
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCore–shell-structured Li3V2(PO4)3–LiVOPO4 nanocomposites cathode for high-rate and long-life lithium-ion batteries†
Pingping Sun a,
Xiuzhen Wanga,
Kai Zhua,
Xiao Chena,
Xia Cuib,
Qingyu Xu*ac,
Dong Sud,
Qi Fan*ab and
Yueming Sunb
a,
Xiuzhen Wanga,
Kai Zhua,
Xiao Chena,
Xia Cuib,
Qingyu Xu*ac,
Dong Sud,
Qi Fan*ab and
Yueming Sunb
aDepartment of Physics, Southeast University, Nanjing 211189, China. E-mail: xuqingyu@seu.edu.cn; fanqi1984@126.com
bCollege of Chemistry and Chemical Engineering, Southeast University, Nanjing 211189, China
cNational Laboratory of Solid State Microstructures, Nanjing University, Nanjing 210093, China
dCenter for Functional Nanomaterials, Brookhaven National Laboratory, Upton, New York 11973, USA
First published on 13th January 2017
Abstract
A facile strategy has been developed to construct unique core–shell-structured Li2.7V2.1(PO4)3 nanocomposites with a Li3V2(PO4)3 core and LiVOPO4 shell by using nonstoichiometric design and high-energy ball milling (HEBM) treatment. The HEBM treatment supplies enough energy to drive the excess V atoms to the surface to form a V-enriched shell. Such kind of cathode can deliver a high reversible capacity of 131.5 mA h g−1 at 0.5C, which is close to the theoretical capacity (133 mA h g−1 in 3.0–4.3 V). Even at 20C, it still delivers an excellent discharge capacity of 116.3 mA h g−1, and a remarkable capacity of 111.0 mA h g−1 after 1000 cycles, corresponding to an ultra-small capacity-loss of 0.0046% per cycle. The significantly improved high-rate electrochemical performance can be attributed to the active shell of LiVOPO4, which not only efficiently facilitates the electron and Li+ ion transport during cycling processes, but also accommodates more Li+ ions to effectively compensate the capacity loss of the core.
1. Introduction
Lithium-ion batteries (LIBs) are important power sources for the increasing demands from portable electronic products, electrical vehicles, hybrid electrical vehicles and so on.1–4 However, developing rechargeable LIBs with a high capacity, fast charge/discharge rate, and long lifespan is still a significant challenge.5–10 Among the various components in LIBs, the cathode materials have attracted intensive attention since they have significant impacts on battery capacity, cycle life, safety and cost.11–17 However, high-rate performance of these cathode materials is still limited due to their low intrinsic electronic and ionic conductivity and fails to meet the necessary demands for LIBs in power electric vehicles.18–20 To circumvent these problems, the conventional effective methods to improve the high-rate performance are coating the nano-sized cathode particles with highly conductive materials to facilitate the transportation of electrons and Li+ ions.21–28 Among them, most of the coating shell is inactive and generally regarded as protecting layer to minimize the direct contact of active material with electrolyte, which has no contribution to the capacity.29 Different from the others, Zheng et al. synthesized phosphate-based composite cathode materials by regulating the contents of each phase, and found that vanadium phosphate polymers as cathode materials exhibit improved electrochemical performance.30,31 B. Kang et al. synthesized nonstoichiometric LiFe1−2yP1−yO4−δ with highly conductive Fe3+-containing Li4P2O7 shell, realizing the ultrahigh-rate capability that full battery discharge in 10–20 s can be achieved.32 Similarly, in our previous work, we synthesized nonstoichiometric Li3V2(PO4)3 (Li2.7V2.1(PO4)3, denoted as LVP) by tuning the Li deficiency and V excess.33 The electrochemical performance is clearly improved while the composites shows no core–shell structure, and the rate performance enhancement has been ascribed to the self-doping effect. High energy ball milling (HEBM) emerged as a efficient technique to produce nanomaterials has been widely used in the preparation of phosphates.34–37 In this work, we further adopt HEBM treatment on LVP (denoted as CSLVP), core–shell structure is clearly formed with excess V locating in the shell. The phase of shell has been confirmed to be LiVOPO4, which provides more space for the storage of Li+ ions during the high-rate charge/discharge processes. Together with the smaller particle size and higher electronic conductivity, the high-rate performance is significantly improved with long cycle stability.2. Experimental section
