
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ruthenium-catalyzed solvent-free conversion of furfural to furfuryl alcohol†
József M. Tukacsa,
Márton Bohusa,
Gábor Dibób and
László T. Mika *a
*a
aDepartment of Chemical and Environmental Process Engineering, Budapest University of Technology and Economics, Budapest, Hungary. E-mail: laszlo.t.mika@mail.bme.hu; Fax: +36 1 463 3197; Tel: +36 1 463 1263
bJános Selye University, Faculty of Education, Department of Chemistry, Bratislavská str. 3322, Komárno SK-94501, Slovakia
First published on 13th January 2017
Abstract
Bidentate phosphine-modified Ru-based homogeneous catalyst systems were developed for solvent-free conversion of furfural to furfuryl alcohol as a versatile biomass-based C5-platform molecule. The effect of the ligand structure and ligand concentration, the operating pressure and the reaction temperature on the catalyst's activity were studied in details. Under optimized conditions complete conversion of furfural to furfuryl alcohol was achieved in the absence of any added solvent and without any by-product formation. The maximum turnover frequency (6273 h−1) and 100% atom economy were obtained by using homogeneous Ru/Ph2P(CH2)4PPh2 catalyst, which was recyclable for twelve consecutive runs without affecting the catalytic performance of the catalyst.
Introduction
The chemical industry, which produces enormous amounts of value-added chemicals, highly depends on fossil carbon resources. The refinery processing of crude oil continuously provides bulk chemicals for both petrochemical and fine chemical industries. Although, it is difficult to estimate the rate of depletion of crude oil, which is the main source of carbon-based building blocks, the environmental and sustainability issues have drawn increasing attention to the identification and application of alternative basic chemicals. Since biomass1 and biomass-based wastes2 are globally available, these renewable resources represent an attractive solution for the production of synthetic building blocks in the future. Recently, the competitive application of biomass-based alternatives could be limited by dramatically changing price of crude oil, after its depletion technologies that could adapt alternative feedstocks will represent one way to produce carbon-based end products.The rapidly expanding research activity on biomass conversion has identified several renewable platform molecules e.g. 5-hydroxymethyl furfural,3 levulinic acid,4 γ-valerolactone (GVL)5,6 which could replace fossil based ones in petrochemical industry. Since furfural (FAL) has also been selected as a biomass-based basic chemical,7 its subsequent transformation has received an utmost importance in the last decade.
One of the most important derivatives of furfural is furfuryl alcohol (FOL), which is an another valuable C5-platform molecule. It can be used for the production of thermostatic resins, plasticizers, adhesives, fragrances, diols, tetrahydrofurfuryl alcohol, 2-methyltetrahydrofuran (Scheme 1).8,9 While its hydrolysis results in the formation of levulinic acid (LA),8a,10,11 ethyl levulinate can be obtained by its ethanolysis.12 It is important to emphasize that LA is a feedstock for the synthesis of GVL that can be used for the production of energy and carbon-based chemicals5 including fuels,13 fuel additives,5 alkanes,14 fine chemicals,15 or used as a green solvent.16 Consequently, an efficient solvent-free conversion of FAL to FOL can be the initial step in the environmentally benign and more sustainable production of biomass-based C5-building blocks.
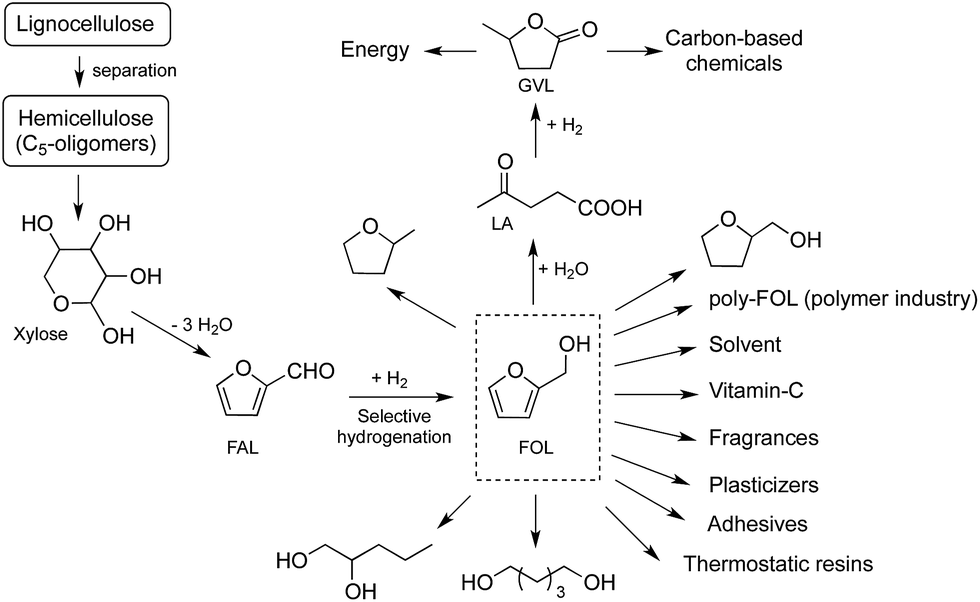 | ||
| Scheme 1 Reaction scheme for conversion of hemicellulose to furfuryl alcohol (FOL) and its selected applications. | ||
