
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Microstructural evolution of regenerated silk fibroin/graphene oxide hybrid fibers under tensile deformation†
Chao Zhang‡
a,
Yaopeng Zhang‡ *a,
Jie Luob,
Jingru Shia,
Huili Shaoa and
Xuechao Hua
*a,
Jie Luob,
Jingru Shia,
Huili Shaoa and
Xuechao Hua
aState Key Laboratory for Modification of Chemical Fibers and Polymer Materials, College of Materials Science and Engineering, Donghua University, Shanghai 201620, China. E-mail: zyp@dhu.edu.cn; Fax: +86-21-67792855; Tel: +86-21-67792954
bSchool of Material Science and Energy Engineering, Foshan University, Foshan 528000, China
First published on 13th January 2017
Abstract
Graphene oxide (GO) with outstanding mechanical properties is a satisfactory filler to reinforce artificial silk. To prepare tough regenerated silk fibroin/graphene oxide (RSF/GO) hybrid fibers, it is important to understand the structural evolution of the hybrid fibers with external deformation. The morphology and microstructural changes of the hybrid fibers at different tensile strains were investigated by SEM, Raman spectra and synchrotron radiation wide angle X-ray diffraction (SR-WAXD). Longer and deeper stripes were found on the surface of RSF/GO fibers with the increase of strain as revealed by SEM. SR-WAXD results revealed that the fraction and orientation of crystals and the mesophase had slight changes in the elastic deformation zone of the fiber. However, in the plastic deformation zone of the fiber with a strain from 5 to 18%, the fraction of crystals gradually increased while the fraction of the mesophase decreased. During this deformation, the orientation of crystals (fc) firstly increased significantly and then increased slightly, while the orientation of the mesophase (fm) increased steadily beyond the yield point. When the strain exceeded 18%, the fraction of crystals decreased while the fraction of mesophase increased. The fc had a rapid increase again at a strain above 18%. A model was proposed to explain how the tensile deformation affects the molecular orientation of the hybrid fibers.
Introduction
Animal silks, especially from spiders and silkworms, have attracted great attention due to theirs fascinating tensile properties, good biocompatibility and biodegradability.1–3 The outstanding mechanical properties of natural silks are believed to be attributed to the microfibril hierarchical structure4,5 with three condensed state phases: β-sheet crystals, amorphous regions and the mesophase. One of the challenges for human beings to fabricate high performance artificial silks is how to construct the hierarchical structures like from animals.A large number of studies showed that strain-induced structure changes in deformed silk materials allowed the manipulation of mechanical properties which have practical importance in the polymer engineering field. Numata et al.6 investigated the microstructural change of a bundle of silkworm cocoon silks during deformation by using synchrotron radiation wide angle X-ray scattering and demonstrated that the stretching process could not induce the β-sheet formation to a significant degree but could promote the orientation of β-sheet crystals. Polarized synchrotron FTIR spectroscopy measurements of A. pernyi silk monofilament revealed that the stretching process did not have a significant effect on the β-sheet content and orientation.7 Sirichaisit et al.8 studied the molecular deformation of both silkworm (Bombyx mori) and spider dragline (Nephila edulis) silks by using a combination of mechanical deformation and Raman spectroscopy. The band shift in the Raman spectra was linearly dependent upon stress for both types of silk fibers. In addition to natural silks, regenerated silk fibroin (RSF) film was also adapted to investigate the structural evolution during drawing process. Huot et al.9 demonstrated that a continuous drawing of RSF film could initially facilitate the conformation transformation from unordered structure to β-sheet, and subsequently improve the orientation of the formed β-sheet. It is found that there are obvious differences between natural and man-made silk materials in terms of the structural evolution during external deformation. Therefore, it is necessary to understand the microstructure evolution of RSF fibers in tensile deformation. Understanding the microstructural change is helpful for us to fabricate excellent performances of artificial silk fibers.
According to some literatures, the maximum breaking strength of RSF fibers, which were obtained from wet- or dry-spinning process could be reached about 350–380 MPa,10,11 and the breaking strength of electrospun fiber mats obtained from RSF aqueous solution were about 8.6 MPa after post-treatment.12 Generally, the post-treatment, for example, immersing in the methanol/water solution and water vapour treatment, is necessary and critical for improving the thermal and mechanical properties of RSF-based materials.13 In order to manipulate the properties of RSF fibers or make spinning process feasible, some synthetic polymers or natural biodegradable polymers, like polyethylene oxide (PEO)14 and poly(L-lactide) (PLLA),15 and other nano-particles, such as metallic oxide,16 carbon-based fillers17 and organic nano-particles,18 have been introduced to RSF spinning dope. Graphene oxide (GO), which exhibits light weight, good dispersion and perfect flexibility, has attracted more and more attentions during the last decade. Many polymer based materials reinforced with GO sheets showed significant improvement in mechanical properties,19 thermal stability20 and dielectric performance.21 In our previous work,22 RSF/GO hybrid fibers with excellent mechanical properties were prepared. The addition of two dimensional (2D) GO sheets could develop a new mesophase (or interphase) caused by the interaction between RSF and GO sheets, which was also found in other GO sheets reinforced polymer systems.23,24 The new mesophase is different from the inherent mesophase in natural silks, which is depicted as: (1) an intermediate connects between crystallites and either amorphous or other crystallites,25,26 and (2) a phase that consists of partially oriented amorphous region or weakly oriented crystalline regions of unaggregated β-sheets.27,28 Since the mesophase is critical for the reinforcement of RSF materials,23,29,30 it is important to study the microstructure development of nanofiller-incorporated silk fibroin materials during tensile deformation.
In this paper, synchrotron radiation wide angle X-ray diffraction (SR-WAXD) and Raman spectra were applied to investigate the microstructure development of RSF/GO hybrid fibers at different stretching states. This may be useful for understanding the deformation mechanism of RSF/GO hybrid fibers, which is interesting for the researchers who work on fabricating high performance composite fibers.
