
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bioisosteric modification of known fucosidase inhibitors to discover a novel inhibitor of α-L-fucosidase†
Chandramohan Bathula‡
a,
Shreemoyee Ghosh‡b,
Santanu Hatia,
Sayantan Tripathyc,
Shailja Singhc,
Saikat Chakrabartib and
Subhabrata Sen*a
aDepartment of Chemistry, School of Natural Sciences, Shiv Nadar University, Tehsil Dadri, Chithera, Uttar Pradesh 201314, India. E-mail: subhabrata.sen@snu.edu.in
bStructural Biology and Bio-Informatics Division, CSIR-Indian Institute of Chemical Biology, 4, Raja SC Mullick Road, Kolkata, West Bengal 700032, India
cDepartment of Life Sciences, School of Natural Sciences, Shiv Nadar University, Tehsil Dadri, Chithera, Uttar Pradesh 201314, India
First published on 13th January 2017
Abstract
Bioisosteric modification of known fucosidase inhibitors A and B, resulted in three new types of molecules, 4b, 5c and 6a (belonging to furopyridinedione, thiohydantoin and hydantoin chemotypes) that could potentially bind to α-L-fucosidase (bovine kidney origin). Molecular docking revealed and compared the putative binding interaction between 4b, 5c and 6a with A and B against the active site of a homology model of α-L-fucosidase. Based on this initial investigation, design and synthesis of a library of small molecules based on furopyridinedione, thiohydantoin and hydantoin, followed by their in vitro screening against α-L-fucosidase (bovine kidney origin) generated a potent inhibitor (compound 4e) with IC50 of ∼0.7 μM. Compound 4e possessed no cytotoxic properties when tested against healthy mammalian COS-1 cells. Reaction kinetics study suggested it to be a mixed inhibitor. Finally compounds 4a, b, e and f, bearing the furopyridinedione motif also exhibited substantial inhibition of the proliferation of MCF 7 breast cancer cells.
Introduction
α-Glycosidases, belonging to a family of glycoside hydrolases, are enzymes that help in the hydrolysis of glycosidic bonds in complex sugars.1 α-L-Fucosidases are glycosidases that are involved in the hydrolysis of the α-1,6-linked fucose joined to the reducing end of N-acetylglucosamine moiety of the glycoproteins.2 Along with glycosyltransferases they form the major catalytic component for the making and breaking of glycosidic bonds.3 α-L-Fucosidases play an important role in the cleaning of the nature by degrading cellulose and hemicellulose.4 They are also involved in normal functioning of cells along with their roles in antibacterial defense mechanisms, and viral pathogenesis.5 Recent reports indicated that the activity of human α-L-fucosidase 2 is critical for the pathogenesis of Helicobacter pylori which in turn has implications in gastric cancer among other diseases.6 α-L-Fucosidase deficiency leads to various metabolic disorders, tumor occurrences and metastasis.7 Inhibition of α-L-fucosidases could lead to the synthesis of disorganized oligosaccharides that in turn could disrupt cell–cell and cell–virus recognition process.8–10 For these reasons, α-L-fucosidases are clinically important targets and the design and synthesis of potent and selective inhibitors remains an attractive goal.10Bioisosteric modification is a replacement strategy for rational design of new drugs that involves substitution of functional groups of a bioactive molecule with chemical moieties that possess similar physical or chemical attributes to generate better biological responses, be it potency, adsorption, dissolution, improvement of selectivity, metabolic stability, reduction of side effects and etc. For example, in drug discovery fluorine is one of the most common bioisostere of hydrogen.11 Other than providing metabolic stability it also influences lipophilicity of the resulting molecule. Carboxylic acid bioisosteres such as oxadiazoles, oxazole, tetrazoles and etc. provide enhancement of potency and increase of lipophilicity.12 Similarly, bioisosteric transformation of amide with trifluoroethylamine in cathepsin K inhibitors generated analogs with improved potency, selectivity and metabolic stability.13
Herein we report discovery of a novel inhibitor of α-L-fucosidase (bovine kidney origin), based on arylidenefuropyridinedione scaffold via bioisosteric modification of known fucosidase inhibitors A and B (Fig. 1).10d,e The initial design resulted in three diverse molecules (4b, 5c and 6a) belonging to different chemotypes such as hydantoin, thiohydantoin and furopyridinedione. The choice of the compounds was rationalized by their molecular docking against a homology model of α-L-fucosidase and comparing the binding interactions with the ones for A and B. A library was synthesized based on the designed scaffolds and enzymatic profiling of the library revealed that one of the compounds, 4e, inhibits α-L-fucosidase (bovine kidney origin) substantially. Mechanistic study revealed that the molecule binds to the enzyme as a mixed inhibitor. Its molecular docking with α-L-fucosidase indicated potential binding pockets of the enzyme. Further screening of the furopyridinedione analogs 4a–f against MCF 7 breast cancer cells indicated promising antiproliferative activity.
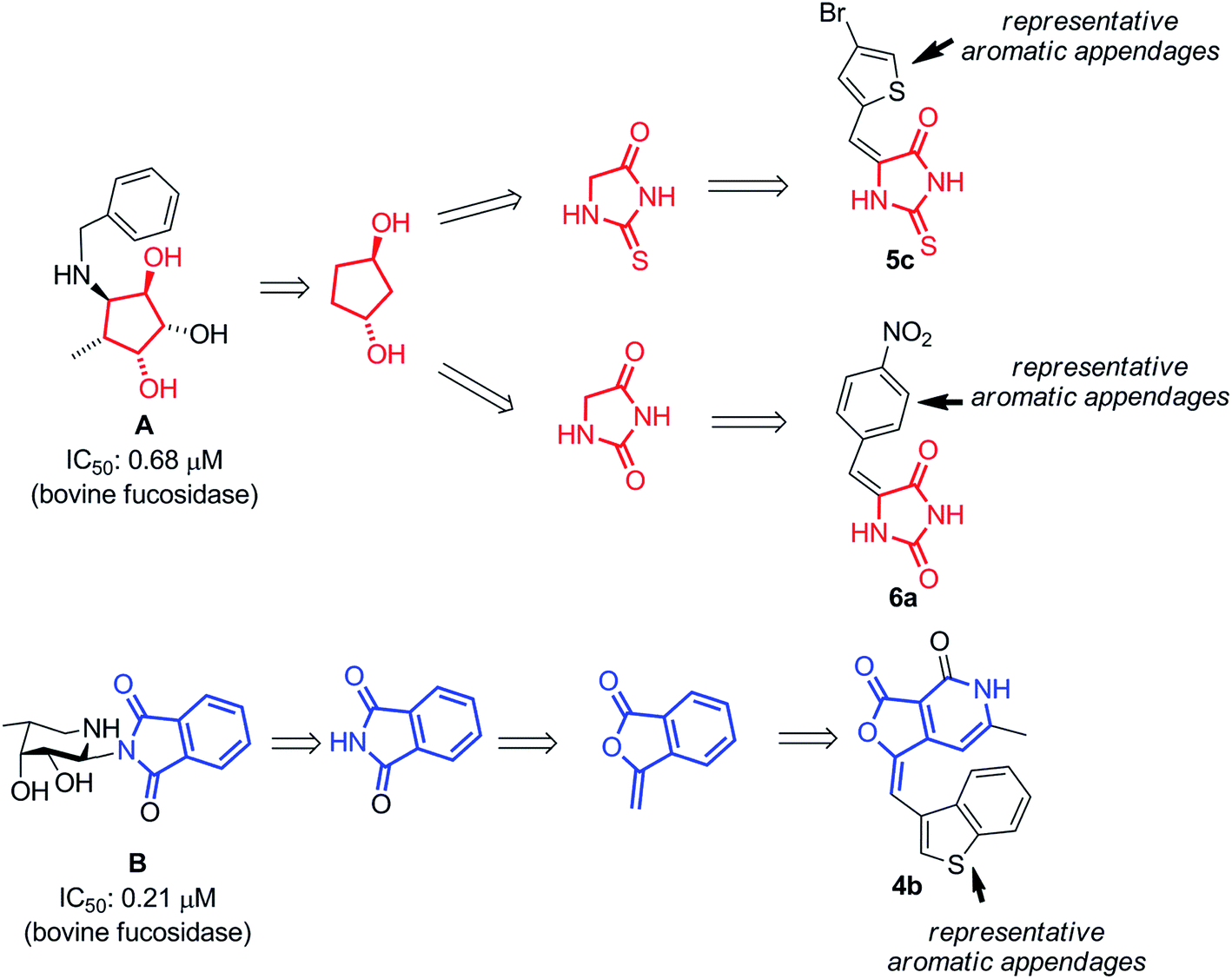 | ||
| Fig. 1 The scheme depicts representative α-L-fucosidase inhibitors (A and B) that inspired the design of compound 4b, 5c and 6a. | ||
Results and discussion
Design
To begin with the design of compounds, we screened the literatures for the available α-L-fucosidase inhibitors. In our search for small molecule heterocycles as suitable inhibitors of α-L-fucosidase we selected A and B that exhibited inhibitory activity in sub μmolar range against bovine α-L-fucosidase (Fig. 1).10d,e We intended to design novel molecules inspired from the heterocyclic as well as the sugar component of these compounds. Hence to mimic the multihydroxylated cyclopentyl benzylamine A an intuitive transformation to the thiohydantoin and hydantoin provided compounds 5c and 6a. We felt it will be interesting to replace the chiral hydroxylated functionalities of A at C2 and C4 with dicarbonyl and thiocarbonyl functionalities. The C2 and C4 in the ring system were replaced with the corresponding nitrogen bioisosteres. In a similar manner the pthalimide inhibitor B, that inhibited bovine-α-L-fucosidase with an IC50 of 0.21 μM, was intuitively replaced with furopyridinedione compound 4b. The N-functionality of the pthalimide was substituted with its corresponding bioisostere oxygen and one of the amide carbonyls was replaced with an arylidene functionality. The arylidene functionalities were introduced with the hope of better binding over the original molecules A and B against the bovine α-L-fucosidase (Fig. 1).Molecular docking and analysis of relevant 2D descriptors
To rationalize our design, compounds A and B along with 4b, 5c and 6a were docked against a homology model created from Bos taurus α-L-fucosidase considering catalytic N-terminal region of Thermotoga maritima α-L-fucosidase (PDB ID: 2ZXD) as the template. FlexX program was used for docking.14Fig. 2A shows the best docking pose of compound A with the homology model. It shows a hydrogen bonding between C3 hydroxy group of A and Q245 residue along with a couple of hydrophobic interactions with W248 (Fig. 2B). A π-stacking was also observed between the N-benzyl functional group of A and W20 residue. Interestingly compound B binds to the homology model via two hydrogen bonding interactions with H24 and L226 residues, two hydrophobic interactions between the imide ring of B with W20 and Y33 residues at the active site of the model. Finally a cation–pi interaction with residues H24 and π-stacking with W20 was also observed.
 | ||
| Fig. 2 2-Dimensional view of the representative poses of A and B with respect to the homology model of Bos taurus α-L-fucosidase. Probable interacting residues within the 5 Å of the docked compounds are marked. | ||
Fig. 3A depicts the docking of 4b, 5c and 6a with Bos taurus α-L-fucosidase. The best FlexX docking pose indicated probable hydrogen bonds between the azobenzofuran ring of compound 4b (green) and K226, K224, R208, D174, Y126 and H83 residues of Bos taurus α-L-fucosidase. A likely pi–pi stacking interaction between benzothiophene ring and W20 is also shown. Fig. 3B provides the best pose of docking of compound 5c to Bos taurus α-L-fucosidase. The 2-thiohydantoin ring of compound 5c (purple) forms 10 hydrogen bonds with K226, W20, H83, Y126, D174 and R208 in the active site. Finally Fig. 3C shows the best FlexX derived pose of docking of compound 6a to Bos taurus α-L-fucosidase. The hydantoin moiety of compound 6a (grey) forms 3 hydrogen bonds with K141 and H83 in the active site. Nitrobenzene ring forms a possible hydrophobic pi-stacking interaction with W20. Nitro group of the nitrobenzene ring forms 4 hydrogen bonds with Y126, H7 and K224.
 | ||
| Fig. 3 Structural views of the representative poses of candidate compounds with respect to Bos taurus α-L-fucosidase. Three-dimensional views of representative docking poses of compounds 4b, 5c and 6a (panel A, B, and C, respectively) derived from FlexX program are shown in cartoon modes. Probable interacting residues within the 5 Å of the docked compounds are marked in orange. Images were generated using the CHIMERA software.15 | ||
A closer look at the docking structure of A with the homology model of Bos taurus α-L-fucosidase indicated multiple interactions of phenyl substitution of the exocyclic amine with several amino acid residues on the enzyme (Fig. 2A). The hydrogen bonding with exocyclic hydroxyl group further strengthens the interaction. Similar interaction was evoked by the arylidene moiety of 5c and 6a (which were inspired from A) as they mimicked the benzylidene moiety of A. Additionally when the cyclopentane ring got replaced with hydantoin and thiohydantoin moieties, the nitrogen functionalities present in the new ring systems enhanced the binding interactions (Fig. 3A). In case of B, the pthalimide functionality generated substantial binding interactions as depicted in Fig. 2B. Hence we felt it was pragmatic to modify it bioisosterically to the novel pyridofuranone moiety containing the arylidene functionality (4b) that could enhance the interactions further. This was supported from the above docking experiments.