Obviously, the most effective protocol to produce furfuryl alcohol is the hydrogenation of furfural by using a selective catalyst. The reduction can be carried out in the presence of either heterogeneous7 or homogeneous catalysts. The heterogeneous copper-chromite catalyst, operating in either gas or liquid phase, has been exclusively used for selective hydrogenation of FAL at industrial scale for over six decades. Its selectivity to FOL is 98% in liquid phase, and 35–98% in gas phase. However, due to the catalyst's toxicity and carcinogenic potential, several attempts have been made to introduce environmentally benign alternatives.17 Accordingly, several heterogeneous systems were invented based on copper,8b,9b,18 cobalt,9a palladium,19 platinum,20 and nickel21 operating in temperature range of 100–200 °C. Expectedly, the transition metal-based homogeneous catalyst systems exhibits significantly higher activity, selectivity, and better mass and heat transfer efficiency under milder conditions as compared to heterogeneous ones. Paganelli et al. reported the aqueous phase reduction of FAL by using a recyclable dihydrothioctic acid-modified Rh-based system with high conversion (70–99%) and outstanding selectivity (>98%). However, the turnover frequencies (TOFs) were rather low (20–60 h−1).22 It has been recently shown that RuCl2(PPh)3, in the presence of acetic acid, can reduce FAL with TOF of 32 h−1 and conversion of 93.6%.23 Similar activity for FAL hydrogenation was detected for Ru-tris(2,2,6,6-tetramethyl-3,5-heptanedionate)3 catalyst with conversion of 86%.24 Rothenberg et al. introduced an NHC-based Ru-catalyst for the conversion of FAL to FOL by using iPrOH as a hydrogen source. Although excellent selectivities were achieved at 60 °C, the addition of base e.g. KOH and KOtBu were necessary, which resulted in separation problems.25
We report here the development of a selective, solvent-free hydrogenation system for conversion of furfural to furfuryl alcohol using a recyclable bidentate phosphine modified ruthenium catalyst (Scheme 2).
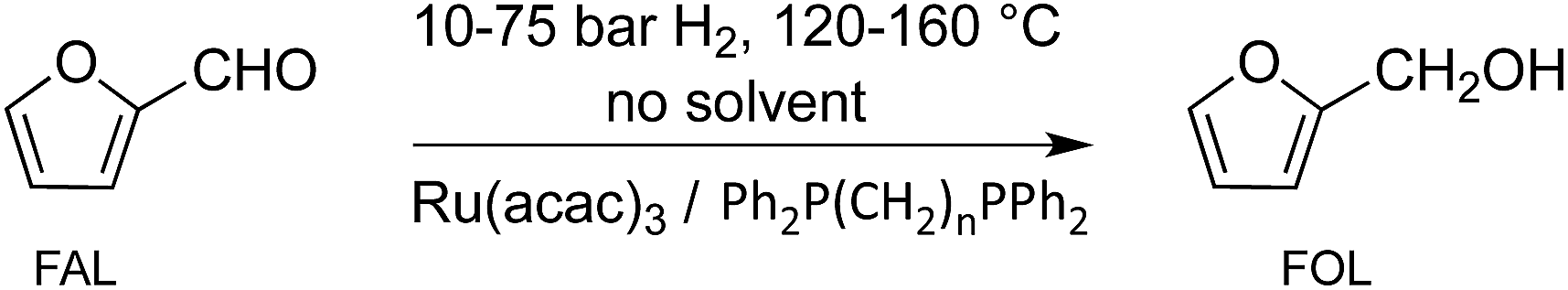 | ||
| Scheme 2 Conversion of FAL to FOL; n = 2–6. | ||
Results and discussion
It has been known that the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond can be selectively reduced in the presence of sulfonated phosphine-modified ruthenium catalysts either under aqueous conditions or without any applied solvent.14,26 It has to be emphasized that solvent-free liquid phase selective homogeneous conversion of FAL to FOL can open a greener and safer way to produce biomass-based furfuryl alcohol. Firstly, we attempted to reduce FAL (30 mL, 0.36 mol) in the presence of a catalyst in situ formed from Ru(acac)3 (0.047 mmol) and TPPTS (0.5 mmol) at 90 bar H2 and 140 °C for 3 h.27 In the absence of any solvent, low conversion and TOF were obtained (Table 1, entry 1). Although, by introducing nBuP(C6H4-m-SO3Na)2 (Bu-DPPDS) ligand, three times higher activity was achieved under identical conditions. However both yields of FOL and TOF values remained moderate (Table 1, entries 2–3). It is important to note that no by-product formation i.e. self-resinification8a,28 was detected. Li et al. revealed that FAL was selectively converted to FOL in the presence of RuCl2(PPh)3 catalyst in EtOAc as a solvent under p(H2) = 23 bar at 70 °C.23 After reproducing the published results (Table 1, entry 4), we evaluated the reduction of FAL in the presence of a bidentate 1,4-bis(diphenylphosphino)butane (DPPB) ligand. Since it was reported that, by replacement of a monodentate P-ligands to bidentate ones, the activity of Ru-based hydrogenation catalyst systems was significantly enhanced.29 While no reaction was detected at 70 °C, full conversion was achieved at 140 °C for 1.3 h with TOF = 2895 h−1 and without by-product formation (Table 1, entry 5). So far, to our best knowledge, no additional solvent-free selective homogeneous reduction of FAL to FOL has been reported yet.
