
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Regioselective Zn(OAc)2-catalyzed azide–alkyne cycloaddition in water: the green click-chemistry†
Maria A.
Morozova
a,
Mekhman S.
Yusubov
b,
Bohumil
Kratochvil
cd,
Václav
Eigner
d,
Alexander A.
Bondarev
e,
Akira
Yoshimura
bf,
Akio
Saito
 g,
Viktor V.
Zhdankin
bf,
Marina E.
Trusova
*a and
Pavel S.
Postnikov
*b
g,
Viktor V.
Zhdankin
bf,
Marina E.
Trusova
*a and
Pavel S.
Postnikov
*b
aDepartment of Biotechnology and Organic Chemistry, National Research Tomsk Polytechnic University, 43 Lenin ave., 634050 Tomsk, Russia. E-mail: trusova@tpu.ru; Tel: +7(3822)563-861
bDepartment of Technology of Organic Substances and Polymer Materials, National Research Tomsk Polytechnic University, 43 Lenin ave., 634050 Tomsk, Russia. E-mail: postnikov@tpu.ru; Tel: +79039136029
cDepartment of Physical and Analytical Chemistry, National Research Tomsk Polytechnic University, 43 Lenin ave., 634050 Tomsk, Russia
dDepartment of Solid State Chemistry, University of Chemistry and Technology, Prague, Technicka 5, 166 28, Czech Republic
eDepartment of Biomedicine, Altai State University, 656049, Barnaul, Lenina av. 61, Russia
fDepartment of Chemistry and Biochemistry, University of Minnesota Duluth, Duluth, MN 55812, USA
gDivision of Applied Chemistry Institute of Engineering Tokyo University of Agriculture and Technology Koganei, Tokyo 184-8588, Japan
First published on 8th February 2017
Abstract
A new method of azide–alkyne cycloaddition (AAC) in the presence of Zn(OAc)2 as an inexpensive and environmentally friendly catalyst in neat water has been developed. The proposed methodology has been applied for the synthesis of 1,4-disubstituted-1,2,3-triazoles from terminal alkynes and 1,4,5-trisubstituted-1,2,3-triazoles from internal alkynes. It has been found that Zn-catalyzed AAC is extremely sensitive to steric hindrance in acetylenes and a method of regioselective triazole ring formation has been proposed. Particularly important is the isolation and characterization of a relatively stable Zn-containing intermediate, which has been characterized by NMR and HRMS.
Introduction
Recently, the azide–alkyne cycloaddition (AAC), originally proposed by Huisgen, has become a powerful basis for the “click-reactions”.1 Since 2001, when the concept of “click-chemistry” was established by Sharpless and co-workers,2 this reaction has been transformed into a unique tool for the design and synthesis of a wide range of products. The modern Cu-catalyzed azide–alkyne cycloaddition (CuAAC) was originally developed by Fokin and Sharpless.3 This method is characterized by high regioselectivity, inexpensive catalytic system, and it can be realized in neat water or alcohols. The procedure has a wide synthetic applicability: the reaction proceeds smoothly with various terminal alkynes and azides which can be generated in situ.4 The CuAAC has been applied in the synthesis of dendrimers,5 rotaxanes and catenanes,6 in the design of new drugs7 and functional polymers,8 and in materials science.9In the last decade, numerous modifications have been suggested for the improvement of the AAC catalysts. Thus, it was found that the addition of nitrogen ligands results in a decreased reaction time and allows the use of a smaller amount of catalyst.10 Furthermore, the development of new catalytic systems based on transition metals (MAAC) has become a new trend in click-chemistry.4a,11 The utilization of new catalysts made it possible to decrease the reaction time, increase the yields of 1,2,3-triazoles, and change the reaction path from the formation of 1,4-disubstituted-1,2,3-triazoles to 1,5-disubstituted-1,2,3-triazoles. For example, the Ag2O nanoparticles have been successfully applied in AgAAC reaction as well as the AgN(CN)2/DIPEA system in aqueous ethylene glycol solution.12 The Cu(111) or Au-assisted AAC has made it possible to carry out the on-surface covalent coupling and significantly improved the applicability of click-reactions in materials science.13 Recently, the highly active catalytic systems based on nickel have been developed.15 However, all catalytic systems listed above have a significant disadvantage: they are applicable only for the preparation of 1,4-disubstituted-1,2,3-triazoles from terminal alkynes.14 The originally proposed Ru-catalyzed AAC based on a Cp*RuCl complex allowed the synthesis of 1,5-disubstituted-1,2,3-triazoles from terminal alkynes and 1,4,5-trisubstituted-1,2,3-triazoles from internal alkynes.16 Later, the replacement of Cp*RuCl with RuH2(CO)(PPh3)3 made it possible to change the reaction path to the regioselective formation of 1,4-disubstituted-1,2,3-triazoles.16,17 Noteworthily, the procedure utilizing Ru-based catalytic systems is quite complicated and requires an inert atmosphere and Schlenk techniques, which significantly decreases the synthetic applicability of this method. Moreover, the Ru-based catalysts are relatively expensive.
