
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lewis acid stabilization and activation of primary N-nitrosamides†
D. Scott Bohle *,
Zhijie Chua,
Inna Perepichka and
Kristopher Rosadiuk
*,
Zhijie Chua,
Inna Perepichka and
Kristopher Rosadiuk
Department of Chemistry, McGill University, 801 Sherbrooke St. W., Montreal, H3A 0B8, Canada. E-mail: scott.bohle@mcgill.ca
First published on 10th January 2017
Abstract
The primary nitrosamides, here exemplified by the N-nitrosoalkylcarbamates, ROC(O)NHNO [R = CH3 (1), R = C2H5 (2)], show a markedly Lewis acid dependent chemistry. Although their alkali metal salts M[ROC(O)NNO] [M = K, R = CH3 (3), M = Na, R = CH3 (3-Na), M = K, R = C2H5 (4), M = Na, R = C2H5 (4-Na)] are stable isolable yellow solids, in solution they undergo pH dependent decomposition to give nitrogen and nitrous oxide as the primary nitrogen containing gaseous products. However chelation to a transition metal, [Re(L)(CO)2(PPh3)2], L = ROC(O)NNO− gives rise to isomeric kinetic and thermodynamic products. Both are thermally stable and together illustrate the stability provided by a chelating low valent transition metal. The preparation, IR, NMR, and X-ray crystallographic characterization of these derivatives are reported. Taken together, these results demonstrate that the Lewis acid identity is an important factor in primary nitrosamide chemistry.
Introduction
In contrast with the well-known and relatively stable N-nitramides, A, the known N-nitrosamides RC(O)N(NO)R′, B, are much less thermally stable.1 As shown in Scheme 1, part of this high reactivity is due to their multiple and varied decomposition pathways. Thus, as a class, the N-nitrosamides have three recognized decomposition pathways2 which range from the most common 1,3-acyl NO shift (path I),3 amide hydrolysis (path II),4 to N–N homolysis (path III).5 The utility of nitrosamides as reagents is also directly related to these facile decomposition pathways. For example, the hydrolysis path II, can be used to generate diazomethane when R′ = Me and R = urea, from the breakdown of the diazotate D product.6 Also the 1,3-acyl shift path I for R′ = alkyl and O(t-Bu) leads to radical pair generation following nitrogen extrusion from the diazo compound C.7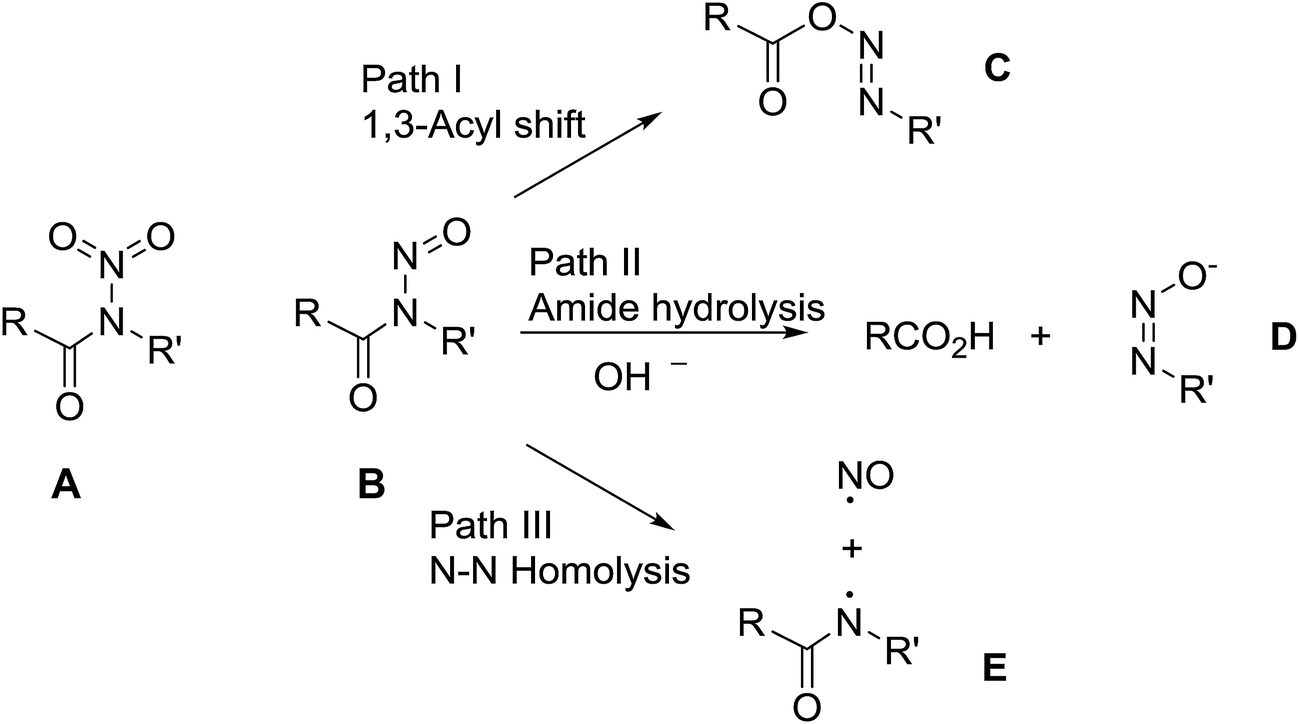 | ||
| Scheme 1 Recognized N-nitrosamide reactivity. | ||
Although the N–N homolysis path III is the more unusual transformation, it can occur either through photolysis2 or thermally as demonstrated by Koenig5 for R = cyclopropyl and R′ = t-butoxy, it is a potential source of biologically useful NO donors.8 All of the chemistry in Scheme 1 pertains to the secondary nitrosamides, in which R = aryl, alkyl, or alkoxy groups.
However not all of the N-nitrosamide reactivity is accounted for with these three pathways. Several reports describe olefins in the products of the N-nitrosamide decomposition.9 In the preparation of N-benzoyl N-nitroso-O-tert-butylhydroxamide, F (Scheme 2), the expected 1,3-acyl migration pathway I is the observed decomposition product when F is kept cold and promptly isolated after its preparation. However, Koenig noted that warming the initial product solution to 0°, or washing with dilute acid, resulted in the rapid release of nitrous oxide and benzoic anhydride (pathway IV).10 In addition, early physical organic mechanistic studies from two groups differed with one finding good first order behaviour across a range of substrates temperatures3a and the other finding an inhibition or competing pathway in half of sixteen cases.11 Assuming mechanistic uniformity, this led one of the studies to conclude, in a strange footnote,3a that the other results were marred by catalytic impurities or faulty apparatus.3a Thus, even for the secondary nitrosamides, there remain open mechanistic questions and uncertainties relating to their decomposition chemistry beyond that depicted in Scheme 1.
 | ||
| Scheme 2 Nitrous oxide releasing decays. | ||
For primary nitrosamides RC(O)N(NO)H, G, the available chemistry is particularly sparse with only two well characterized examples to study.9a,12 Even so, species like G are important intermediates in the nitric oxide and nitrous acid mediated deamination of primary amides, Scheme 3.13 For the class of primary nitrosamides, as with primary nitrosamines, their instability relates in part to the ionisable and tautomerizable proton and it has been incorrectly stated1 that the O–H tautomer H, is the more stable in spite of compelling crystallographic and spectroscopic evidence for the related MeN(NO)H.4,14 The scarcity of primary nitrosamines stems not only from their inherent reactivity, but that their synthetic access is currently limited to a zinc reduction of the corresponding primary nitroamides (eqn (1)).
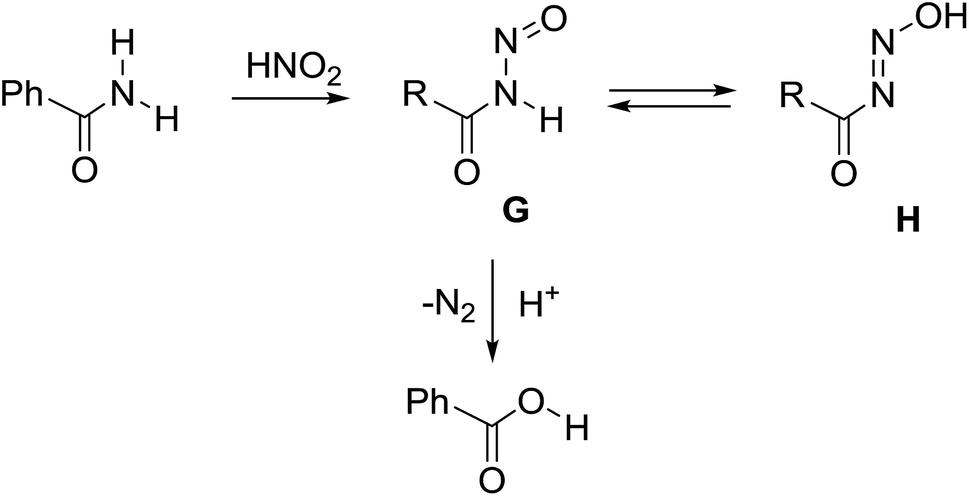 | ||
| Scheme 3 Primary nitrosamines in nitrosative deamination. | ||
Our interest in these compounds stems from the general utility of the nitroamides and secondary nitrosamides as synthetic reagents. For example, the secondary nitrosamides are excellent transition metal nitrosylation precursors, and the N-methyl analogs may be good reagents for methylene addition to transition metals, a reaction of diazomethane.15 We note that the extrusion or elimination of nitrogen is often used to generate high energy reactive intermediates and that, in principle, this strategy could be easily applied to the elimination of nitrous oxide.
 | (1) |
Nevertheless, nitrous oxide elimination remains a rarely used synthetic strategy. In part this may be due to the preparative difficulties for the small number of substrates containing XNNOY or XNN(Y)O, fragments which are poised to eliminate X–Y and generate NNO. We were intrigued by the reports of the potential nitrous oxide donors, the N-nitrosocarbamates, by Thiele and Lachman from 1893–1895 (ref. 9a and 12) eqn (1). In these seminal reports the isolation of these unusual reactive species and investigation into the facile low temperature elimination of a gas was attributed to being nitrogen and not nitrous oxide. In a recent series of reports Benin and coworkers described a number of new N-nitrosocarbamates.16 What remains unclear is why these should develop such robust N–N bonds, and yet have such labile N–O bonds. Furthermore, why are NO and N2O not seen in these product streams as is found in the decomposition of many other N-nitroso species?5
In their work with the N-nitrosocarbamates, the N-nitro analogs and the parent nitro-amine, nitramide (H2NNO2), Thiele and Lachman laid a significant foundation to this chemistry.9a,12,17 White and co-workers reported on the synthesis of a series of secondary N-nitrosocarbamates and carried out mechanistic studies with the assumption that the decomposition pathway of these compounds were similar to the primary N-nitrosocarbamates through the loss of N2.9b,13b,18 They proposed a series of possible mechanisms initiated by N-nitroso rearrangement. More recently, Benin et al. reported an in-depth kinetic study on the decomposition and alkylation of a tetrabutylammonium salt of N-nitrosoethylcarbamate coupled with DFT calculations, all on the basis of nitrogen gas evolution.16a
We have previously reported the first structurally characterised metal complexes of an N-nitrosocarbamate19 and have an interest in this class of compounds for their unusual electronic properties as ligands. While working with the N-nitrosocarbamates, we noticed a discrepancy in one of the decomposition products which had been assumed to be carbon dioxide and dinitrogen gas. Herein we report: (1) the structural and spectroscopic characterization of two N-nitrosocarbamates and their salts; (2) the observation of nitrous oxide as a significant minor component of their decomposition products; (3) analysis of their electronic spectra, and the use of UV-spectroscopy to follow the kinetics of their decomposition; (4) a new set of Re(I) chelates of the N-nitrosocarbamates form where an initial kinetic product with (N,O)-N-nitroso coordination rearranges to a thermodynamic (O,N)-acylnitroso isomer. These latter complexes represent new coordination modes for this class of ligands, the nitrosamides, and are only the second example of how transition metal Lewis acid coordination is able to stabilize this class of compounds, as their nitrosocarbamate.