O bond can be selectively reduced in the presence of sulfonated phosphine-modified ruthenium catalysts either under aqueous conditions or without any applied solvent.14,26 It has to be emphasized that solvent-free liquid phase selective homogeneous conversion of FAL to FOL can open a greener and safer way to produce biomass-based furfuryl alcohol. Firstly, we attempted to reduce FAL (30 mL, 0.36 mol) in the presence of a catalyst in situ formed from Ru(acac)3 (0.047 mmol) and TPPTS (0.5 mmol) at 90 bar H2 and 140 °C for 3 h.27 In the absence of any solvent, low conversion and TOF were obtained (Table 1, entry 1). Although, by introducing nBuP(C6H4-m-SO3Na)2 (Bu-DPPDS) ligand, three times higher activity was achieved under identical conditions. However both yields of FOL and TOF values remained moderate (Table 1, entries 2–3). It is important to note that no by-product formation i.e. self-resinification8a,28 was detected. Li et al. revealed that FAL was selectively converted to FOL in the presence of RuCl2(PPh)3 catalyst in EtOAc as a solvent under p(H2) = 23 bar at 70 °C.23 After reproducing the published results (Table 1, entry 4), we evaluated the reduction of FAL in the presence of a bidentate 1,4-bis(diphenylphosphino)butane (DPPB) ligand. Since it was reported that, by replacement of a monodentate P-ligands to bidentate ones, the activity of Ru-based hydrogenation catalyst systems was significantly enhanced.29 While no reaction was detected at 70 °C, full conversion was achieved at 140 °C for 1.3 h with TOF = 2895 h−1 and without by-product formation (Table 1, entry 5). So far, to our best knowledge, no additional solvent-free selective homogeneous reduction of FAL to FOL has been reported yet.
| Entry | [Ru] × 103 (mol dm−3) | Ligand | [Ligand] × 103 (mol dm−3) | T (°C) | p(H2) (bar) | Time (h) | Conversion (%) | TON | TOF (h−1) |
|---|---|---|---|---|---|---|---|---|---|
| a Solvent EtOAc (20 mL), catalyst precursor: RuCl2(PPh3). | |||||||||
| 1 | 1.56 | TPPTS | 15.6 | 140 | 90 | 3 | 13.8 | 1054 | 351 |
| 2 | 1.56 | Bu-DPPDS | 15.6 | 140 | 90 | 3 | 38.7 | 2956 | 985 |
| 3 | 1.56 | Bu-DPPDS | 15.6 | 140 | 90 | 5 | 65.8 | 5027 | 1005 |
| 4 | 3.07a | PPh3 | — | 70 | 30 | 3 | 98.0 | 33 | 11 |
| 5 | 3.10 | DPPB | 31.0 | 140 | 30 | 1.3 | >99.9 | 3764 | 2895 |
| 6 | 3.10 | DPPB | 31.0 | 140 | 75 | 1.3 | >99.9 | 3764 | 2895 |
| 7 | 3.10 | DPPB | 31.0 | 140 | 50 | 1.3 | >99.9 | 3764 | 2895 |
| 8 | 3.10 | DPPB | 31.0 | 140 | 25 | 1.3 | >99.9 | 3764 | 2895 |
| 9 | 3.10 | DPPB | 31.0 | 140 | 15 | 1.3 | 89.0 | 3350 | 2576 |
| 10 | 3.10 | DPPB | 31.0 | 140 | 10 | 1.3 | 59.7 | 2220 | 1708 |
| 11 | 3.10 | DPPB | 31.0 | 140 | 25 | 0.8 | >99.9 | 3764 | 4705 |
| 12 | 3.10 | DPPE | 31.0 | 140 | 25 | 1.5 | <1 | <1 | <1 |
| 13 | 3.10 | DPPPr | 31.0 | 140 | 25 | 1.5 | 6.3 | 237 | 182 |
| 14 | 3.10 | DPPPe | 31.0 | 140 | 25 | 1.3 | 90.4 | 3402 | 2617 |
| 15 | 3.10 | DPPH | 31.0 | 140 | 25 | 1.3 | 24.1 | 903 | 695 |
| 16 | 3.10 | DPPB | 31.0 | 160 | 25 | 0.6 | >99.9 | 3764 | 6273 |
| 17 | 3.10 | DPPB | 31.0 | 120 | 25 | 1.3 | 3.2 | 113 | 86 |
| 18 | 3.10 | DPPB | 31.0 | 130 | 25 | 1.3 | 6.9 | 263 | 202 |