For the better understanding of the suitability of the new scaffolds 4b, 5c and 6a over A and B, we have quantified the binding strength of the molecules against Bos taurus α-L-fucosidase as depicted in Table 1. As the value suggests compounds, 4b and 6a exhibited much stronger binding against the enzyme compared to A and B (Table 1).
| Compound # | Compound 4b | Compound 5c | Compound 6a | Compound A | Compound B |
|---|---|---|---|---|---|
| Bos taurus α-L-fucosidase-docking score | −18.9486 | −9.4218 | −22.1022 | −14.6481 | −10.2268 |
The 2D structures of the compounds were built and various 3D conformation was searched using conformational search tool. The conformation having least energy was selected for docking studies. For docking studies both the modelled structure and template structure (PDB ID: 2ZX5) were aligned and the complex ligand in the crystal structure (PDB ID: 2ZX5) were used as the binding site. Protein preparation was performed using FlexX prepare receptor wizard and finally the docking was done using FlexX program with default parameters.
Next, in a bid to understand the physicochemical indications rudimentary to the design we computed few relevant medicinal chemistry descriptors of 4b, 5c and 6a along with A and B (Table 2).
| Compounds | log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P P |
Mol. wt | HBD | HBA | TPSA | No. of rotatable bonds | Solubility (logSw) [mol L−1] |
Lipinski drug like |
|---|---|---|---|---|---|---|---|---|
| A | 0.54 | 237.30 | 4 | 4 | 72.72 | 3 | −1.07014 | 1 |
| B | 0.18 | 276.29 | 3 | 5 | 89.87 | 1 | −1.7297 | 1 |
| 4b | 3.61 | 309.35 | 2 | 3 | 55.4 | 1 | −5.7326 | 1 |
| 5c | 1.869 | 289.18 | 2 | 2 | 73.22 | 1 | −4.6118 | 1 |
| 6a | 1.17 | 233.18 | 2 | 2 | 104.0 | 2 | −3.2700 | 1 |
From the information above it was revealed that all the compounds adhere to the Lipinsky's rules (Table 1). The TPSA of all the compounds are <140 Å2 thereby indicating good permeability of cell membranes. Interestingly the designed compounds 4b, 5c and 6a has logP in the range of 1.1–3.6 (Table 2) which indicated better membrane absorption and binding ability to the enzyme and poor aqueous solubility when compared with A and B. Low logSw of compounds of 4b, 5c and 6a indicated that substituting the aryl or heteroaryl appendages to the central motif of these compounds with polar functionalities may improve the overall solubility (Table 2).
The analyses and comparison of the binding results and the physicochemical properties of A/B and 4b, 5c and 6a encouraged us to synthesize the library of compounds with hydantoin, thiohydantoin and furopyridinedione motifs and subsequently screen them against bovine-α-L-fucosidase.
Chemistry
With the molecular design rationalized through docking experiments, we decided to obtain a library based on 4b, 5c and 6a. We envisioned Knoevenagel condensation reaction between the central motifs i.e. hydantoin, thiohydantoin and furopyridinedione with appropriate aldehydes, as a suitable strategy to afford the desired compounds. Accordingly diverse bases viz. morpholine, piperidine, triethyl amine, diisopropyl ethylamine and pyrrolidine in variety of solvents such a methanol, ethanol, isopropanol, dimethyl formamide and acetonitrile were screened in the reaction of hydantoin, thiohydantoin and furopyridinedione with 3,5-difluorobenzaldehyde. Ethanol with piperidine provided the best condition for the Knoevenagel reaction yielding 4c, 5b and 6c in 75–92% yield. Incidentally the final product precipitated from the reaction mixture and could be purified by multiple diethyl ether wash. Hence, this facile optimized protocol enabled us to generate the library of molecules (4, 5 and 6a–f) through combinatorial synthesis.Accordingly, the Knoevenagel reaction was conducted in parallel in custom made reaction chambers with pre-tared vials with ethanol (EtOH) solution of furopyridinedione (0.24 M), thiohydantoin (0.34 M) and hydantoin (0.4 M) at 80 °C (Scheme 1). On completion of the reaction final products precipitated and the supernatant ethanol was removed. The precipitate was subsequently washed (twice) with diethyl ether. The whole operation was conducted in parallel using Tecan Evo Freedom liquid handling system with robotics arms. The vials were dried in genevac and were weighed to determine the final yield. A mother plate was prepared with the samples at 50 μM concentration in DMSO, for in vitro screening (Scheme 1). The yield of the compounds ranged from 75–98%. The compounds were thoroughly characterized by 1H, 13C and high resolution mass spectroscopy.