Experimental section
Preparation of RSF/GO dry-spun hybrid fibers
B. mori silk fibers (produced in 2014 and in Tongxiang city, Zhejiang Province, China) were boiled twice in an aqueous solution of 0.5 wt% Na2CO3 for 30 min to remove the sericin and rinsed with distilled water. After drying at room temperature, the degummed silks were dissolved in 9.3 mol L−1 LiBr aqueous solution at 40 °C for 2 h and dialyzed against (molecular weight cutoff 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) distilled water for 3 days to obtain the RSF aqueous solution. Then the RSF aqueous solution of 20 wt% was mixed with GO suspension (0.5 mg mL−1, the Sixth Elements HiTech Development Co., Ltd., China). The thickness of GO sheets was about 1.1 nm (Fig. S1†). The size distribution of the GO sheets were mainly concentrated in the range of 160–1440 nm and 3700–6430 nm, and the intensity ratio (area ratio) of the two peaks was about 98.5/1.5 (Fig. S2†). The mass ratio of GO/RSF was set 1/1000. Then the blend solution was condensed in forced airflow at 8 °C and 60 ± 5% relative humidity, according to the method reported in our previous works.31–34 The RSF/GO hybrid fibers were dry-spun from a 45 wt% spinning dope in air by using a custom-made equipment at 25 °C and 45% relative humidity. The speed of take-up roller was set as 3 cm s−1 and the distance between spinneret and take-up roll was 10 cm. The as-spun fibers were kept in a sealed dryer for 48 h. Then the as-spun fibers were stretched in 80 vol% ethanol aqueous solution and immersed in it for 2 h. The draw ratio was set as 2× and determined as the ratio of the final length of fibers in the ethanol aqueous solution to the initial length of as-spun fibers in the ethanol aqueous solution. The RSF fibers without GO fillers were fabricated at the same conditions.
000) distilled water for 3 days to obtain the RSF aqueous solution. Then the RSF aqueous solution of 20 wt% was mixed with GO suspension (0.5 mg mL−1, the Sixth Elements HiTech Development Co., Ltd., China). The thickness of GO sheets was about 1.1 nm (Fig. S1†). The size distribution of the GO sheets were mainly concentrated in the range of 160–1440 nm and 3700–6430 nm, and the intensity ratio (area ratio) of the two peaks was about 98.5/1.5 (Fig. S2†). The mass ratio of GO/RSF was set 1/1000. Then the blend solution was condensed in forced airflow at 8 °C and 60 ± 5% relative humidity, according to the method reported in our previous works.31–34 The RSF/GO hybrid fibers were dry-spun from a 45 wt% spinning dope in air by using a custom-made equipment at 25 °C and 45% relative humidity. The speed of take-up roller was set as 3 cm s−1 and the distance between spinneret and take-up roll was 10 cm. The as-spun fibers were kept in a sealed dryer for 48 h. Then the as-spun fibers were stretched in 80 vol% ethanol aqueous solution and immersed in it for 2 h. The draw ratio was set as 2× and determined as the ratio of the final length of fibers in the ethanol aqueous solution to the initial length of as-spun fibers in the ethanol aqueous solution. The RSF fibers without GO fillers were fabricated at the same conditions.
Scanning electron microscopy (SEM)
Surface morphology of RSF fibers with/without GO filler at different tensile strains were observed by SEM (Hitachi S-3000N) at an voltage of 10 kV.Mechanical test
The mechanical properties of RSF/GO fibers and RSF fibers were examined by Instron 5565 at (25 ± 2) °C and (45 ± 5) % relative humidity. The extension rate and gauge length were 2 mm min−1 and 1 cm, respectively.Raman spectroscopy
Raman spectra of the RSF/GO fibers with various strains were obtained by using Renishaw Raman spectrometer with a resolution of 2 cm−1. A He–Ne laser at 633 nm was used to generate an intensity of 0.51–0.68 mW and the data were recorded from 1000 to 3000 cm−1.SR-WAXD experiments
SR-WAXD measurements were performed at BL15U1 beamline (micro-focus beamline) of Shanghai Synchrotron Radiation Facility (SSRF), China using an X-ray with energy of 16 keV. The energy ring was 3.5 GeV. The beam current was 220 mA. The wavelength (λ) of the X-ray was 0.07746 nm and the spot size was 2.1 × 3.1 μm2. The sample-to-detector distance during WAXD experiment was 196.7 mm. The exposure time for each sample was 30 seconds. All the WAXD patterns were recorded with a Rayonix SX-165 detector. The obtained two dimensional (2D) WAXD patterns of RSF/GO fibers and RSF fibers were converted to the corresponding one dimensional (1D) WAXD diffractograms by using a software of Fit2D (V 12.077).The tensile deformation for single fiber was achieved by carrying out stress-control test and strain-control test, respectively.
 | ||
| Fig. 1 Schematic diagram of (a) stress-control, (b) strain-control test for RSF/GO fiber. | ||
cosθ, where K = 0.9, λ = 0.07746 nm, β is the full width at half-maximum (FWHM) of the diffraction peak and θ is the corresponding Bragg angle.
 | ||
| Fig. 2 WAXD data processing to obtain the fraction of crystal, mesophase, amorphous and the azimuthal profiles of degummed silk. (a) and (b) Radius integration and (c) azimuth integration were performed on the 2D-WAXD pattern of degummed silk in different areas to obtain the corresponding (d) and (e) 1D-WAXD diffractogram and (f) azimuthal profiles at d-spacing of 0.45 nm. The red circular area in (a), ellipse area of (020) and (210) diffractions in (b), and arc area in (c) represent the different integration areas. (g), (h) and (i) are the corresponding deconvolutions of (d), (e) and (f), respectively. | ||
It is considered that crystals and amorphous regions are interlinked by an intermediate, which is called mesophase.26,27 For RSF/GO fibers, due to the complex interaction between GO sheets and RSF molecules, such as hydrogen bonding and electrostatic interaction, some RSF molecules can be absorbed on GO sheets, thus forming an interphase zone. For composite fibers, it is generally difficult to clearly distinguish the interphase zones and the mesophase between crystalline regions and amorphous regions, because there is no straight method to calculate the fractions of the two parts. Therefore, the interphase was also included in mesophase in this work. The fraction of mesophase (Am) was obtained according to literatures.27,28
A region containing the main reflections of (210) and (020) lattice planes were selected firstly. The 1D WAXD profiles (Fig. 2h) were then fitted with three Gaussian peaks including two sharp reflection peaks for (210) and (020) lattice planes (crystalline component) and one peak for mesophase. The area of the mesophase peak was used to estimate the fraction of mesophase. Finally, the fraction of amorphous (Aa) was estimated according to Aa = 100% − Ac − Am.