It has been shown that hydrogenation of CO double bonds, by using homogeneous mono or bidentate phosphine-modified Ru-catalysts, showed saturation kinetics for hydrogen pressure.27,30 Therefore, the influence of the hydrogen pressure on the conversion of FAL was studied in the range of 10–100 bar using Ru/DPPB system (Table 1, entries 6–10). Expectedly, significant pressure dependence was revealed below 20 bar and the conversion became independent over 25 bar (Fig. 1). These results are in good agreement with the reported kinetics for the Ru/PPh3 system,23 or for reduction of LA.27 It should be noted that full conversion was achieved under 25 bar of H2 at 140 °C for 50 min (Table 1, entry 11 and ESI Fig. S1†), as well.
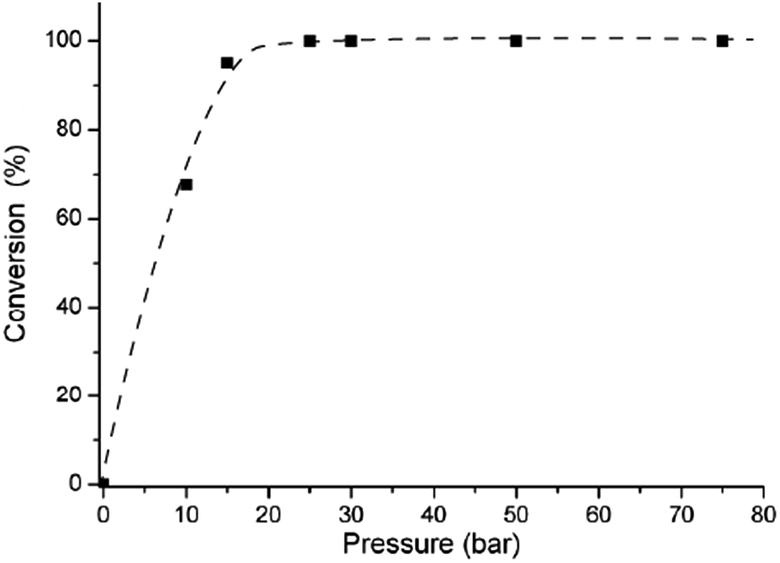 | ||
| Fig. 1 Effect of p(H2) on the conversion of FAL. [Ru] = 3.1 × 10−3 mol dm−3 (0.16 mol%), [DPPB] = 3.1 × 10−2 mol dm−3 (1.6 mol%), 140 °C, t = 1.3 h. | ||
It has been shown for bidentate ligand modified Ru-based catalyst that the size of the chelate ring on the catalytically active hydride species has a significant influence on the activity.29 By varying the number of methylene spacers in the ligand, in case of 1,2-bis(diphenylphoshino)ethane (DPPE) and 1,3-bis(diphenylphoshino)propane (DPPPr) resulted in significantly lower conversion under identical conditions (Table 1, entries 12, 13). 1,5-Bis(diphenylphoshino)pentane (DPPPe) showed similar activity compared to DPPB; however, in case of the incorporation of six methylene groups between the phosphorus atoms (DPPH), moderate conversion was detected (Table 1, entries 14, 15). It is well established that reaction of Ru(acac)3 and BINAP leads to the formation of Ru(BINAP)(acac)2 complexes under hydrogen.31 Since the chelating ring of Ru–BINAP and Ru–DPPB complexes are similar, it is reasonable that the initial step of catalyst activation is formation Ru(DPPB)(acac)2, which turns to hydride form Ru(DPPB)2(H)2 under reaction conditions.32 Due to the six CH2 units, it can also be assumed that DPPH partially acts as a monodentate ligand in the Ru-center.33 In addition, electronic properties of the ligands could have significant effect on the activity as well as an electron donating monodentate trialkyl phosphine could react with FAL as an aldehyde to form an adduct.34 To establish this interaction, 10 μL (0.04 mmol) of PBu3 was mixed with 500 μL (6 mmol) of FAL at room temperature. The 31P-NMR of the reaction mixture established the interaction between PBu3 and FAL by a broad peak at 35.2 ppm. When DPPB (0.038 mmol) was dissolved in 500 μL (6 mmol) of FAL, this interaction cannot be detected by 31P-NMR. Thus, reaction of FAL with a diphenylalkylphosphine subunit is seemingly retarded. To compare the activity of PBu3 modified Ru catalyst with the DPPB analogue, we attempted to reduce 30 mL of FAL in the presence of catalyst system in situ formed from 0.095 mmol Ru(acac)3 and 1.86 mmol of PBu3 under 25 bar at 140 °C. Expectedly, the PBu3 modified system has significantly lower activity (TOF = 684 h−1, ESI Fig. S2†) than DPPB containing one.