Experimental
General
Unless noted, all reagents and solvents are used as supplied commercially. Dry CH3OH is distilled from freshly generated magnesium methoxide, dry CHCl3 is distilled from CaCl2, dry CH2Cl2 is distilled from CaH2, dry hexanes, pentanes and benzene are distilled from sodium benzophenone ketyl. 1H and 31P spectra are recorded on Varian Mercury 200, 400, 500 MHz or Bruker AVANCE 400, 500 MHz instruments. All chemical shifts are recorded in δ (ppm) relative to residual solvent signals for 1H spectra and external H3PO4 for 31P spectra. Melting/decomposition points are measured by using a TA-Q2000 differential scanning calorimeter. The IR spectra are recorded in KBr disks by using ABB Bomem MB Series spectrometer with a spectral resolution of 4 cm−1. Elemental analyses are performed in the Elemental Analyses Laboratory at University of Montreal. The presence of formulated solvates were confirmed by NMR and/or crystallography.Nitramide,20 Re(O)(Cl)3(PPh3)2,21 Re(Cl)(CO)3(PPh3)2,22 and [Re(CH3CN)2(CO)2(PPh3)2](ClO4)23 are synthesized by literature methods. [Re(CH3CN)2(CO)2(PPh3)2](BF4) is synthesized using AgBF4 instead of AgClO4.
X-ray crystallography
Crystals are mounted on glass fibres or Mitegen mounts using Paratone-N from Hampton Research and single-crystal X-ray diffraction experiments are carried out with a BRUKER APEX-II CCD diffractometer by using graphite-monochromated MoKα radiation (λ = 0.71073 Å) and KRYOFLEX for low temperature experiments. SAINT24 is used for integration of the intensity reflections and scaling and SADABS25 for absorption correction. Direct methods are used for structures with only light elements while Patterson maps are used to generate the initial solution for the potassium salts and Re complexes. For large and unusual unit cell parameters, CELL_NOW26 was used to check the viability of the suggested lattice and subsequent space group solution. Location of non-hydrogen atomic are carried out using Fourier difference maps with the refinements solved by full-matrix least-squares method on F2 of all data using SHELXTL24 software. The hydrogen atoms positions are placed in calculated positions. The program SQUEEZE27 is used to estimate and remove the residual electron density associated with disordered solvent molecules within the large solvent channels in the crystals of 5.Diffraction quality crystals of N-nitrosocarbamates are obtained from vacuum sublimation of the compounds using ice-water cold finger condenser and heating the crude material between 23 to 30 °C. The crystals of the potassium salts of the N-nitrosocarbamates were grown from CH3OH/ether at −21 °C. Diffraction quality crystals of the Re complex 5 is obtained from slow diffusion of either pentanes or petroleum ether into CH2Cl2 solutions at 5 °C over a week, while 6 and 8 are from layered solutions of dichloromethane and pentanes. Large solvent channels in the crystals of 5 require low temperature diffraction data collection to prevent the loss of solvates. The crystal structure of 6 is a co-crystal of two non-identical independent molecules. Crystallographic data and data collection parameters are shown in Table S5.†
Ice (∼10 g) is added and cold conc. aqueous ammonia (4.1 mL, 59.5 mmol) is added in excess and stirred for 5 min to give a yellow solution (ensure there is still some ice floating in solution, if not add more ice). The solution is filtered through a plug of Celite to give a yellow filtrate. If the residue is yellow, it is washed with dilute aqueous ammonia (0.5 mL conc. NH4OH in 2 mL deionised H2O) and deionised H2O until the washings are colourless. Ice (∼50 g) is added into the yellow filtrate followed by a solution of AgNO3 (1.76 g, 10.4 mmol) with rapid stirring. Acetic acid is added slowly, forming initially a white suspension, which dissolves on further addition of acid (if no ice is visible during the addition, add more ice). A yellow suspension slowly forms and acetic acid is added till the solution is acidic to pH paper (total volume ∼ 150 mL). The reaction mixture is left to stand between 10–15 min during which substantial yellow precipitate appears. The yellow precipitate, Ag[CH3OC(O)NNO] (assumed 2.18 g, 10.3 mmol), is filtered as rapidly as possible and washed consecutively with deionised H2O, C2H5OH and CH3OH and processed immediately as it rapidly decomposes on prolonged exposure to moisture (the sintered filter may get clogged due to the fine particulate nature of the Ag salt, an alternative would be to centrifuge the suspension and wash with C2H5OH). The filtrate is observed to be colourless.
NaCl (0.610 g, 10.4 mmol) is dissolved in an ice-water solution and added to Ag[CH3OC(O)NNO] (assumed 2.18 g, 10.3 mmol) and stirred/shaken vigourously for 5 min to give a yellow suspension. The yellow suspension is filtered through a pad of Celite and washed successively with cold deionised H2O till the filtrate is colourless. The filtrate is added into a separatory funnel containing ethyl acetate (20 mL), ice (∼2 g) and conc. H2SO4 (0.500 g, 5.10 mmol). The organic layer is separated, kept, and the yellow aqueous layer further extracted with ethyl acetate (4 × 20 mL) or till the aqueous layer is colourless. The organic extracts are combined and dried over anhydrous CaCl2, kept cold and under a N2 atmosphere for 30 min. The yellow ethyl acetate suspension is filtered and ethyl acetate is removed using a rotary evaporator without heating to give bright yellow solids of N-nitrosomethylcarbamate which is further purified by vacuum sublimation (ice-water condenser and heating at 22–23 °C) to give bright yellow crystals of N-nitrosomethylcarbamate (0.315 g, 3.03 mmol, 29% yield) (1) (note: to minimize the loss of 1 due to its low melting point, the sublimator is cooled to around 6–7 °C before the vacuum is applied). Decomposition point: 60.5 °C (−83.3 kJ mol−1). IR (cm−1): 3427vs, 3199s, 2969m, 1759vs, 1707s, 1619s, 1529s, 1470s, 1459s, 1432m, 1385w, 1330w, 1250vs, 1089vs, 1061s, 929s, 766m, 717m, 560m, 444w. 1H NMR (200 MHz, CDCl3) ppm: δ = isomer α (minor) 3.68 (s, CH3), isomer β (minor) 3.91 (s, CH3), isomer γ (major) 4.06 (s, CH3), 10.65 (NH, broad). Ratio of 0.03![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.05:1 respectively. 1H NMR (200 MHz, CD3OD) ppm: δ = isomer α (minor) 3.60 (s, CH3), isomer β (minor) 3.78 (s, CH3), isomer γ (major) 3.97 (s, CH3). Ratio of 0.02:0.04:1 respectively. UV (CH3OH λmax, nm (ε, M−1 cm−1)): 366sh (12.6), 385sh (34.1), 401 (61.6), 417 (88.5), 436 (76.7). UV (CHCl3 λmax, nm (ε, M−1 cm−1)): 368sh (21.3), 383sh (43.0), 396 (75.0), 412 (105.0), 430 (88.7).
:0.05:1 respectively. 1H NMR (200 MHz, CD3OD) ppm: δ = isomer α (minor) 3.60 (s, CH3), isomer β (minor) 3.78 (s, CH3), isomer γ (major) 3.97 (s, CH3). Ratio of 0.02:0.04:1 respectively. UV (CH3OH λmax, nm (ε, M−1 cm−1)): 366sh (12.6), 385sh (34.1), 401 (61.6), 417 (88.5), 436 (76.7). UV (CHCl3 λmax, nm (ε, M−1 cm−1)): 368sh (21.3), 383sh (43.0), 396 (75.0), 412 (105.0), 430 (88.7).
:0.04:1. UV: (CH3OH λmax, nm (ε, M−1 cm−1)) = 395 (37.9).N-Nitrosomethylcarbamate (1) (0.315 g, 3.03 mmol) is dissolved in dry THF (15 mL) under an N2 atmosphere. The yellow solution is cooled in an ice-bath and NaH (0.117 g, 2.92 mmol) is added in portions during which some bubbling is observed. The suspension is allowed to warm to room temperature and stirred overnight. The solvent is removed in vacuo to give pale yellow solids. Dry pentanes is added and the solids sonicated to remove the mineral oil and decanted. The pale yellow residue is washed with diethylether twice and the solids collected into a sample vial as a suspension in ether and subsequently dried under vacuum to give pale yellow powder of Na[CH3OC(O)NNO] (3-Na) (0.319 g, 2.53 mmol, 87% yield). Note: the yellow crystals are hygroscopic, sensitive to friction and can detonate! IR (cm−1): 2952w, 1695s, 1443m, 1328m, 1274s, 12333vs, 1221vs, 1210vs, 1194vs, 1113m, 1075m, 967m, 880m, 834w, 790m, 461w. 1H NMR (400 MHz, CD3OD) δ = isomer B (minor) 3.66 (s, CH3), isomer A (major) 3.83 (s, CH3), relative ratio of 0.52:1. UV: (CH3OH λmax, nm (ε, M−1 cm−1)) = 397 (39.4). HRMS (ESI): m/z [M − Na]− calcd for C2H3O3N2: 103.0149; found: 103.0149.
Room temperature reaction. [Re(CH3CN)2(CO)2(PPh3)2](ClO4) (0.030 g, 0.032 mmol) is dissolved in CH2Cl2 (2 mL) and added to a solution of potassium N-nitrosomethylcarbamate (3) (0.006 g, 0.038 mmol) in CH3OH or C2H5OH (6 mL) in a 50 mL flask. The white suspension was stirred overnight during which time the suspension turned from a white to a bright yellow. The solvent is removed and the residue extracted with CH2Cl2 (10 mL) followed by filtration through a sintered glass filter. The solvent is removed to give yellow solids containing isomers of Re(η2-CH3OC(O)NNO)(CO)2(PPh3)2 (5) (0.020 g, 0.023 mmol, 72% yield). IR (cm−1): 3055w, 2964wv, 2924w, 1942vs (CO), 1864vs (CO), 1750m, 1633m (broad), 1481m, 1434s, 1384m, 1311m, 1261m, 1181m, 1094s, 1029m, 910m, 804w, 746m, 695s, 518s. 1H NMR (200 MHz, CDCl3) ppm: 3.00 (s, CH3, major isomer), 3.46 (s, CH3, minor isomer), 7.38 (m, 18H-PPh3), 7.48, (m, 12H-PPh3). 1H NMR (500 MHz, CDCl3) ppm: 3.07 (s, CH3, minor isomer = 0.21), 3.41 (s, CH3, major isomer = 0.79), 7.39 (m, 18H-PPh3), 7.52 (m, 12H-PPh3). 31P[1H] NMR (81 MHz, CDCl3) ppm: 23.74 (s, minor isomer = 0.24), 29.01 (s, major isomer = 0.76). 31P[1H] NMR (202 MHz, CDCl3) ppm: δ = 23.74 (s, minor isomer = 0.19), 29.03 (s, major isomer = 0.81).
Elevated temperature reaction. [Re(CH3CN)2(CO)2(PPh3)2](ClO4) (0.030 g, 0.032 mmol) and potassium N-nitrosomethylcarbamate (3) (0.006 g, 0.038 mmol) were placed in 50 mL flask. Degassed CH3OH or C2H5OH (6 mL) is added and the reaction heated to 60 °C for 45 min. The white suspension turned into a bright yellow suspension. The solvent is removed and the residue extracted with CH2Cl2 (10 mL) and filtered through a glass frit. The CH2Cl2 solvent is removed in vacuo to give orange solids containing isomers of Re(η2-CH3OC(O)NNO)(CO)2(PPh3)2 (6) (0.021 g, 0.024 mmol, 75% yield). IR (cm−1): 3054w, 1952vw, 1950vs (CO), 1870vs (CO), 1748m, 1556m, 1481m, 1434s, 1419m, 1327s, 1266m, 1185m, 1158m, 1093s, 1066s, 998m, 995m, 744s, 695vs, 634m, 515vs. 1H NMR (200 MHz, CDCl3) ppm: δ = 3.07 (s, CH3, major isomer = 0.83), 3.41 (s, CH3, minor isomer = 0.17), 7.40 (m, 18H-PPh3), 7.48 (m, 12H-PPh3). 1H NMR (500 MHz, CDCl3) ppm: δ = 3.07 (s, CH3, major isomer = 0.83), 3.41 (s, CH3, minor isomer = 0.17), 7.38 (m, 18H-PPh3), 7.47 (m, 12H-PPh3). 31P[1H] NMR (81 MHz, CDCl3) ppm: δ = 23.73 (s, major isomer = 0.78), 29.03 (s, minor isomer = 0.22). 31P[1H] NMR (202 MHz, CDCl3) ppm: δ = 23.74 (s, major isomer = 0.82), 29.03 (s, minor isomer = 0.18).
A sample containing a mixture of both isomers together was sent for elemental analysis. Anal. calcd. for C40H33N2O5P2Re·0.8CH2Cl2 (937.83 g mol−1) % C, 52.25; H, 3.72; N, 2.99. Found% C, 52.49; H, 3.75; N, 2.68. Dichloromethane was confirmed by 1H NMR and X-ray crystallography.
IR monitoring experiments
CH3OH was added into a gas-sealed IR cell and the gas-phase IR absorption was collected as the background. Compound 3-Na (0.020 g, 1.59 mmol) was added into a gas-sealed IR cell and monitored over time at room temperature. The procedure was carried out similarly for 4-Na.Quantification of N2O released from the decay of 3-Na
Nitrous oxide was measured by means of a Bruker SCION-GCMS device, using EI and a quadrapole ion trap. Vials (16–19 mL) containing 3-Na (10–30 mg) were flushed with argon, sealed with a septum, and 500 μL of water was introduced with a syringe. The water present is assumed to reach the saturation point with regards to nitrous oxide, using Henry's law constants.29 The majority of the gas produced is trapped in the liquid, with never more 4% of the total volume produced, so the pressure change is assumed to be insignificant. The mixture of argon and nitrous oxide is assumed to behave ideally. The gas is collected with a sealable 1 mL SGE gas tight syringe and injected manually into the GC-MS system. Gas samples were taken after 3.5 hours and after 24 hours of decay. The trial was repeated using methanol in place of water, using Henry's law.30Results and discussion
Synthesis and structures of the primary nitrosocarbamates and their salts
The N-nitrosocarbamates 1 and 2 are synthesized by the reduction of the corresponding N-nitrocarbamates using Zn/acetic acid (Scheme 4).9a,17 The synthetic procedures described by Thiele had a precaution stipulating that the synthesis has to be carried out precisely ‘Die Reindarstellung dieses sehr zersetzlichen Körpers gelingt nur unter Einhaltung nachstehender Vorshcrift’. In our hands this preparation has proved to be particularly troublesome as some of the steps were difficult to follow and used unusual techniques and devices even by 1895 standards. A more recent description of these synthesis only goes to the isolation of the silver salt with no further steps towards conversion to the neutral form.16a We have included in the experimental section detailed procedures that will consistently give high yields and good purity of these compounds. Temperature, pH, salt identity, and concentration are all factors optimized here. In addition, the silver salts are modestly photosensitive and need to be shielded from direct light. Finally, the N-nitrosocarbamates 1 and 2 are hygroscopic solids with high vapour pressures; leaving these compounds in the open results in their eventual disappearance and/or deliquescence. For example, in humid air 1 and 2 will dissolve to give yellow solutions. It is best to store these compounds in a cold, dry and sealed environment as they are difficult to desiccate once coming into contact with moisture as per conditions described before.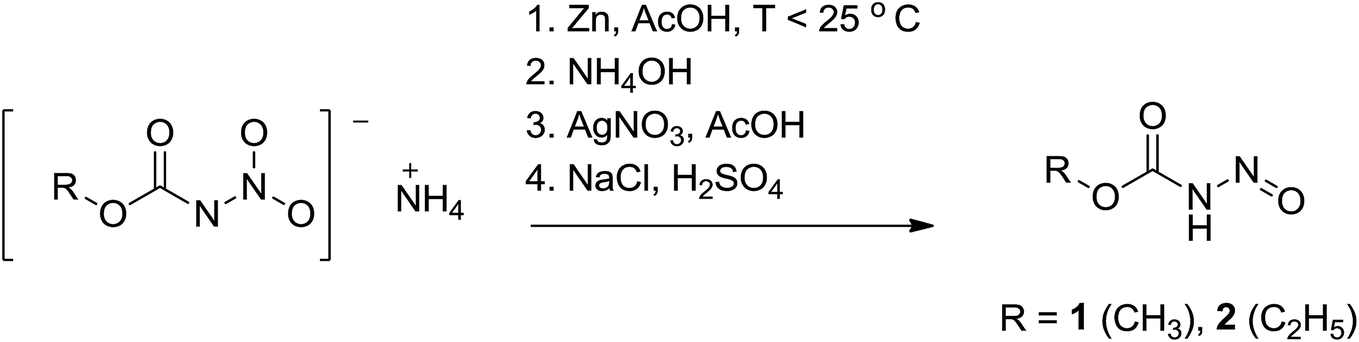 | ||
| Scheme 4 Synthesis of N-nitrosocarbamates from reduction of N-nitrocarbamates. | ||
The solid state and solution structures of the N-nitrosocarbamates provide a significant insight into the reaction and coordination chemistry that follows. Diffraction grade crystals of 1 and 2 are readily grown by sublimation at low temperature. The resulting solid state structures (Fig. 1) contain well-ordered strongly hydrogen bonded, N(1)–H⋯O(2) layers (Fig. S7†) of the molecular acids with the single (Z,E) conformation shown in Fig. 1. The key experimentally derived metric parameters for 1–3 are listed in Table 1, while Fig. 2 and Table 2 have the theoretically calculated DFT values for the gas phase species. For the nitrogen acids 1 and 2 the solid state structure is best described as an N(1)–H⋯O(2) hydrogen bond with a planar amide geometry at N(1), and single N(1)–N(2) bond and a nitroso like double bond for N(2)–O(1). This solid state configuration, Z around the C(1)–N(1) bond E around the N(1)–N(2) bond, is one of four possible N–H containing configurations, Fig. 2. DFT, B3LYP/6-311++g**, indicates that in the gas phase the E/Z geometry is 3.7 kcal mol−1 more stable than the crystallographically observed Z/E configuration.
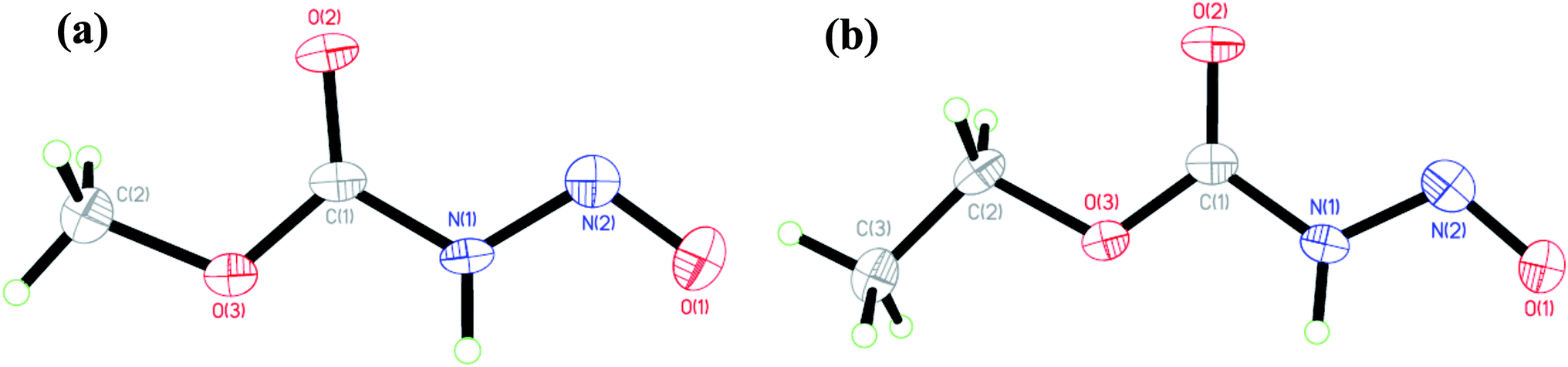 | ||
| Fig. 1 Solid state crystallographically determined molecular structures of (a) 1 and (b) 2 at 50% thermal ellipsoids. | ||
| Compound | 1 | 2 | 3 |
|---|---|---|---|
| N(1)–N(2) | 1.354(3) | 1.350(3) | 1.321(3) |
| N(1)–C(1) | 1.385(3) | 1.383(3) | 1.397(3) |
| O(1)–N(2) | 1.217(3) | 1.212(7) | 1.260(2) |
| C(1)–O(2) | 1.206(2) | 1.198(3) | 1.215(3) |
| C(1)–O(3) | 1.326(3) | 1.321(3) | 1.349(3) |
| N(2)–N(1)–C(1) | 118.64(17) | 119.05(18) | 109.06(18) |
| O(1)–N(2)–N(1) | 112.76(19) | 113.4(2) | 113.13(17) |
| O(1)–N(2)–N(1)–C(1) | 180.0(0) | 180.0(1) | 177.45(19) |
| O(2)–C(1)–N(1)–N(2) | 0.0(0) | 0.0(1) | −1.9(3) |
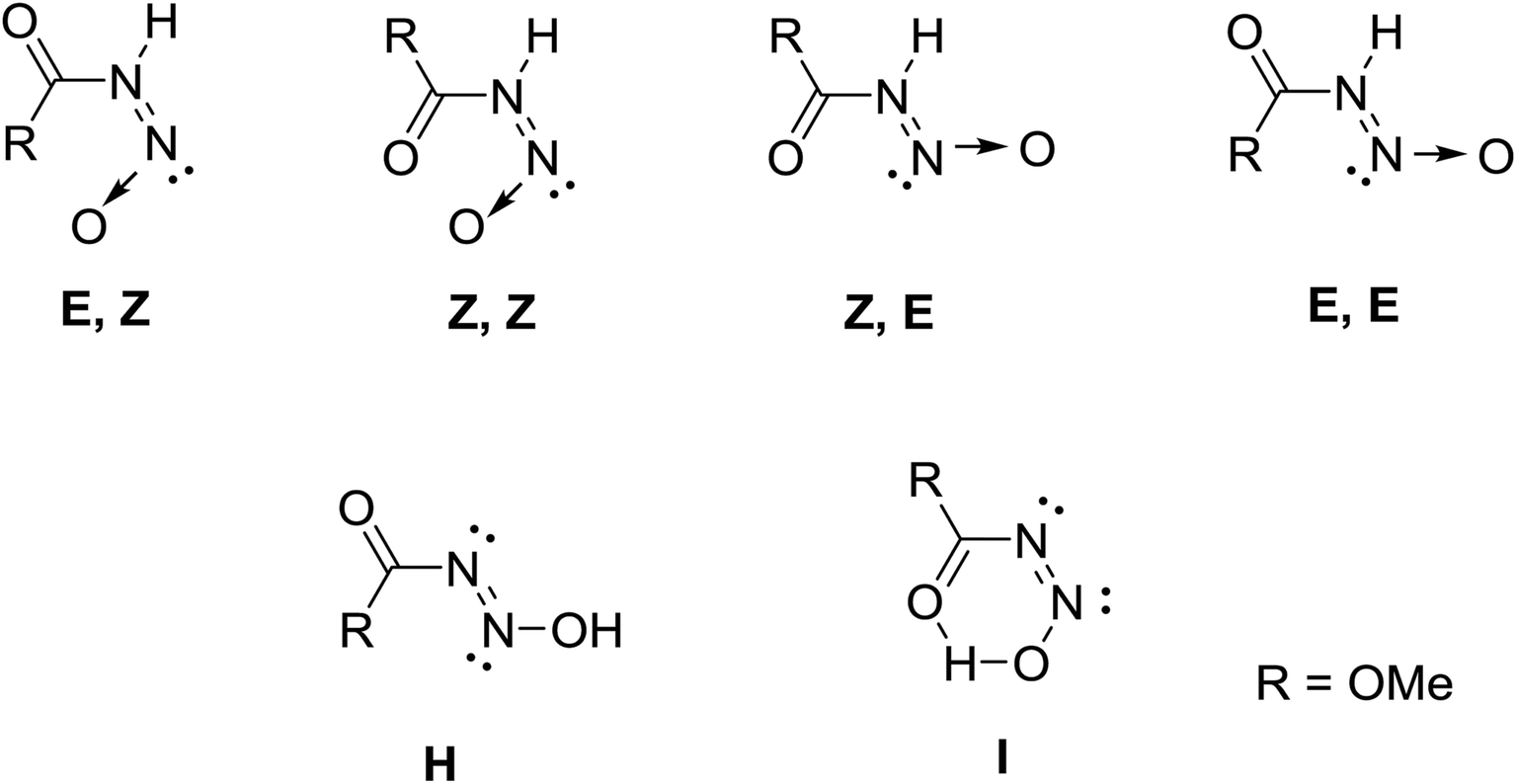 | ||
| Fig. 2 Conformers of MeOC(O)NH(NO) and two of its OH tautomers. | ||
| Structure | E/Z | Z/Z | Z/E | E/E | H | I |
|---|---|---|---|---|---|---|
| a Density functional theory, B3LYP/6-311++g**, gas phase results.b Oscillator strength f × 104. | ||||||
| E (A.U.) | −413.8472362 | −413.846855 | −413.8532021 | −413.8537858 | −413.842355 | −413.83512 |
| Rel. E (kcal mol−1) | 0 | 0.3 | 3.7 | 4.1 | 3.06 | 7.6 |
| C–N (Å) | 1.41268 | 1.40572 | 1.40274 | 1.40261 | 1.43668 | 1.42791 |
| N–N (Å) | 1.38681 | 1.38496 | 1.35889 | 1.36056 | 1.22323 | 1.25138 |
| N–O (Å) | 1.19576 | 1.19476 | 1.2057 | 1.20613 | 1.38423 | 1.35777 |
| O–C–N–N (°) | 0.01 | 0.02 | −0.01 | 179.96 | 86.01 | −0.05 |
| UV-vis (nm)/[fb] | 435.48[13] | 441.87[12] | 415.13[9] | 416.04[9] | 289.86[5] | 371.39[26] |
| 235.36[0] | 242.09[1] | 241.16[2] | 236.28[0] | 216.78[245] | 237.71[0] | |
| 212.07[1406] | 214.07[624] | 211.97[1726] | 211.73[1230] | 205.43[454] | 207.04[30] | |
This assignment of solid state backbone planar geometry Z/E and the metric parameters are strong support for an N–H intermolecular bonding. Significantly, the azohydroxymate tautomer, MeOC(O)N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NOH, H optimizes to a nonplanar backbone, τOCNN = 86°, with longer N–N and shorter N–O bond lengths than experimentally observed. Attempts to localize planar NOH tautomers, those with a crystallographically observed planar backbone, all correspond to transition state stationary states rather than minima. The only exception to this stability trend is the conformation I with an internal hydrogen bond, but even here this ground state structure is 7.60 kcal mol−1 higher in energy than E/Z and 3.54 kcal mol−1 higher in energy than the nonplanar azooxime structure H shown in Fig. 2. These factors, as well as the short N(1)–O(3) separations of 2.869(3) and 2.836(2) Å in 1 and 2 respectively, clearly indicate the correct solid state primary nitrosamide conformation is G rather than the suggested form H.1 Thus, the nitrosocarbamates are N–H containing nitrogen acids.
NOH, H optimizes to a nonplanar backbone, τOCNN = 86°, with longer N–N and shorter N–O bond lengths than experimentally observed. Attempts to localize planar NOH tautomers, those with a crystallographically observed planar backbone, all correspond to transition state stationary states rather than minima. The only exception to this stability trend is the conformation I with an internal hydrogen bond, but even here this ground state structure is 7.60 kcal mol−1 higher in energy than E/Z and 3.54 kcal mol−1 higher in energy than the nonplanar azooxime structure H shown in Fig. 2. These factors, as well as the short N(1)–O(3) separations of 2.869(3) and 2.836(2) Å in 1 and 2 respectively, clearly indicate the correct solid state primary nitrosamide conformation is G rather than the suggested form H.1 Thus, the nitrosocarbamates are N–H containing nitrogen acids.
In solution, 1 and 2 rapidly equilibrate to a mixture of conformers. The alkyl group on the carbamates gives rise to slightly downfield resonance signals when compared to alkoxy groups. In the 1N NMR spectra of the N-nitrosocarbamates 1 and 2 multiple sets of resonance signals are observed for the alkyl groups. These resonance signals and their relative intensities are also observed when crystallographically characterized samples are re-dissolved in solution. Thus, in solution, conformational equilibria for both primary nitrosamides 1 and 2 are established under a slow exchange regime in the 1H NMR experiment. This suggests relatively high interconversion barriers between at least three of the conformers that results from extensive pi-delocalized bonding over the whole framework. Finally in the 1H NMR the nitrogen acids have a downfield broad signal around 10.5 ppm which can be assigned to the proton bound to the nitrogen of the amide. This signal's chemical shift and line broadening are solvent dependent. The calculated metric parameters in Table 2 reflect this with all conformers having comparatively short C–N and long N–N bond lengths.
Although the vibrational modes for 1 and 2 correspond to coupled framework modes, which cannot be assigned to specific group modes, the observed IR solid state bands are consistent with a Z/E conformation in the G tautomer. In solution the UV spectra of 1 (Fig. S1†) and 2 (Fig. S3†) indicate that the bright yellow color of the primary nitrosamides are due to a vibronically split peak at ∼400 nm. This low lying band common to many nitroso species is assigned to a n → π* band for the nitrogen lone pair.31 To rule out the coupling of the N–H group into this band the spectrum was also measured in CH3OD, and as shown in Fig. S2† there is little H/D isotope dependence for this peak or in the vibronic splitting and so the n → π* assignment holds. Support for this is also found in the vibronic splitting, Table S2,† where the average 1100 cm−1 splitting suggests N–O π* acceptance. As with other nitrosamines,32 the energy of the absorbance envelope is also solvent dependent and is red-shifted in non-polar solvents. Although solvent polarity has been proposed to cause these shifts,33 subsequent studies suggest that hydrogen bonding is the more important factor.34 It has also been shown that this n → π* transition is sensitive to variation in the ratio of the isomers of the nitrosamines,35 but in these spectra the well resolved vibronic bands suggests either a major species, or spectral similarity for the conformers at equilibria. Note that the time dependent DFT/TD predictions for excitation energies (Table 2) predict the energies of these bands being Z/Z ∼ E/Z < E/E ∼ Z/E but with similar absorptivities.
Alkali metal salts of the primary nitrosamides
The acidic proton in the primary nitrosamides is readily exchanged and new salts isolated. The identity of the cation is an important variable in this chemistry due the strong cation/anion interactions. The un-optimised conversion of 2 into alkali salts are only mentioned by Thiele9a from the silver salt in the aqueous solutions, but we have found good conversion of these compounds into 3/3-Na and 4/4-Na from 1 and 2 respectively under anhydrous conditions (Scheme 5).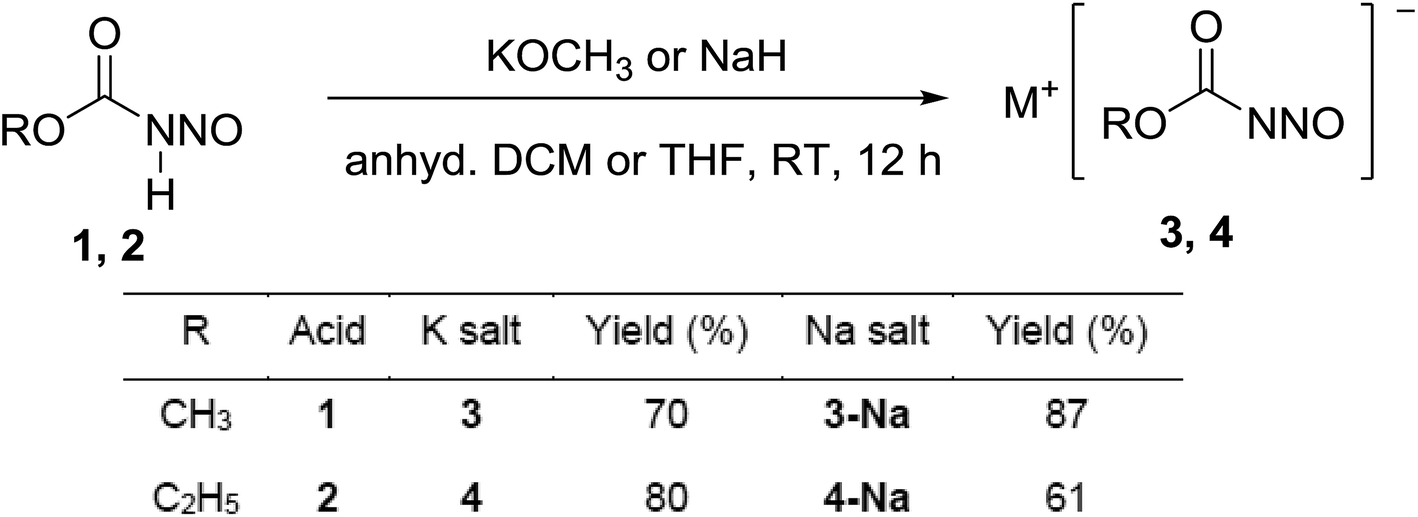 | ||
| Scheme 5 Synthesis of alkali salts 3/3-Na and 4/4-Na. | ||
The alkali salts 3/3-Na and 4/4-Na are relatively stable in the solid state but the potassium salts are exceedingly hygroscopic. Care must be taken when handling them as they are friction sensitive and explosive! Prior attempts to obtain satisfactorily C, H, and N elemental analyses for these salts have been hampered by their instability and hygroscopicity.16a Although our results are an improvement on these efforts, they too are invariably low in C, H, and N as result of their instability.
The solid state anionic structure of the potassium salt of the N-nitrosocarbamates 3 (Fig. 3) are observed to take up the (Z,E) configuration similar to the parent neutral acid conformer. Note that these stereochemical assignments are irrespective of the cation/anion interactions. Comparison with the MP2/DFT gas-phase calculations by Benin et al.16a using N-nitrosocarbamic acid anion [HOC(O)NNO]− as reference gives the (Z,E) configuration as the ground state isomer similar to the structure observed in the single crystal diffraction experiment. In the solid state the vibrational spectra for these salts are surprisingly simple compared to 1 and 2. They do share a band around 900 to 1000 cm−1 in the Raman spectrum which is usually difficult to observe with IR. Similar combination bands due to coupling of the carbamate and nitroso groups can be observed.
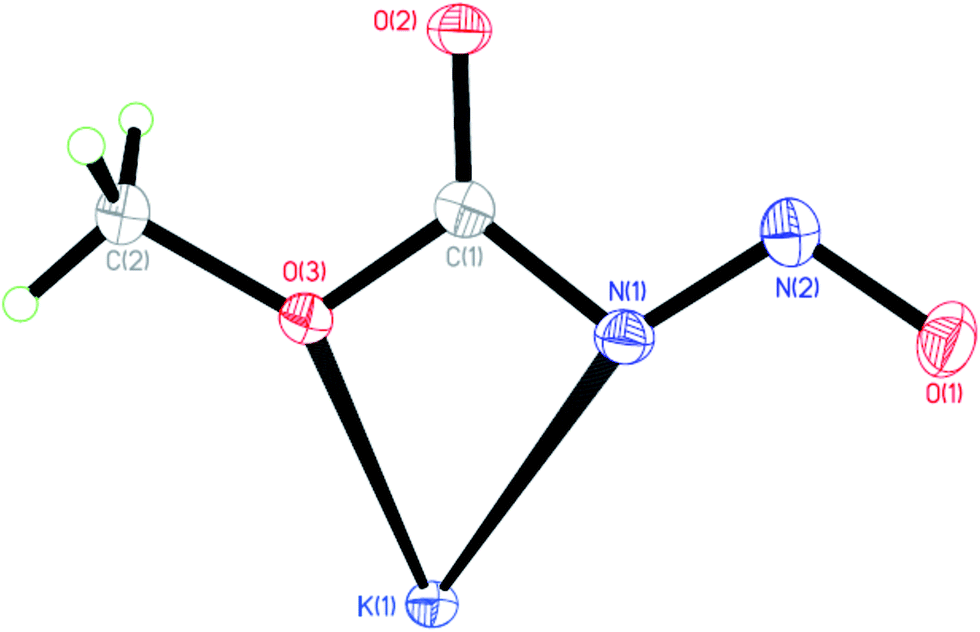 | ||
| Fig. 3 Molecular structure of 3 at 50% thermal ellipsoids, other K atoms have been omitted. | ||
The anions of the potassium and sodium salts 3/3Na and 4/4-Na have similar 1H NMR spectra which indicate the presence of conformers with similar ratios in CD3OD as was found for the parent acids. There is however a cation dependent equilibrium for the observed conformers as three possible resonance signal sets are observed for the potassium salts 3 and 4 compared to two for the sodium salts 3-Na and 4-Na. For the tetra-n-butylammonium salts, Benin and coworkers report only one set of NMR peaks for the anion.16a This could be due to fast conformational exchange in the absence of a coordinating cation, to the presence of only a single major conformer, or to the isomer's resonance being obscured by the cation's alkyl peaks. As both a solid, and in solution, the potassium salt 3 is pale yellow and has only a weak broad band in the UV-vis spectrum at 400 nm with lower absorptivity (Fig. S4†). The vibronic coupling absorption feature in 1 is absent in 3 and broadened in 4-Na (Fig. S5†). Again there is a clear cation dependence in the solution chemistry of these primary nitrosamide salts.
Decomposition of sodium N-nitrosomethylcarbamate (3-Na)
The decomposition of the N-nitrosocarbamates have been reported to give several products depending on the decomposition conditions.9 In general, the products that are suggested are carbon dioxide (CO2), alcohol (ROH) or alkene (R2CCR2) and dinitrogen (N2). Thiele et al. had carried out some careful analytical analysis to trap and identify the gases that evolve and from their results suggested that N2 is the only nitrogen containing product due to its inert nature towards reduction by H2 with Pd to give ammonia. Since Thiele's work, this analytical method36 has been examined and has been shown that nitrous oxide is either not a substrate for H2/Pd reduction under these conditions or that ammonia is not the expected product.37 Thus the assumption that N2 is the sole nitrogen containing product of the decomposition is incorrect. Subsequent reports of the N-nitrosocarbamates, regardless whether they are primary [ROC(O)NHNO] or secondary [ROC(O)N(R′)NO] forms have assumed that N2 is evolved and incorporated the loss of this gas into proposed mechanistic schemes but have not experimentally verified it.
In our endeavours towards a developing a nitrousate anion, [N2O]2−, precursor, we have found that when 3-Na is placed in an alcohol solution and left standing at room temperature in a sealed vessel, a gas that exhibits IR bands between 2240 and 2210 cm−1 was observed to evolve over time (Fig. 4). The bands are located within a region that is characteristic of nitrous oxide (N2O) and this is confirmed with an experiment using base-catalysed decomposition of nitramide to generate N2O and H2O in the same sealed vessel (Fig. S8†). The symmetric bands for N2O at 1300 and 1275 cm−1 are broad and weak and are partially overlapped with some bands from the decomposition products (Fig. S9†). The same observation was found to occur in 4-Na (Fig. S10†) but the amount of N2O evolved from the decomposition is much less. The results from this experiment clearly demonstrates that nitrous oxide is formed in the decomposition of 3-Na and to a much smaller extent for 4-Na. Analysis of the crude residue by IR indicate the presence of a carbonate salt (ν = 1631 vs. cm−1) as part of the decomposition product. This carbonate species suggests some hydrolysis of an amide. The related class of compounds N-nitro amides/carbamates (RC(O)NHNO2, R = alkyl, alkoxy) have been reported to decompose to give N2O, CO2 and alcohol or alkene.38
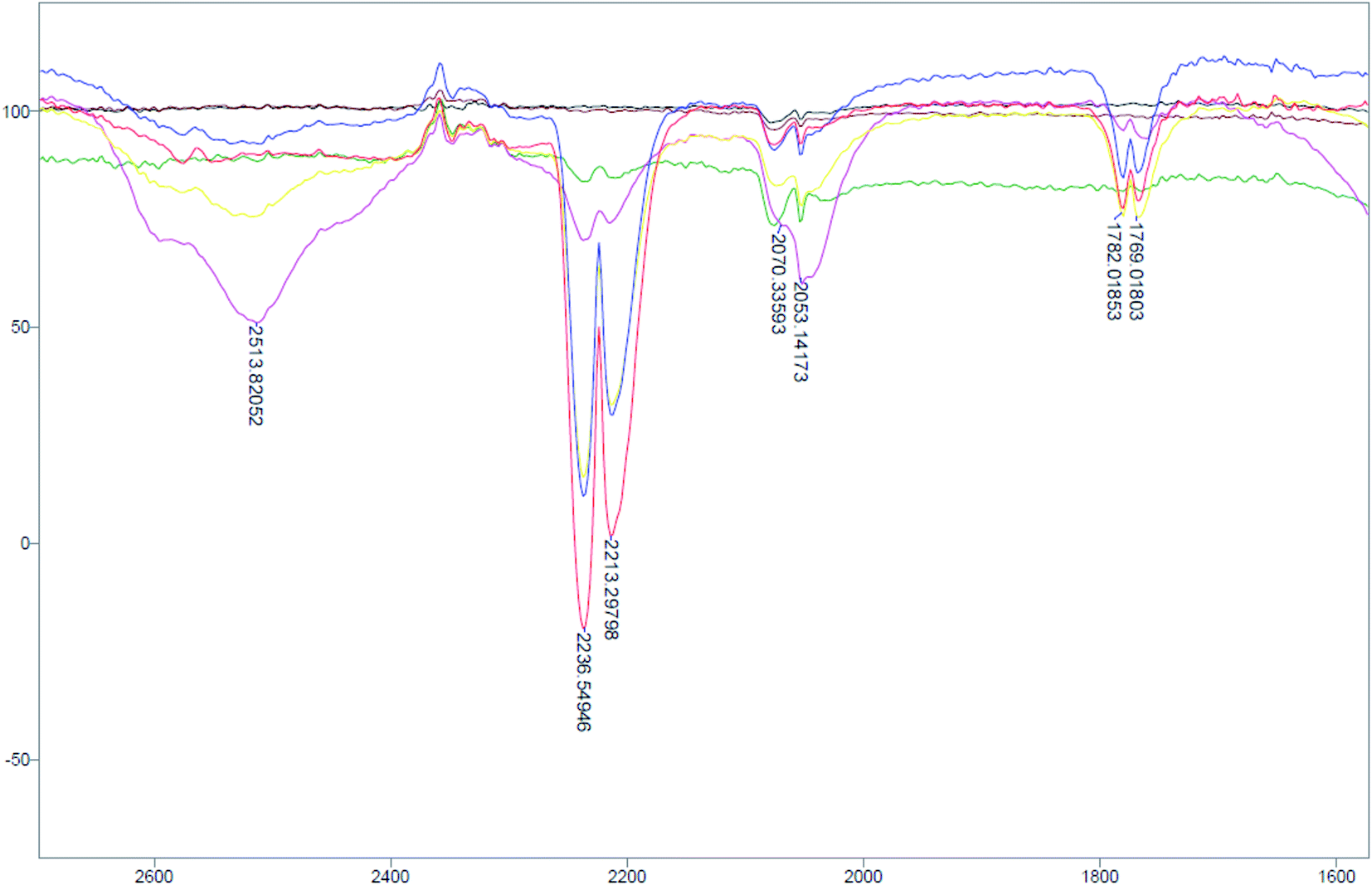 | ||
| Fig. 4 Gas-phase IR monitoring of 3-Na in CH3OH between 2700 to 1600 cm−1. Bands (ν: cm−1) 2237, 2213 are from N2O (g). Black: CH3OH; brown: CH3OH with 3-Na initial; green: after 45 min; pink: after 1.5 h; yellow: after 5 h; blue: after 6 h; red: after 12 h. | ||
The unexpected observation of N2O from decomposition of 3-Na prompted us to quantify the amount of N2O released from this process. Using calibrated GC-MS analysis, p. 20 in the ESI,† we find that N2O from the decomposition of 3-Na has a maximum yield (based on the assumption of HNO/NO− (ref. 39) production, in which case 2 equivalents of 3-Na are needed for 1 equivalent of N2O) of 5 ± 5% in water, and a 10 ± 3% yield in methanol. Thus N2O is a minor product in the decay of 3-Na in methanol.
The UV-vis spectra of 3-Na has an absorbance at 397 nm which decreases over time in methanol (Fig. S12†). The decomposition follows first order decay kinetics with an approximate half-life (t1/2) of ln2/k = 112 min, where k = 0.0062(4) min−1. During the later stages of the reaction a fine suspension of one of the products, most likely a carbonate salt, forms, and thus there is general increase in background due to scattering. This adds to the systematic errors in fitting this plot to a single exponential.
An alternative less sensitive method to monitor the rate of decomposition of 3-Na is by 1H NMR (Fig. 5). We can determine that there are at least two isomers (isomer A: major; isomer B: minor) of 3-Na in solution. Of interest is the resonance signal of non-deuterated CH3OH at 3.37 ppm. From the spectra, this signal is observed to increase over time, while that of isomer A and B decrease. More importantly, the decomposition of isomer A is much more rapid then that of B. This means that isomer A and B do not interconvert rapidly on the NMR times scale but are in equilibrium, and that their decomposition either follows two separate pathways or requires the conversion of B to A. It may be possible that the decomposition of isomer B goes through the same pathway as A but the conversion rate of B is slow. It is also important to note that small amounts of CH3OH is already observed in the initial spectrum and can only be due to the decomposition of 3-Na as a clean sample of CD3OD is not found to have a signal at 3.37 ppm. This observation gives additional support to the fact that 3-Na is relatively unstable and this in turn may contribute to the difficulty in our, and all prior attempts, to obtain a reliable elemental analyses. The presence of isomers with different stabilities also implies that simply fitting these decay reactions to limiting first order rate laws requires care in assigning the rate determining step.
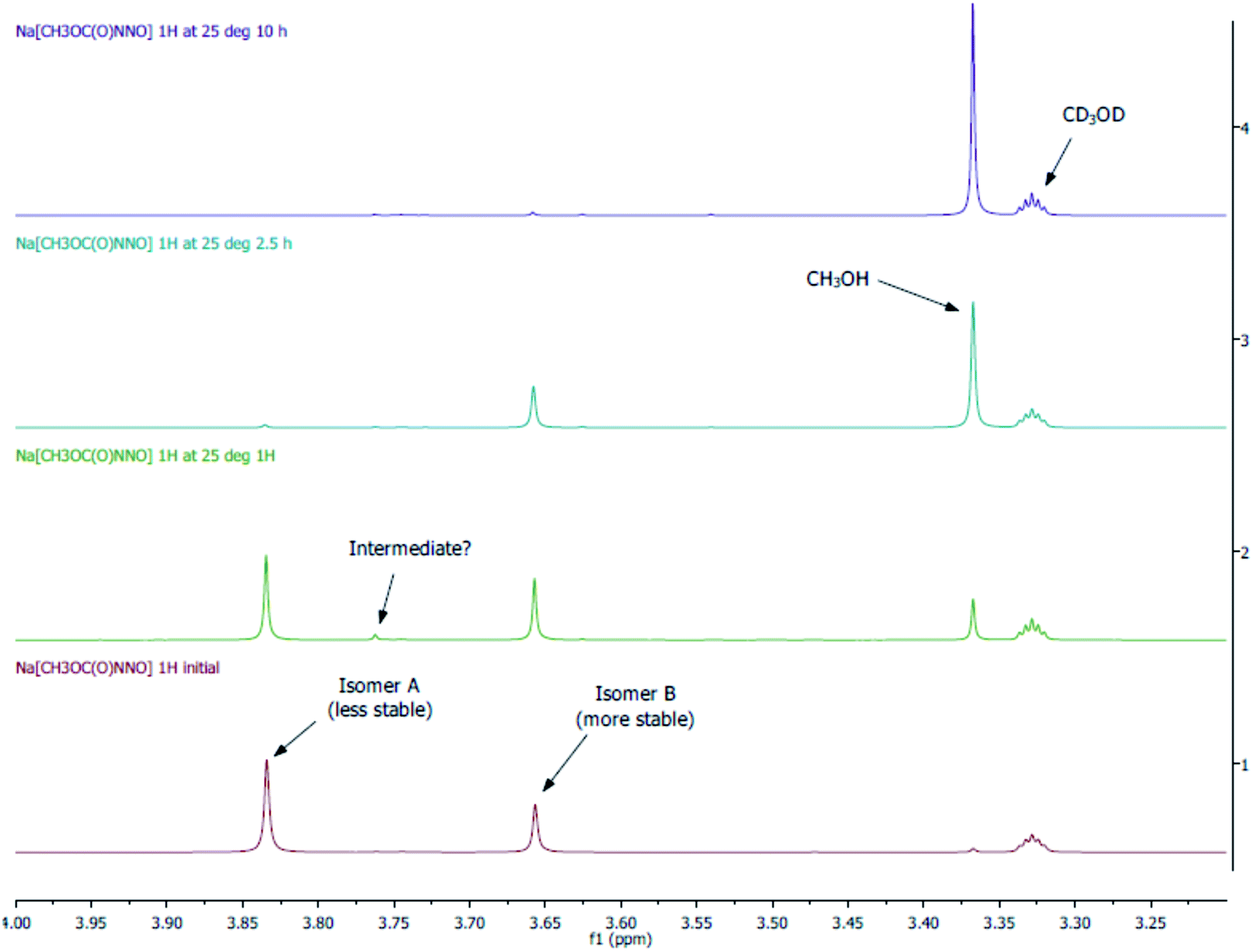 | ||
| Fig. 5 1H NMR monitoring decomposition of 3-Na in CD3OD. | ||
The detection of N2O coupled with its quantification and limited decomposition profiles from UV and 1H NMR of 3-Na means there are competing decomposition pathways for the N-nitrosamides. Among the possibilities are the generation of NO−/HNO from Hoffman rearrangement40 or a direct rearrangement to give N2O and a very unstable intermediate which rapidly degrades to methanol. Evidence of HNO/NO− generation from the decomposition of 3-Na can be seen from the absence of N2O when thiol (dodecanethiol) was present in solution (thiols act as potent HNO scavengers)41 whereas added thiol had no effect on N2O generation from a nitramide source (which proceeds without an HNO/NO− intermediate). The amount of N2O observed from the degradation of 4-Na (Fig. S10†) is significantly less than that of 3-Na which indicates the substituent dependence of these pathways. In addition, there is a role for the Lewis acid or counter cation in the N-nitrosamides decomposition pathways. Clearly before any mechanistic deductions can be made for the decay on the nitrosamides additional fundamental experimental data needs to be determined first: (1) how is anion stability in solution affected by the counter cation? (2) What are the full range of products from these decompositions? (3) Could there be separate decomposition pathways for the isomers or only a preferred pathway that is determined by interconversion of the isomers? (4) Are there alternate decomposition pathways generating other decomposition products? These factors clearly need to be established to provide a proper framework for further mechanistic work. In addition it is pertinent to re-examine the mechanistic studies for the nitro derivatives38 of these compounds.
Lewis acid stabilization of N-nitrosamides through coordination
The meta-stability, cation dependency, and fluxionality of the primary N-nitrosamides in solution makes a stable complex desirable for clarifying many of these questions. Transition-metal complexation provides an attractive option to study ligands which disproportionate or rearrange easily. As a starting point kinetically inert d6 complexes with two cis coordination sites were selected so that it might be possible to chelate a single nitrosamide anion. Carbonyl and triphenylphosphine ligands were selected as reporter groups to gauge purity, identity, and electronic structure. The four conformers for the [ROC(O)NNO]− shown in Fig. 2 allow for at least eight different chelating modes and distinguishing between the observed geometries requires care, even for well-ordered structures, as C, N, and O have similar X-ray scattering factors and thus not unequivocally discernable in electron density maps. For these studies the use of the ethyl derivatives, 2 and 4 are particularly important because of the extra heavy atom in the chain aid in the unambiguous assignment of atoms in the resulting model. Apart from our recent work with iridium19 the only prior coordination chemistry for this class of ligands is a briefly mentioned binuclear Pd complex of N-nitrosoethylcarbamate, but the spectroscopic properties, purity and identity of the complex were unfortunately not followed up in subsequent work.42The reaction of [Re(CH3CN)2(CO)2(PPh3)2]+ with 3 and 4 gives rise to slightly different reaction products which depend upon the reaction conditions (Scheme 6). The reaction of [Re(CH3CN)2(CO)2(PPh3)2]+ with 3 and 4 at room temperature gives mostly complex 5 and 7. The colourless suspension is observed to turn yellow over 10 h. The 31P NMR spectrum of the product of 3 with [Re(CH3CN)2(CO)2(PPh3)2]+ shows a major resonance signal at 29.03 ppm with other minor resonance signals some of which are broad. The 1H NMR spectra of these reaction mixtures is quite complicated with a major signal at 3.41 ppm which is assigned to the methyl group of the coordinated N-nitrosomethylcarbamate ligand. The IR spectrum has two bands at 1942 and 1864 cm−1 for ν(CO) which is similar to that observed for Re(CH3OC(O)NNO2)(CO)2(PPh3)2 (1936, 1860 cm−1).43 Additional bands at 1750, 1326 and 1070 cm−1 are assigned to ν(CO2)asym, ν(NNO)sym, ν(NNO)asym and ν(N–N) respectively.44 These spectroscopic parameters (Table 3) are assigned to 5 (Fig. 6a).
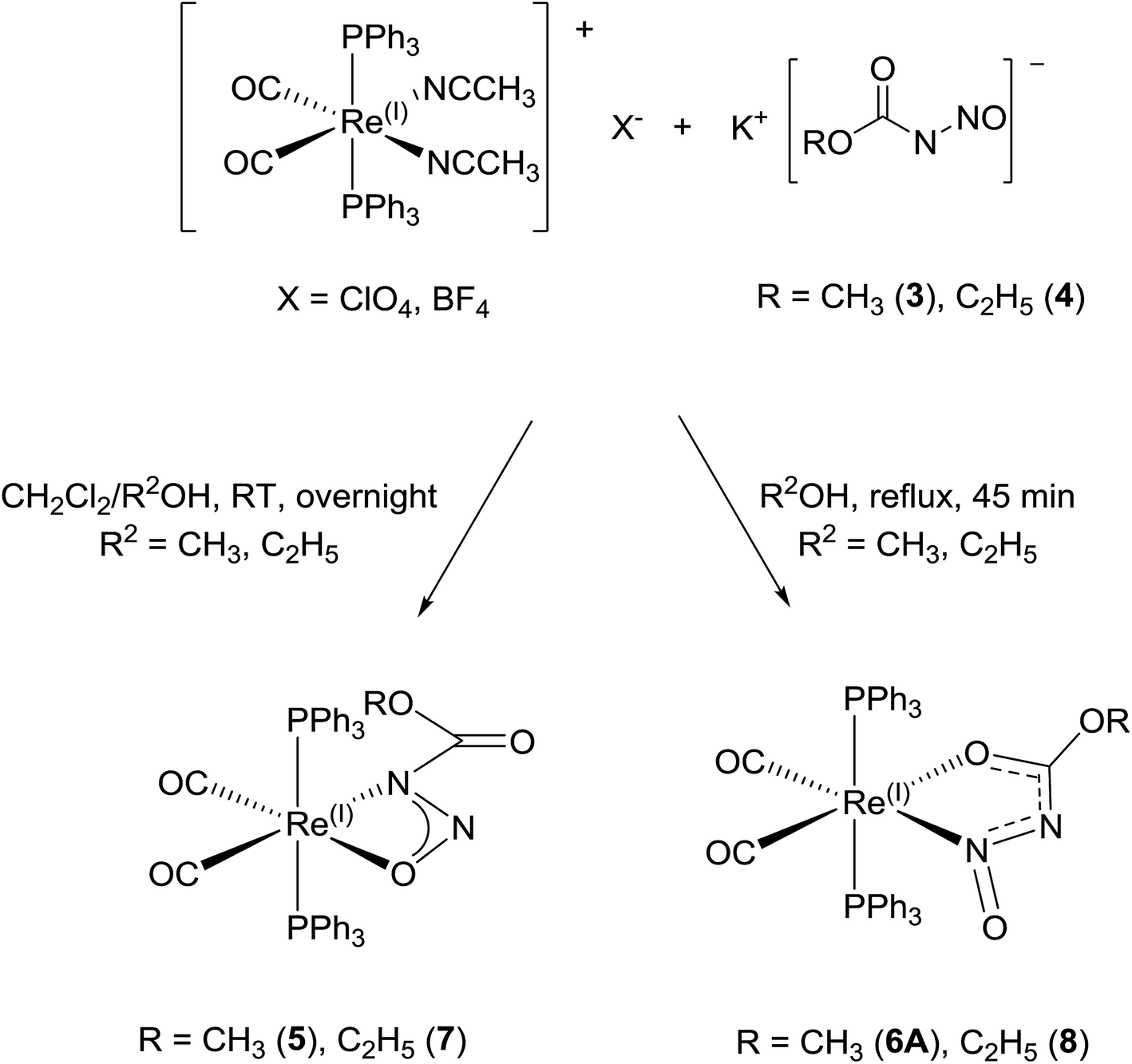 | ||
| Scheme 6 Reaction of [Re(CH3CN)2(CO)2(PPh3)2](X), (X = ClO4, BF4) with 3 and 4 under different reaction conditions to give different products. | ||
| Compound | 5 MeOC(O)NNO | 6A MeOC(O)NNO | 6Bd MeOC(O)NNO | 8 EtOC(O)NNO |
|---|---|---|---|---|
| a O(2) instead of O(3).b O(7) instead of O(4), O(8) instead of O(5), C(5) instead of C(3), C(6) instead of C(4).c N(2) instead of N(1), O(2) instead of O(1).d Identical isomer of 5 with similar label sequence. Re(2) instead of Re(1), O(6) instead of O(1), O(7) instead of O(2), O(8) instead of O(3), O(12) instead of O(7), O(13) instead of O(8), N(3) instead of N(1), N(4) instead of N(2), C(5) instead of C(1), C(6) instead of C(2), C(9) instead of C(5), C(10) instead of C(6). For a discussion of the vibrational modes see the text.e C(4) instead of C(3), C(5) instead of C(4). | ||||
| ν(CO) | 1942, 1864 | 1950, 1870 | 1947, 1870 | |
| Re(1)–N(1) | 2.088(9) | 2.132(6)c | 2.154(15) | 2.144(3)c |
| Re(1)–O(1) | 2.206(9) | 2.177(5)c | 2.165(11) | 2.157(3)c |
| N(1)–N(2) | 1.412(14) | 1.331(9) | 1.38(2) | 1.333(5) |
| N(1)–C(1) | 1.551(17) | 1.377(10) | 1.45(3) | 1.333(5) |
| N(2)–O(1) | 1.358(14) | 1.173(8) | 1.34(2) | 1.239(4) |
| C(1)–O(3) | 1.365(17)a | 1.252(9)a | 1.27(3) | 1.229(6)a |
| Re(1)–C(3) | 1.887(8)b | 1.919(8) | 1.884(7) | 1.920(5)e |
| Re(1)–C(4) | 1.902(7)b | 1.880(7) | 1.898(7) | 1.887(4)e |
| C(3)–O(4) | 1.151(7)b | 1.157(9) | 1.167(8) | 1.153(5)e |
| C(4)–O(5) | 1.163(8)b | 1.172(8) | 1.162(8) | 1.163(5)e |
| Re(1)–N(1)–N(2) | 93.7(7) | 107.7(10) | ||
| N(2)–O(1)–Re(1) | 90.2(7) | 108.4(10) | ||
| N(1)–N(2)–O(1) | 111.7(11) | 90.7(11) | ||
| O(1)–Re(1)–N(1) | 64.5(3) | 73.8(2)c | 53.2(6) | 71.69(13)c |
| N(1)–C(1)–O(2) | 123.8(8) | 126.1(4) | ||
| N(2)–N(1)–C(1) | 112.4(7) | 108.8(4) | ||
| Re(1)–N(2)–N(1) | 117.2(5) | 119.5(3) | ||
| C(1)–O(2)–Re(1) | 112.7(6) | 114.0(3) | ||
| O(1)–N(2)–N(1)–C(1) | 178.2(9) | −179.9(6) | −173.7(15) | 178.4(4) |
| O(3)–C(1)–N(1)–N(2) | 174.2(10)a | 1.2(10)a | −4(2) | −0.7(6)a |
 | ||
| Fig. 6 Molecular structure of (a) 5, (b) 6A and (c) 8 at 50% thermal ellipsoids. Hydrogen atoms have been omitted for clarity. (d) Molecular structure of 6B with four-membered ring coordination. Phenyl rings of PPh3, disordered five-membered ring isomer, hydrogen atoms and solvent molecules have been omitted for clarity. | ||
When the same reaction is carried out at elevated temperature for an hour, a similar colour change is observed. For the reaction with 3, a new major resonance signal at 23.73 ppm is now found in the 31P NMR spectrum along with other minor resonances present, one of which is at 29.03 ppm (approx. 0.20 fraction) which corresponds to 5. In the 1H NMR spectrum a new major singlet at 3.07 ppm is also observed. These signals are assigned to 6A. The IR bands for ν(CO) in 6 is observed at 1950 and 1870 cm−1, which is different from that in 5. However there is still a much weaker intensity ν(CO2)asym band at 1748 cm−1 which can still be observed from 5 which may be assigned to the isomer 6B (Fig. 6d and 7). New bands at 1556, 1419, 1158 and 1066 cm−1 are assigned to the ν(CO2), ν(NNO)sym, ν(NNO)asym and ν(N–N) respectively for the new coordination isomer 6A. The verification of minor 31P NMR signal at 23.73 ppm and 1H NMR signal at 3.41 ppm allowed us to note that small amounts of 6A was also observed (approx. 0.2 fraction) in the room temperature reaction. When 4 is the reactant, the thermodynamic isomer 8 is also mostly observed.
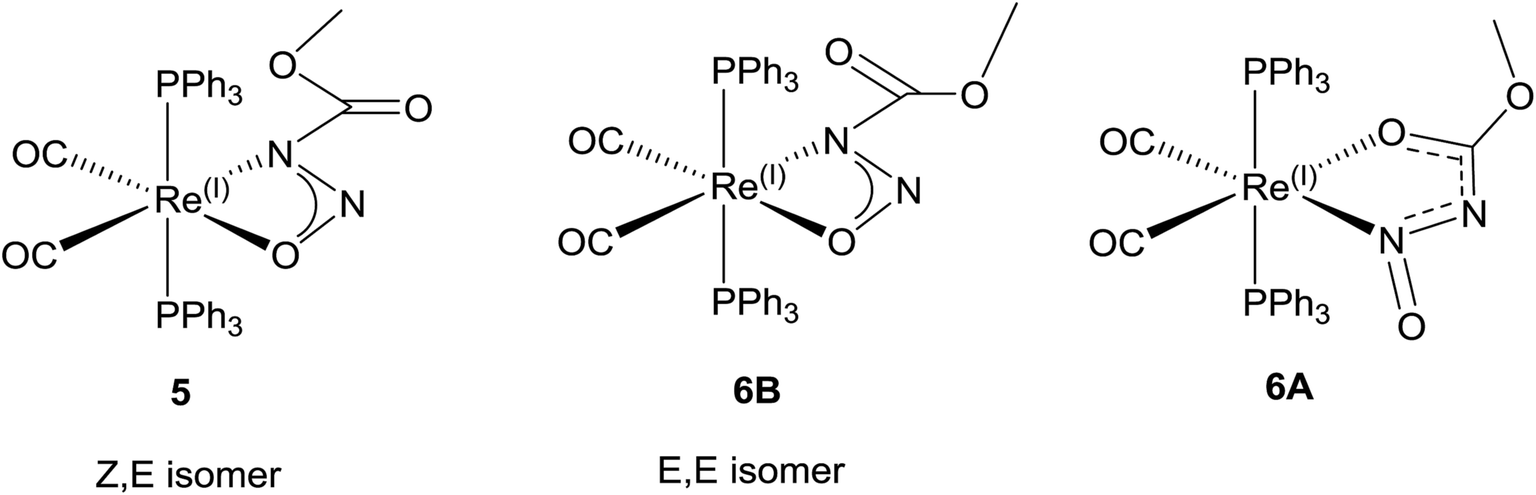 | ||
| Fig. 7 Coordination isomers of 1 in Re(I)(η2-CH3OC(O)NNO)(CO)2(PPh3)2 complexes. | ||
The conversion of the ethyl derivative 7 to 8 occurs more rapidly than the conversion of the methyl derivative 5 to 6A. Within the same reaction time of 12 h for the reaction of Re(CH3CN)2(CO)2(PPh3)2]+ with either 3 or 4, a larger fraction of 8 was observed in the 1H and 31P NMR spectra. With the increased solubility of the ethyl group, the outcome was that the solid state structure of 7 could not be obtained as in this case the kinetic isomer converts into the thermodynamic isomer 8 which then crystallizes preferentially. Complex 7 however has similar IR, 1H, and 31P spectra as 5, which validates its structure assignment. The origin of the differences in the stabilities of the carbamate ester complexes may simply be due to steric effects, but solubility differences may also allow the ethyl complexes to stay in solution and continue to react longer.
Due to the disorder present in the kinetic isomer complex 5 and minor component of the thermodynamic isomer equivalent of 6A in 5 which will be discussed, no suitable structural comparisons can be made. For the thermodynamic isomers 6A and 8, the N(1)–N(2) bond lengths have increased while the N(1)–O(1) nitrosyl bond lengths have decreased upon coordination in the Re complexes. The general structural trend for the free acids 1, 2 and their conjugates in the Ir(I) and Re complexes is a decrease in the N–N bond length upon deprotonation and coordination. Although the thermodynamic rhenium complexes isomer has a different binding mode compared to the Ir(I) system (longer), the C–N bond distance are found to be mostly unchanged, but the O(2)–C(1) bond lengths of the coordinated acyl group are observed to have increased in length on coordination compared to the anion or the Ir(I) system.19 Similar trends of the torsion angles between the NNO fragment and the acyl fragment are observed between the neutral ligand and the complexes which mirror the N-nitroamides.
Kinetic and thermodynamic isomers of Re (N-nitrosocarbamate) complexes and their possible transformation
The X-ray diffraction solution for 6 contains two independent Re complexes that are different isomers of 1 coordinated to the Re center as shown in (Fig. 7). The structure of 6A shows an unprecedented coordination isomer for N-nitroso/N-nitrocarboxy compounds, the first demonstration of this geometry in the coordination chemistry of the nitrogen acids. Structure 6A has the same distorted octahedral coordination around the Re center with cis carbonyl and trans triphenylphosphine ligands. The N-nitrosocarbamate ligand is still bidentate but coordinated to the Re center through the oxygen atom of the acyl group and the nitrogen of the nitroso group. In normal circumstances, this form of coordination is expected for ligands with acyl functional groups as reported before.20,43,45 However in the other examples for the Ir(III) and Re(I) N-nitro nitrogen acids complexes, the four-membered η2-NNO configuration of 5 is preferred.20,43,45 This five-membered acyl-N-nitroso structural isomer is unprecedented with only an example with a related ligand,46 in this case a urea derivative, having been reported. In a case of serendipity, the second Re complex in 6 contains disordered isomers of both 6A and the (E,E) ligand isomer of 5, 6B (Fig. 7).The model for the diffraction data of 6 indicates approximately 11:9 ratio of either structure 6A or structure 6B respectively in the second disordered Re complex (Fig. S15†). Due to the severe disorder encountered, the structural parameters for the ligand in 6B are not precise enough for a meaningful discussion. Nevertheless the stability and quality of the final refinement indicates this is an excellent model for the electron density. Significantly, the model is in accord with both the IR and the NMR spectroscopic data.
The solid state structural observations of 6 could be a ‘frozen frame’ of the transformations occurring in the reaction of [Re(CH3CN)2(CO)2(PPh3)2]+ with 3 over time. From the observations in the 31P, 1H NMR, IR spectroscopy and X-ray diffraction data, we propose a possible sequence of transformations occurring between 1 over time where the kinetic product is 5 while the final thermodynamic product is 6A. Thus 6B does not accumulate in detectable quantities by NMR and suggests a fast second isomerization step, if it follows the same mechanism (Scheme 7).
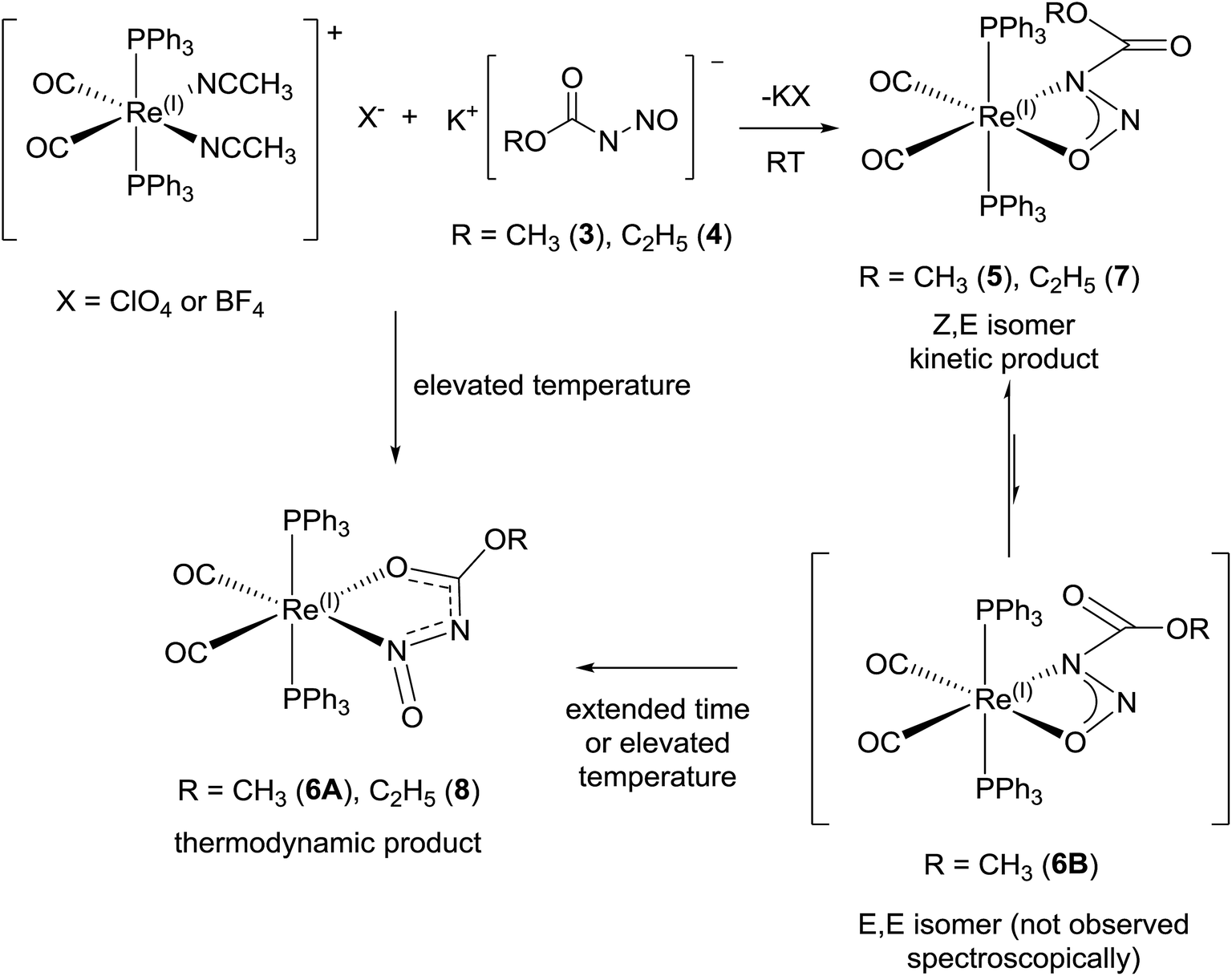 | ||
| Scheme 7 Proposed transformations in reaction of [Re(CH3CN)2(CO)2(PPh3)2](X), (X = ClO4 or BF4) with 3 and 4. | ||
Some evidence of the proposed transformations is observed in the 31P NMR and 1H NMR spectra of the room temperature and reflux condition reactions where the resonance signals for 5 and 6A are observed in fractional amounts corresponding to reaction conditions in the reaction mixtures as well as in the crystal dissolution experiments of both products of these reaction conditions. There were other minor resonance signals present especially in the 31P spectra, however these signals are not observed in both reaction conditions which would allow the assignment of signals to 6B. It is possible that 6B is in equilibrium with 5 in solution but it is not observed due to rapid exchange and just happens to co-crystallize with 6A. In this case, once the crystals of 6 are dissolved, the signals of 6B appear as 5.
This isomerization is an unusual transformation in an usual complex. We note the prior coordination chemistry of N-nitrosocarbamates is restricted to a brief mention of a binuclear Pd complex of 2.42 Apart from the elemental analysis, and the shifts of two resonances in the 1H NMR, the reported spectroscopic details were limited to only two IR bands.42 A ‘simplified’ pathway of conversion from 5 to 6A may involve total ligand dissociation from one side of the complex followed by coordination of the ligand from the opposite side of the N-nitrosomethylcarbamate ligand. This obviously is unlikely to occur. The rearrangement may follow instead a similar pathway as Re(I)(η2-NO2)(CO)2(PPh3)2,23 where CO addition creates N and O coordination isomers of the nitrite ligand either by one ended dissociation followed by coordination or a dimeric intermediate. This type of transformation is known for transition metal nitrite complexes;47 the reaction of Re(I)(η2-NO2)(CO)2(PPh3)2 with HCl gives two products, Re(I)Cl(CO)3(PPh3)2 and Re(I)Cl2(NO)(CO)(PPh3)2 which seems to indicate that the reaction proceeds by a dimer formation of the Re complexes to allow the formal transfer of one of the carbonyl ligands to the other Re complex.23
The isolation of the Re N-nitrosamide complexes provides a glimpse of the potential for stabilisation by suitable Lewis acids. For this class of anionic compounds, treating H+ as the simplest Lewis acid, the stability of the N-nitrosamide anions follow the general series: H+ < alkaline salts (K+, Na+), Ag+ < Ir(I), Re(I). Coordination stability from metal centers that are able to delocalize the negative charge of these anions seem to be advantageous. In addition, the presence of a labile proton or hydride plays a role as our attempts to prepare Ir(III) analogues20,45,48 of these class of compounds have been unsuccessful due to the possible rearrangement/reductive/elimination processes that can occur in the presence of a Ir–H ligand.