2.1 Synthesis
A sol–gel method combined with HEBM treatment was used to prepare CSLVP nanoparticles. Typically, 10.5 mmol NH4VO3 (AR, ≥99.0%), 13.5 mmol LiOH·H2O (AR, ≥99.0%), and 15 mmol NH4H2PO4 (AR, ≥99.0%) were separately dissolved in 50 ml distilled water to prepare NH4VO3, LiOH and NH4H2PO4 solutions. The above solutions were sequentially added to 0.2 M citric acid solution dropwisely and then stirred vigorously at room temperature for 12 h. The mixture was heated in a thermostatic water bath at 80 °C with constant stirring for 6 h to evaporate excessive water until a blue gel was formed. The gel was dried in a vacuum oven at 80 °C for 12 h, and then put into a stainless steel cylindrical container with zirconia ball and ground in a GB-50 (power of 180 W, speed of 1200 rpm) high-speed vibrating ball mill for 1 h. The mixtures were annealed at 300 °C for 6 h under Ar atmosphere to obtain the powders. Then the precursors were ground in a GB-50 high-speed vibrating ball mill for 1 h again and calcined at 800 °C for 4 h in a tube furnace under flowing Ar atmosphere with the heating/cooling rate of 5 °C min−1.2.2 Structural characterizations
Powder X-ray diffraction (XRD) was performed on an X-ray diffractometer (Rigaku Smartlab3) with Cu Kα radiation. The particle-size distribution was measured using a laser particle-size analyzer. Morphologies were studied by a scanning electron microscope (SEM, FEI Inspect F50). The valence states of samples were studied by X-ray photoelectron spectroscopy (XPS, Thermo Fisher Scientific) with Al Kα X-ray source (hν = 1486.6 eV). Transmission electron microscope (TEM) work was taken using a JEOL 2100F TEM/STEM at 200 kV. High angle annular dark-field scanning TEM ((HAADF)-STEM) imaging and spatially resolved electron energy loss spectroscopy (EELS) were performed with a probe-corrected Hitachi 2700C dedicated STEM with an attached Enfina ER spectrometer.2.3 Electrochemical measurements
Electrochemical measurements were performed using CR2025 coin cells. The electrodes were prepared by blending the LVP with acetylene black, carbon nanotube (CNT) and polyvinylidene fluoride (PVDF) binder with weight ratio of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:5:5 in N-methylpyrrolidone (NMP). The slurry of mixture was coated onto an aluminum foil using a manually adjustable film-coating equipment and dried in a vacuum oven at 120 °C for 12 h. The dried positive electrode sheet was cut into discs with diameter of 12 mm, and then pressed under a fixed pressure. The assembly of coin cells was performed in an Ar-filled glove box by using CR2025 coin-type cells. Pure lithium foils were used as counter electrodes, Celgard 2400 polypropylene membranes were used as separators, and 1 M LiPF6 in a solution of ethylene carbonate and dimethyl carbonate (EC and DMC, 1:1 by volume) served as electrolyte. Galvanostatic charge/discharge tests were taken in a voltage cut-off of 3.0–4.3 V (vs. Li/Li+) at different C rates on a multichannel battery testing system (LAND CT2001A, Wuhan) at room temperature. Cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) measurements were performed on an electrochemical workstation (Gamry Reference 600) with an AC voltage amplitude of 5 mV on the fresh cells in a frequency range from 100 KHz to 10 MHz. The specific capacity was calculated based on the weight of active materials.
:10:5:5 in N-methylpyrrolidone (NMP). The slurry of mixture was coated onto an aluminum foil using a manually adjustable film-coating equipment and dried in a vacuum oven at 120 °C for 12 h. The dried positive electrode sheet was cut into discs with diameter of 12 mm, and then pressed under a fixed pressure. The assembly of coin cells was performed in an Ar-filled glove box by using CR2025 coin-type cells. Pure lithium foils were used as counter electrodes, Celgard 2400 polypropylene membranes were used as separators, and 1 M LiPF6 in a solution of ethylene carbonate and dimethyl carbonate (EC and DMC, 1:1 by volume) served as electrolyte. Galvanostatic charge/discharge tests were taken in a voltage cut-off of 3.0–4.3 V (vs. Li/Li+) at different C rates on a multichannel battery testing system (LAND CT2001A, Wuhan) at room temperature. Cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) measurements were performed on an electrochemical workstation (Gamry Reference 600) with an AC voltage amplitude of 5 mV on the fresh cells in a frequency range from 100 KHz to 10 MHz. The specific capacity was calculated based on the weight of active materials.