The influence of temperature on the activity was monitored by the use of DPPB ligand (Table 1, entries 16–18). Below 130 °C negligible conversions were detected after 1.3 h. Expectedly, at 160 °C complete conversion was obtained for 0.6 h exhibiting TOF of 6273 h−1 as the highest TOF value published for homogeneous reduction of furfural so far.
To achieve further improvement in the TOF by optimizing the ligand concentration in the reaction mixture, we investigated the reduction of FAL by using a Ru/DPPB catalyst in the concentration range of [DPPB] = 15.5 × 10−3 to 37.2 × 10−3 at a constant Ru content of 3.1 × 10−3 mol dm−3. Expectedly, the ligand content had a strong influence on the formation of FOL as shown in Fig. 2. It is in accordance with results published for Ru-catalyzed reduction of CO bond.27
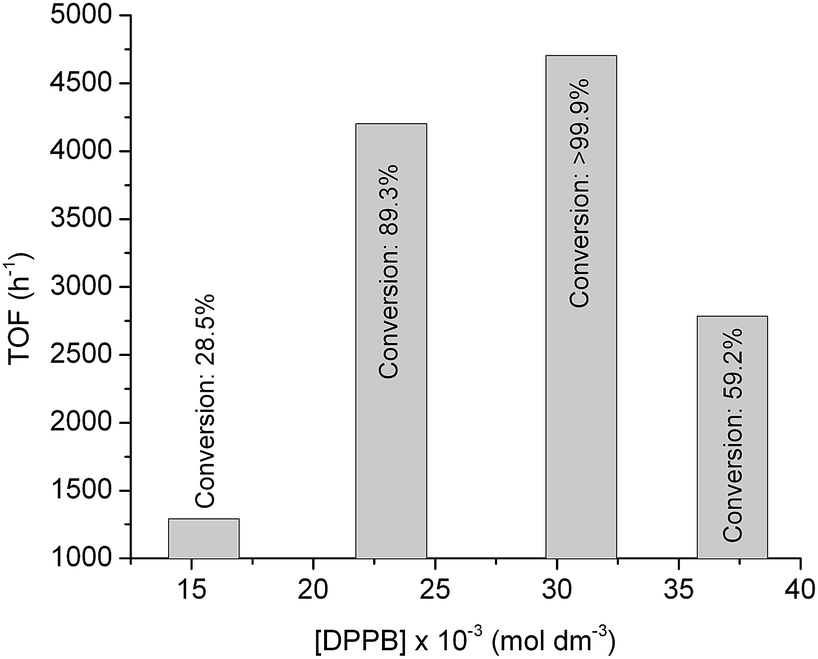 | ||
| Fig. 2 Turnover frequencies of the conversion of conversion of FAL to FOL under 25 bar of H2 at 140 °C for 50 min. [Ru] = 3.1 × 10−3 mol dm−3 (0.16 mol%). | ||
The recyclability is one of the most important issues of a homogeneous catalyst system. Any other solvent-free conversion opens a facile way to separate the homogeneous catalyst from the product by a simple distillation, supposing that the catalyst is stable enough. Therefore we evaluated the possible separation of the catalyst in situ generated form 0.191 mmol Ru(acac)3 and ten-fold excess of DPPB in 30 mL of FAL. Under 25 bar of H2 at 140 °C, a yield of 62.4% was obtained for 0.5 h. When the reaction was completed after 1.3 h, all volatile compounds were removed from the dark brown solution by vacuum distillation (3–5 mm Hg) performed on a 20 cm glass column resulting in a colorless liquid, which was identified as 99.99% FOL by GC-MS. In addition, no phosphorus species was detected by 31P-NMR spectroscopy. The dark brown glue-like distillation residue containing the catalyst, the excess amount of ligands, and the rest of furfuryl alcohol could be completely dissolved in 30 mL of FAL at room temperature. The brown solution was transferred into the reactor followed by pressurizing to 25 bar of H2. The reaction was initiated by increasing the temperature up to 140 °C. Similarly to the workup procedures of the initial run, the second yield was also >99.9% FOL which was unchanged in the next 10 runs (Fig. 3, ESI Table S1†). The conversions were checked by sampling at 0.5 h and it can be stated that no significant changes in the catalyst's activity were observed in the first 12 runs (Fig. 2). The results evidenced, that the improved Ru/BDPP catalyst system was stable enough for successful catalyst recycling including a low-pressure separation step (Fig. 4).
