supercomputing Center.References
- G. Brumfiel, Nature, 2003, 422, 104 CrossRef CAS PubMed.
- V. Dusastre, Nature, 2001, 414, 331 CrossRef.
- M. Z. Jacobson, W. G. Colella and D. M. Golden, Science, 2005, 308, 1901–1905 CrossRef CAS PubMed.
- F. Zhao, F. Harnisch, U. Schrorder, F. Scholz, P. Bogdanoff and I. Herrmann, Environ. Sci. Technol., 2006, 40, 5193–5199 CrossRef CAS PubMed.
- J. K. Norskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef CAS.
- H. A. Gasteiger, J. E. Panels and S. G. Yan, J. Power Sources, 2004, 127, 162–171 CrossRef CAS.
- C. H. Cui, L. Gan, M. Heggen, S. Rudi and P. Strasser, Nat. Mater., 2013, 12, 765–771 CrossRef CAS PubMed.
- J. Greeley, I. E. L. Stephens, A. S. Bondarenko, T. P. Johansson, H. A. Hansen, T. F. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. K. Norskov, Nat. Chem., 2009, 1, 552–556 CrossRef CAS PubMed.
- D. S. Su, J. Zhang, B. Frank, A. Thomas, X. C. Wang, J. Paraknowitsch and R. Schlogl, ChemSusChem, 2010, 3, 169–180 CrossRef CAS PubMed.
- T. Xing, J. Sunarso, W. R. Yang, Y. B. Yin, A. M. Glushenkov, L. H. Li, P. C. Howlett and Y. Chen, Nanoscale, 2013, 5, 7970–7976 RSC.
- K. P. Gong, F. Du, Z. H. Xia, M. Durstock and L. M. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- L. T. Qu, Y. Liu, J. B. Baek and L. M. Dai, ACS Nano, 2010, 4, 1321–1326 CrossRef CAS PubMed.
- M. D. Stoller, S. J. Park, Y. W. Zhu, J. H. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498–3502 CrossRef CAS PubMed.
- T. O. Wehling, K. S. Novoselov, S. V. Morozov, E. E. Vdovin, M. I. Katsnelson, A. K. Geim and A. I. Lichtenstein, Nano Lett., 2008, 8, 173–177 CrossRef CAS PubMed.
- A. A. Balandin, S. Ghosh, W. Z. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902–907 CrossRef CAS PubMed.
- Z. H. Sheng, H. L. Gao, W. J. Bao, F. B. Wang and X. H. Xia, J. Mater. Chem., 2012, 22, 390–395 RSC.
- C. Z. Zhang, N. Mahmood, H. Yin, F. Liu and Y. L. Hou, Adv. Mater., 2013, 25, 4932–4937 CrossRef CAS PubMed.
- Z. Yang, Z. Yao, G. F. Li, G. Y. Fang, H. G. Nie, Z. Liu, X. M. Zhou, X. Chen and S. M. Huang, ACS Nano, 2012, 6, 205–211 CrossRef CAS PubMed.
- I. Y. Jeon, M. Choi, H. J. Choi, S. M. Jung, M. J. Kim, J. M. Seo, S. Y. Bae, S. Yoo, G. Kim, H. Y. Jeong, N. Park and J. B. Baek, Nat. Commun., 2015, 6, 8 Search PubMed.
- Y. Zheng, Y. Jiao, L. Ge, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2013, 52, 3110–3116 CrossRef CAS PubMed.
- J. Liang, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2012, 51, 11496–11500 CrossRef CAS PubMed.
- L. P. Zhang and Z. H. Xia, J. Phys. Chem. C, 2011, 115, 11170–11176 CAS.
- X. L. Zhang, Z. S. Lu, Z. M. Fu, Y. A. Tang, D. W. Ma and Z. X. Yang, J. Power Sources, 2015, 276, 222–229 CrossRef CAS.
- L. P. Zhang, J. B. Niu, M. T. Li and Z. H. Xia, J. Phys. Chem. C, 2014, 118, 3545–3553 CAS.
- M. Kaukonen, A. V. Krasheninnikov, E. Kauppinen and R. M. Nieminen, ACS Catal., 2013, 3, 159–165 CrossRef CAS.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864–B871 CrossRef.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- B. Delley, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 19 CrossRef PubMed.
- Y. Sha, T. H. Yu, Y. Liu, B. V. Merinov and W. A. Goddard, J. Phys. Chem. Lett., 2010, 1, 856–861 CrossRef CAS.
- S. Zuluaga and S. Stolbov, J. Chem. Phys., 2011, 135, 134702 CrossRef PubMed.
- Y. Shang, J.-x. Zhao, H. Wu, Q.-h. Cai, X.-g. Wang and X.-z. Wang, Theor. Chem. Acc., 2010, 127, 727–733 CrossRef CAS.
- R. Faccio, L. Fernández-Werner, H. Pardo, C. Goyenola, O. N. Ventura and Á. W. Mombrú, J. Phys. Chem. C, 2010, 114, 18961–18971 CAS.
- L. Yu, X. Pan, X. Cao, P. Hu and X. Bao, J. Catal., 2011, 282, 183–190 CrossRef CAS.
- http://webbook.nist.gov/chemistry/, accessed Feb 9, 2015.
- M. T. Li, L. P. Zhang, Q. Xu, J. B. Niu and Z. H. Xia, J. Catal., 2014, 314, 66–72 CrossRef CAS.
- X. W. Bai, E. J. Zhao, K. Li, Y. Wang, M. G. Jiao, F. He, X. X. Sun, H. Sun and Z. J. Wu, Carbon, 2016, 105, 214–223 CrossRef CAS.
- P. Zhang, X. L. Hou, J. L. Mi, Y. Q. He, L. Lin, Q. Jiang and M. D. Dong, Phys. Chem. Chem. Phys., 2014, 16, 17479–17486 RSC.
- Z. Duan and G. Wang, J. Phys. Chem. C, 2013, 117, 6284–6292 CAS.
- Z. Duan and G. Wang, Phys. Chem. Chem. Phys., 2011, 13, 20178 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 shows all the possible configurations for each adsorbed species involved in the ORR on Sn doped graphene. Fig. S2 shows the free energy diagrams for the reaction pathway I (a → b → c → d → e) on Sn–Gra and Fig. S3 shows the free energy diagrams for the reaction pathway II (a → b → f → e) on Sn–Gra. See DOI: 10.1039/c6ra25118h |
| This journal is © The Royal Society of Chemistry 2017 |