3. Results and discussion
The XRD pattern of CSLVP nanocomposites clearly indicates the formation of highly crystalline phase, as shown in Fig. 1(a). It can be seen that most diffraction peaks can be indexed to the monoclinic Li3V2(PO4)3 phase (P21/n, JCPDS No. 01-072-7074), which is consistent with our previous work.33 However, by carefully investigating the XRD pattern, some weak diffraction peaks marked by arrows can be observed, which cannot be indexed to Li3V2(PO4)3 phase. These peaks coincide quite well with the standard pattern of LiVOPO4, indicating the possible impurity phase, which has been further confirmed by other characterization methods and will be discussed later. Inset of Fig. 1(a) is a SEM image, showing that CSLVP are composed of particles with homogeneous morphology. Both particle sizes and particle-size distribution of the samples were further measured by a laser particle-size analyzer (Fig. S1, ESI†). HEBM treatment significantly decreased the particle size and narrowed the size distribution. The main fraction of CSLVP particles has size of about 6 μm, which is significantly smaller than that of LVP without HEBM treatment (about 100 μm).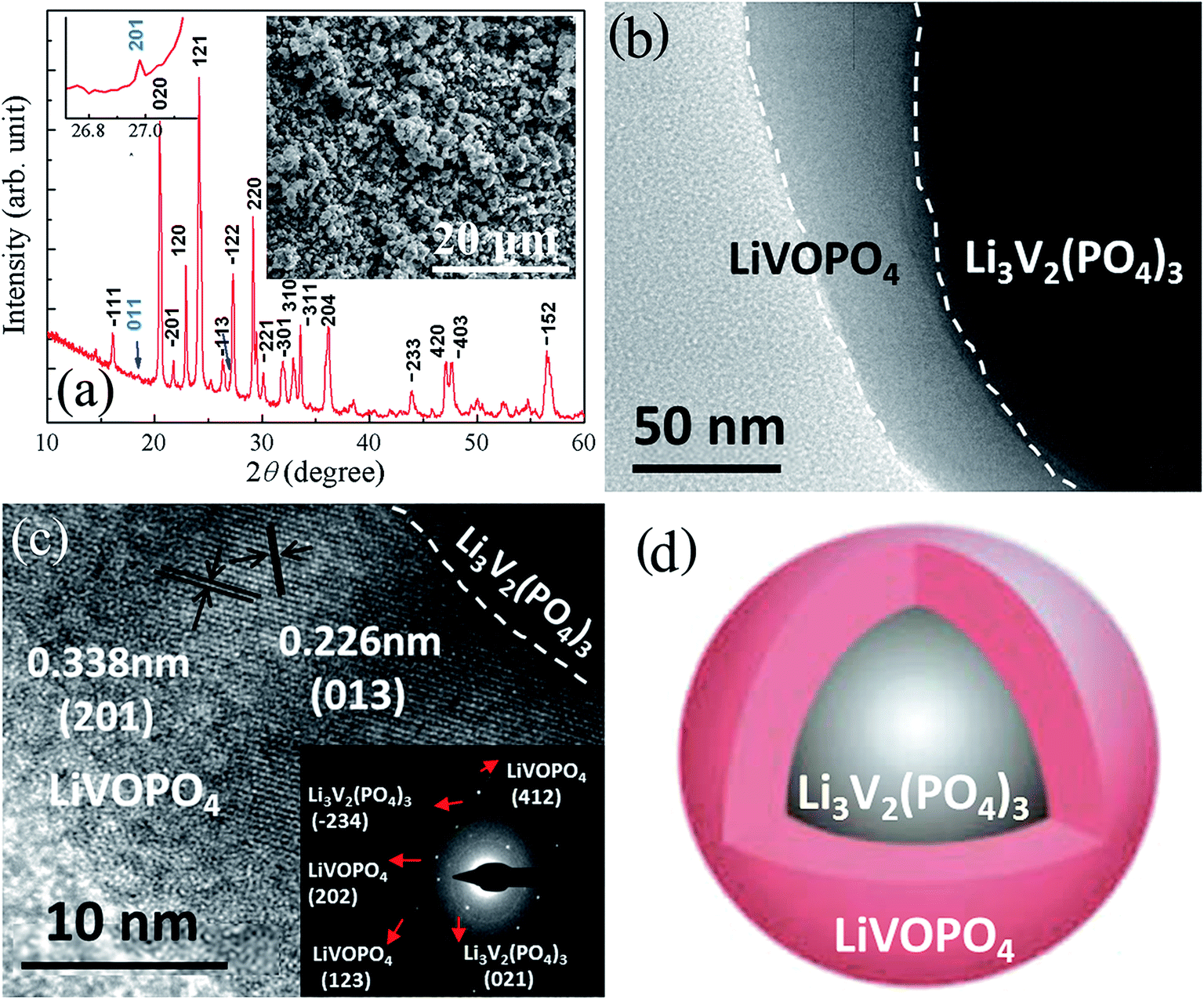 | ||
| Fig. 1 (a) XRD (b) TEM, (c) HRTEM, (d) Schematic structure of CSLVP, left inset in (a) is the enlarged view of XRD pattern, showing the peak at around 27°, right inset in (a) is the SEM image, and inset in (c) is the corresponding SAED pattern. | ||
The detailed structure was further examined by TEM investigations. It can be clearly seen from Fig. 1(b) that the CSLVP particles have a core–shell structure with significant contrast in the low magnified TEM image. It is notable that the thickness of the shell varies from particle to particle. In order to investigate the detailed composites of the surface phase, we select the particles with much thick shell for the TEM investigation. To determine the phase of shell, high resolution TEM (HRTEM) was performed and the image of shell is shown in Fig. 1(c). The d spacing of lattice fringes marked in the image was determined to be 0.226 nm and 0.338 nm, together with the angle between the crystal planes from the Fast Fourier Transformation (FFT) image (Fig. S2, ESI†), which can be indexed to the (013) and (201) planes of orthorhombic LiVOPO4. To confirm the shell phase, selected-area electron diffraction (SAED) was performed, and the SAED pattern from the CSLVP particle is inserted in Fig. 1(c). Besides the strong diffraction spots from Li3V2(PO4)3, some much weaker spots can be observed, which can be indexed by LiVOPO4. These results suggest that the impurity phase of LiVOPO4 detected in the XRD pattern forms the shell covered on the Li3V2(PO4)3 particles. Thus, a core–shell structure with core of Li3V2(PO4)3 and shell of LiVOPO4 has been formed for CSLVP, as shown the schematic structure in Fig. 1(d).
We further performed the elemental analysis of CSLVP in a STEM with EELS by line scan from edge to center of a particle, as shown in inset of Fig. 2(a). The exact concentration of each element is not easy to be determined by this technique. Here we use the evolution of relative spectrum intensity to characterize the concentration variation of each element. As can be seen from Fig. 2(a), the EELS line-profile reveals that the intensity ratio of V K edge and O K edge is almost constant from 0 to 13 nm away from the edge, and then gradually decreases into the core, confirming the formation of a shell with higher V concentration. The smaller V/O intensity ratio becomes nearly constant beyond ∼18 nm away from the edge. Considering the main phase of Li3V2(PO4)3 determined from XRD, a extra phase with higher V/O ratio has been formed at the surface of the particles. This is consistent with the above determined shell phase of LiVOPO4 by HRTEM and SAED (V/O atomic ratio of 1:5 is higher than that of 1:6 for Li3V2(PO4)3).
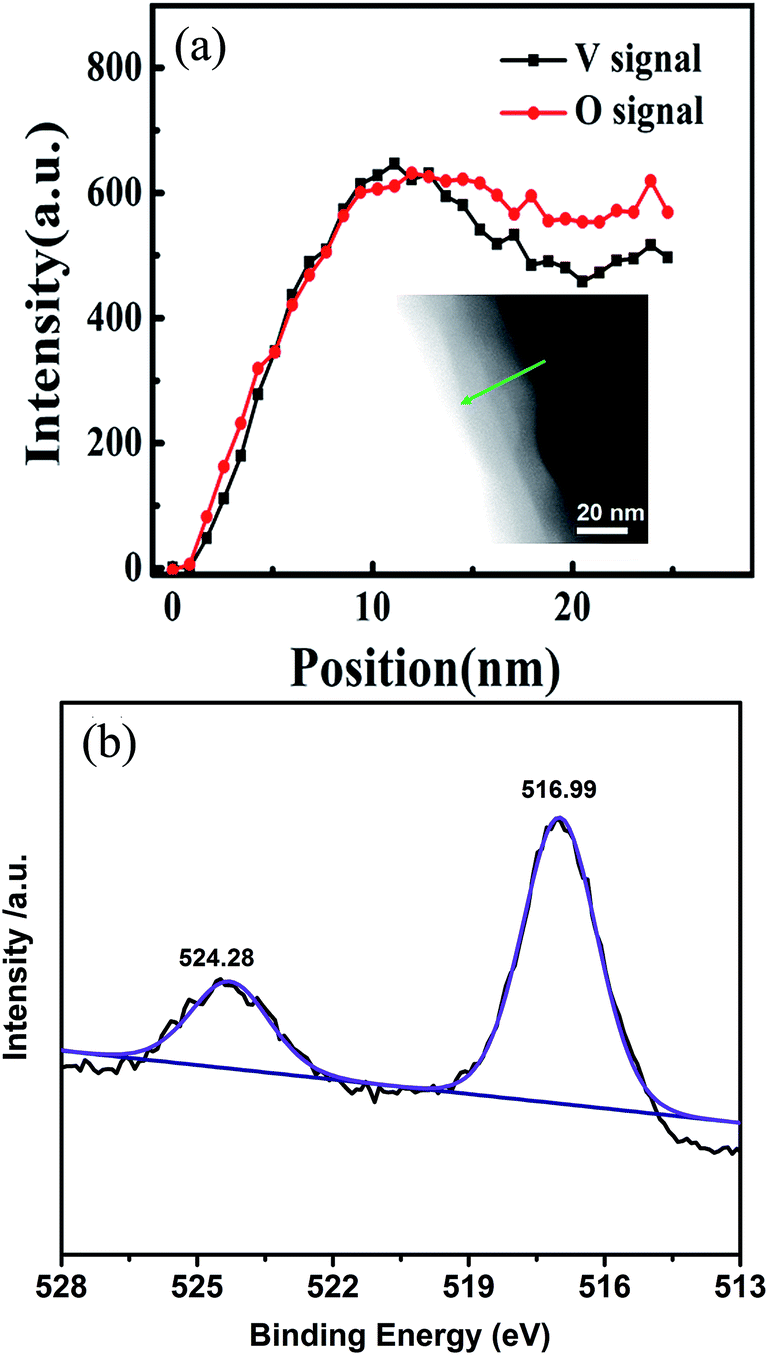 | ||
| Fig. 2 (a) EELS intensity of V and O for CSLVP, inset shows the STEM image with an arrow indicating the line scan of EELS. (b) XPS spectrum of V2p in CSLVP. | ||
Since XPS is a surface sensitive method and penetrates only into a depth of around 5 nm, the elemental information in the shell can be mainly detected by XPS. Further evidence for the existence of a LiVOPO4 shell is provided by XPS. V2p XPS peaks of CSLVP are shown in Fig. 2(b). It is obvious that V2p peaks of CSLVP significantly shifts to higher binding energy, compared with that of LVP.33 Two peaks at 516.99 eV and 524.28 eV are assigned to V2p3/2 and V2p1/2, respectively. The binding energies are consistent with those of V4+ in LiVOPO4.38 XPS spectra of C1s, Li1s, O1s, P2p are shown in Fig. S3,† similar to those of LVP.33 The similar XPS spectra of C1s for CSLVP and LVP indicates the similar structure of residual carbon, which is not influenced by the HEBM treatment. Thus, the possible contribution to the electrochemical performance from the different residual carbon can be safely excluded. Combined with all the structural characterizations, we can conclude that a shell of LiVOPO4 is formed around the core of Li3V2(PO4)3, and the schematic structure of CSLVP particles is shown in Fig. 1(d).
Here, we'd like to give a short explanation on the formation mechanism of such core–shell structure by nonstoichiometric design and HEBM treatment. B. Kang et al. has introduced the nonstoichiometric strategy on preparation of LiFePO4.32 At low sintering temperature, only the XRD pattern corresponding to LiFePO4 can be observed. With increasing the sintering temperature, Fe2P, Li3PO4, Li4P2O7 impurity phases can be observed and form the shell covering the LiFePO4 particles.32 These results suggest that stoichiometric cores will be formed, and the excess atoms tend to be driven out and locate at the surface of particles to form the shell, if the atoms acquire enough energy. In our nonstoichiometric Li2.7V2.1(PO4)3, the atoms didn't acquire enough energy at the sintering temperature of 800 °C and only pure phase without core–shell structure is formed in our previous study.33 HEBM has the advantage of forming ultrafine particles, increasing the chemical activity and promoting the atom diffusivity by providing the high mechanical energy, which can significantly lower the chemical reaction temperature. Thus, after HEBM, the excess V atoms acquire enough energy and diffuse to the surface during the sintering process, and Li3V2(PO4)3 cores are formed. To balance the total valence state, higher valence state V (here is V4+) atoms has to be formed at shell, similar to nonstoichiometric LiFePO4.32 Thus, LiVOPO4 shell is formed.