 | ||
| Fig. 3 Effect of p(H2) on the conversion of FAL. [Ru] = 3.1 × 10−3 mol dm−3 (0.16 mol%), [DPPB] = 3.1 × 10−2 mol dm−3 (1.6 mol%), 140 °C, t = 1.3 h. | ||
 | ||
| Fig. 4 Catalyst recycling set-up. | ||
Experimental
Chemicals (ESI†) were purchased from Sigma-Aldrich Ltd., Budapest, Hungary and used as received. TPPTS ((3,3′,3′′-phosphanetriyltris(benzenesulfonic acid) trisodium salt), P(C6H4-m-SO3Na)3)35 and BuP(C6H4-m-SO3Na)2 (Bu-DPPDS)36 ligands were prepared by published methods.In a typical high pressure hydrogenation experiment, a 120 mL Parr HP reactor (ESI Fig. S3†) was charged with 30 mL (34.6 g, 360 mmol) furfural followed by the addition of Ru(acac)3 and the corresponding phosphine ligand resulting in a dark brown solution. The reaction mixture was pressurized to the desired values and heated up to desired temperature to initiate the reaction. Samples were taken for off-line GC analysis via a dip-leg into a sample holder. After the given reaction time, the autoclave was cooled to ambient temperature and stirring was stopped.
The configuration of catalyst recycling experiments is depicted on ESI Fig. S4.†
GC-FID sample analysis were performed on a HP 5890 instrument equipped with a HP-5 capillary column (15 m) using H2 as a carrier gas. For the analysis, 10 μL of the reaction mixture was added to 1 mL of methylene chloride followed by the addition of 10 μL toluene as an internal standard.
Conclusions
We have demonstrated that the catalyst in situ generated from Ru(III)–acetylacetonate and 1,4-bis(diphenylphoshino)butane can be used for the efficient conversion of furfural to furfuryl alcohol as a potential biomass-based platform chemicals under solvent-free conditions, which is one of the main goals of green chemistry with very high selectivity and activity (TOF ≥ 6270 h−1). The catalyst was found to be recyclable for twelve consecutive runs without significant changes in catalyst's activity and selectivity. The maximum reduction rate was achieved by applying 10-fold excess of DPPB ligand to the Ru precursor under 25 bar of H2 at 160 °C.Acknowledgements
This work was founded by the National Research, Development and Innovation Office – NKFIH (PD 116559). L. T. Mika is grateful to the support of János Bolyai Research Scholarship of the Hungarian Academy of Sciences.Notes and references
- (a) G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044 CrossRef CAS PubMed; (b) A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411 CrossRef CAS PubMed.
- (a) R. Sheldon, Green Chem., 2014, 16, 950 RSC; (b) C. O. Tuck, E. Perez, I. T. Horváth, R. A. Sheldon and M. Poliakoff, Science, 2012, 337, 695 CrossRef CAS PubMed.
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538 RSC.
- (a) J. J. Bozell, L. Moens, D. C. Elliot, Y. Wang, G. G. Neuenscwander, S. W. Fitzpatrik, R. J. Biliski and J. L. Jarnefeld, Resour., Conserv. Recycl., 2000, 28, 227 CrossRef; (b) B. Girisuta, L. P. B. M. Janssen and H. J. Heeres, Chem. Eng. Res. Des., 2006, 84, 339 CrossRef CAS; (c) V. M. Ghorpade and M. A. Hanna, Industrial Application for levulinic acid, Cereals: Novel Uses and Processes, ed. G. M. Campbell, C. Webb and S. L. McKee, Plenum, New York, 1997, pp. 49–57 Search PubMed.