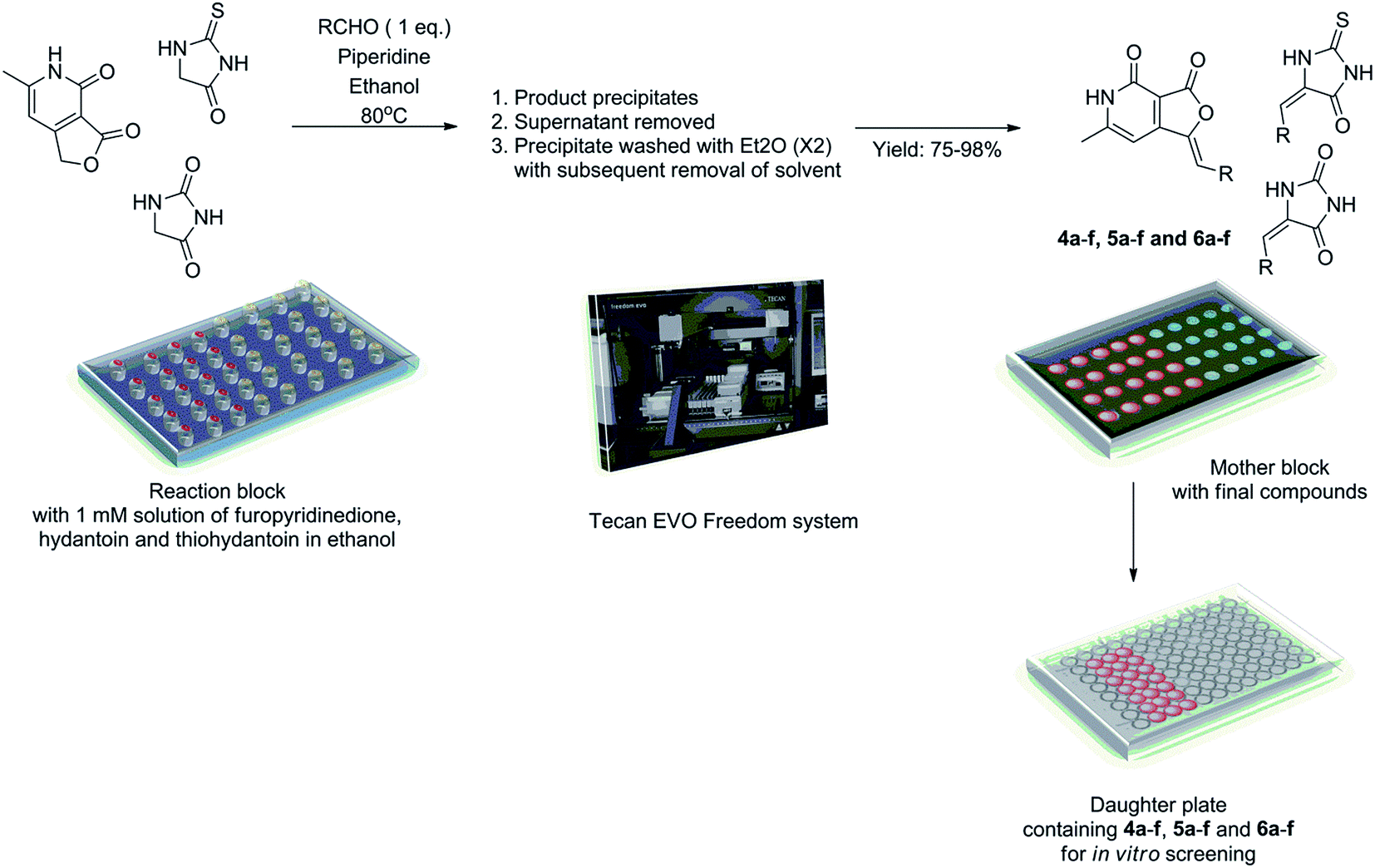 | ||
| Scheme 1 Combinatorial synthesis of the library of compounds 4a–f, 5a–f and 6a–f. | ||
Pharmacological studies
| Entry | Compound # | Compounds | IC50 against α-fucosidasea (bovine kidney) in μM |
|---|---|---|---|
| a Concentration of compound that reduced enzyme activity by 50%.b The concentration was observed in our laboratory assay method. | |||
| 1 | 4a |  |
24.9 |
| 2 | 4b | 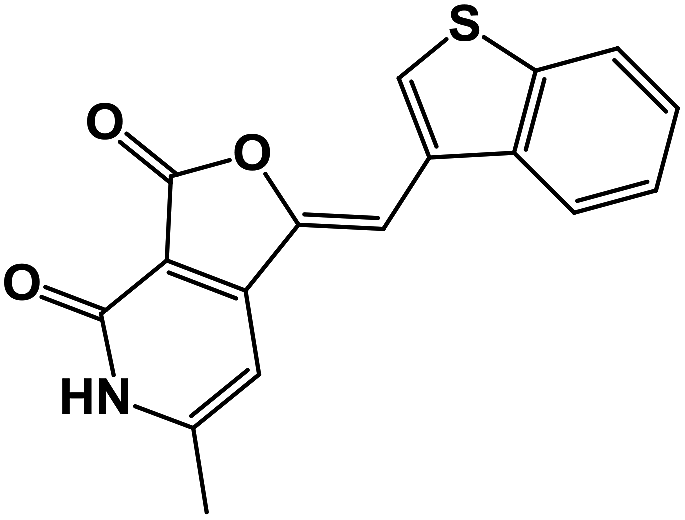 |
21.3 |
| 3 | 4c |  |
>100 |
| 4 | 4d |  |
1.8 |
| 5 | 4e | 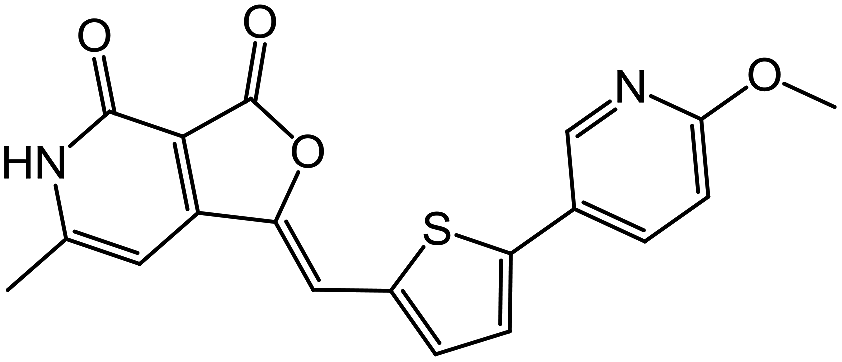 |
0.68 |
| 6 | 4f |  |
2.9 |
| 7 | 5a |  |
18.7 |
| 8 | 5b |  |
43.6 |
| 9 | 5c |  |
24.1 |
| 10 | 5d |  |
28.3 |
| 11 | 5e | 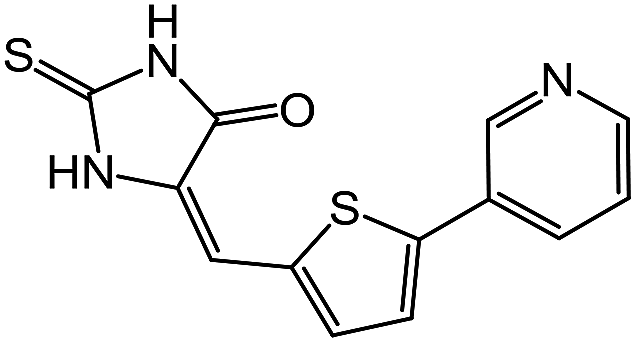 |
24.2 |
| 12 | 5f |  |
31.2 |
| 13 | 6a |  |
6.4 |
| 14 | 6b | 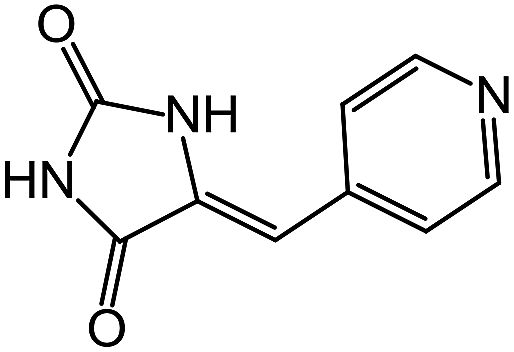 |
48.2 |
| 15 | 6c |  |
50.8 |
| 16 | 6d |  |
>100 |
| 17 | 6e |  |
>100 |
| 18 | 6f |  |
19.1 |
| 19b | Swainsonine | 27.5 | |
Structure activity relationship
α-L-Fucosidase inhibitory activity results obtained for our compounds revealed that the central moiety A and the aromatic or heteroaromatic appendage B influenced the primary structure activity relationship within the library (Fig. 4). The methylene spacer between segment A and B apparently induce no influence in the activity of the molecule. From the inhibition results in Table 2, it is quite clear that: (a) the molecules with furopyridinedione in segment A i.e. 4a–4f (other than 4c), are significantly more active than their hydantoin and thiohydantoin counterparts; (b) the substituted thiophene derivatives (in segment B) such as 4d–4f demonstrated substantially improved activity over their other aromatics and heteroaromatic analogs (4a–4c). Compound 4e demonstrated the highest inhibitory effect against α-L-fucosidase with IC50 concentration of 0.68 μM. This is far better than the reference standard inhibitor, swainsonine (27.5 for α-L-fucosidase) (Table 3).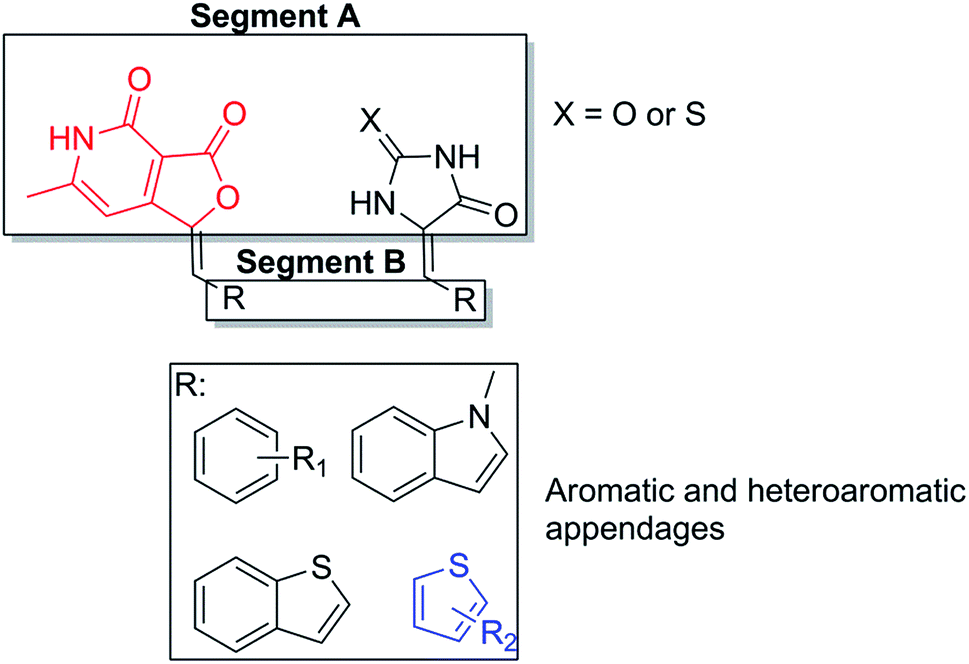 | ||
| Fig. 4 Structure activity relationship studies of the library of molecules against α-L-fucosidase (bovine kidney origin). | ||
Interestingly similar trend involving substituted thiophene was also observed with the thiohydantoin compounds, where 5c–5e exhibited better inhibitory potency over 5a, b and f.
Among the hydantoin analogs, 6a–6f (Table 3) there was no substantial inhibitory activity. Only p-nitrophenyl derivative 6a, was most active (IC50 of 6.4 μM). However the toxic legacy of nitro functionalities in drug discovery made it less promising. The fact that the hydantoin analogs did not possess the thiophene moiety in segment B, could be a reason for their inactivity.
Thorough analysis of the structure activity relationship further emphasized that the pyridone moiety and the substituted thiophene component complement each other in strengthening the binding with the enzyme. Interestingly the thiophene analogs of thiohydantoin scaffold exhibited better potency over the other compounds, though their overall activity was ∼20 times lesser than the thiophene analogs of pyridone scaffold. This further emphasized the importance of substituted thiophene and the pyridone scaffold in the SAR of our library.