Core–shell structure has been widely studied in the cathode materials of LIBs, but the shell material is generally inactive, such as carbon materials, conductive polymers, etc., which only facilitates the fast transfer of electrons and Li+ ions.39,40 In our novel core–shell-structured CSLVP, the shell material of LiVOPO4 is also an electrochemical active material.41 Thus, the shell can also contribute to the Li+ ions storage and capacity of the cathode, and improved electrochemical performance can be expected.
The electrochemical performances of CSLVP were examined by galvanostatic cycling in CR2025 coin-type cells. Metallic lithium was used as the counter electrode. Fig. 3(a) depicts the first charge/discharge profiles of CSLVP at different current rates (1C = 133.0 mA h g−1) in the voltage range of 3.0–4.3 V at room temperature. When the rate is set at 0.5C, the CSLVP exhibits three pairs of charge/discharge plateaus locating at around 3.60/3.57, 3.68/3.65, and 4.08/4.05 V, owing to a series of reversible phase transitions during electrochemical reactions. The initial discharge capacity of CSLVP at the rate of 0.5C is 131.5 mA h g−1 (close to the theoretical capacity of 133.0 mA h g−1 for Li3V2(PO4)3), which is higher than that of LVP (129.2 mA h g−1).33 The specific capacity is calculated based on the weight of active materials. When the current rate is stepwisely increased from 0.5C to 20C, CSLVP preserves stable capacities during the entire process (Fig. 3(b)), demonstrating its excellent high-rate capability. When the current rate increases to 20C, the discharge capacity of 116.3 mA h g−1 (87.4% of the theoretical capacity) is still delivered for CSLVP. About 97.3% of the initial capacity for CSLVP is recovered after the current rate is turned back to 0.5C, indicating very good electrochemical reversibility. Long-term cycling performance of CSLVP at a high rate of 20C is shown in Fig. 3(c). The CSLVP cathode delivers an initial discharge capacity of 116.3 mA h g−1 and still maintains a remarkable capacity of 111.0 mA h g−1 after 1000 cycles, corresponding to capacity retention as high as 95.4%. These are among the best reported performances of Li3V2(PO4)3 cathode materials.40 LVP cathode only delivers an initial discharge capacity of 92.5 mA h g−1 at 20C and maintains a capacity of 85.1 mA h g−1 after 1000 cycles (Fig. S4, ESI†).33
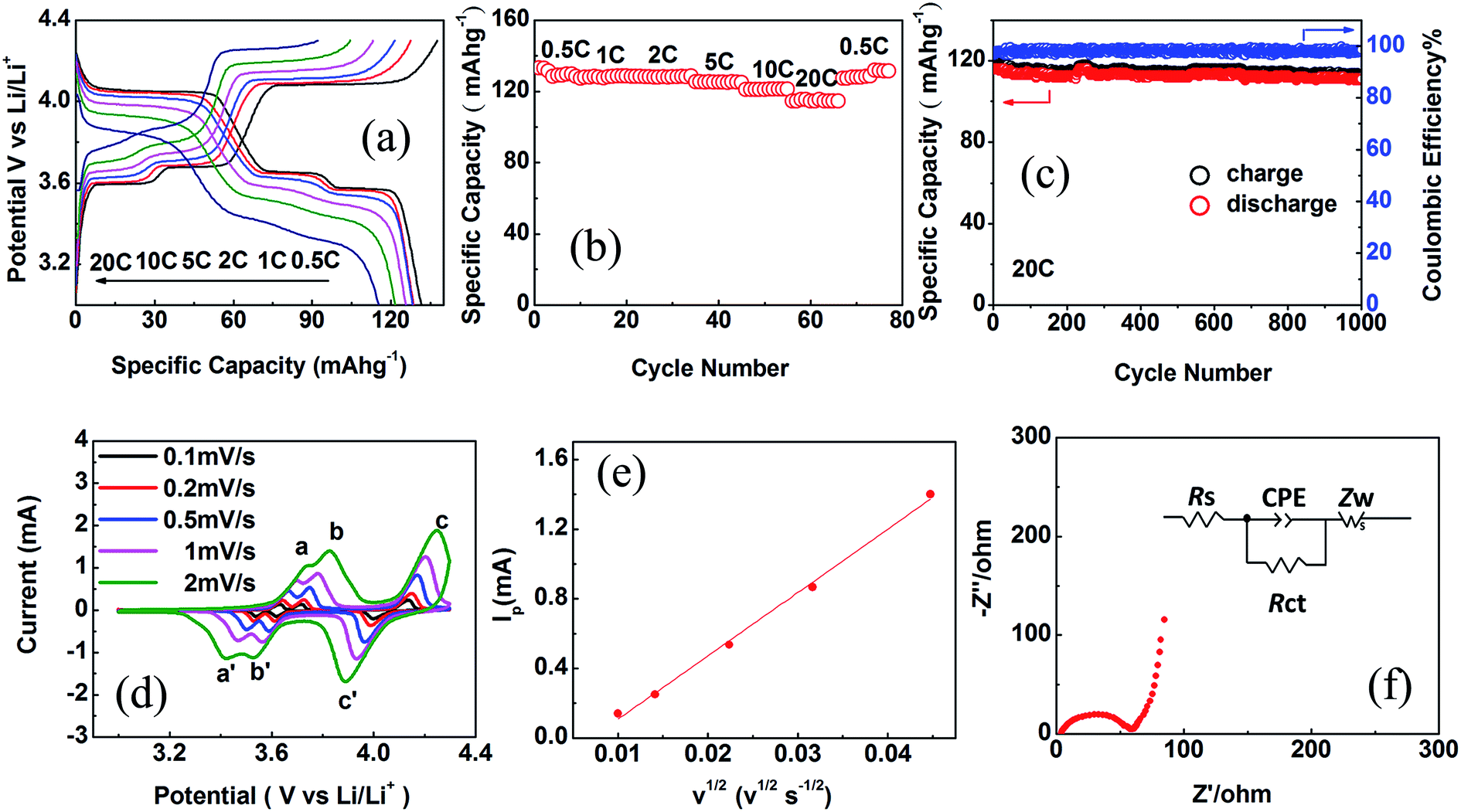 | ||
| Fig. 3 (a) Charge/discharge profiles at different current rates, (b) rate performance, (c) long-term cycling performance, (d) CV at different scanning rates. (e) Linear fitting of Ip vs. v1/2 curves for the peak c, and (f) Nyquist plots of CSLVP cathode in fresh cell, the inset in (f) shows the corresponding equivalent circuit. | ||
To further investigate the effect of active shell on the electrochemical performance of CSLVP cathode, CV and EIS measurements were performed on CSLVP. The peak current increases with increasing scanning rate, meanwhile the cathodic and anodic peaks shift to lower and higher potentials, respectively (Fig. 3(d)). Compared with Li3V2(PO4)3, CSLVP cathode undergoes three complete phase transitions with higher current, indicating higher reversibility and faster kinetics during electrochemical reaction.33 The Li+ ion diffusion coefficients are calculated, based on Randles Sevcik equation for semi-infinite diffusion of Li+ into LVP:42
| Ip = 2.69 × 105n3/2AC0D1/2v1/2 | (1) |
Finally, we will concentrate our discussion on the core–shell structure composed of both active materials for the further improvement of high-rate performance. The energy storage of LIBs depends on the intercalation and extraction of Li+ ions in the cathode materials, which decides the capacity of cathode. A schematic diagram is drawn in Fig. 4 to show the mechanism for the high-rate charge/discharge processes. For conventional cathode without any coverage, only the outer region of particles takes part in the Li+ intercalation and extraction and large part in the center remains intact, due to the limited Li+ ion and electron conductivities, as shown in Fig. 4(a). The volume of active region decreases drastically with increasing rate, leading to the drastic decrease of high-rate capacity. With carbon or conductive polymers covering the cathode particles and improved Li+ ion and electron conductivities, more active materials take part in the charge/discharge processes, leading to the improved capacity, as shown in Fig. 4(b). For CSLVP, based on the above structural characterizations, core–shell structure with both active materials has been confirmed, as shown the schematic diagram in Fig. 1(d). Though the penetration depth of Li+ ions is still limited during high-rate charge/discharge processes, the active material at shell might further provides an extra capacity. It has been reported that LiVOPO4 offers the theoretical capability of 166 mA h g−1 with charge/discharge plateau of 4.0 V, which is higher than that of 133 mA h g−1 for Li3V2(PO4)3 working under 4.3 V.41 By optimizing the LiVOPO4 shell thickness, the diffusion coefficient of Li+ ions will not be obviously influenced, and the similar intact part in the center remains. This active shell can accommodate more Li+ ions at high charge/discharge rate, which can compensate the unreached cathode material in the center of particles during the Li+ ion intercalation/extraction processes and further improve the capacity, as shown in Fig. 4(c).
 | ||
| Fig. 4 Schematic diagrams of the high-rate charge/discharge processes for (a) cathode particle without shell, (b) cathode particle with conductive shell, and (c) core–shell-structured cathode particle with active shell. The grey and black colors mark the active parts that take part in the charge/discharge processes, and the black color of shell in (c) indicates the higher capacity of shell than that of the core. | ||
LiVOPO4–Li3V2(PO4)3 core–shell structure has been reported previously, but the high-rate performance was deteriorated.38 This is due to the formation of LiVOPO4 shell from Li3V2(PO4)3 particles by partial oxidation at low temperature (350 °C), and an amorphous layer between LiVOPO4 and Li3V2(PO4)3 is formed. The amorphous layer may block the transport of Li+ ions, leading to worse performance at high rate.38 In contrast, the core–shell structure with LiVOPO4 shell and Li3V2(PO4)3 core is formed simultaneously at high temperature in our case, and high crystallinity of both core and shell is achieved without such amorphous interface layer, as is confirmed from the HRTEM image of CSLVP shown in Fig. 1(c), which guarantees the high transfer rate of Li+ ions.