An azimuthal scan was performed at d-spacing of 0.45 nm on the 2D-WAXD pattern of degummed silks. The corresponding 1D azimuthal intensity profile was fitted with two Gaussian peaks (Fig. 2i). According to literatures,28,39,40 the narrower peak reflected crystalline materials and the broader peak indicated oriented amorphous materials. The inherent mesophase in silks was defined as partially oriented amorphous regions,27,28 and the interface zones between RSF and GO sheets can also be oriented since the oriented GO sheets surrounded by the interface could compel their environment to be oriented during the spinning and post-treatment process. Thus it may be reasonable to consider that the broader peak reflects the orientation of mesophase in the RSF/GO hybrid fibers.16 The crystalline orientation factor (fc) and mesophase orientation factor (fm) were estimated from the corresponding fitted peaks by using Herman's orientation factor as below:
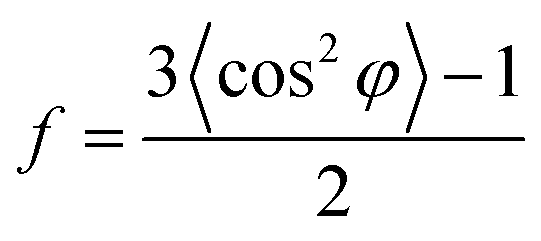 | (1) |
φ can be obtained using the following equation:
 | (2) |
Result and discussion
Raman spectra of GO and RSF/GO hybrid fiber
Raman spectroscopy experiments were performed on single RSF/GO hybrid fiber, since Raman spectroscopy was a useful tool to analyze the structure change of carbon-based materials. Typical Raman spectra of RSF/GO fibers with a mass ratio of 1000/1 are shown in Fig. 3. For comparison purpose, the Raman spectrum of GO powder was also given. In spite of the low mass ratio of GO/RSF, the GO bands (D band (circa 1335 cm−1) and G band (circa 1596 cm−1)) were dominate in the Raman spectra of RSF/GO hybrid fibers. Normally, D band usually refers to the defects in graphite structure, and G band relates to in-plane vibration of sp2 hybridized carbon atoms.41,42 The intensity ratio of the D to G bands (ID/IG) is a parameter to evaluate disorder degree of GO sheets.43 For RSF/GO hybrid fiber prior to tensile deformation (0% strain), the value of ID/IG was about 1.06. When the strain increased to 5% and 15%, the value of ID/IG was about 1.19 and 1.20, respectively. This indicated that during the deformation process, GO structure could be affected and the disorder degree of GO sheets gradually increased. However, at the strain of 25%, the value of ID/IG decreased to 1.09. Since some GO sheets with larger particle size existed in RSF/GO fibers and the stress concentration around large GO sheets could accelerate them to produce cracks during the deformation process, the initial increase of ID/IG could be attributed to the structural damage of large GO sheets. After that, as the strain further increased, the preferred orientation of GO sheets in the fibers could be gradually predominant since the stretching process can lead to the alignment of GO sheets. The value of ID/IG thus decreased when the strain was increased from 15% to 25%. According to some literatures,44–46 the deformation process of some carbon-based materials can be analyzed by studying G or D Raman bands-shifts. However, in this work, D band or G bands-shifts were not observed for RSF/GO fibers during the deformation process. To further analyze the microstructural change of RSF/GO fibers under different deformation states, SR-WAXD was carried out for the fibers. | ||
| Fig. 3 Raman spectra of GO powder and RSF/GO hybrid fiber at different tensile strains. | ||
Morphological and microstructural change of RSF/GO hybrid fibers during tensile deformation
The morphology of RSF and RSF/GO hybrid fibers with different strains are shown in Fig. 4. It can be seen that initial RSF fibers were smooth, with a diameter of about 11 μm. The RSF/GO fibers without tensile deformation also showed a uniform morphology with similar diameter to RSF fibers. Some stripes were observed on the surface of the initial RSF/GO fibers, which may be owing to the stretching process in the post-treatment. At strain 5%, these stripes along the stretching direction became obvious and some small stripes gradually formed on the surface of RSF/GO fibers. When strain further increased, the longer and deeper stripes can be observed (red arrows indicates the positions). These results suggested that the fibrillar structure (microfibrils) of RSF/GO fibers might gradually be oriented along the stretching direction during the tensile deformation. In general, stripes developed at the strain ranging from 0 to 18%. The RSF/GO hybrid fibers did not show any evident signs of cracks in the outer layer at a strain between 0 and 18%. The morphology change of the RSF/GO hybrid fibers during the stretching process was similar to the single tri-layered polystyrene (PS)–thermoplastic polyurethane (TPU)–polystyrene (PS) fibers fabricated by electrospinning,47 as the long and deep stripes can be firstly observed on the surface of the latter followed by formation of the cracks from the SEM pictures. Since there was lower orientation degree in RSF/GO fibers, the produce of cracks for RSF/GO fibers might need higher strain than 18%. | ||
| Fig. 4 Morphologies of (a) RSF fiber and RSF/GO hybrid fiber at different strains: (b) 0, (c) 5%, (d) 12% and (e) 18%. The scale bars in the picture represent 5 μm. | ||
The microstructural changes of RSF and RSF/GO fibers during the tensile deformation were investigated by the synchrotron radiation wide angle X-ray diffraction (SR-WAXD). In order to obtain clear WAXD patterns by collecting sufficient diffraction intensities, the draw ratio of 2× was selected to study the microstructural changes of RSF/GO fibers during the deformation since the average diameter of fibers decreased with the increasing draw ratio. Furthermore, compared with the traditional X-ray diffractometers, the SR-WAXD has high spatial resolution and high sensitivity for materials with micro or nano scale. Therefore, SR-WAXD technique was introduced for single RSF and RSF/GO fibers in this study. Generally, stress–strain curve of fiber materials can be mainly classified into two parts by yield point: elastic deformation zone and plastic deformation zone. In order to investigate the structure evolution of RSF/GO fibers in the elastic deformation zone and plastic deformation zone, we carried out stress-control test and strain-control test, respectively. Fig. 5 and S3† show the 2D-WAXD patterns of RSF/GO fibers and RSF fibers at different strains in the stress–strain curves, respectively. It was found that the elastic deformation zone and plastic deformation zone of RSF/GO and RSF fibers were separated by the yield point of 3.6% and 2.2%, respectively. For RSF/GO fibers in Fig. 5, A0–A3 were the 2D-WAXD patterns of the elastic deformation zone, while B1–B4 were the 2D-WAXD patterns of the plastic deformation zone. Similarly, for RSF fiber in Fig. S3,† C0–C3 and D1–D4 represented 2D-WAXD patterns of the elastic and plastic deformation zones, respectively. The uniform amorphous halo in A0 and C0 indicated that the fraction of crystals and molecular orientation degree in RSF/GO and RSF hybrid fibers at initial states were both low.