Reaction kinetics study
Inhibitory kinetic assay of the most potent compound 4e was performed to identify the mode of inhibition of the most potent compound 4e against α-L-fucosidase. These inhibition kinetic studies were determined by graphical means applying primary (Lineweaver–Burk) and secondary plots. Fig. 5 illustrates the α-L-fucosidase inhibition kinetic plots for compound 4e (also please refer ESI†). In Fig. 5 it was observed that all the data lines on the Lineweaver–Burk plots of 4e intersected in the second quadrant indicating a mixed type of inhibition. The inhibition mechanism involves the binding of these inhibitors either to α-L-fucosidase alone or the glycosidases + substrate complex. Hence, this sort of inhibition subsidizes two inhibition constants viz. K1 and K2. The constants were determined from the slope of 2° plots and Y intercepts from LB-plot against the concentration of each inhibitor (data not shown in this paper). For compound 4e, the K1 and K2 constants against α-L-fucosidase were observed as 0.63 μM and 0.81 μM. The smaller dissociation constants demonstrate stronger inhibition of enzyme. Such mixed inhibition indicates the presence of an allosteric binding site in the enzyme where 4e may bind to, instead of the primary binding site. One of the ways to confirm this is co-crystalizing 4e with the enzyme followed by single crystal X-ray analysis. This is presently ongoing in our lab.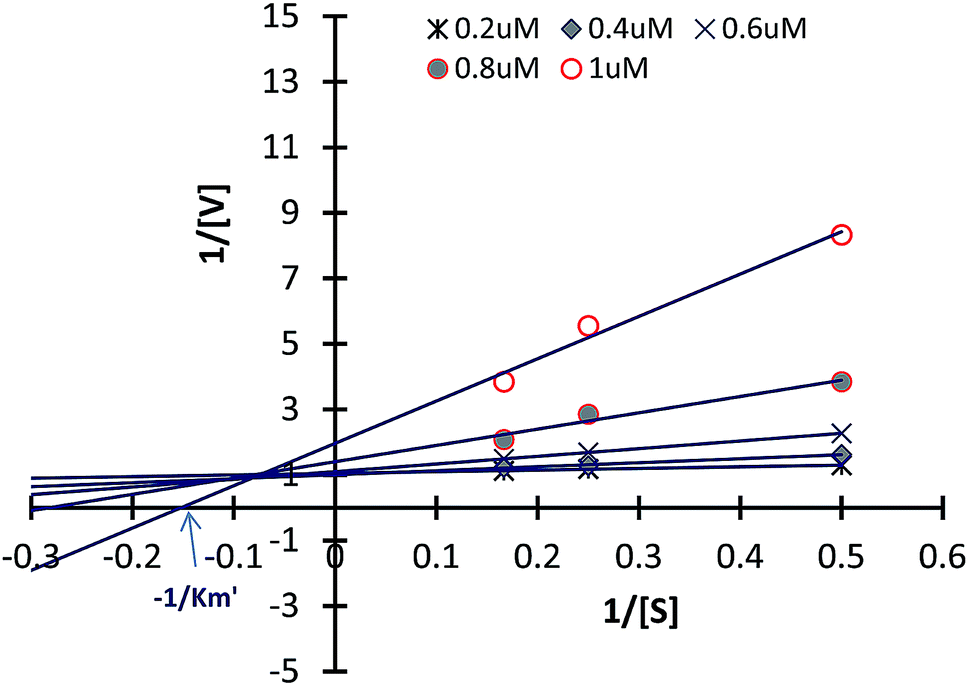 | ||
| Fig. 5 Double-reciprocal (Lineweaver–Burk) plots of α-L-fucosidase (bovine kidney) inhibition kinetics exhibited by compound 4e; α-L-fucosidase (0.5 IU mL−1) was exposed to compound 4e at 37 °C for 10 min, followed by varying concentrations of 4-nitrophenyl α-L-fucoside (PNP-Fuc). | ||
Investigating anti-breast cancer properties of α-L-fucosidase inhibitors
The evolution of glycobiology in last thirty years revealed the role of carbohydrates in many human cancers. Very recently Listinsky et al. reported that fucose is responsible in various activities in breast cancer cells such as haematogenesis, metastasis, tumor invasion and epithelial cell migration.16 Fucose has also been found in few breast cancer biomarkers namely E-selectin.17 Such reports contained illustrative examples, in vitro study against breast cancer cells and clinical data from human screenings as evidence to support the hypothesis.These reports prompted us to investigate the anti-breast cancer properties of our α-L-fucosidase inhibitors. Accordingly we subjected the most potent inhibitors 4a–f, to in vitro cytotoxicity assay against MCF 7 breast cancer cells. To our utmost gratification, among the six compounds screened, the heteroaryl substituted analogs 4a, 4b, 4e and 4f exhibited 50–85% inhibition of the proliferation of MCF 7 (Table 4, entry 2, 3, 6 and 7). Interestingly the most active α-L-fucosidase inhibitor 4e was only second best in deterring the proliferation of MCF 7 cells [62.02%] (Table 4, entry 5) to compound 4f, which exhibits ∼85% inhibition (at par with the reference drug etoposide). The half maximal inhibitory concentration (IC50) of 4f is 2.3 μM.
| Entry | Compound number | MCF7 (MTT, 48 h)a | |
|---|---|---|---|
| %ge inhibition | EC50bc (μM) | ||
| a All data is the mean of one experiment conducted in triplicates.b EC50 of 4f is the mean ± SD of three experiments.c Unless otherwise indicated, the differences were considered to be statistically significant at P < 0.05. The analyses were performed using GraphPad Prism Software version 5.02 (GraphPad Inc., La Jolla, CA, USA). | |||
| 1 | Etoposide | 84.30 | — |
| 2 | 4a | 49.50 | — |
| 3 | 4b | 60.39 | — |
| 4 | 4c | — | — |
| 5 | 4d | — | — |
| 6 | 4e | 62.02 | — |
| 7 | 4f | 84.92 | 2.33 |
Conclusion
Herein we have described discovery of a small heterocycles 4e and 4f, based on furopyridinedione that inhibits α-L-fucosidase (bovine kidney origin) cells with low micromolar IC50. The original library comprising of the 6-methylfuro[3,4-c]pyridine-3, thiohydantoin and hydantoin molecules was designed by intuitive scaffold hopping and bioisosteric modification of known fucosidase and fucosyltransferase inhibitors. Molecular docking exercise among these molecules along with the enzymes rationalized the design. A small library of compounds containing all the three chemotypes was synthesized. They were then screened against α-L-fucosidase (bovine kidney origin) with swainsonine as the reference standard. Furopyridinedione compound 4e, with IC50 ∼ 0.7 μM emerged as the most active compound with potency nearly 40 fold more than the standard inhibitor. Reaction kinetics study indicated it to be a mixed inhibitor. Screening of 4e against healthy mammalian COS cells indicated no cytotoxic effect. Correlation between fucose and breast cancer prompted further screening that resulted in 4a, b, e and f exhibiting substantial inhibition of proliferation of MCF 7 breast cancer cells. All these results indicate that 6-methylfuro[3,4-c]pyridine-3,4(1H,5H)-dione molecules can be considered as promising lead against α-L-fucosidase which in turn can be utilized to develop candidate for breast cancer.Methods
Experimental
Homology modeling
Homology modeling of Bos taurus α-L-fucosidase and subsequent loop modelling were performed using the MODELLER 9.11 package. Final models were subjected to energy minimization cycles in the GROMACS online energy minimization server and/or Schrodinger's restrained minimization program.18,19 Validation of the final models (Fig. S1†) was done using the PROCHECK and Verify_3D structure validation tools.20,21 Root mean square deviations (RMSD) between the models and template structures were calculated using the CHIMERA structure visualization software.15Identification of inhibitor binding domain of α-L-fucosidase
The N- and C-terminal domains of α-L-fucosidase are formed of α + β secondary structure and all beta secondary structure, respectively. Known inhibitors bind to the N-terminal (α/β) domain (residues 1-357) of α-L-fucosidase.In order to ascertain the inhibitor binding domain of α-L-fucosidase, a blind docking approach using PatchDock was employed for docking the dihydrofuro[3,4-c]pyridinone derivative (compound 4b) to full-length bacterial (Thermotoga maritima) α-L-fucosidase (PDB ID: 2ZX5).22 Clustering analysis of the resultant solutions revealed that maximum number of solutions prefers binding of compound 4b at the N terminal domain of 2ZX5 (Fig. S2 in ESI†).
A second blind docking of compound 4b with only the N-terminal domain of 2ZX5 was performed to determine the binding pocket within the N-terminal binding domain. Clustering analysis revealed maximum number of solutions prefer binding of 4b to that pocket of N-terminal 2ZX5, which is known to bind other conventional inhibitor compounds. Thus, blind docking experiments provide confidence to the assumption that 4b and perhaps other similar derivatives may bind to the known inhibitor binding pocket located at N-terminus of α-L-fucosidase.
Molecular docking of candidate compounds to inhibitor binding pocket of α-L-fucosidase
The candidate compounds were docked to the known inhibitor binding pocket of N-terminal 2ZX5 using molecular docking software, viz., FlexX.14FlexX was used to predict the geometry of protein–ligand complex using a hybrid approach for placing ligand fragments in the active site. This hybrid approach combines the best of classic triangle matching and single interaction scan. A more negative value of FlexX score indicates better fit of ligand in the protein active site.
In vitro inhibition screening assays of α-L-fucosidase
The effectiveness of the compounds on inhibition of α-L-fucosidase (bovine kidney origin) was assessed in 96-well plates using the respective substrate and buffering system according to the procedure reported by Ferreres et al.23 Prior to assays, all the test compounds were dissolved in dimethylsulfoxide (DMSO) and eventually diluted to attain the desired concentration. The absorbance was measured spectrophotometrically at 400 nm (Epoch reader; version 2.00.18). The decrease in absorbance (ΔA) was compared with that of control (buffer instead of test compound) to compute the inhibitory profile of enzyme. The data used for the determination of IC50 concentrations were fitted by non-linear regression fitting and the variance analysis was carried out by using MINITAB 15 software (trial version). The concentration of inhibition required for 50% of α-glucosidase activity under the assay conditions was defined as the IC50 value. The half maximal inhibitory (IC50) concentrations were determined from two independent assays, performed in duplicate.| Inhibition (%) = (ΔAcontrol − ΔAsample)/ΔAcontrol × 100%. |
One unit (IU) is defined as the amount of enzyme (α-glucosidase/α-mannosidase/α-L-fucosidase) which produces 1 μmol of PNP (p-nitro phenol) per min at 37 °C under the respective reaction conditions described below.
For AFI, 100 μL of p-nitrophenol-α-L-fucoside substrate (2 mM, PNP-Fuc dissolved in 2 mM sodium citrate buffer at a pH of 4.2) and different concentrations (10–50 μM) of the test compounds were taken in 96-well plates. Then the final volume of the reaction mixture was made up to 200 μL with 2 mM sodium citrate buffer (pH 4.2). The hydrolytic reaction was commenced by the addition of α-L-fucosidase enzyme (0.5 IU mL−1) (obtained from Sigma Aldrich, Bangalore) and the plates were incubated at 37 °C for 15 min. The reaction was terminated by the addition of 50 μL of 2 N Na2CO3 solution.
Inhibition-kinetics
The kinetic mode of inhibition of the highest active compounds against three α-L-fucosidase was determined as said above by preparing a series of test solutions in which the concentration of the respective substrate was varied in the presence of different concentrations of the inhibitors (10–50 μM). The mode of inhibition (i.e. competitive, non-competitive, uncompetitive or mixed-type) of the screened compounds was determined on the basis of the inhibitory effects on Km (dissociation constant) and Vmax (maximum reaction velocity) of the enzyme.24 These studies were computed using the primary (Lineweaver–Burk plot) plots, which are the double reciprocal plots of enzyme reaction velocities (1/V) versus substrate concentrations (1/[S]). Analysis of the same data by secondary plots of slope versus [inhibitor] and Y-intercept versus [inhibitor] were also performed. The Lineweaver–Burk (LB) equation follows as: 000 units per mL, streptomycin-10000 μg mL−1, Gibco USA). Growth medium (pH 7.4) was prepared by adding 5% heat inactivated FBS (Fetal Bovine Serum, Gibco, USA) and stored at 4 °C. The medium was changed 10–18 h prior to experiment, and cells were confluent at the time of experimentation. Cells were dislodged from flasks by gentle trypsinization containing 0.25% trypsin. MCF7 cells were regularly sub-cultured thrice a week in a seeding density of 40000–50000 cells per cm2 area.000 cells/100 μL of media were seeded per well and allowed to stretch and adhere overnight at 37 °C. Following day media was removed and 100 μL fresh media was added to the grown cells. Adhered cells were then treated with the compounds 4a–f dissolved in DMSO in triplicates at a standard concentration of 50 μM for 48 hours. Cytotoxic effect was evaluated using ability of live cells to cleave MTT ((3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide)) (Sigma-Aldrich, St. Louis, MO, USA), into formazan crystals. Post 48 hours of treatment, 10 μL of MTT (5 mg mL−1 in PBS) was added to the cells and incubated for 4 hours in dark at 37 °C. The violet colored crystals formed were then dissolved in 100 μL DMSO solvent (dimethyl sulfoxide, Sigma-Aldrich, St. Louis, MO, USA). The colorimetric assay was read measured by a MultiMode Plate Reader (Bio-Rad) at 595 nm. Percentage of inhibition of drug was calculated by following formula:
000 units per mL, streptomycin-10000 μg mL−1, Gibco USA). Growth medium (pH 7.4) was prepared by adding 5% heat inactivated FBS (Fetal Bovine Serum, Gibco, USA) and stored at 4 °C. The medium was changed 10–18 h prior to experiment, and cells were confluent at the time of experimentation. Cells were dislodged from flasks by gentle trypsinization containing 0.25% trypsin. MCF7 cells were regularly sub-cultured thrice a week in a seeding density of 40000–50000 cells per cm2 area.000 cells/100 μL of media were seeded per well and allowed to stretch and adhere overnight at 37 °C. Following day media was removed and 100 μL fresh media was added to the grown cells. Adhered cells were then treated with the compounds 4a–f dissolved in DMSO in triplicates at a standard concentration of 50 μM for 48 hours. Cytotoxic effect was evaluated using ability of live cells to cleave MTT ((3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide)) (Sigma-Aldrich, St. Louis, MO, USA), into formazan crystals. Post 48 hours of treatment, 10 μL of MTT (5 mg mL−1 in PBS) was added to the cells and incubated for 4 hours in dark at 37 °C. The violet colored crystals formed were then dissolved in 100 μL DMSO solvent (dimethyl sulfoxide, Sigma-Aldrich, St. Louis, MO, USA). The colorimetric assay was read measured by a MultiMode Plate Reader (Bio-Rad) at 595 nm. Percentage of inhibition of drug was calculated by following formula:| (Average of control O.D595 − average of treated O.D595)/control O.D595 × 100. |
Chemistry
All reactions were carried out in flame-dried pre-tared vials magnetic stirring. Unless otherwise noted, all experiments were performed under argon atmosphere. All reagents were purchased from Sigma Aldrich, Acros or Alfa Aesar. Solvents were treated with 4 Å molecular sieves or sodium and distilled prior to use. 1H NMR and 13C NMR spectra were recorded with tetramethylsilane (TMS) as internal standard at ambient temperature unless otherwise indicated on a Varian 300/400 and JEOL JNM-ECX500 MHz at 500 MHz for 1H NMR and 100 MHz for 13C NMR. Chemical shifts are reported in parts per million (ppm) and coupling constants are reported as Hertz (Hz). Splitting patterns are designated as singlet (s), broad singlet (bs), doublet (d), triplet (t). Splitting patterns that could not be interpreted or easily visualized are designated as multiple (m). The Mass Spectrometry analysis was done on the 6540 UHD Accurate-Mass Q-TOF LC/MS system (Agilent Technologies) equipped with Agilent 1290 LC system obtained by the Dept. of Chemistry, School of Natural Sciences, Shiv Nadar University, Uttar Pradesh 201314, India.Acknowledgements
We thank Shiv Nadar University, Department of Biotechnology (Breast Cancer-Pilot Grant) and CSIR-Indian Institute of Chemical Biology for the financial support. SC acknowledges CSIR network project (BSC0121) and ST acknowledges DBT for funding. SG is supported by UGC fellowship grant.Notes and References
-
(a) P. E. Goss, M. A. Baker, J. P. Carver and J. W. Dennis, Clin. Cancer Res., 1995, 1, 935–944 CAS
; (b) A. Herscovics, Biochim. Biophys. Acta, 1999, 1473, 96–107 CrossRef CAS PubMed
- B. Winchester, Glycobiology, 2005, 15, 1R–15R CrossRef CAS PubMed
- J. Van den Brink and R. P. de Vries, Appl. Microbiol. Biotechnol., 2011, 91, 1477–1492 CrossRef PubMed
- M. Günl, L. Neumetzler, F. Kraemer, A. de Souza, A. Schultink, M. Pena, W. S. York and M. Pauly, Plant Cell, 2011, 23, 4025–4040 CrossRef PubMed
- T. K. Tolle, D. Glebe, M. Linder, D. Linder, S. Schmitt, R. Geyer and W. H. Gerlich, J. Virol., 1998, 72, 9978–9985 CAS
- T. W. Liu, C. W. Ho, H. H. Huang, S. M. Chang, S. D. Popat, Y. T. Wang, M. S. Wu, Y. J. Chen and C. H. Lin, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14581–14586 CrossRef CAS PubMed
- J. C. Michalski and A. Klein, Biochim. Biophys. Acta, Mol. Basis Dis., 1999, 1455, 69–84 CrossRef CAS
- D. G. Hildebrand, S. Lehle, A. Borst, S. Haferkamp, F. Essmann and K. Schulze-Osthoff, Cell Cycle, 2013, 12, 1922–1927 CrossRef CAS PubMed
- J. Intra, et al., Gene, 2007, 392, 34–46 CrossRef CAS PubMed
-
(a) L. V. Hooper, et al., Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9833–9838 CrossRef CAS PubMed
- G. A. Patani and E. J. LaVoie, Chem. Rev., 1996, 96, 3147–3176 CrossRef CAS PubMed
- C. Ballatore, D. M. Huryn and A. B. Smith, ChemMedChem, 2011, 8, 385–395 CrossRef PubMed
- E. Isabel, et al., Bioorg. Med. Chem. Lett., 2011, 21, 920–923 CrossRef CAS PubMed
- B. Kramer, M. Rarey and T. Lengauer, Proteins: Struct., Funct., Genet., 1999, 37, 228–241 CrossRef CAS
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed
- J. J. Listinsky, G. P. Siegal and C. M. Listinsky, Am. J. Transl. Res., 2011, 3, 292–322 CAS
- Schrödinger Release 2013-3: LigPrep, version 2.8, Schrödinger, LLC, New York, NY, 2013 Search PubMed
- E. Lindahl, C. Azuara, P. Koehl and M. Delarue, Nucleic Acids Res., 2006, 34, W52–W56 CrossRef CAS PubMed
- Schrödinger Release 2013-3: Maestro, version 9.6, Schrödinger, LLC, New York, NY, 2013 Search PubMed
- R. A. Laskowski, M. W. MacArthur, D. S. Moss and J. M. Thornton, J. Appl. Crystallogr., 1993, 26, 283–291 CrossRef CAS
- D. Eisenberg, R. Luthy and J. U. Bowie, Methods Enzymol., 1997, 277, 396–404 CAS
- D. Schneidman-Duhovny, Y. Inbar, R. Nussinov and H. J. Wolfson, Nucleic Acids Res., 2005, 33, W363–W367 CrossRef CAS PubMed
- F. Ferreres, A. Gil-Izquierdo, J. Vinholes, S. T. Silva, P. Valentao and P. B. Andrade, Food Chem., 2012, 134, 894–904 CrossRef CAS PubMed
- B. T. Burlingham and T. S. Widlanski, J. Chem. Educ., 2003, 80, 214–218 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra24939f |
| ‡ Equal contributors. |
| This journal is © The Royal Society of Chemistry 2017 |