In 2010 Chen and co-workers have proposed the procedure for azide–alkyne cycloaddition catalyzed by zinc on charcoal.18 This method has an excellent tolerance to functional groups: the cycloaddition readily proceeds with aryl or alkyl azides and alkynes with a broad range of substituents. This catalytic system can be easily recycled with a slight decrease of catalytic activity. The main advantage of ZnAAC is in the possibility of cycloaddition of di-substituted alkynes and appropriate azides with the formation of 1,4,5-trisubstituted-1,2,3-triazoles. However, the authors could not provide the mechanistic rationalization and explanation of the observed reactivity. This catalytic system does not allow the use of neat water; the authors have obtained satisfactory yields of the targeted 1,2,3-triazoles only in DMF. It should be noted that the steric hindrance has a strong effect on the product yields, and this method is not suitable for the synthesis of 1,2,3-triazoles from o-substituted aromatic azides. Later, Greaney and Smith have developed an alternative ZnAAC methodology via interactions of terminal alkynes with azides in the presence of an equimolecular amount of ZnEt2.19 The ZnEt2-assisted AAC led to the formation of 1,5-disubstituted-1,2,3-triazoles or 1,4,5-trisubstituted-1,2,3-triazoles via the interaction of the intermediate triazolyl-zinc derivative with an electrophile. The authors proposed the generation of the corresponding zinc-acetylides via the direct interaction between ZnEt2 and acetylenes. The regioselectivity of this reaction was explained by Lan through the computational studies of the transition states of the proposed Zn-intermediates.20 Unfortunately, a clear explanation of the ZnAAC mechanism for the reactions of internal alkynes has not been presented yet. Moreover, the known procedures of ZnAAC are not in full agreement with the basic principles of “click” methodology. Therefore, there is a strong need for a mechanistic understanding of zinc-catalysed AAC for controlling the regioselectivity in the reactions of internal alkynes. Moreover, in comparison with Ru- and Cu-catalyzed AAC, whose mechanisms have been clearly explained,16c,21 the ZnAAC mechanism remains a blind spot in the MetAAC chemistry.
Results and discussion
In this work, we present a new method for Zn-promoted AAC with substrate-controlled regioselectivity in neat water using Zn(OAc)2 as an inexpensive and environmentally friendly catalyst. This procedure is applicable to the cycloaddition of alkyl or aryl azides with internal and terminal alkynes. Also, our procedure can be applied to the synthesis of aryl-substituted triazoles directly from aromatic amines. Moreover, we have isolated and characterized the key Zn-containing intermediate, which provides an insight into the mechanism of ZnAAC.The optimization of Zn-catalyzed alkyne–azide cycloaddition was conducted using various Zn-containing catalytic systems in neat water and at various temperatures in the reaction between 4-azidonitrobenzene 1a and phenylacetylene 2a (Table 1). In agreement with the previous observation of Chen and co-workers, Zn on charcoal demonstrates low activity in neat water and the formation of the desired 1,2,3-triazoles is not observed (entry 1).18 In contrast, the reaction in the presence of the water-soluble Zn(OTf)2 affords 1,2,3-triazole 3aa with quantitative yield (entry 2). Similar activity has been demonstrated by Zn(OAc)2 with product 3aa being isolated in excellent yield (entry 6). Increasing the temperature to 75 °C resulted in a significant reduction of the reaction time with slightly lower product yields (entries 3 and 7). Furthermore, the addition of ascorbic acid allowed the achievement of full conversion of the starting materials in 16 hours with high product yields (entries 4 and 8).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst, mol% | T (°C) | t (h) | Yieldb (%) |
| a Reactions conditions: 1a (1 equiv.), 2a (1 equiv.), water (10 ml). b Isolated yield. c Described previously.18 d Reaction was carried out under microwave irradiation (75 °C, constant power 80 W). e Full conversion of the starting materials was not achieved. | ||||
| 1 | Zn/C, 10 mol%c | 50 | 15 | 0 |
| 2 | Zn(OTf)2, 10 mol% | rt | 320 | 95 |
| 3 | Zn(OTf)2, 10 mol% | 75 | 30 | 90 |
| 4 | Zn(OTf)2, 10 mol%, ascorbic acid, 20 mol% | 75 | 16 | 86 |
| 5d | Zn(OTf)2, 10 mol%, ascorbic acid, 20 mol% | 75 | 5.5 | 87 |
| 6 | Zn(OAc)2, 10 mol% | rt | 320 | 96 |
| 7 | Zn(OAc)2, 10 mol% | 75 | 28 | 88 |
| 8 | Zn(OAc)2, 10 mol%, ascorbic acid, 20 mol% | 75 | 16 | 85 |
| 9d | Zn(OAc)2, 10 mol%, ascorbic acid, 20 mol% | 75 | 6 | 97 |
| 10d | Zn(OAc)2, 5 mol%, ascorbic acid, 10 mol% | 75 | 8 | 85 |
| 11d | Zn(OAc)2, 20 mol%, ascorbic acid, 40 mol% | 75 | 5 | 94 |
| 12d | ZnCl2, 10 mol%, ascorbic acid, 20 mol% | 75 | 6 | 50e |
| 13d | ZnSO4, 10 mol%, ascorbic acid, 20 mol% | 75 | 6 | 48e |

Finally, we have found that the target 1,2,3-triazole 3aa is formed in high yield at 75 °C in a microwave reactor. Surprisingly, the inexpensive and readily available Zn(OAc)2 demonstrates slightly higher activity than Zn(OTf)2 (entries 5 and 9). We have also tested ZnCl2 and ZnSO4, but the full conversion of the starting materials has not been achieved after 6 hours in a microwave reactor and product 3aa was isolated only in 48–50% yield (entries 12 and 13).
In order to exclude the possible formation of triazoles under simple heating we carried out the experiment under similar conditions without the addition of the Zn catalyst (Scheme 1). The full conversion of 1a and 2a was not achieved. We observed the formation of both the isomers of the triazole (1,4- and 1,5-triazoles) with 37% total yield and an isomer ratio of 81![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :19. These results confirm the crucial role of Zn(OAc)2 for the regioselectivity of the process.
:19. These results confirm the crucial role of Zn(OAc)2 for the regioselectivity of the process.
 | ||
| Scheme 1 AAC reaction of 1-azido-4-nitrobenzene 1a and phenylacetylene 2a without the catalyst. | ||
Using optimized reaction conditions, we have investigated the synthesis of 1,4-disubstituted-1,2,3-triazoles. The optimized catalytic system allowed the preparation of a wide range of 1,2,3-triazoles from aromatic and benzylic azides as well as aromatic and aliphatic acetylenes in good to excellent yields (Table 2).
|
|
||||
|---|---|---|---|---|
| Entry | Azide, R = | Acetylene, R′ = | Triazole, yieldb (%) | t (h) |
| a Reaction conditions: azide (1 equiv.), acetylenes (1 equiv.), water (10 ml), microwave irradiation (75 °C, constant power 80 W). b Isolated yield. | ||||
| 1 | 1a, 4-O2NC6H4 | 2a, Ph | 3aa, 97 | 6 |
| 2 | 1b, 3-O2NC6H4 | 2a, Ph | 3ba, 94 | 7 |
| 3 | 1c, 4-NCC6H4 | 2a, Ph | 3ca, 92 | 7 |
| 4 | 1d, 4-MeC6H4 | 2a, Ph | 3da, 90 | 9 |
| 5 | 1e, 2-MeC6H4 | 2a, Ph | 3ea, 61 | 7 |
| 6 | 1f, 4-MeOC6H4 | 2a, Ph | 3fa, 92 | 5 |
| 7 | 1g, 2-MeOC6H4 | 2a, Ph | 3ga, 59 | 7 |
| 8 | 1h, 2-(PhCO)-5-ClC6H3 | 2a, Ph | 3ha, 70 | 9 |
| 9 | 1i, 2,4,6-Br3C6H2 | 2a, Ph | 3ia, 78 | 7 |
| 10 | 1j, 4-PhC6H4 | 2a, Ph | 3ja, 75 | 8 |
| 11 | 1k, β-C14H9 | 2a, Ph | 3ka, 71 | 6 |
| 12 | 1l, C6H5CH2 | 2a, Ph | 3la, 87 | 6 |
| 13 | 1m, C4H9 | 2a, Ph | 3ma, 66 | 6 |
| 14 | 1a, 4-O2NC6H4 | 2b, 4-O2NC6H4 | 3ab, 77 | 5 |
| 15 | 1a, 4-O2NC6H4 | 2c, pentyl | 3ac, 92 | 6 |
| 16 | 1d, 4-MeC6H4 | 2c, pentyl | 3dc, 80 | 5 |
| 17 | 1a, 4-O2NC6H4 | 2d, OHCH2 | 3ad, 93 | 6 |
| 18 | 1f, 4-MeOC6H4 | 2d, OHCH2 | 3fd, 61 | 7 |

In the case of 2-azidonitrobenzene, the desired 1,2,3-triazole was not obtained. We observed the formation of benzofuroxan (Scheme 2). According to a published procedure, the microwave heating of the 2-azidonitrobenzene derivatives led to the formation of appropriate benzofuroxans with good yields.22 In order to exclude the influence of the Zn catalyst, we carried out the experiment without any additives of ascorbic acid or Zn(OAc)2.
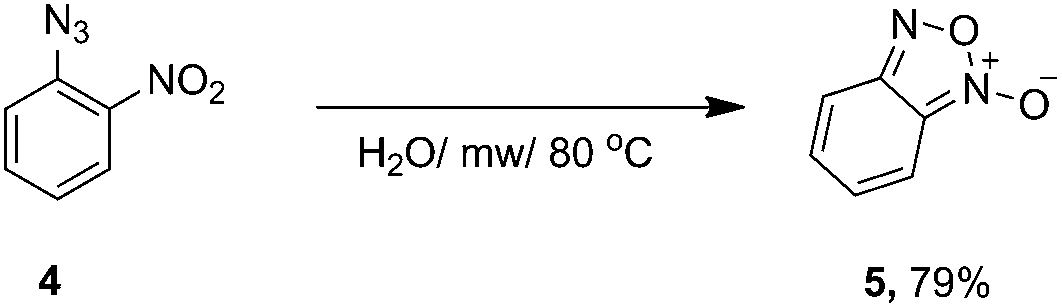 | ||
| Scheme 2 Formation of benzofuroxan from 2-azidonitrobenzene. | ||
Simple heating of 2-azidonitrobenzene under microwave irradiation in neat water led to the formation of benzofuroxan in 79% yield.
Previously, Chen and co-workers have demonstrated that Zn on charcoal allows the AAC reaction of tolane as an example of an internal alkyne.18 In order to evaluate the reactivity of the Zn(OAc)2–ascorbic acid catalytic system, we conducted experiments with a range of disubstituted acetylenes (Table 3).
|
|
|||||
|---|---|---|---|---|---|
| Entry | 1,2,3-Triazole | Yieldb (%) | Entry | 1,2,3-Triazole | Yieldb (%) |
| a Reaction conditions: azide (1.1 equiv.), acetylenes (1 equiv.), water (10 ml), microwave irradiation (130 °C, constant power 150 W). b Isolated yield. c Reaction conditions: azide (2 equiv.), acetylenes (1 equiv.), water (10 ml), microwave irradiation (130 °C, constant power 150 W). d Reaction conditions: azide (2 equiv.), acetylenes (1 equiv.), water (10 ml), microwave irradiation (80 °C, constant power 90 W). | |||||
| 1 |

|
96 | 6 |

|
80 |
| 2 |

|
51c | 7 |
|
79 |
| 3 |

|
47c | 8 |

|
70 |
| 4 |

|
60 | 9 |

|
69d |
| 5 |

|
65 | 10 |
|
69 |

In agreement with literature data, internal acetylenes exhibited lower reactivity than terminal acetylenes. In order to reduce the reaction time, all experiments were carried out at higher temperatures and under an increased power of the MW-source.
Cycloaddition of the highly reactive 4-azidonitrobenzene 1a and tolane 2e afforded 1,2,3-triazole 3ae in almost quantitative yield (Table 3, entry 1). Arylazides 1g and 1e with electron-donating substituents showed significantly lower reactivity producing the corresponding 1,2,3-triazoles 3ge and 3ee in moderate yields even after using a double excess of the azide (entries 2 and 3). It should be noted that the Zn/C catalyst was completely unreactive in the reaction between o-substituted azides and tolane.18 The high reactivity of our catalytic system is best demonstrated by the reaction of 4-azidonitrobenzene 1a with the electron-poor acetylenedicarboxylic acid 2f, which produced 1,2,3-triazole 3af in 60% yield (entry 4).
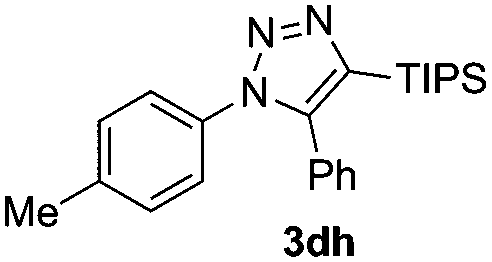
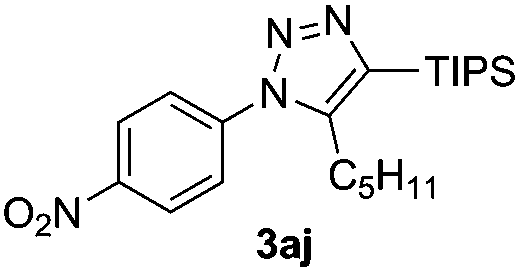
The high reactivity of the Zn(OAc)2 catalyst allowed us to develop a substrate-determined regioselective method for the synthesis of fully decorated 1,4,5-trisubstituted-1,2,3-triazoles (entries 5–10). We have found that the Zn-catalyzed AAC reaction is very sensitive to steric hindrance in the acetylene moiety. In particular, the reaction of aromatic azides 1a, 1d, and 1n and TIPS-substituted phenylacetylene 2h led to the formation of 1,5-diaryl derivatives of 1,2,3-triazoles 3ah, 3dh, and 3nh (entries 6–8). The TIPS-derivative of 1-hexyne 2j reacted in a similar way (entry 10). Similar regioselectivity was observed in the reactions of TMS-substituted acetylenes 2g and 2i (entries 5 and 9). The structures of triazoles 3ai, 3aj and 3dh were established by X-ray analysis (Fig. 1; X-ray structures of 3aj and 3dh are provided in the ESI†).
 | ||
| Fig. 1 The X-Ray structure of 1,2,3-triazole 3ah. | ||
The relatively high temperatures can cause the formation of desired triazoles without the Zn catalyst. In order to clarify this question, we carried out the reaction between 1-azido-4-nitrobenzene 1a and the TIPS-derivative of phenylacetylene 2h in the absence of Zn(OAc)2. After 6 hours of microwave heating we observed the formation of black tar without any traces of desired triazole 3ah.
The observed high regioselectivity of Zn(OAc)2-catalyzed AAC requires a suitable mechanistic explanation. Two main approaches to the regioselective synthesis of 1,2,3-triazoles are currently known: the RuAAC with the formation of 1,5-disubstituted-1,2,3-triazoles17 and the CuAAC for the preparation of 1,4-disubstituted-1,2,3-triazoles.10e,23 The mechanisms of both reactions were thoroughly investigated by experimental and theoretical studies.3,4a,5a,17 A substrate-determined regioselectivity has not been previously observed in the ZnAAC reactions. In fact, the previously published ZnEt2-mediated cycloaddition is not applicable to the reactions of disubstituted alkynes because its mechanism involves the initial formation of Zn-acetylides from terminal alkynes.19
Chen and co-workers have proposed that Zn-catalyzed AAC can proceed through the intermediate formation of a Zn-containing six-membered cycle.18 Based on this suggestion, we propose the mechanism of Zn(OAc)2-catalyzed AAC shown in Scheme 3.
 | ||
| Scheme 3 The proposed mechanism for the Zn-catalyzed formation of (A) 1,4-disubstituted-1,2,3-triazoles and (B) 1,4,5-trisubstituted-1,2,3-triazoles. | ||
We suggest that the initial step of ZnAAC is the formation of a π-complex between Zn(OAc)2 and acetylene. In the next step, the resulting complex interacts with the azide giving the metallacycle I. In the final stage the Zn2+ is regenerated via extrusion from metallacycle I. We may speculate that the presence of ascorbic acid accelerates the reaction due to its participation in the redox processes involving zinc species, or due to the general acid catalysis.
We suggest that the regioselectivity of the formation of the intermediate is determined by the conformation of a six-membered metallacycle. In the case of a mono-substituted acetylene, the hydrogen does not restrict the formation of metallacycle I. Moreover, the R1 group makes unfavorable the addition of the azide with opposite orientation.
Surprisingly, the alkyl-substituents in terminal acetylenes 2c and 2d also have a strong effect on regioselectivity. The formation of the regioisomeric 1,5-disubstituted-1,2,3-triazoles in the reaction of azides 1a, 1d, and 1f with terminal acetylenes 2c and 2d was not observed (Table 2, entries 15–18). In contrast, the presence of a large (L) group such as TIPS or TMS makes entirely unfavorable the formation of intermediate I. Only one possible reaction path is possible through the generation of intermediate II.
Obviously, the steric hindrance of the R2-group affects the reaction conditions, and the AAC requires the increased temperature and microwave power and longer reaction time (Table 3). Even under these conditions we did not observe the formation of 1,4-diarylsubstituted-1,2,3-triazole derivatives in the reaction with TIPS-acetylenes.
In order to prove the formation of intermediate I we carried out a comprehensive study of the reaction between azide 1i and acetylene 2a. The GCMS analysis of the reaction mixture after full conversion of the starting materials showed that the reaction mixture contains a compound with retention time and mass-spectra different from product 3ia (the chromatogram and mass-spectrum are presented in ESI Fig. S1†). Moreover, in the mass-spectra we observed the presence of a Zn-cation, which made possible the identification of the Zn-containing intermediate Iia. The HPLC analysis with qTOF detection confirmed the presence of intermediate Iia as a complex with MeCN (Fig. S2†). The isotopic pattern of the isolated intermediate Iia was in agreement with the presence of a tribromobenzene ring and Zn in the molecular ion. In particular, the peak with m/z 560 indicates the presence of 64Zn. Moreover, the discrepancy in the intensity of typical bromine isotopes (m/z 562 and 564) can be explained by the appearance of two isotopes of Zn: 64Zn and 66Zn. It should be noted that intermediate Iia decomposes in the process of ionization with the formation of triazole 3ia.
We succeeded in the isolation of the pure intermediate Iia in 18% yield after full conversion of the starting azide and acetylene (Scheme 4). In order to improve the yield of compound Iia, we carried out the reaction at room temperature using four equivalents of phenylacetylene and Zn(OAc)2 (Scheme 4).
 | ||
| Scheme 4 The synthesis and reactivity of intermediate Iia. | ||
This procedure allows the isolation of intermediate Iia in 48% yield after 2 hours. Interestingly the addition of phenylacetylene to the reaction mixture significantly increased the yield of Iia (Scheme 4). It should be noted that simple heating of the isolated intermediate Iia in acetone affords 1,2,3-triazole 3ia in quantitative yield after simple filtration of acetone solution through the silica pad for removing of the Zn-containing byproduct.
The NMR study of compound Iia demonstrated a significant difference of chemical shifts of azide 1i, phenylacetylene 2a, and isolated 1,2,3-triazole 3ia (Fig. 2). The protons of the phenyl ring (H3–H5) shifted downfield in comparison with the starting phenylacetylene. The most pronounced effect was observed for o-protons (H3): from 7.46 to 7.95 ppm. A similar change was observed for the singlet of protons H1 (from 7.98 to 8.31 ppm).
 | ||
| Fig. 2 The comparison of the 1H NMR spectra of intermediate Iia, 1,2,3-triazole 3ia and a mixture azide 1i with alkyne 2a. | ||
The presence of a signal from proton H2 clearly indicates that the formation of intermediate Iia proceeds without the involvement of an acetylenic C–H bond. However, the signal of H-2 protons shifted downfield by 4.83 ppm (from 4.19 to 9.01 ppm). We explain this fact by the formation of a metallacycle (Fig. 2). It should be noted that HMBS data demonstrated a correlation between proton H2 and carbon C2 (Fig. S5†). The 13C NMR spectra for intermediate Iia in a mixture with the starting materials were in agreement with these observations (Fig. S6†).
The signals of acetylenic carbons shifted from 80.7 ppm (β-carbon of the acetylenic bond) to 129.07 ppm and from 83.4 ppm (α-carbon of the acetylenic bond) to 123.4 ppm.
These shifts can be explained by the formation of intermediate Iia with the disappearance of the acetylenic C–C bond. In order to exclude the possible formation of 1,2,3-triazole 3ia, we compared the NMR spectra of intermediate Iia with the spectra of product 3ia (Fig. 2). The differences of chemical shifts were found for all characteristic protons and also carbons (Fig. S6†).
Thus, the mechanistic studies confirm the hypothesis about the formation of a six-membered metallacycle as the reaction intermediate. The regioselectivity of AAC is controlled by steric hindrance of the substituent in the structure of the internal acetylene.
Taking into account the availability of silyl acetylenes and the possibility of a straightforward desilylation, our method offers a good approach to the general synthesis of 1,4- and 1,5-disubstituted-1,2,3-triazoles. Moreover, compared to the Ru- or Ir-based systems, Zn(OAc)2 in water represents a much cheaper and greener catalytic system.
Conclusions
In conclusion, we have developed a new method for the azide–alkyne cycloaddition reaction catalyzed by Zn(OAc)2–ascorbic acid in neat water. The proposed method has a good tolerance to various functional groups and allows the preparation of 1,4-disubstituted-1,2,3-triazoles and 1,4,5-trisubstituted-1,2,3-triazoles from aromatic or aliphatic azides and terminal or internal acetylenes. Based on the developed ZnAAC, we have proposed a regioselective procedure for the synthesis of silylated triazole derivatives with controlled regioselectivity from available silyl-acetylenes. In order to prove the proposed mechanism of ZnAAC, we have isolated the reaction intermediate and identified its structure by HRMS and NMR studies. We believe that the developed method has great potential in organic synthesis.Experimental
General procedure for the preparation 1,4-disubstituted-1,2,3-triazoles from azides (1a–m) and terminal alkynes (2a–d)
The mixture of azides (1a–m) (0.5 mmol) and alkynes (2a–d) in water (10 ml) was vigorously stirred for 2–3 minutes. After that, 10 mol% Zn(OAc)2 (0.05 mmol, 0.011 g) and 20 mol% ascorbic acid (0.10 mmol, 0.017 g) were added to the reaction mixture. The reaction vessel was placed in a microwave reactor (t = 75 °C and power = 80 W) and heated until full conversion of the starting materials and intermediate (TLC, hexane:EtOAc = 7:3). After completion of the reaction, the reaction mixture was extracted with CH2Cl2 (3 × 20 mL), washed with water and brine and then dried with anhydrous Na2SO4. The solvent was removed in vacuo to give the crude 1,4-disubstitituted-1,2,3-triazoles (3aa–ma, 3ab, 3ac, 3dc, 3ad, and 3fd) which were purified by column chromatography (silica gel, eluent: hexane:EtOAc = 9:1).
The crystal suitable for the X-ray study was obtained by slow evaporation of ether solution of 3ia.
General procedure for the preparation 1,4,5-trisubstituted-1,2,3-triazoles from aromatic azides (1a, 1d, 1g, 1e, and 1n) and disubstituted acetylenes (2e–j)
The mixture of azides (1a, 1d, 1g, 1e, and 1n) (0.5 mmol) and disubstituted acetylenes (2e–j) in water (10 ml) was vigorously stirred for 2–3 minutes. After that, 10 mol% Zn(OAc)2 (0.05 mmol, 0.011 g) and 20 mol% ascorbic acid (0.10 mmol, 0.017 g) were added to the reaction mixture. The reaction vessel was placed in a microwave reactor (t = 130 °C and power = 150 W) and heated until full conversion of the starting materials and intermediate (TLC, hexane:EtOAc = 7:3). After completion of the reaction, the reaction mixture was extracted with CH2Cl2 (3 × 20 mL), washed with water and brine and then dried with anhydrous Na2SO4. The solvent was removed in vacuo to give the crude 1,4,5-trisubstituted-1,2,3-triazoles (3ae, 3ee, 3ge, 3af, 3ng, 3ah, 3dh, 3nh, 3ai, and 3aj) which were purified by column chromatography (silica gel, eluent: hexane:EtOAc = 9:1).
The crystal suitable for the X-ray study was obtained by slow evaporation of ether solution of 3ah.
The full characterization of all the prepared compounds, including the NMR-spectra, HRMS data and X-ray structures is provided in the ESI.†
Acknowledgements
This work was supported by the Russian Ministry of Education and Science (Scientific Program no. 4.5924.2017). The authors greatly acknowledge the Laboratory of Physical–Chemical Analytical Methods of Tomsk State University for the NMR and HRMS analyses.References
- (a) R. Huisgen, Angew. Chem., Int. Ed., 1963, 2, 565 CrossRef; (b) R. Huisgen, Angew. Chem., Int. Ed., 1963, 2, 633 CrossRef.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS PubMed.
- (a) C. Wang, D. Ikhlef, S. Kahlal, J.-Y. Saillard and D. Astruc, Coord. Chem. Rev., 2016, 316, 1 CrossRef CAS; (b) D. Wang, N. Li, M. Zhao, W. Shi and B. Chen, Green Chem., 2010, 12, 2120 RSC; (c) L. Liang and D. Astruc, Coord. Chem. Rev., 2011, 255, 2933 CrossRef CAS; (d) J. E. Hein, J. C. Tripp, L. B. Krasnova, K. B. Sharpless and V. V. Fokin, Angew. Chem., Int. Ed., 2009, 48, 8018 CrossRef CAS PubMed; (e) P. M. E. Gramlich, C. T. Wirges, A. Manetto and T. Carell, Angew. Chem., Int. Ed., 2008, 47, 8350 CrossRef CAS PubMed; (f) R. Hudson, C. Li and A. Moores, Green Chem., 2012, 14, 622 RSC; (g) M. F. Debets, S. S. van Berkel, S. Schoffelen, F. P. J. T. Rutjes, J. C. M. van Hest and F. L. van Delft, Chem. Commun., 2010, 46, 97 RSC; (h) V. Hong, S. I. Presolski, C. Ma and M. G. Finn, Angew. Chem., Int. Ed., 2009, 48(52), 9879 CrossRef CAS PubMed.
- (a) G. Franc and A. K. Kakkar, Chem. Soc. Rev., 2010, 39, 1536 RSC; (b) M. V. Walter and M. Malkoch, Chem. Soc. Rev., 2012, 41, 4593 RSC.
- (a) K. D. Hanni and D. A. Leigh, Chem. Soc. Rev., 2010, 39, 1240 RSC; (b) N. H. Evans and P. D. Beer, Chem. Soc. Rev., 2014, 43, 4658 RSC.
- (a) X. Wang, B. Huang, X. Liu and P. Zhan, Drug Discovery Today, 2016, 21, 118 CrossRef CAS PubMed; (b) S. G. Agalave, S. R. Maujan and V. S. Pore, Chem. – Asian J., 2011, 6, 2696 CrossRef CAS PubMed; (c) J.-F. Lutz and Z. Zarafshani, Adv. Drug Delivery Rev., 2008, 60, 958 CrossRef CAS PubMed.
- (a) M. R. Ramdas, K. S. S. Kumar and C. P. R. Nair, Mater. Lett., 2016, 172, 216 CrossRef CAS; (b) A. P. Vogt, Macromolecules, 2010, 43, 1 CrossRef.
- (a) W. Xi, T. F. Scott, C. J. Kloxin and C. N. Bowman, Adv. Funct. Mater., 2014, 24, 2572 CrossRef CAS; (b) W.-B. Zhang, X. Yu, C.-L. Wang, H.-J. Sun, I.-F. Hsieh, Y. Li, X.-H. Dong, K. Yue, R. van Horn and S. Z. D. Cheng, Macromolecules, 2014, 47, 1221 CrossRef CAS.
- (a) S. Díez-González, E. C. Escudero-Adan, J. Benet-Buchholz, E. D. Stevens, A. M. Z. Slawin and S. P. Nolan, Dalton Trans., 2010, 39, 7595 RSC; (b) J. D. Egbert, C. S. J. Cazin and S. P. Nolan, Catal. Sci. Technol., 2013, 3, 912 RSC; (c) J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302 CAS; (d) S. I. Presolski, V. Hong, S. H. Cho and M. G. Finn, J. Am. Chem. Soc., 2010, 132, 14570 CrossRef CAS PubMed; (e) L. S. Campbell-Verduyn, L. Mirfeizi, R. Dierckx, P. H. Elsinga and B. L. Feringa, Chem. Commun., 2009, 2139 RSC; (f) V. O. Rodionov, S. I. Presolski, S. Gardinier, Y. Lim and M. G. Finn, J. Am. Chem. Soc., 2007, 129, 12696 CrossRef CAS PubMed.
- J. Totobenazara and A. J. Burke, Tetrahedron Lett., 2015, 56, 2853 CrossRef CAS.
- (a) A. M. Ferretti, A. Ponti and G. Molteni, Tetrahedron Lett., 2015, 56, 5727 CrossRef CAS; (b) A. A. Ali, M. Chetia, B. Saikia, P. J. Saikia and D. Sarma, Tetrahedron Lett., 2015, 56, 5892 CrossRef CAS.
- (a) F. Bebensee, C. Bombis, S.-R. Vadapoo, J. R. Cramer, F. Besenbacher, K. V. Gothelf and T. R. Linderoth, J. Am. Chem. Soc., 2013, 135, 2136 CrossRef CAS PubMed; (b) O. D. Arado, H. Mönig, H. Wagner, J.-H. Franke, G. Langewisch, P. A. Held, A. Studer and H. Fuchs, ACS Nano, 2013, 7, 8509 CrossRef PubMed.
- S. Díez-González, A. Correa, L. Cavallo and S. P. Nolan, Chem. – Eur. J., 2006, 29, 7558 CrossRef PubMed.
- H. S. P. Rao and G. Chakibanda, RSC Adv., 2014, 4, 46040 RSC.
- (a) J. S. Oakdale, R. K. Sit and V. V. Fokin, Chem. – Eur. J., 2014, 20, 11101 CrossRef CAS PubMed; (b) L. Zhang, X. Chen, P. Xue, H. H. Y. Sun, I. D. Williams, K. B. Sharpless and G. Jia, J. Am. Chem. Soc., 2005, 127, 15998 CrossRef CAS PubMed; (c) B. C. Boren, S. Narayan, L. K. Rasmussen, L. Zhang, H. Zhao, Z. Lin and V. V. Fokin, J. Am. Chem. Soc., 2008, 130, 8923 CrossRef CAS PubMed; (d) J. R. Johansson, E. Hermansson, B. Nordén, N. Kann and T. Beke-somfai, Eur. J. Org. Chem., 2014, 2703 CrossRef CAS; (e) L. K. Rasmussen, B. C. Boren and V. V. Fokin, Org. Lett., 2007, 4, 5 Search PubMed; (f) T. Biet, T. Cauchy and N. Avarvari, Chem. – Eur. J., 2012, 18, 16097 CrossRef CAS PubMed.
- P. N. Liu, J. Li, F. H. Su, K. D. Ju, L. Zhang, C. Shi, H. H. Y. Sung, I. D. Williams, V. V. Fokin, Z. Lin and G. Jia, Organometallics, 2012, 31, 4904 CrossRef CAS.
- X. Meng, X. Xu, T. Gao and B. Chen, Eur. J. Org. Chem., 2010, 5409 CrossRef CAS.
- C. D. Smith and M. F. Greaney, Org. Lett., 2013, 15, 4826 CrossRef CAS PubMed.
- Y. Li, X. Qi, Y. Lei and Y. Lan, RSC Adv., 2015, 5, 49802 RSC.
- (a) B. T. Worrell, J. A. Malik and V. V. Fokin, Science, 2013, 340, 457 CrossRef CAS PubMed; (b) L. Jin, E. A. Romero, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2015, 137, 15696 CrossRef CAS PubMed; (c) E. Boz and N. S. Tüzün, J. Organomet. Chem., 2013, 724, 167 CrossRef CAS.
- E. Leyva, S. Leyva, R. M. Gonzalez-Balderas, D. de Loera and R. Jimenez-Catano, Mendeleev Commun., 2013, 23, 217 CrossRef CAS.
- (a) A. Kolarovic, M. Schnurch and M. D. Mihovilovic, J. Org. Chem., 2011, 76, 2613 CrossRef CAS PubMed; (b) A. Hubbard, T. Okazaki and K. K. Laali, J. Org. Chem., 2008, 73, 316 CrossRef CAS PubMed; (c) Y. Wang, J. Liu and C. Xia, Adv. Synth. Catal., 2011, 353, 1534 CrossRef CAS; (d) A. S. Kumar, M. A. Reddy, M. Knorn, O. Reiser and B. Sreedhar, Eur. J. Org. Chem., 2013, 4674 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1513614–1513617. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00787b |
| This journal is © the Partner Organisations 2017 |