 | ||
| Fig. 5 The stress–strain curve of the RSF/GO hybrid fibers at a 2× draw ratio and the corresponding 2D-WAXD patterns at different strains. | ||
It was observed that in the elastic deformation, there was no obvious difference among A0–A3 and C0–C3, which indicated that the elastic deformation had slight effect on the crystalline structure and orientation degree of RSF/GO and RSF fibers. The reason may be that only chemical bond lengths and bond angles were changed in this deformation zone. It has also been reported that the initial tensile deformation (stretching deformation ratio of 1.1) upon spider dragline silk could misaligned the orientation of β-sheet crystals along the fiber axis.6 However, when RSF/GO fibers came into the plastic deformation zone, the 2D-WAXD patterns of them began to change with the increase of the strain. Specifically, the intensity of diffraction ring in the patterns for B1 and B2 increased, while diffraction arcs appeared in the 2D-WAXD patterns for B3 and B4 and gradually became clearer with the increasing strain. This indicated that crystalline structures in the fibers were influenced by the stretching beyond the yield point, and the molecular chain segments started to orientate along the direction of stretching, which led to change of the orientation degree. On the other hand, the intensity of diffraction ring in the 2D-WAXD patterns of RSF fibers also gradually increased in the plastic deformation zone (Fig. S3†).
The corresponding 1D-WAXD diffractograms of RSF/GO fibers and RSF fibers at different strains are shown in Fig. 6a and S4a.† There was still no obvious changes for the 1D-WAXD diffractograms of A0–A3 and C0–C3. However, the diffraction peaks of (021), (002), (040) and (023) lattice plains in B1–B4 become gradually broader, which suggests that β-sheet crystals in RSF/GO hybrid fibers became more tightly packed with the increasing of strain.6
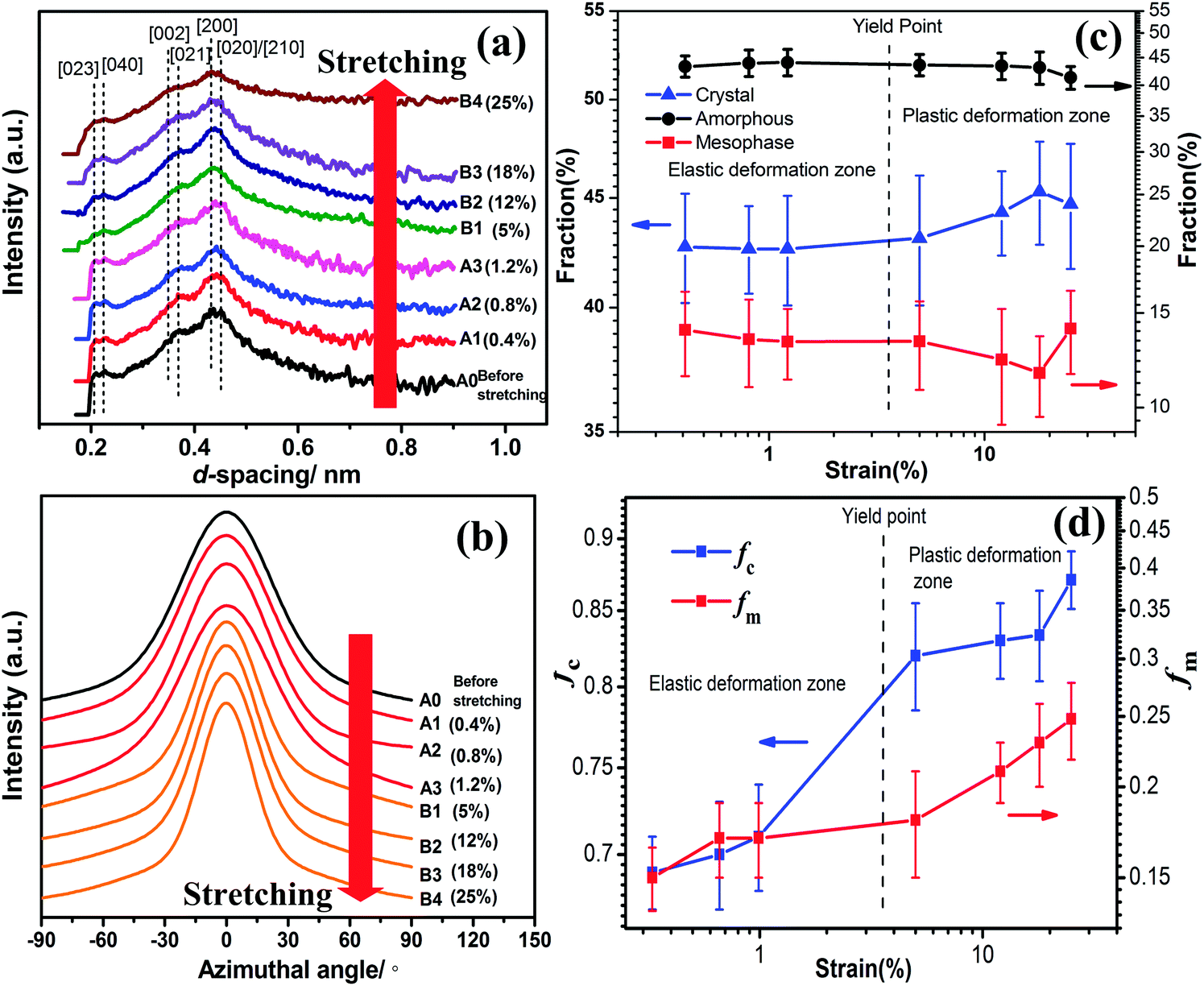 | ||
| Fig. 6 Analysis of (a) the 1D-WAXD diffractograms of RSF/GO hybrid fiber in different stretching states: (b) azimuthal profiles at d-spacing of 0.45 nm, (c) the variation of fraction of crystallinity, amorphous and mesophase, and (d) the variation of Herman's orientation factor of crystalline region and mesophase. | ||
Using the methods mentioned earlier, the evolution of amorphous, crystal and mesophase for RSF/GO hybrid fibers and RSF fibers were determined from the corresponding 1D-WAXD data, and the results were illustrated in Fig. 6c and S4c.† It was found that in the elastic deformation zone, the fractions of amorphous, crystal and mesophase of both RSF and RSF/GO hybrid fibers changed slightly, which suggested that the microstructure of the fibers almost remained constant until to the limited strain. In addition, the fraction of mesophase in RSF/GO hybrid fibers was higher than that of RSF fibers, which could be due to the formation of interphase between RSF and GO sheets in the hybrid fibers (Fig. S4c†).
In the plastic deformation zone, for RSF/GO fibers at the strain ranging from 5 to 18%, the fraction of mesophase decreased from 13.3 to 11.6, while the crystal content increased from 43.1 to 45.3. This indicated that under the larger tensile deformation, a part of mesophase in the fiber could be converted to crystals. In other words, the mesophase in the fibers was not a stable (or metastable) phase and it could be evolved to the crystalline structure during the stretching process. On the basis of the result of variation of amorphous, mesophase and crystals, it can be