- I. T. Horváth, H. Mehdi, V. Fábos, L. Boda and L. T. Mika, Green Chem., 2008, 10, 238 RSC; K. Yan, Y. Yang, J. Chai and Y. Lu, Appl. Catal., B, 2015, 179, 292 CrossRef CAS.
- K. Yan, Y. Yang, J. Chai and Y. Lu, Appl. Catal., B, 2015, 179, 292 CrossRef CAS.
- R. Mariscal, P. Maireles-Torres, M. Ojeda, I. Sádaba and J. M. López Granados, Energy Environ. Sci., 2016, 9, 1144 CAS.
- (a) H. E. Hoydonickx, W. M. Van Rhijn, W. Van Rhijn, D. E. De Vos and P. A. Jacobs, Furfural and Derivatives, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2012, DOI:10.1002/14356007.a12_119.pub2; (b) M. M. Villaverde, N. M. Bertero, T. F. Garetto and A. J. Marchi, Catal. Today, 2013, 213, 87 CrossRef CAS.
- (a) M. Audemar, C. Ciotonea, K. De Oliveira Vigier, S. Royer, A. Ungurenau, B. Dragoi, E. Dumitriu and F. Jérome, ChemSusChem, 2015, 8, 1885 CrossRef CAS PubMed; (b) D. Vargas-Hernández, J. M. Rubio-Caballero, J. Santamaría-Gonzales, R. Moreno-Tost, J. M. Mérida-Robles, M. A. Pérez-Cruz, A. Jiménez-López, R. Hernández-Huesca and P. Maireles-Torres, J. Mol. Catal. A: Chem., 2014, 383–384, 106 CrossRef; (c) R. F. Perez and M. A. Fraga, Green Chem., 2014, 16, 3942 RSC.
- R. H. Lock and K. Reynolds, US 2763665, 1952.
- (a) B. Girisuta, L. P. B. M. Janssen and H. J. Heeres, Chem. Eng. Res. Des., 2006, 84, 339 CrossRef CAS; (b) J. J. Bozell, L. Moens, D. C. Elliott, Y. Wang, G. G. Neuenschwander, W. W. Fitzpatrick, R. J. Bilski and J. L. Jarnefeld, Resour., Conserv. Recycl., 2000, 28, 227 CrossRef.
- J.-P. Lange, W. D. van de Graaf and R. J. Haan, ChemSusChem, 2009, 2, 437 CrossRef CAS PubMed.
- J. Q. Bond, J. A. Dumesic, D. M. Alonso, D. Wang and R. M. West, Science, 2010, 327, 1110 CrossRef CAS PubMed.
- H. Mehdi, V. Fábos, R. Tuba, A. Bodor, L. T. Mika and I. T. Horváth, Top. Catal., 2008, 48, 49 CrossRef CAS.
- (a) J. M. Tukacs, B. Fridrich, G. Dibó, E. Székely and L. T. Mika, Green Chem., 2015, 17, 5189 RSC; (b) V. Fábos, M. Y. Lui, Y. F. Mui, Y. Y. Wong, L. T. Mika, L. Qi, E. Cséfalvay, V. Kovács, T. Szūcs and I. T. Horváth, ACS Sustainable Chem. Eng., 2015, 3, 1899 CrossRef; (c) D. Fegyverneki, L. Orha, G. Láng and I. T. Horváth, Tetrahedron, 2010, 66, 1078 CrossRef CAS; (d) A. Strádi, M. Molnár, M. Óvári, G. Dibó, F. U. Richter and L. T. Mika, Green Chem., 2013, 15, 1857 RSC; (e) M. Chalid, H. J. Heeres and A. A. Broekhuis, J. Appl. Polym. Sci., 2011, 123, 3556 CrossRef; (f) F. M. A. Geilen, B. Engendahl, A. Harwardt, W. Marquardt, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2010, 49, 5510 CrossRef CAS PubMed.
- (a) P. Pongrácz, L. Kollár and L. T. Mika, Green Chem., 2016, 18, 842 RSC; (b) A. Strádi, M. Molnár, P. Szakál, D. Gáspár, G. Dibó and L. T. Mika, RSC Adv., 2015, 5, 72529 RSC; (c) G. Strappaveccia, L. Luciani, E. Bartollini, A. Marrocchi, F. Pizzo and L. Vaccaro, Green Chem., 2015, 17, 1071 RSC; (d) G. Strappaveccia, E. Ismalaja, C. Petrucci, D. Lanari, A. Marrocchi, M. Drees, A. Facchetti and L. Vaccaro, Green Chem., 2015, 17, 365 RSC; (e) E. Ismalaj, G. Strappaveccia, E. Ballerini, F. Elisei, O. Piermatti, D. Gelman and L. Vaccaro, ACS Sustainable Chem. Eng., 2014, 2, 2461 CrossRef CAS; (f) L. Qui and I. T. Horváth, ACS Catal., 2012, 2, 2247 CrossRef; (g) L. Qi, Y. F. Mui, S. W. Lo, M. Y. Lui, G. R. Akien and I. T. Horváth, ACS Catal., 2014, 4, 1470 CrossRef CAS.