Based on the above results, we suggest that core–shell-structured cathode material with active shell can be an efficient route to achieve the high-rate electrochemical performance. To realize such objective, several points should be emphasized. First, the active shell material should have higher capacity and similar voltage plateau as the core material. This is the prerequisite for the selection of active materials as shell, which guarantees to achieve higher capacity than the core materials and compensate the capacity loss of core. Second, the shell should be thin enough. Otherwise, the shortcomings of shell materials, such as low electrical conductivity, low Li+ ion diffusion coefficient, etc., may limit its electrochemical performance, since the shell may take greater part at high-rate charge/discharge processes. For the thin shell with thickness smaller than the Li+ ion diffusion length, the capacity of shell will not be influenced much, since Li+ ions can easily transport through the shell. Thus, the full capacity of shell at high rate can be maintained. And last, the crystallinity should be high. The intercalation/extraction of Li+ ions should not be blocked by the worse crystal structure between the shell and core, otherwise, the capacity of core will be significantly deteriorated. With these points all satisfied, the core–shell-structured cathode material may exhibit higher capacity, especially at high-rate charge/discharge processes. It should be pointed out that the core–shell structure at this moment cannot be finely controlled by nonstoichiometry and HEBM treatment in the solid state reaction process, for example, the shell thickness is not optimized and varies from particle to particle in CSLVP. Novel methods, such as atomic layer deposition, might be applied for the deposition of active shell in the future experiments, thus the composition and structure can be controlled more accurately, and further improvement of electrochemical performance at ultra-high rate can be achieved.
4. Conclusion
In summary, we have developed a cost-efficient strategy to synthesize core–shell-structured CSLVP cathode of superior high-rate and long-life performance by nonstoichiometry and HEBM treatment. The excess V has been confirmed to locate at the shell to form the phase of LiVOPO4. The active shell can accommodate more Li+ ions during the high-rate charge/discharge processes. Together with the significantly decreased particle size and improved electronic conductivity, CSLVP exhibits much improved high-rate electrochemical performance. When cycling at a rate of 0.5C in the range of 3–4.3 V, CSLVP exhibits an initial discharge capacity of 131.5 mA h g−1. Even at 20C, an initial discharge capacity of 116.3 mA h g−1 is delivered, with a remarkable capacity of 111.0 mA h g−1 after 1000 cycles, corresponding to capacity retention of 95.4%. Our strategy is simple, and can be easily integrated into large-scale industrial production processes. Through further optimization, CSLVP cathode material can be applied to high power applications such as electric vehicles and energy storage for smart grids.Acknowledgements
This work is supported by the Scientific Research Foundation of Graduate School of Southeast University and National Natural Science Foundation of China (51172044, 51471085, 51407029), the Natural Science Foundation of Jiangsu Province of China (BK20151400), China Postdoctoral Science Foundation (2012M520968), the International Postdoctoral Exchange Fellowship Program 2014 by the Office of China Post-doctoral Council, and the open research fund of Key Laboratory of MEMS of Ministry of Education, Southeast University. Partial electron microscopy work was performed at the Center for Functional Nanomaterials, Brookhaven National Laboratory, supported by U.S. Department of Energy, Office of Basic Energy Sciences, under contract no. DE-SC-00112704. We thank Xiaofeng Chang and Prof. Peng Wang from Nanjing University for their assistance in the TEM investigation, Prof. Guangbin Ji from Nanjing University of Aeronautics and Astronautics for the particle-size distribution measurements.References
- J.-M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- C. K. Chan, H. Peng, G. Liu, K. Mcilwrath, X. F. Zhang, R. A. Huggins and Y. Cui, Nat. Nanotechnol., 2008, 3, 31–35 CrossRef CAS PubMed.
- J. B. Goodenough and K.-S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS PubMed.
- M. J. Armstrong, C. O'Dwyer, W. J. Macklin and J. D. Holmes, Nano Res., 2014, 7, 1–62 CrossRef CAS.
- J. J. Wang and X. L. Sun, Energy Environ. Sci., 2012, 5, 5163–5185 CAS.
- Q. L. Wei, Q. Y. An, D. D. Chen, L. Q. Mai, S. Y. Chen, Y. L. Zhao, K. M. Hercule, L. Xu, A. M. Khan and Q. J. Zhang, Nano Lett., 2014, 14, 1042–1048 CrossRef CAS PubMed.
- Y. G. Wang, Y. R. Wang, E. Hosono, K. X. Wang and H. S. Zhou, Angew. Chem., Int. Ed., 2008, 47, 7461–7465 CrossRef CAS PubMed.
- S.-Y. Chung, J. T. Bloking and Y.-M. Chiang, Nature, 2002, 1, 123–128 CrossRef CAS PubMed.
- D. Mohanty, S. Kalnaus, R. A. Meisner, K. J. Rhodes, J. L. Li, E. A. Payzant, D. L. Wood III and C. Daniel, J. Power Sources, 2013, 229, 239–248 CrossRef CAS.
- D.-C. Li, T. Muta, L.-Q. Zhang, M. Yoshio and H. Noguchi, J. Power Sources, 2004, 132, 150–155 CrossRef CAS.
- X. K. Zhi, G. C. Liang, L. Wang, X. Q. Ou, J. P. Zhang and J. Y. Cui, J. Power Sources, 2009, 189, 779–782 CrossRef CAS.
- E. Hosono, T. Kudo, I. Honma, H. Matsuda and H. S. Zhou, Nano Lett., 2009, 9, 1045–1051 CrossRef CAS PubMed.
- D. K. Kim, P. Muralidharan, H. Lee, R. Ruffo, Y. Yang, C. K. Chan, H. Peng, R. A. Huggins and Y. Cui, Nano Lett., 2008, 8, 3948–3952 CrossRef CAS PubMed.
- H. Huang, S.-C. Yin, T. Kerr, N. Taylor and L. F. Nazar, Adv. Mater., 2002, 14, 1525–1528 CrossRef CAS.
- J. Kim, J.-K. Yoo, Y. S. Jung and K. Kang, Adv. Energy Mater., 2013, 3, 1004–1007 CrossRef CAS.
- Y. P. Zhou, X. H. Rui, W. P. Sun, Z. C. Xu, Y. Zhou, W. J. Ng, Q. Y. Yan and E. Fong, ACS Nano, 2015, 9, 4628–4635 CrossRef CAS PubMed.
- R. V. Hagen, A. Lepcha, X. F. Song, W. Tyrra and S. Mathur, Nano Energy, 2013, 2, 304–313 CrossRef.
- Y. Wang and G. Z. Cao, Adv. Mater., 2008, 20, 2251–2269 CrossRef CAS.
- M. S. Whittingham, Chem. Rev., 2004, 104, 4271–4301 CrossRef CAS PubMed.
- X. F. Zhang, N. Bçckenfeld, F. Berkemeier and A. Balducci, ChemSusChem, 2014, 7, 1710–1718 CrossRef CAS PubMed.
- X.-L. Wu, L.-Y. Jiang, F.-F. Cao, Y.-G. Guo and L.-J. Wan, Adv. Mater., 2009, 21, 2710–2714 CrossRef CAS.
- Y. Z. Luo, X. Xu, Y. X. Zhang, Y. Q. Pi, Y. L. Zhao, X. C. Tian, Q. Y. An, Q. L. Wei and L. Q. Mai, Adv. Energy Mater., 2014, 4, 1–8 Search PubMed.
- H. D. Liu, P. Gao, J. H. Fang and G. Yang, Chem. Commun., 2011, 47, 9110–9112 RSC.
- C. Wang, H. M. Liu and W. S. Yang, J. Mater. Chem., 2012, 22, 5281–5285 RSC.
- C. Li, H. P. Zhang, L. J. Fu, H. Liu, Y. P. Wu, E. Rahm, R. Holze and H. Q. Wu, Electrochim. Acta, 2006, 51, 3872–3883 CrossRef CAS.
- L.-L. Zhang, G. Liang, G. Peng, F. Zou, Y.-H. Huang, M. C. Croft and A. Ignatov, J. Phys. Chem. C, 2012, 116, 12401–12408 CAS.
- R. Y. Zhang, Y. Q. Zhang, K. Zhu, F. Du, Q. Fu, X. Yang, Y. H. Wang, X. F. Bie, G. Chen and Y. J. Wei, ACS Appl. Mater. Interfaces, 2014, 6, 12523–12530 CAS.
- H. Li, Z. X. Wang, L. Q. Chen and X. J. Huang, Adv. Mater., 2009, 21, 4593–4607 CrossRef CAS.
- W. J. Ge, X. Li, H. Wang, W. Li, A. J. Wei, G. C. Peng and M. Z. Qu, J. Alloys Compd., 2016, 684, 594–603 CrossRef CAS.
- J. C. Zheng, Y. D. Han, L. B. Tang and B. Zhang, Electrochim. Acta, 2016, 198, 195–202 CrossRef CAS.
- J. C. Zheng, Y. D. Han, B. Zhang, C. Shen, L. Ming and J. F. Zhang, ACS Appl. Mater. Interfaces, 2014, 6, 13520–13526 CAS.
- B. Kang and G. Ceder, Nature, 2009, 458, 190–193 CrossRef CAS PubMed.
- P. P. Sun, S. Qin, X. Z. Wang, R. Y. An, Q. Y. Xu, X. Cui, Y. M. Sun, S. B. Wang, P. Wang and Q. Fan, J. Power Sources, 2015, 293, 922–928 CrossRef CAS.
- J. Zheng, X. Li, Z. Wang, H. Guo, Q. Hu and W. Peng, J. Power Sources, 2009, 189, 476–479 CrossRef CAS.
- J. Zheng, X. Li, Z. Wang, S. Niu, D. Liu, L. Wu, L. Li, J. Li and H. Guo, J. Power Sources, 2010, 195, 2935–2938 CrossRef CAS.
- J. Wang, Z. Wang, X. Li, H. Guo, X. Wu, X. Zhang and W. Xiao, Electrochim. Acta, 2013, 87, 224–229 CrossRef CAS.
- J. Wang, X. Li, Z. Wang, B. Huang, Z. Wang and H. Guo, J. Power Sources, 2014, 251, 325–330 CrossRef CAS.
- B. Zhang, C. Shen, J.-C. Zheng, Y.-D. Han, J.-F. Zhang, L. Ming, J.-L. Wang, S. Qin and H. Li, J. Electrochem. Soc., 2014, 161, A748–A752 CrossRef CAS.
- M. Kim, H. Kim, S. Lee, D. Kim, D. Ruan, K. Y. Chung, S. H. Lee, K. C. Roh and K. Kim, Sci. Rep., 2016, 6, 26686 CrossRef CAS PubMed.
- C. F. Liu, R. Massé, X. H. Nan and G. Z. Cao, Energy Storage Materials, 2016, 4, 15–58 CrossRef.
- M. M. Ren, Z. Zhou, L. W. Su and X. P. Gao, J. Power Sources, 2009, 189, 786–789 CrossRef CAS.
- H. Wang, Y. Li, C. Huang, Y. Zhong and S. Liu, J. Power Sources, 2012, 208, 282–287 CrossRef CAS.
- X. H. Rui, N. Ding, J. Liu, C. Li and C. H. Chen, Electrochim. Acta, 2010, 55, 2384–2390 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra26790d |
| This journal is © The Royal Society of Chemistry 2017 |