speculated that the strain-induced mesophase mainly refer to the phase between the crystalline and amorphous region, not the interphase between GO sheets and RSF. Ran et al.48,49 also found that the mesophase in Kevlar fibers could be converted to crystals during deformation. However, this phenomenon is different from that studied from B. mori and A. yamamai silks6,50 in which the stretching deformation did not facilitate the crystallization of silk fibroin. When the strain exceeded 18%, the fraction of crystal and amorphous in the RSF/GO fibers decreased, while that of mesophase increased. It illustrated that once the tensile deformation exceeded a certain point (18%), some nano β-sheet crystals in the fibers could be damaged and broke into small fragments of mesophase, and at the same time, the chains in the amorphous regions could be also oriented and packed with a certain degree to form the mesophase. After that, the defects of the crystal structure further increased with the increasing strain until the fiber rupture. For RSF fibers, after the yield point, it can also be seen that the fraction of crystals increased and the fraction of amorphous phase decreased (Fig. S4c†). With regard to the structural evolution of RSF and RSF/GO fibers induced by strain upon stretching, we confirmed that the tensile deformation could affect the crystalline structure of artificial silk fibers to some degree.
For silkworm cocoon silk fibers, β-sheet crystals were more highly oriented with the increase of tensile deformation.6 In order to study the effect of deformation on the molecular orientation of RSF/GO and RSF fibers, an azimuthal scan at d-spacing of 0.45 nm were performed, which are shown in Fig. 6b and S4b.† It was observed that for both RSF/GO and RSF fibers, the full-width at half-maximum (FWHM) of main diffraction peaks had little change in the elastic deformation zone, while they gradually became narrow in the plastic deformation. This indicated that during the whole stretching deformation process, the alignment of silk fibroin chains along the fiber axis occurred only in the plastic deformation zone. By deconvolution of 1D azimuthal intensity profiles, we further estimated the orientation degree of crystalline region and mesophase, respectively, which are shown in Fig. 6d and S4d.† When RSF/GO fibers were at small strain (0 < ε < 1%), fc and fm were around 0.70 and 0.16, respectively. As the strain increased (1.2% < ε < 5%), fc exhibited a sudden increase, while fm increased slightly at this moment, suggesting that the orientation of mesophase was lagged behind the orientation of crystalline region during the tensile deformation. When the strain was in the range from 5% to 18%, fm exhibited a steady and rapid growth, while fc reached a plateau value. It indicated that the orientation of mesophase was predominant at this stage. When the strain was beyond 18%, fc increased significantly again. fc and fm of RSF/GO hybrid fibers at the strain of 25% were 0.87 and 0.25, respectively. It was also found that fc at high strain increased faster (ε > 18%) than at low strain (1.2% < ε < 5%). This is consistent with the previous report9 about the structural change of RSF film under tensile deformation, in which a lower rate of molecular orientation at lower strain and a higher one at higher strain were observed. Interestingly, the orientation factor of crystalline regions and mesophase of RSF/GO fibers before fracture were still higher than that of RSF fibers, which may account for the better mechanical properties of RSF/GO fibers.