Conclusion
The updated synthetic preparation of the N-nitrosocarbamate 1, 2 and the corresponding salts: potassium, 3, 4 and sodium: 3-Na, 4-Na allows for more comprehensive exploration of their chemistry. Clearly the spectroscopy of these anions in solution indicates the presence of distinct conformation isomers which do not interconvert on the NMR timescale. The vibrational spectra of these compounds are unique due to the conjugated functional groups. Electronic spectra of the N-nitrosocarbamates have similar properties with nitroso compounds where vibronic coupling in the n to π* absorption is observed. Single crystal diffraction experiments of the compounds indicate the (Z,E) isomer to be preferred. The anionic salts of the N-nitrosocarbamate have slight structural differences compared to their parent neutral analogs. The evolution of N2O from the decomposition of 3-Na and 4-Na in methanol is unexpected as previous reports have assumed the loss of N2 to be one of the products. This new discovery puts into question the results of previous attempts at mechanistic and kinetic studies. A reinvestigation of the solution chemistry of the N-nitrosamides is required. New Re N-nitrosamides complexes which are stable have been synthesized and characterised. The isolation of kinetic and thermodynamic isomers of this class of ligands is unprecedented and extolls the flexible coordination modes of these ligands. The stabilisation of the anionic N-nitrosamides by the Re complexes presents possible avenues to probe their reactivity.Of the two isolable N-nitrosamides the carbamate system is now the best characterized and clearly indicates a cation, or Lewis acid, dependent set of conformational dynamics and stability. Many of these considerations probably apply to the closely related guanidine and urea based N-nitrosamines, and choosing the right counter cation may be an important consideration for extending this chemistry. Alternatively, in a heterogenous environment such as in physiological fluids any individual N-nitrosamide may undergo markedly different reactions. Mapping out this range of reactivity is an important prelude to understanding nitrosamide biochemistry.
Acknowledgements
We gratefully acknowledge helpful discussions with Dr K. Kirschbaum for X-ray diffraction structure analysis for 5, CRC, CFI, and NSERC for support in the forms of a CRC, CFI, and discovery grants to DSB.References
- J.-P. Anselme, in N-Nitrosamines, ed. J.-P. Anselme, ACS Sympossium Series, Washington (D.C.), 1979, ch. 1, vol. 47, pp. 1–21 Search PubMed.
- Y. L. Chow, in N-Nitrosamines, ACS Sympossium Series, 1979, ch. 2, vol. 101, pp. 13–37 Search PubMed.
- (a) R. Huisgen and H. Reimlinger, Justus Liebigs Ann. Chem., 1956, 599, 161–182 CrossRef CAS; (b) R. Huisgen, Justus Liebigs Ann. Chem., 1951, 573, 163–181 CrossRef CAS.
- E. Mueller, W. Hoppe, H. Hagenmaier, H. Haiss, R. Huber, W. Rundel and H. Suhr, Chem. Ber., 1963, 96, 1712–1719 CrossRef CAS.
- T. Koenig, J. A. Hoobler and W. R. Mabey, J. Am. Chem. Soc., 1972, 94, 2514–2515 CrossRef CAS.
- (a) H. Meerwein, Chem. Zentralbl., 1933, 104, 1758 Search PubMed; (b) F. W. Bollinger, F. N. Hayes and S. Siegel, J. Am. Chem. Soc., 1950, 72, 5592–5594 CrossRef CAS.
- T. W. Koenig and M. Deinzer, J. Am. Chem. Soc., 1968, 90, 7014–7019 CrossRef CAS.
- T. Ohwada, M. Miura, H. Tanaka, S. Sakamoto, K. Yamaguchi, H. Ikeda and S. Inagaki, J. Am. Chem. Soc., 2001, 123, 10164–10169 CrossRef CAS PubMed.
- (a) J. Thiele and A. Lachman, Liebigs Ann., 1895, 288, 267–311 CrossRef CAS; (b) E. H. White, J. Am. Chem. Soc., 1955, 77, 6014–6022 CrossRef CAS.
- T. W. Koenig, M. Deinzer and J. A. Hoobler, J. Am. Chem. Soc., 1971, 93, 938–944 CrossRef CAS.
- (a) K. Heyns and W. V. Bebenburg, Chem. Ber., 1953, 86, 278–286 CrossRef CAS; (b) K. Heyns and O. F. Woyrsch, Chem. Ber., 1953, 86, 76–83 CrossRef CAS; (c) K. Heyns and W. V. Bebenburg, Justus Liebigs Ann. Chem., 1955, 595, 55–68 CrossRef CAS; (d) K. Heyns and W. V. Bebenburg, Justus Liebigs Ann. Chem., 1955, 595, 69–80 CrossRef CAS; (e) K. Heyns and W. V. Bebenburg, Chem. Ber., 1956, 89, 1303–1306 CrossRef CAS; (f) K. Heyns and A. Heins, Justus Liebigs Ann. Chem., 1960, 634, 29–49 CrossRef CAS.
- J. Thiele, Liebigs Ann., 1893, 273, 133–144 CrossRef.
- (a) L. Kong, E. Saavedra Joseph, S. Buzard Gregory, X. Xu, L. Hood Brian, P. Conrads Thomas, D. Veenstra Timothy and K. Keefer Larry, Nitric Oxide, 2006, 14, 144–151 CrossRef CAS PubMed; (b) E. H. White, J. Am. Chem. Soc., 1955, 77, 6011–6014 CrossRef CAS.
- E. Muller, H. Haiss and W. Rundel, Chem. Ber., 1960, 93, 1541–1552 CrossRef CAS.
- (a) D. S. Bohle, G. R. Clark, C. E. F. Rickard, W. R. Roper, W. E. B. Shepard and L. J. Wright, J. Chem. Soc., Chem. Commun., 1987, 563–565 RSC; (b) M. A. Gallop and W. R. Roper, in Adv. Organometal. Chem., ed. F. G. A. Stone and R. West, Elsevier, New York, 1986, vol. 25, pp. 121–198 Search PubMed.
- (a) V. Benin, P. Kaszynski and J. G. Radziszewski, J. Am. Chem. Soc., 2002, 124, 14115–14126 CrossRef CAS PubMed; (b) V. Benin, J. Mol. Struct.: THEOCHEM, 2006, 764, 21–31 CrossRef CAS; (c) H. Hedian and V. Benin, J. Chem. Crystallogr., 2011, 41, 1976–1980 CrossRef CAS; (d) A. Thakkalapally and V. Benin, Tetrahedron, 2005, 61, 4939–4948 CrossRef CAS; (e) S. Venkata, E. Shamo and V. Benin, Synth. Commun., 2009, 39, 1406–1414 CrossRef CAS.
- J. Thiele and F. Dent, Liebigs Ann., 1898, 302, 245–272 CrossRef CAS.
- (a) E. H. White, J. Am. Chem. Soc., 1955, 77, 6008–6010 CrossRef CAS; (b) E. H. White and C. A. Aufdermarsh, J. Am. Chem. Soc., 1961, 83, 1174–1178 CrossRef CAS; (c) E. H. White and C. A. Aufdermarsh, J. Am. Chem. Soc., 1961, 83, 1179–1190 CrossRef CAS.
- D. S. Bohle and Z. Chua, Organometallics, 2015, 34, 1074–1084 CrossRef CAS.
- D. S. Bohle and Z. Chua, Inorg. Chem., 2011, 50, 3135–3140 CrossRef CAS PubMed.
- G. W. Parshall, L. W. Shive and F. A. Cotton, in Inorg. Synth., John Wiley & Sons, Inc., 1977, vol. 17, pp. 110–112 Search PubMed.
- X. L. Luo, H. Liu and R. H. Crabtree, Inorg. Chem., 1991, 30, 4740–4742 CrossRef CAS.
- K. L. Leighton, K. R. Grundy and K. N. Robertson, J. Organomet. Chem., 1989, 371, 321–328 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, Sadabs, Twinabs, University of Göttingen, Germany, 1996 Search PubMed.
- G. M. Sheldrick, CELL NOW, program for unit cell determination, University of Göttingen, Germany & Bruker AXS Inc, Madison (WI), USA, 2005 Search PubMed.
- A. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef CAS PubMed.
- O. A. Luk'yanov, O. V. Anikin, V. P. Gorelik and V. A. Tartakovsky, Russ. Chem. Bull., 1994, 43, 1457–1461 CrossRef.
- R. F. Weiss and B. A. Price, Mar. Chem., 1980, 8, 347–359 CrossRef CAS.
- E. Sada, S. Kito and Y. Ito, Ind. Eng. Chem. Fundam., 1975, 14, 232–237 CAS.
- (a) D. R. Battiste, L. P. Davis and R. V. Nauman, J. Am. Chem. Soc., 1975, 97, 5071–5078 CrossRef CAS PubMed; (b) W. S. Layne, H. H. Jaffe and H. Zimmer, J. Am. Chem. Soc., 1963, 85, 435–438 CrossRef CAS.
- R. N. Haszeldine and B. J. H. Mattinson, J. Chem. Soc., 1955, 4172–4185, 10.1039/jr9550004172.
- H. McConnell, J. Chem. Phys., 1952, 20, 700–704 CrossRef CAS.
- (a) G. J. Brealey and M. Kasha, J. Am. Chem. Soc., 1955, 77, 4462–4468 CrossRef CAS; (b) A. K. Chandra and S. Basu, Trans. Faraday Soc., 1960, 56, 632–637 RSC.
- C. J. Michejda, N. E. Davidson and L. K. Keefer, J. Chem. Soc., Chem. Commun., 1976, 633–634, 10.1039/c39760000633.
- (a) P. Sabatier and J. B. Senderens, Compt. rend., 1895, 120, 1212–1215 CAS; (b) P. Sabatier and J. B. Senderens, Compt. rend., 1902, 135, 278–281 Search PubMed.
- (a) Y. Yoshida, S. Ogata, S. Nakamatsu, H. Inoue and C. Iwakura, Electrochim. Acta, 1999, 44, 3585–3587 CrossRef CAS; (b) D. McCalman, K. Kelley, C. Werth, J. Shapley and W. Schneider, Top. Catal., 2012, 55, 300–312 CrossRef CAS.
- E. H. White and L. A. Dolak, J. Am. Chem. Soc., 1966, 88, 3790–3795 CrossRef CAS.
- (a) V. Shafirovich and S. V. Lymar, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 7340–7345 CrossRef CAS PubMed; (b) V. Shafirovich and S. V. Lymar, J. Am. Chem. Soc., 2003, 125, 6547–6552 CrossRef CAS PubMed; (c) D. L. H. Williams, in Nitrosation Reactions and the Chemistry of Nitric Oxide, Elsevier Science, Amsterdam, 2004, ch. 13, pp. 221–229, DOI:10.1016/b978-044451721-0/50014-1.
- J. Poisson, I. S. Butler and D. S. Bohle, J. Mol. Struct., 2016, 1116, 30–36 CrossRef CAS.
- (a) X. Sha, T. S. Isbell, R. P. Patel, C. S. Day and S. B. King, J. Am. Chem. Soc., 2006, 128, 9687–9692 CrossRef CAS PubMed; (b) M. E. Shoman, J. F. DuMond, T. S. Isbell, J. H. Crawford, A. Brandon, J. Honovar, D. A. Vitturi, C. R. White, R. P. Patel and S. B. King, J. Med. Chem., 2011, 54, 1059–1070 CrossRef CAS PubMed.
- R. Robson, Inorg. Chim. Acta, 1984, 85, 195–198 CrossRef CAS.
- D. S. Bohle and Z. Chua, ChemistrySelect, 2016, 1, 2096–2101 CrossRef CAS.
- (a) R. G. Coombes, Nitro and nitroso compounds, in Compr. Org. Chem., Pergamon, 1979, pp. 305–381 Search PubMed; (b) G. G. Suchkova and L. I. Maklakov, Vib. Spectrosc., 2009, 51, 333–339 CrossRef CAS.
- D. S. Bohle and Z. Chua, Inorg. Chem., 2014, 53, 11160–11172 CrossRef CAS PubMed.
- (a) R. K. Murmann, R. Glaser and C. Barnes, J. Chem. Crystallogr., 2005, 35, 317–325 CrossRef CAS; (b) R. K. Murmann, R. Glaser and C. L. Barnes, J. Coord. Chem., 2005, 58, 279–294 CrossRef CAS.
- M. A. Hitchman and G. L. Rowbottom, Coord. Chem. Rev., 1982, 42, 55–132 CrossRef CAS.
- Z. Chua, Dissertation, McGill University, 2013.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details for the ethyl derivative of the N-nitrosocarbamates and Re complexes, UV-vis spectra of 1 in CD3OD, 2, 3-Na and 4-Na, gas-phase IR spectra of the decomposition of nitramide, 3-Na and 4-Na. UV-monitoring decomposition of 3-Na. Tables of the crystallographic metric parameters for 1–3, 5–6, 8, disordered isomer of 5, the two asymmetric molecules of 7 and disordered structure of 7B. The calibration of the GC-MS for N2O quantification and brief discussion of SQUEEZE results are included. Diagram of IR gas-cell. CCDC 1416401–1416403, 1435727, 1435728, 1435738. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra24421a |
| This journal is © The Royal Society of Chemistry 2017 |