- A. Corma, S. Iborra and A. Welty, Chem. Rev., 2007, 107, 2411 CrossRef CAS PubMed.
- (a) B. M. Nagaraja, A. H. Padmasri, B. D. Raju and K. S. R. Rao, J. Mol. Catal. A: Chem., 2007, 265, 90 CrossRef CAS; (b) B. M. Nagaraja, V. S. Kumar, V. Shasikala, A. H. Padmasri, B. Sreedhar, B. D. Raju and K. S. R. Rao, Catal. Commun., 2003, 4, 287 CrossRef CAS; (c) N. Merat, C. Godawa and A. Gaset, J. Chem. Technol. Biotechnol., 1990, 48, 145 CrossRef CAS.
- Y. Zhao, Environ. Chem. Lett., 2014, 12, 185 CrossRef CAS.
- M. T. Taylor, L. J. Durndell, M. A. Isaacs, C. M. A. Parlet, K. Wilson, A. F. Lee and G. Kyriakou, Appl. Catal., B, 2016, 180, 580 CrossRef CAS.
- H. Li, S. Zhang and H. Luo, Mater. Lett., 2004, 58, 2741 CrossRef CAS.
- S. Paganelli, O. Piccolo, P. Pontini, R. Tassini and V. D. Rathod, Catal. Today, 2015, 247, 64 CrossRef CAS.
- F. Huang, W. Li, Q. Lu and X. Zhu, Chem. Eng. Technol., 2010, 33, 2082 CrossRef CAS.
- M. D. Bhor, A. G. Panda, N. S. Nandurkar and B. M. Bhanage, Tetrahedron Lett., 2008, 49, 6475 CrossRef CAS.
- (a) Z. Strassenberger, M. Mooijman, E. Ruijte, A. H. Alberts, C. de Graaf, R. V. A. Orru and G. Rothenberg, Appl. Organomet. Chem., 2010, 24, 142 Search PubMed; (b) Z. Strassenberger, M. Mooijman, E. Ruijte, A. H. Alberts, A. G. Maldonado, R. V. A. Orru and G. Rothenberg, Adv. Synth. Catal., 2010, 352, 2201 CrossRef PubMed.
- F. Joó, Z. Tóth and M. T. Beck, Inorg. Chim. Acta, 1977, 25, L61 CrossRef.
- J. M. Tukacs, D. Király, A. Strádi, G. Novodárszki, Z. Eke, G. Dibó, T. Kégl and L. T. Mika, Green Chem., 2012, 14, 2057 RSC.
- (a) T. Kim, R. S. Assary, H. Kim, C. L. Marshall, D. J. Gosztola, L. A. Curtiss and P. C. Stair, Catal. Today, 2013, 205, 60 CrossRef CAS; (b) T. Kim, R. S. Assary, C. L. Marshall, D. J. Gosztola, L. A. Curtiss and P. C. Stair, ChemCatChem, 2011, 3, 1451 CrossRef CAS.
- O. Kröcher, R. A. Köppel and A. Baiker, Chem. Commun., 1997, 453 RSC.
- (a) S. I. Fujita, Y. Sano, B. M. Bhanage and M. Arai, J. Chem. Eng. Jpn., 2003, 36, 155 CrossRef CAS; (b) M. Chalid, A. A. Broekhius and H. J. Heeres, J. Mol. Catal. A: Chem., 2011, 341, 14 CrossRef CAS.
- T. Manimaran, T.-C. Wu, W. D. Klobucar, C. H. Kolich and G. P. Stahly, Organometallics, 1993, 12, 1467 CrossRef CAS.
- J. A. A. A. Gervaux, S. Sabo-Etienne and B. Chaudret, Organometallics, 1997, 16, 2000 CrossRef.
- G. Kiss and I. T. Horváth, Organometallics, 1991, 10, 3798 CrossRef CAS.
- D. Lantos, M. Contel, S. Sanz, A. Bodor and I. T. Horváth, J. Organomet. Chem., 2007, 692, 453 CrossRef.
- W. A. Hermann and C. W. Kohlpainter, Inorg. Synth., 1998, 32, 8 CrossRef.
- L. T. Mika, L. Orha, N. Farkas and I. T. Horváth, Organometallics, 2009, 28, 1593 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra24723g |
| This journal is © The Royal Society of Chemistry 2017 |