Deformation mechanism of RSF/GO hybrid fibers
Huot et al.9 reported that tensile deformation upon RSF film did not cause the molecular alignment immediately, but firstly imposed a conformational transformation of silk fibroin. So it is reasonable to consider that only bond lengths and bond angles are changed when the RSF/GO fibers are at small strain. In such a case, fc and fm had no obvious changes at 0–1.2% strain. Compared with the less change of orientation degree in the elastic deformation zone, the variation of both fc and fm in the plastic deformation zone were quite complicated. In order to explain it, a schematic model for RSF/GO fiber was put forward in this work, which was shown in Fig. 7. On the whole, it can be classified into three stages. Firstly, fc exhibited a significant increase and fm increased slightly at a strain between 1.2 and 5%. It could be attributed to 2D GO sheets which influenced the mobility of RSF molecular chains due to the interfacial interaction between them. So the unconstrained crystals which were far away from GO sheets were firstly oriented along the stretching direction. Due to the strong interaction between RSF and GO sheets,23,29,51 the GO sheets could directly affect the orientation of mesophase which was around them. However, the GO sheets which had the larger size than crystals, were not easy to move in the fibers at small strain. Hence, fm increased slightly and the orientation of mesophase was lagged behind the crystalline orientation at this stage. With the increase of strain (5% < ε < 18%), the fiber came into the second stage, in which the orientation of mesophase was predominant over that of crystals. From the analysis of Raman spectral of RSF/GO fibers at the strain of 5% and 15%, we learned that GO sheets in the fibers could have obvious deformation. Thus it is reasonable to deduce that a part of GO sheets start to move along the stretching direction at large strain. A certain degree of alignment of GO sheets in the RSF matrix could induce the preferred orientation of mesophase, so the significant increase of fm could result from the orientation of GO sheets. On the other hand, due to the better mobility of molecular chains in mesophase than that in crystalline regions, the orientation of crystals was not enhanced obviously at this stage. Finally, as the strain exceeded 18%, fc increased obviously again. Considering that the orientation degree of GO sheets could increase with the increasing strain, and oriented GO sheets surrounded by mesophase could compel their environment to be oriented, those β-sheet crystals which were confined or restricted by GO sheets were rearranged and aligned along the fiber axis. In brief, the structural evolution of RSF/GO fibers during the tensile deformation can be summarized as follows. The initial stretching (ε < 1%) applied on the fiber did not affect the microstructure, while with the increase of strain, the parts of mesophase in the fiber were gradually converted to the crystals, which resulted in the increase of fraction of crystal (Ac) and the decrease of fraction of mesophase (Am). During the stretching process, the orientation of mesophase was lagged behind the orientation of crystals. When the applied strain was beyond 18%, Ac decreased, while Am and the orientation of crystals and mesophase still increased.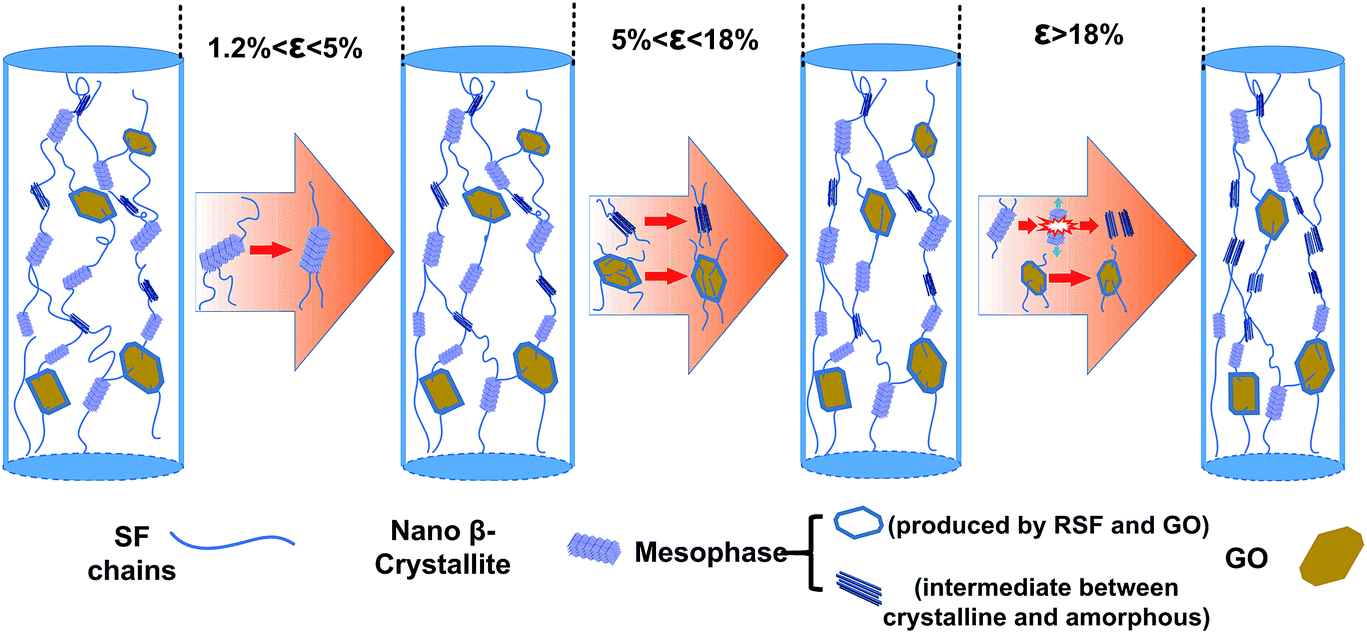 | ||
| Fig. 7 A schematic model demonstrating microstructural changes of RSF/GO fibers in different strain ranges. | ||
Conclusion
Raman spectroscopy, SR-WAXD techniques and SEM were applied to extensively study microstructural change and morphology of the dry-spun RSF/GO hybrid fibers at different stretching deformations. The surface of RSF/GO fibers after being stretched exhibited some stripes, and these stripes became longer and deeper with increasing strains. The Raman spectra confirmed that the structure of GO sheets was affected by the fiber deformation, and the disorder degree of GO sheets significantly increased at the initial stretching process. When the single hybrid fiber was stretched in the elastic deformation zone, the fraction of crystal, mesophase and amorphous, and the orientation degree of the fiber almost remained the constant. In the plastic deformation zone with a strain ranging from 5 to 18%, the fraction of crystal gradually increased while the fraction of mesophase decreased. A further strain larger than 18% led to the destruction of nano β-sheet crystallites and formation of mesophase. Both fc and fm changed slightly in the initial stretching step (ε < 1.2%). With the increase of deformation, fc increased first rapidly then slowly beyond the yield point, while fm exhibited a significant increase at a strain over 5%. When the strain exceeded 18%, fc increased significantly again.Since the drawing process could affect the mechanical properties of fiber materials through improving their crystalline structure and molecular orientation, understanding the microstructure development of composites during the process is useful to fabricate high performance materials. This work systemically and thoroughly investigated the effect of tensile deformation upon the structural development of RSF/GO hybrid fibers, which could not be obtained from a simple analysis. It may serve as a reference for studying the microstructure evolution of other polymer/nanofiller hybrid fibers during tensile deformation, and be helpful to analyze and unravel the enhancement mechanism and deformation mechanism of them.
Acknowledgements
This work is sponsored by the National Natural Science Foundation of China (21274018, 21674018), the National Key Research and Development Program of China (2016YFA0201702), “Shuguang Program” supported by Shanghai Education Development Foundation and Shanghai Municipal Education Commission (15SG30), DHU Distinguished Young Professor Program (A201302), the Fundamental Research Funds for the Central Universities, and the Programme of Introducing Talents of Discipline to Universities (No. 111-2-04).References
- N. Bhardwaj, W. T. Sow, D. Devi, K. W. Ng, B. B. Mandal and N. J. Cho, Integr. Biol., 2015, 7, 53–63 RSC.
- B. B. Mandal, A. Grinberg, E. S. Gil, B. Panilaitis and D. L. Kaplan, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7699–7704 CrossRef CAS PubMed.
- B. D. Lawrence, M. Cronin-Golomb, I. Georgakoudi, D. L. Kaplan and F. G. Omenetto, Biomacromolecules, 2008, 9, 1214–1220 CrossRef CAS PubMed.
- T. Seydel, K. Kolln, I. Krasnov, I. Diddens, N. Hauptmann, G. Helms, M. Ogurreck, S. G. Kang, M. M. Koza and M. Muller, Macromolecules, 2007, 40, 1035–1042 CrossRef CAS.
- I. Krasnov, I. Diddens, N. Hauptmann, G. Helms, M. Ogurreck, T. Seydel, S. S. Funari and M. Muller, Phys. Rev. Lett., 2008, 100, 4 Search PubMed.
- K. Numata, H. Masunaga, T. Hikima, S. Sasaki, K. Sekiyama and M. Takata, Soft Matter, 2015, 11, 6335–6342 RSC.
- S. J. Ling, Z. M. Qi, D. P. Knight, Y. F. Huang, L. Huang, H. Zhou, Z. Z. Shao and X. Chen, Biomacromolecules, 2013, 14, 1885–1892 CrossRef CAS PubMed.
- J. Sirichaisit, V. L. Brookes, R. J. Young and F. Vollrath, Biomacromolecules, 2003, 4, 387–394 CrossRef CAS PubMed.
- A. Huot, T. Lefevre, J. F. Rioux-Dube, F. Paquet-Mercier, A. P. Nault, M. Auger and M. Pezolet, Appl. Spectrosc., 2015, 69, 689–698 CrossRef CAS PubMed.
- M. J. Sun, Y. P. Zhang, Y. M. Zhao, H. L. Shao and X. C. Hu, J. Mater. Chem., 2012, 22, 18372–18379 RSC.
- G. R. Plaza, P. Corsini, E. Marsano, J. Perez-Rigueiro, M. Elices, C. Riekel, C. Vendrely and G. V. Guinea, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 455–465 CrossRef CAS.
- S. N. Fan, Y. P. Zhang, H. L. Shao and X. C. Hu, Int. J. Biol. Macromol., 2013, 56, 83–88 CrossRef CAS PubMed.
- A. Motta, L. Fambri and C. Migliaresi, Macromol. Chem. Phys., 2002, 203, 1658–1665 CrossRef CAS.
- M. Wang, H. J. Jin, D. L. Kaplan and G. C. Rutledge, Macromolecules, 2004, 37, 6856–6864 CrossRef CAS.
- L. Peng, S. H. Jiang, M. Seuss, A. Fery, G. Lang, T. Scheibel and S. Agarwal, Macromol. Mater. Eng., 2016, 301, 48–55 CrossRef CAS.
- H. Pan, Y. P. Zhang, H. L. Shao, X. C. Hu, X. H. Li, F. Tian and J. Wang, J. Mater. Chem. B, 2014, 2, 1408–1414 RSC.
- G. Q. Fang, Z. K. Zheng, J. R. Yao, M. Chen, Y. Z. Tang, J. J. Zhong, Z. M. Qi, Z. Li, Z. Z. Shao and X. Chen, J. Mater. Chem. B, 2015, 3, 3940–3947 RSC.
- L. Liu, X. G. Yang, H. Y. Yu, C. Ma and J. M. Yao, RSC Adv., 2014, 4, 14304–14313 RSC.
- Y. Q. He, N. N. Zhang, Q. J. Gong, H. X. Qiu, W. Wang, Y. Liu and J. P. Gao, Carbohydr. Polym., 2012, 88, 1100–1108 CrossRef CAS.
- H. Z. Ke, Z. Y. Pang, Y. F. Xu, X. D. Chen, J. P. Fu, Y. B. Cai, F. L. Huang and Q. F. Wei, J. Therm. Anal. Calorim., 2014, 117, 109–122 CrossRef CAS.
- Y. H. Li, M. Fan, K. Wu, F. L. Yu, S. G. Chai, F. Chen and Q. Fu, Composites, Part A, 2015, 73, 85–92 CrossRef CAS.
- C. Zhang, Y. P. Zhang, H. L. Shao and X. C. Hu, ACS Appl. Mater. Interfaces, 2016, 8, 3349–3358 CAS.
- K. S. Hu, M. K. Gupta, D. D. Kulkarni and V. V. Tsukruk, Adv. Mater., 2013, 25, 2301–2307 CrossRef CAS PubMed.
- C. Y. Wan and B. Q. Chen, J. Mater. Chem., 2012, 22, 3637–3646 RSC.
- X. Hu, D. Kaplan and P. Cebe, Macromolecules, 2008, 41, 3939–3948 CrossRef CAS.
- S. A. Fossey and S. Tripathy, Int. J. Biol. Macromol., 1999, 24, 119–125 CrossRef CAS PubMed.
- G. R. Plaza, J. Perez-Rigueiro, C. Riekel, G. B. Perea, F. Agullo-Rueda, M. Burghammer, G. V. Guinea and M. Elices, Soft Matter, 2012, 8, 6015–6026 RSC.
- S. Sampath, T. Isdebski, J. E. Jenkins, J. V. Ayon, R. W. Henning, J. Orgel, O. Antipoa and J. L. Yarger, Soft Matter, 2012, 8, 6713–6722 RSC.
- L. Huang, C. Li, W. J. Yuan and G. Q. Shi, Nanoscale, 2013, 5, 3780–3786 RSC.
- Y. Yin, K. Hu, A. M. Grant, Y. H. Zhang and V. V. Tsukruk, Langmuir, 2015, 31, 10859–10870 CrossRef CAS PubMed.
- J. Luo, L. L. Zhang, Q. F. Peng, M. J. Sun, Y. P. Zhang, H. L. Shao and X. C. Hu, Int. J. Biol. Macromol., 2014, 66, 319–324 CrossRef CAS PubMed.
- W. Wei, Y. P. Zhang, Y. M. Zhao, J. Luo, H. L. Shao and X. C. Hu, Mater. Sci. Eng., C, 2011, 31, 1602–1608 CrossRef CAS.
- J. X. Zhu, Y. P. Zhang, H. L. Shao and X. C. Hu, Fibers Polym., 2014, 15, 540–546 CrossRef CAS.
- Y. Jin, Y. C. Hang, Q. F. Peng, Y. P. Zhang, H. L. Shao and X. C. Hu, RSC Adv., 2015, 5, 62936–62940 RSC.
- L. F. Drummy, D. M. Phillips, M. O. Stone, B. L. Farmer and R. R. Naik, Biomacromolecules, 2005, 6, 3328–3333 CrossRef CAS PubMed.
- Y. Takahashi, M. Gehoh and K. Yuzuriha, Int. J. Biol. Macromol., 1999, 24, 127–138 CrossRef CAS PubMed.
- I. C. Um, C. S. Ki, H. Y. Kweon, K. G. Lee, D. W. Ihm and Y. H. Park, Int. J. Biol. Macromol., 2004, 34, 107–119 CrossRef CAS PubMed.
- Y. Shen, M. A. Johnson and D. C. Martin, Macromolecules, 1998, 31, 8857–8864 CrossRef CAS.
- A. H. Simmons, C. A. Michal and L. W. Jelinski, Science, 1996, 271, 84–87 CAS.
- D. T. Grubb and L. W. Jelinski, Macromolecules, 1997, 30, 2860–2867 CrossRef CAS.
- S. Bose, T. Kuila, M. E. Uddin, N. H. Kim, A. K. T. Lau and J. H. Lee, Polymer, 2010, 51, 5921–5928 CrossRef CAS.
- J. Gao, F. Liu, Y. L. Liu, N. Ma, Z. Q. Wang and X. Zhang, Chem. Mater., 2010, 22, 2213–2218 CrossRef CAS.
- X. J. Shen, Y. Liu, H. M. Xiao, Q. P. Feng, Z. Z. Yu and S. Y. Fu, Compos. Sci. Technol., 2012, 72, 1581–1587 CrossRef CAS.
- R. J. Young, L. Gong, I. A. Kinloch, I. Riaz, R. Jalil and K. S. Novoselov, ACS Nano, 2011, 5, 3079–3084 CrossRef CAS PubMed.
- Z. L. Li, R. J. Young and I. A. Kinloch, ACS Appl. Mater. Interfaces, 2013, 5, 456–463 CAS.
- Y. Gao, L. Q. Liu, S. Z. Zu, K. Peng, D. Zhou, B. H. Han and Z. Zhang, ACS Nano, 2011, 5, 2134–2141 CrossRef CAS PubMed.
- S. H. Jiang, G. G. Duan, E. Zussman, A. Greiner and S. Agarwal, ACS Appl. Mater. Interfaces, 2014, 6, 5918–5923 CAS.
- S. Ran, D. Fang, X. Zong, B. S. Hsiao, B. Chu and P. M. Cunniff, Polymer, 2001, 42, 1601–1612 CrossRef CAS.
- S. F. Ran, X. H. Zong, D. F. Fang, B. S. Hsiao, B. Chu, P. M. Cunniff and R. A. Phillips, J. Mater. Sci., 2001, 36, 3071–3077 CrossRef CAS.
- K. Numata, R. Sato, K. Yazawa, T. Hikima and H. Masunaga, Polymer, 2015, 77, 87–94 CrossRef CAS.
- B. Liang, L. Fang, Y. C. Hu, G. Yang, Q. Zhu and X. S. Ye, Nanoscale, 2014, 6, 4264–4274 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Figure for AFM images of the GO nanosheets as well as the particle size and particle size distribution of GO dispersion solution. Table for the parameters of SR-WAXD on RSF/GO and RSF fiber in elastic deformation zone. 2D WAXD patterns and microstructural evolution of RSF fibers during tensile deformation. See DOI: 10.1039/c6ra22544f |
| ‡ Chao Zhang and Yaopeng Zhang contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |