DOI:
10.1039/C6QO00713A
(Research Article)
Org. Chem. Front., 2017,
4, 986-994
Mechanistic studies for dirhodium-catalyzed ring expansion reactions†
Received
15th November 2016
, Accepted 6th February 2017
First published on 9th February 2017
Abstract
The mechanisms for dirhodium-catalyzed ring expansion reactions of azide tethered methylenecyclopropanes and their analogues were systematically investigated by DFT calculations. The calculation results indicate that the dirhodium catalyst is essential for generating a reactive Rh2–nitrene intermediate having radical character; however, it is not essential for controlling final product selectivities. For substrates involving a three-membered ring, the experimentally obtained C–N bond formation product is a thermodynamically favored product. In contrast, for substrates having a larger ring, the kinetically favored product is the main product. The DFT calculations presented here account for previous experimental findings, and throw light on other dirhodium-catalyzed reactions involving nitrene or carbene intermediates.
Introduction
The rhodium-catalyzed reactions involving rhodium–carbenoid chemistry, represent one of the most commonly used and efficient methods in organic synthesis.1 The dirhodium complexes (Rh2L4) as catalysts have been widely utilized in a variety of transformations, including a saturated C–H bond-activation/C–N bond-forming reaction,2 cascade cyclopropanation/skeletal rearrangement reactions,3 ylide formations,4etc. It has become widely identified that the nature of the rhodium–carbenoid intermediates generated in these transformations has a major impact on the reaction outcome. Besides commonly recognized rhodium–carbenoid intermediates, some Rh2–nitrene active intermediates can undergo single-electron-transfer (SET) to generate Rh2(III,II)–nitrene radical intermediates, which exhibit different reactive properties. Du Bois's group2b,e and Berry's group5 reported that the SET can take place between nitrene and a dirhodium(II) dimer, resulting in Rh2(III,II) species which is the catalyst resting state in intramolecular amination reactions. Zhang's group have carried out theoretical studies on the mechanism for the Rh2-catalyzed intramolecular C–H amination involving a triplet mixed-valent Rh2(III,II) nitrene radical via the SET oxidation of the Rh2(II,II) dimer.6 However, these Rh2(III,II) nitrene radical intermediates are scarcely reported and studied in other types of reactions.
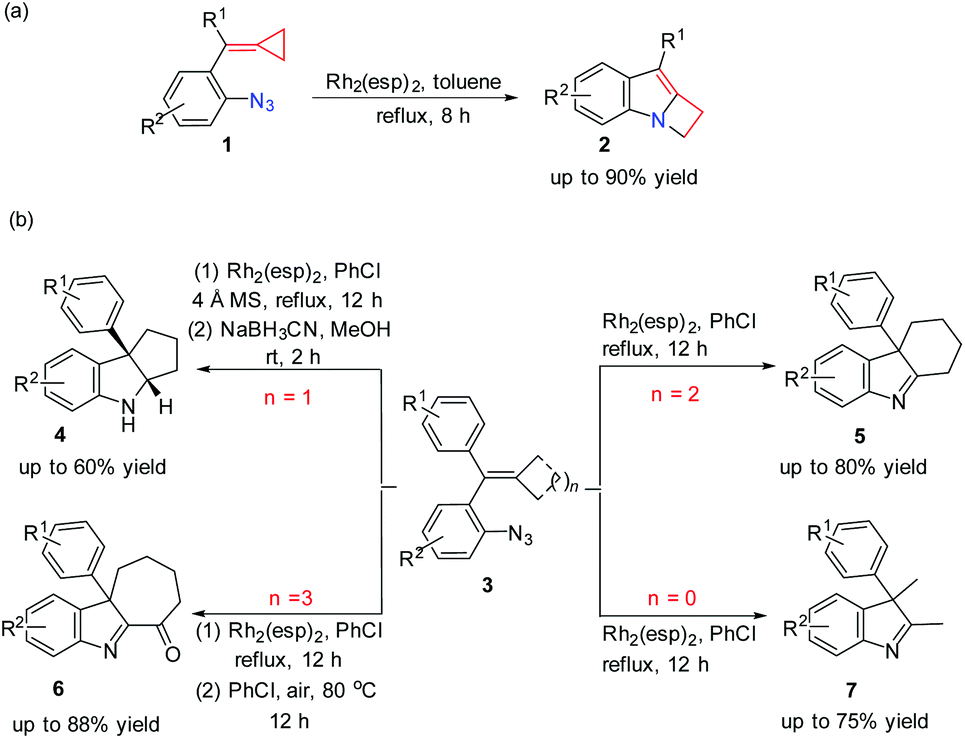
Very recently, our group reported a ring expansion of azide tethered methylenecyclopropanes (MCPs) 1 catalyzed by a dirhodium complex, giving indole fused azetidines 2 (Scheme 1(a)); moreover, we also found that the reactions of 3 having different sized rings or without rings under rhodium catalysis would go different directions to give related products 4–7 (Scheme 1(b)).7 Preliminary DFT calculations on the key reaction steps indicated that the cyclization and SET pathways are probably controlled by a designed radical clock with respect to the reactions of substrate 1.7
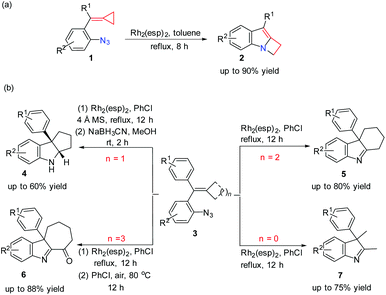 |
| | Scheme 1 Dirhodium-catalyzed ring expansion reactions of azide tethered MCPs and azide-styrenes. | |
Although previous experimental and preliminary theoretical results indicated that Rh2(III,II) nitrene radical intermediates play a role in these reactions, several mechanistic questions still remain unanswered: How is the Rh2(III,II) nitrene radical intermediate formed? What is the role of the catalyst in each of the reaction steps? What is the origin for the product selectivity? With the aim to improve our understanding of the reaction mechanism and to account for the influence of the ring size in the substrate for product selectivity in this dirhodium-catalyzed reaction, we carried out more detailed theoretical studies via DFT calculations. Herein, we would like to report our theoretical results on the reaction mechanism of the dirhodium-catalyzed reaction of azide tethered MCPs.
Computational methods
All of the calculations were performed with the Gaussian 09 software package.8 The geometries of all minima and transition states have been optimized using the BPW91 pure functional,9 with the 1997 Stuttgart relativistic small-core effective core potential (Stuttgart RSC 1997 ECP)10 for the Rh atom, augmented with a 4f function [ζf(Rh) = 1.350],11 and the 6-31G(d) basis set for other atoms (denoted as BSI). The BPW91 pure functional used here was tested previously and demonstrated the reliable prediction of the singlet–triplet energy difference (Est) of the dirhodium–nitrene species after comparison with the more accurate CCSD(T) method.12 The dispersion corrections were considered in affecting the overall energy profile, however, they did not influence the relative stabilities for all species in the test reaction (for details, see the ESI†).13 The stabilities of the Kohn–Sham wave functions were confirmed by stability analyses14 for all of the stationary points along the reaction pathways at the BPW91/BS1 level. The subsequent frequency calculations on the stationary points were carried out at the same level of theory to ascertain the nature of the stationary points as minima or first-order saddle points on the respective potential energy surfaces. All transition states were characterized by one and only one imaginary frequency pertaining to the desired reaction coordinate. The intrinsic reaction coordinate (IRC) calculations15 were carried out at the same level of theory to further authenticate the transition states. Thermochemical corrections to 298.15 K have been calculated for all minima from unscaled vibrational frequencies obtained at this same level. The solvent effect was estimated by the IEFPCM method with radii and non-electrostatic terms for the SMD solvation model16 in toluene (ε = 2.3741) or chlorobenzene (ε = 5.6968). Solvation single-point computations utilized a basis set (denoted as BSII) consisting of the 6-311++G(d,p) basis set for C, H, N and O atoms and the same Stuttgart basis set as in BSI for the Rh atoms were performed based on the gas phase optimized structures.
The solvation Gibbs free energies for all stationary points shown in Schemes 3–9, were estimated as Gsolv = Esolv(SMD-calculated) + ΔGcorr_gas, where Esolv(SMD-calculated) refers to the solvation single point energy and ΔGcorr_gas refers to the thermal correction to the free energy of the solute in the gas phase.
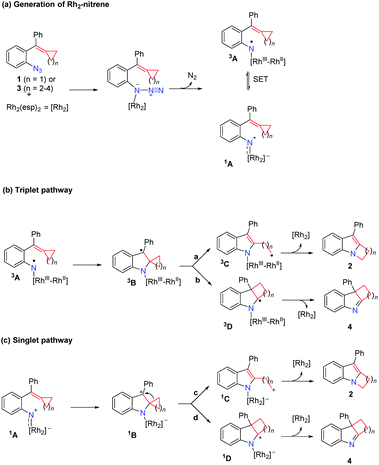 |
| | Scheme 2 Proposed reaction mechanism. | |
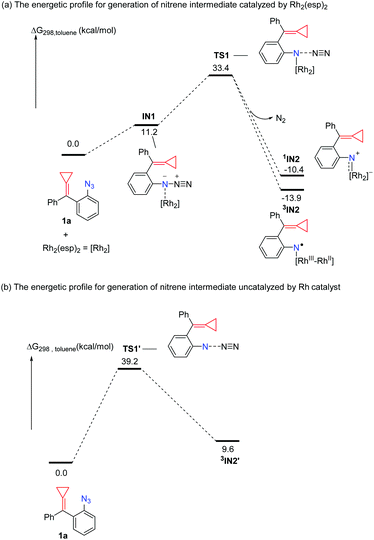 |
| | Scheme 3 Solvation Gibbs free energy profiles for catalyzed and uncatalyzed N2 extrusion. | |
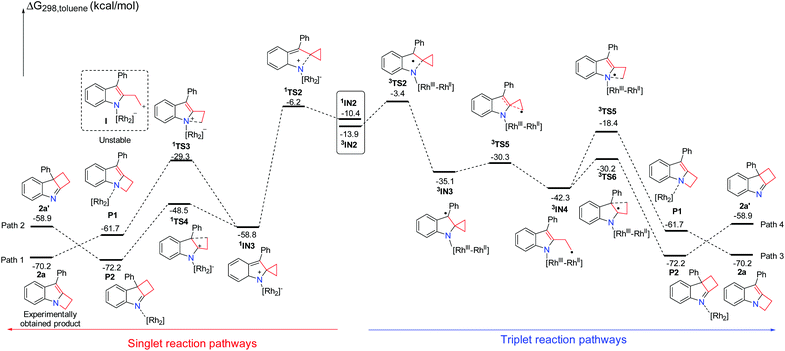 |
| | Scheme 4 Solvation Gibbs free energy profiles for the reaction of 1a having a three-membered ring. | |
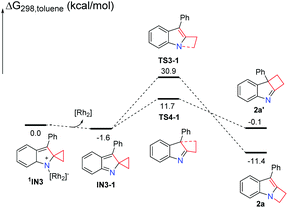 |
| | Scheme 5 Solvation Gibbs free energy profiles for generation of product 2a without the Rh2(esp)2 catalyst. | |
 |
| | Scheme 6 Solvation Gibbs free energy profiles for the reaction of 3a having a four-membered ring. | |
 |
| | Scheme 7 Solvation Gibbs free energy profiles for generation of product 4a without the Rh2(esp)2 catalyst. | |
 |
| | Scheme 8 Solvation Gibbs free energy profiles for the reaction of 3b having a five-membered ring. | |
 |
| | Scheme 9 Solvation Gibbs free energy profiles for generation of product 5a without the Rh2(esp)2 catalyst. | |
Results and discussion
Proposed reaction pathways
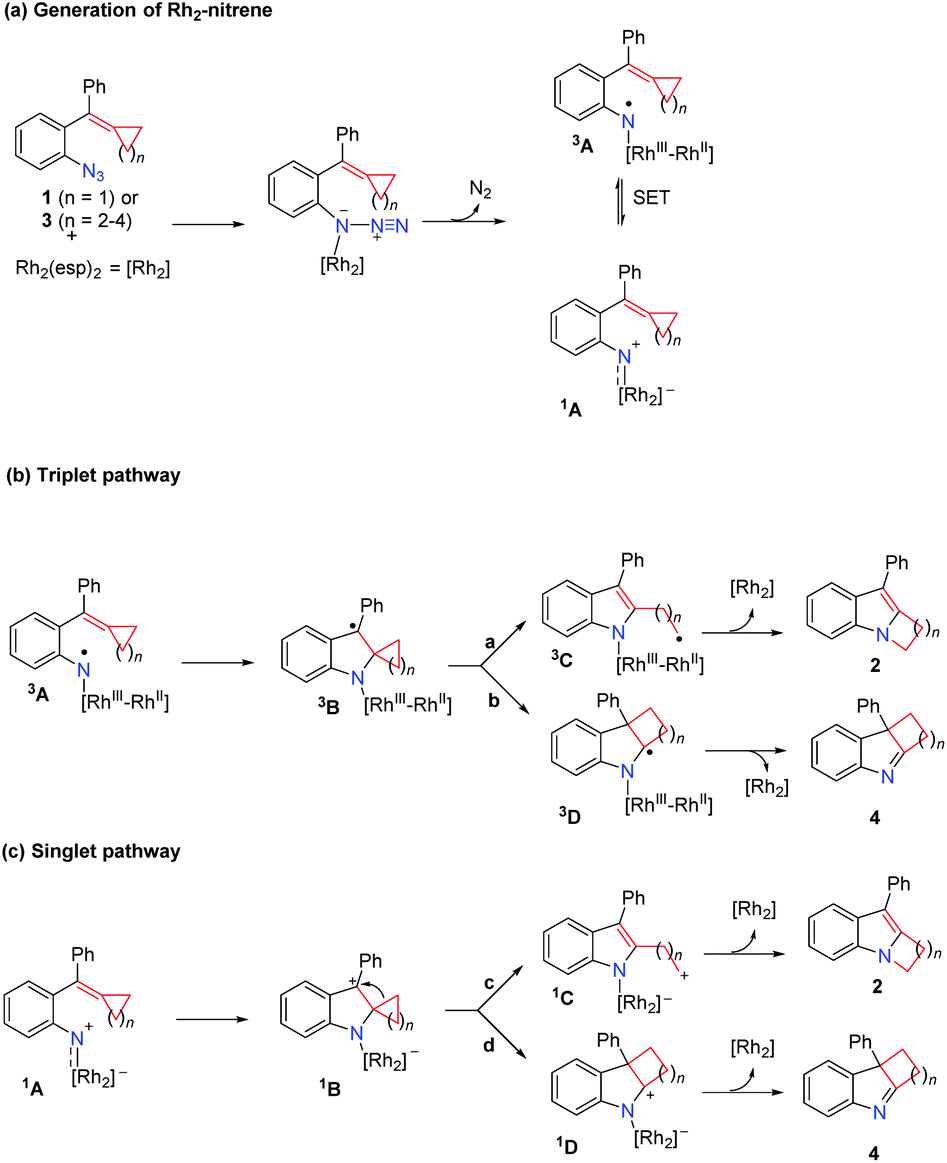
A plausible reaction mechanism is proposed in Scheme 2. Coordination of azide 1 or 3 to the Rh2(esp)2 complex and extrusion of N2 gives the Rh2–nitrene 1A; intramolecular single electron transfer (SET) can take place, resulting in the species 3A which has the nitrogen centered radical's character (Scheme 2(a)). The following reaction can undergo the triplet pathway starting from the species 3A (Scheme 2(b)) or the singlet pathway starting from the species 1A (Scheme 2(c)). For substrate 1 (n = 1), the reaction is proposed to proceed via the triplet pathway (path a) to give rise to product 2 together with the regeneration of the Rh2(esp)2 catalyst. As for substrate 3 (n = 2–4), the reactions are proposed to proceed via the singlet pathway, and do not undergo ring-opening to give intermediate 1C (path c) and the intermediate 1B undergoes a concerted 1,2-alkyl shift via path d to afford the corresponding 3H-indole product 4 and regenerates the Rh2(esp)2 catalyst.
DFT calculations have been frequently used to investigate the mechanisms for Rh-catalyzed reactions and their related reactions.17 Herein, DFT calculations are employed to investigate the detailed mechanism and the influence of ring size on product selectivity. Two parts are presented by the following mechanistic studies. First, employing typical substrates and Rh2(esp)2, the formation of key Rh2–nitrene intermediate and its character are investigated and discussed in detail. Then, we investigated close-shell singlet and open-shell triplet pathways for the Rh2-catalyzed ring expansion of azide tethered MCPs and azide-styrene, and rationalized the product selectivity affected by the ring size of the substrate.18
Generation of Rh2–nitrene intermediate and its character
As for a variety of Rh-catalyzed reactions, the first generation of nitrene intermediate is important, and how the Rh catalyst plays its role in the formation of a nitrene intermediate also attracts a lot of interest. We first examined both Rh-catalyzed and uncatalyzed N2 extrusion to generate a nitrene intermediate (Scheme 3). We focus our initial mechanistic studies using the substrate 1a. As shown in Scheme 3, the Rh-catalyzed pathway is initiated from the formation of the complex IN1. Passing through the transition state TS1, the nitrenoid intermediate IN2 is generated, where 1IN2 and 3IN2 are denoted as singlet and triplet multiplicity, respectively. The computed barrier for N2 extrusion is 22.2 kcal mol−1 for the Rh2(esp)2 catalyzed pathway (Scheme 3(a)), which is consistent with other Rh-catalyzed N2 extrusion processes for nitrene intermediate generation.19 The transition state TS1′ on the pathway in the absence of the Rh2(esp)2 catalyst is located 5.8 kcal mol−1 above transition state TS1 (Scheme 3(b)). It should be noted here that without the bound Rh2(esp)2, only triplet nitrene 3IN2′ is stable. In the Rh-catalyzed process, the formation of a nitrene intermediate is an exothermic process; in contrast, the formation of a nitrene intermediate is an endothermic process for the pathway without the Rh catalyst. The calculation results indicate that the rhodium catalyst plays a critical role to decrease the energy barrier and makes the generation of nitrene intermediate thermodynamically favorable. For other substrates 3 bearing a four-membered ring or five-membered ring, we obtained similar results (for details, see the ESI†). Thus, the rhodium catalyst is an essential factor for the generation of the Rh2–nitrene intermediate.
Once the Rh2–nitrene intermediate IN2 was formed, its character attracted our attention since several experimental and theoretical reports show that some types of Rh2–nitrene intermediates had radical character.2b,e,5,6 In this case, the 3IN2 with a triplet state is more stable than the 1IN2 with a singlet state by 3.5 kcal mol−1, thus, the triplet state is preferred for the Rh2–nitrene IN2. The optimized structure of 3IN2 at the BS1 level is shown in Fig. 1 and the spin densities on nitrogen and rhodium atoms are also shown in Fig. 1. Based on spin densities and MO analysis, two unpaired electrons reside in the N atom and dirhodium centers, respectively, which indicates that the Rh2+/Rh2+ dimer tends to undergo a facile one-electron oxidation when combined with related reagents,20 leading to the Rh2–nitrene 3IN2 having a mixed-valent Rh2+/Rh3+ dimer and a N radical character.
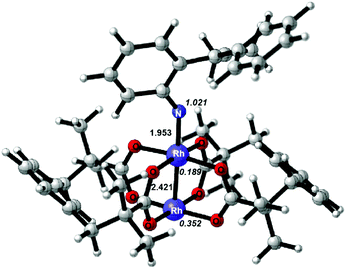 |
| | Fig. 1 Optimized structure at the BS1 level for the Rh2–nitrene 3IN2; key bond lengths (Å) and spin densities on nitrogen and rhodium atoms (in italics) are indicated. | |
Theoretical investigations on the origins of product selectivity
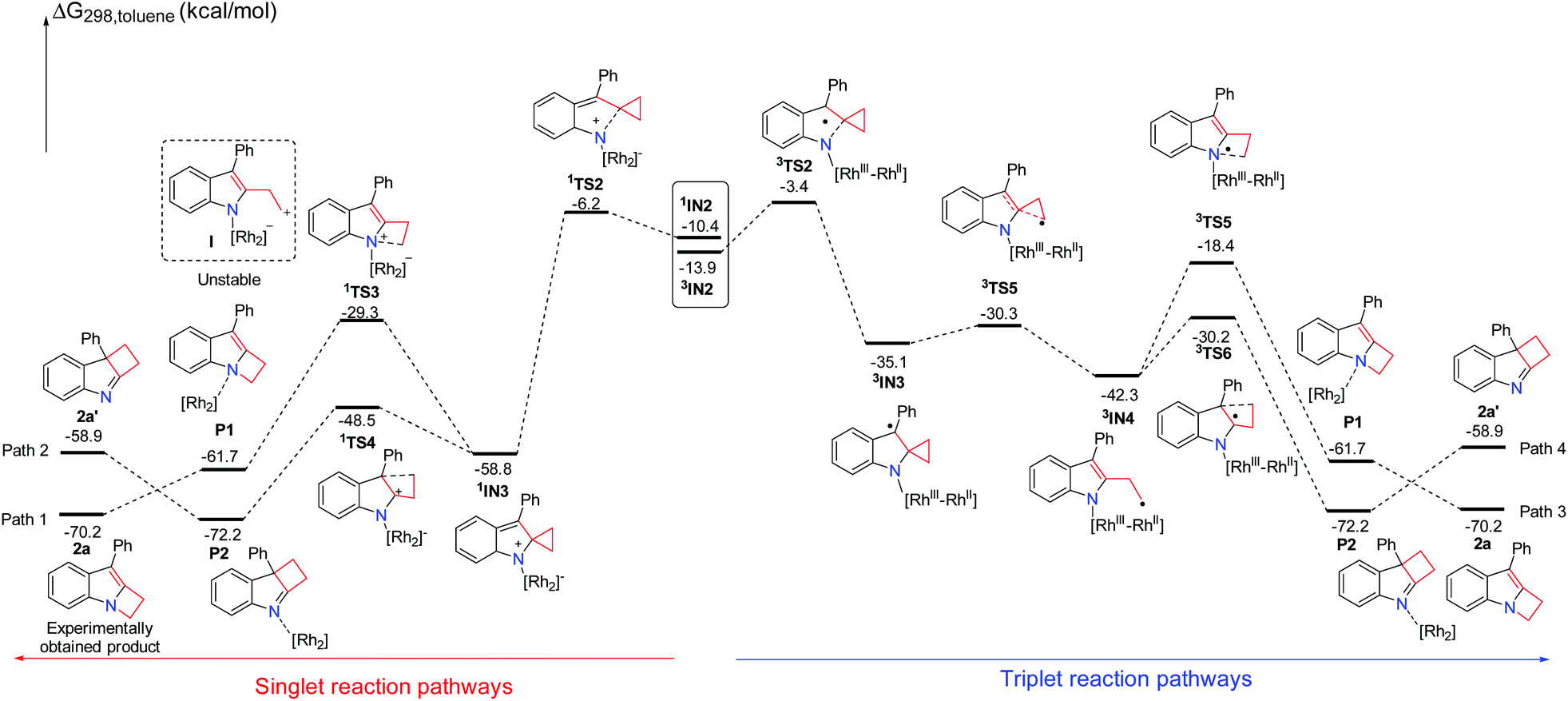
A plausible mechanism for this reaction is suggested in Scheme 2. In order to understand the detailed mechanism and rationalization of product chemoselectivity, we first investigated the possible singlet reaction pathways and triplet pathways using substrate 1a by DFT calculations, and the computed solvation free energy profiles for both singlet and triplet pathways are shown in Scheme 4, respectively. As depicted in Scheme 4, the singlet pathways (paths 1 and 2) are generally favored over the triplet pathways (paths 3 and 4). Although the triplet state 3IN2 is more favored, the ring-closure singlet transition state 1TS2 is 2.8 kcal mol−1 lower than that of the corresponding transition state 3TS2. Passing through the ring-closure singlet transition state 1TS2, the formed intermediate IN3 is also a preferred singlet state, since the singlet state 1IN3 is much more stable than its corresponding triplet state 3IN3 by 23.7 kcal mol−1. Once the intermediate 1IN3 is generated, it can directly undergo a concerted 1,2-alkyl shift either via1TS3 to form a C–N bond or via1TS4 to form a C–C bond, resulting in a product complex P1 or P2. The C–N bond formation transition state 1TS3 is located 19.2 kcal mol−1 above the C–C bond formation transition state 1TS4, which indicates that the C–C bond formation step is kinetically favored. Cleavage of product complex P1 or P2 generates the corresponding product 2a or 2a′. The product 2a is 11.3 kcal mol−1 lower than product 2a′, and it is also an experimentally obtained product. The results presented in Scheme 4 predict that the kinetic controlled product 2a′ should be obtained, which cannot account for the experimental product formation. We have tried to locate the intermediate I shown in Scheme 4, however, it is unstable and always collapses to intermediate 1IN3. This result shows that the intermediate 1IN3 directly undergoes a concerted 1,2-alkyl shift and does not pass the ring-opening step and then cyclization. However, we indeed used TEMPO to trap an intermediate in previous experiments.7 We investigated the ring-opening step on the triplet pathway, and located the stable ring-opening intermediate 3IN4. Starting from 1IN3, a barrier of 28.5 kcal mol−1 is required to generate the intermediate 3IN4, which is slightly lower than a barrier of 29.5 kcal mol−1 for the formation of product 2a. Thus, the generation of the intermediate 3IN4 is a competitive step with the formation of product 2a, and the intermediate 3IN4 could be trapped by TEMPO. From the intermediate 3IN4, the following cyclization triplet pathways are calculated as shown in Scheme 4. The triplet transition states 3TS5 and 3TS6 are located above 1TS3 and 1TS4 by 10.9 kcal mol−1 and 18.3 kcal mol−1, respectively. Therefore, the singlet pathways (paths 1 and 2) are still more favored.
We have demonstrated that the rhodium catalyst is the essential factor for generation of the Rh2–nitrene intermediate. After the Rh2–nitrene intermediate is formed and undergoes ring-closure to form a stable intermediate, the question is whether the rhodium catalyst plays a dominant role in the following steps. We sequentially calculated the reaction pathways without the rhodium catalyst starting from the intermediate 1IN3 (Scheme 5). Releasing the rhodium catalyst, the intermediate IN3-1 is formed. The intermediate IN3-1 without the bound rhodium catalyst can also undergo a 1,2-alkyl shift to yield corresponding products. The C–N bond formation transition state TS3-1 is located 19.2 kcal mol−1 above the C–C bond formation transition state TS4-1, which is similar to the situation for the reaction steps with the rhodium catalyst. A barrier of 32.5 kcal mol−1 for the formation of product 2a is higher than the barrier of 29.5 kcal mol−1 for this same step with the rhodium catalyst by 3.0 kcal mol−1. This result indicates that the rhodium catalyst is not essential for the following ring expansion step once the indole moiety is formed. These results are consistent with our previous experimental observations that the product 2a could be obtained at a higher reaction temperature in the absence of the rhodium catalyst.7 We noticed that the formation of product 2a′ is an endothermic process, which is a reversible step. Thus, the thermodynamic favored product 2a can be obtained at high temperature, which can explain the experimental product formation. Overall, the rhodium catalyst is more crucial for generating the important reactive intermediates, but it does not play a dominant role in controlling the final product selectivity.
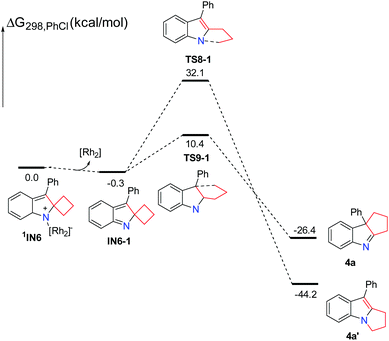
Next, we focus our attention on this Rh-catalyzed ring expansion reaction with respect to the substrates having a larger ring. As for the substrate 3a having a four-membered ring, the possible singlet reaction pathways and triplet pathways are investigated theoretically, and the computed solvation free energy profiles for both singlet and triplet pathways are shown in Scheme 6. Generally speaking, the singlet pathways are favored over the triplet pathways, which is similar to the reaction employing substrate 1a having a three-membered ring. In this case, the Rh2–nitrene intermediate 3IN5 with the triplet state is also more stable. The free energy difference (ΔGst = Gsinglet(1IN5) − Gtriplet(3IN5)) between the singlet and triplet is 2.1 kcal mol−1 which is smaller than that of their analogues having a three-membered ring (ΔGst = Gsinglet(1IN2) − Gtriplet(3IN2) = 3.5 kcal mol−1) by 1.4 kcal mol−1. In a similar manner, the ring-closure singlet transition state 1TS7 is 4.9 kcal mol−1 lower than the corresponding transition state 3TS7. Passing through the ring-closure singlet transition state 1TS7, the generated intermediate IN6 is also a preferred singlet state, since the singlet state 1IN6 is much more stable than its corresponding triplet state 3IN6 by 21.5 kcal mol−1. After the intermediate 1IN6 is yielded, it can directly undergo a concerted 1,2-alkyl shift either via1TS8 to form a C–N bond or via1TS9 to form a C–C bond, resulting in a product complex P3 or P4, respectively. The C–N bond formation transition state 1TS8 is located 22.7 kcal mol−1 above the C–C bond formation transition state 1TS9, which is higher than the energy gap (19.2 kcal mol−1) between 1TS3 and 1TS4, indicating that the barrier for C–N bond formation is more difficult to overcome with respect to the substrate 3a having a four-membered ring. Cleavage of the product complex P3 or P4 generates the corresponding product 4a or 4a′. Although the product 4a is 18.1 kcal mol−1 higher than product 4a′, the product 4a is the experimentally obtained product. Probably, the energy barrier of 33.6 kcal mol−1 for the C–N bond formation step is relatively high due to the steric hindrance between the increased carbon chain and the Rh2(esp)2 catalyst, which is difficult to overcome under the standard reaction conditions. This probably accounts for why only product 4a is obtained experimentally. Thus, the formation of product 4a is a kinetically controlled process. We also calculated the four-membered ring-opening step on the triplet pathway, and located the stable ring-opening intermediate 3IN7.21 However, the barrier of 36.8 kcal mol−1 to generate the intermediate 3IN7 is much higher than the barrier of 10.9 kcal mol−1 for producing 4a, and it is also higher than a barrier of 33.6 kcal mol−1 for the formation of product 4a′. Based on the calculation results, the generation of the radical intermediate 3IN7 requires to overcome a barrier of 36.8 kcal mol−1 starting from the intermediate 1IN6, which is not a competitive step with the formation of product 4a. This result also agrees with the experimental findings that the radical intermediate 3IN7 could not be trapped by TEMPO. From the radical intermediate 3IN7, the following cyclization triplet pathways are also calculated as shown in Scheme 6. The triplet transition states 3TS11 and 3TS12 are located above 1TS8 and 1TS9 by 10.4 kcal mol−1 and 15.6 kcal mol−1, respectively. Therefore, the singlet pathways are still more favored with respect to the formation of product 4a. For comparison, we sequentially calculated the reaction pathways without the rhodium catalyst starting from the intermediate 1IN6 (Scheme 7). Releasing the rhodium catalyst, the intermediate IN6-1 is formed. The intermediate IN6-1 without the bound rhodium catalyst can also undergo a 1,2-alkyl shift to yield the corresponding products. The C–N bond formation transition state TS8-1 is located 21.7 kcal mol−1 above the C–C bond formation transition state TS9-1, which is similar to the situation for the reaction step with the rhodium catalyst. A barrier of 10.7 kcal mol−1 for the formation of the experimentally obtained product 4a is slightly lower than the barrier of 10.9 kcal mol−1 for this same step with the rhodium catalyst. This result also indicates that the rhodium catalyst is not essential for the ring expansion step once the indole moiety is formed. In this case, the formation of product 4a is highly exothermic, which cannot be reversible. Thus, the kinetic controlled product 4a should be obtained, which agrees with the experimental findings.
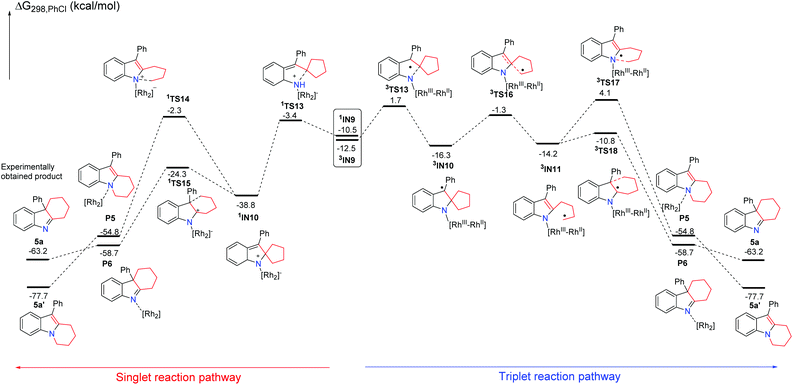
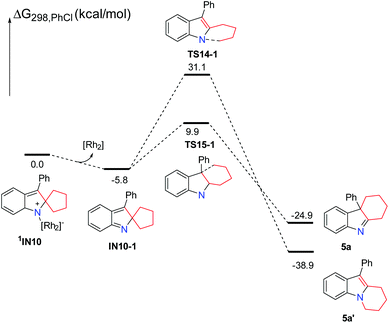
Subsequently, using the substrate 3b having a five-membered ring, the possible singlet reaction pathways and triplet pathways are also investigated theoretically, and the computed solvation free energy profiles for both singlet and triplet pathways are shown in Scheme 8. The singlet pathways are favored over the triplet pathways again, which is similar to the reaction employing the other substrates. For the substrate 3b, the corresponding Rh2–nitrene intermediate 3IN9 with the triplet state is also more stable than its singlet state. The free energy difference (ΔGst = Gsinglet(1IN9) − Gtriplet(3IN9)) between the singlet and triplet is 2.0 kcal mol−1 which is slightly smaller than that of their analogues having a four-membered ring (ΔGst = Gsinglet(1IN5) − Gtriplet(3IN5) = 2.1 kcal mol−1) by 0.1 kcal mol−1. The ring-closure singlet transition state 1TS13 is 5.1 kcal mol−1 lower than the corresponding transition state 3TS13. Passing through the ring-closure singlet transition state 1TS13, the generated intermediate IN10 is also a preferred singlet state, since the singlet state 1IN10 is much more stable than its corresponding triplet state 3IN10 by 22.5 kcal mol−1. After the intermediate 1IN10 is yielded, it can directly undergo a concerted 1,2-alkyl shift either via1TS14 to form a C–N bond or via1TS15 to form a C–C bond, resulting in a product complex P5 or P6, respectively. The C–N bond formation transition state 1TS14 is located 22.0 kcal mol−1 above the C–C bond formation transition state 1TS15, which is almost same as the energy gap (22.7 kcal mol−1) between 1TS8 and 1TS9 and higher than the energy gap (19.2 kcal mol−1) between 1TS3 and 1TS4, indicating that the barrier for the C–N bond formation is also difficult to overcome with respect to the substrate 3b having a five-membered ring. Cleavage of product complex P5 or P6 generates the corresponding product 5a or 5a′. Although the product 5a is 14.5 kcal mol−1 higher than product 5a′, the product 5a is the experimentally obtained product. It is notable that the energy barrier of 36.5 kcal mol−1 for the C–N bond formation step is even higher than that (33.6 kcal mol−1) of its analogue with the four-membered ring. The calculated results reveal that the longer carbon chain makes the C–N bond formation step more difficult to occur, probably due to the steric hindrance between the increased carbon chain and the Rh2(esp)2 catalyst. These results probably explain why only product 5a was obtained and the thermodynamically stable product 5a′ was not obtained in previous experiments. Thus, the formation of product 5a is also a kinetically controlled process. As for the substrate 3b having a five-membered ring, the stable ring-opening intermediate 3IN11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 22 with the triplet state can be located. However, the barrier of 37.5 kcal mol−1 to generate the intermediate 3IN11 is much higher than the barrier of 14.5 kcal mol−1 for producing 5a, and it is slightly higher than a barrier of 36.5 kcal mol−1 for the formation of product 5a′. Based on the calculation results, the generation of the radical intermediate 3IN11 requires to overcome a barrier of 37.5 kcal mol−1 starting from intermediate 1IN10, which is not a competitive step with the formation of product 5a. This result can also explain the experimental finding that the radical intermediate 3IN11 could not be trapped by TEMPO. From the radical intermediate 3IN11, the following cyclization triplet pathways are also calculated as shown in Scheme 8. The triplet transition states 3TS17 and 3TS18 are located above 1TS14 and 1TS15 by 6.4 kcal mol−1 and 13.5 kcal mol−1, respectively. Therefore, the singlet pathways are more favored with respect to yielding product 5a again. In a similar manner, we calculated the reaction pathways without the rhodium catalyst starting from the intermediate 1IN10 (Scheme 9). Upon dissociation of the rhodium catalyst, the intermediate IN10-1 is formed. The intermediate IN10-1 without the bound rhodium catalyst can also undergo a 1,2-alkyl shift to yield the corresponding products. The C–N bond formation transition state TS14-1 is located 21.2 kcal mol−1 above the C–C bond formation transition state TS15-1, which is similar to the situation for the reaction step with the rhodium catalyst. A barrier of 15.7 kcal mol−1 for the formation of experimentally obtained product 5a is slightly higher than the barrier of 14.5 kcal mol−1 for the same step with the rhodium catalyst. This result again indicates that the rhodium catalyst is not essential for the ring expansion step once the indole moiety is formed. In this case, the formation of product 5a is also highly exothermic, which cannot be reversible. Thus, the kinetic controlled product 5a should be accessed, which is in line with the experimental findings.
22 with the triplet state can be located. However, the barrier of 37.5 kcal mol−1 to generate the intermediate 3IN11 is much higher than the barrier of 14.5 kcal mol−1 for producing 5a, and it is slightly higher than a barrier of 36.5 kcal mol−1 for the formation of product 5a′. Based on the calculation results, the generation of the radical intermediate 3IN11 requires to overcome a barrier of 37.5 kcal mol−1 starting from intermediate 1IN10, which is not a competitive step with the formation of product 5a. This result can also explain the experimental finding that the radical intermediate 3IN11 could not be trapped by TEMPO. From the radical intermediate 3IN11, the following cyclization triplet pathways are also calculated as shown in Scheme 8. The triplet transition states 3TS17 and 3TS18 are located above 1TS14 and 1TS15 by 6.4 kcal mol−1 and 13.5 kcal mol−1, respectively. Therefore, the singlet pathways are more favored with respect to yielding product 5a again. In a similar manner, we calculated the reaction pathways without the rhodium catalyst starting from the intermediate 1IN10 (Scheme 9). Upon dissociation of the rhodium catalyst, the intermediate IN10-1 is formed. The intermediate IN10-1 without the bound rhodium catalyst can also undergo a 1,2-alkyl shift to yield the corresponding products. The C–N bond formation transition state TS14-1 is located 21.2 kcal mol−1 above the C–C bond formation transition state TS15-1, which is similar to the situation for the reaction step with the rhodium catalyst. A barrier of 15.7 kcal mol−1 for the formation of experimentally obtained product 5a is slightly higher than the barrier of 14.5 kcal mol−1 for the same step with the rhodium catalyst. This result again indicates that the rhodium catalyst is not essential for the ring expansion step once the indole moiety is formed. In this case, the formation of product 5a is also highly exothermic, which cannot be reversible. Thus, the kinetic controlled product 5a should be accessed, which is in line with the experimental findings.
Summary and conclusions
Based on systematic theoretical studies, we have disclosed the detailed reaction mechanism for the dirhodium-catalyzed ring expansion reactions of azide tethered MCPs and azide-styrenes. The mechanistic scheme we favor on the basis of the results described above can be generally summarized as the following steps: (1) complexation of the azide substrate by the dirhodium catalyst, (2) extrusion of N2 to form a Rh2–nitrene intermediate which has radical character, (3) electrocyclic ring closure to form the indoline ring, (4) concerted 1,2-alkyl shift to form ring expansion products and (5) dissociation of the catalyst. The dirhodium catalyst plays an important role in step 2 to generate the critical Rh2–nitrene intermediate, and, it is also clearly to catalyze the following steps, however, it is not as important as that in step 2. For substrate 1a involving a three-membered ring, the experimentally obtained C–N bond formation product 2a is a thermodynamically favored product; moreover, the three-member-ring-opening step on the triplet pathway is a competitive process with the formation of the main product, thus a radical reaction intermediate can be trapped by TEMPO. In contrast, for substrates 3a and 3b having a larger ring, the reaction barriers for the formation of favored thermodynamic products are pretty high, thus the kinetically favored C–C bond formation products are obtained. In these cases, the ring-opening step on the triplet pathway is not a competitive process with the formation of the main product; therefore, the corresponding radical reaction intermediates cannot be trapped by TEMPO. Our findings are in line with the recent report by Driver and Tantillo,19b which also demonstrates that the rhodium catalyst is crucial for generating the initial reactive intermediate, but not essential for controlling product selectivities in the Rh2(II)-catalyzed reaction. The theoretical studies presented here deepened our understanding of the reaction mechanism of dirhodium-catalyzed ring expansion reactions of azide tethered MCPs and azide-styrenes, which are helpful for understanding other related dirhodium-catalyzed reactions involving nitrene or carbene intermediates.
Acknowledgements
We thank the financial support from the National Basic Research Program of China (973)-2015CB856603, and the National Natural Science Foundation of China (21372250, 20472096, 21372241, 21361140350, 20672127, 21421091, 21121062, 21302203, 20732008 and 21572052). We are grateful for the facility support from Shanghai Supercomputer Center.
Notes and references
- For recent reviews on rhodium-catalyzed transformations, see:
(a) H. M. L. Davies and S. J. Hedley, Chem. Soc. Rev., 2007, 36, 1109–1119 RSC;
(b) H. M. L. Davies and Y. Lian, Acc. Chem. Res., 2012, 45, 923–935 CrossRef CAS PubMed;
(c) A. Padwa, Chem. Soc. Rev., 2009, 38, 3072–3081 RSC;
(d) Y. Jiang, R. Sun, X.-Y. Tang and M. Shi, Chem. – Eur. J., 2016, 22, 17910–17924 CrossRef CAS PubMed.
- For selected reports on Rh-catalyzed C–H bond-activation/C–N bond-forming reaction, see:
(a) C. G. Espino, K. W. Fiori, M. Kim and J. Du Bois, J. Am. Chem. Soc., 2004, 126, 15378–15379 CrossRef CAS PubMed;
(b) K. W. Fiori and J. Du Bois, J. Am. Chem. Soc., 2007, 129, 562–568 CrossRef CAS PubMed;
(c) J. J. Fleming and J. Du Bois, J. Am. Chem. Soc., 2006, 128, 3926–3927 CrossRef CAS PubMed;
(d) D. N. Zalatan and J. Du Bois, J. Am. Chem. Soc., 2008, 130, 9220–9221 CrossRef CAS PubMed;
(e) D. N. Zalatan and J. Du Bois, J. Am. Chem. Soc., 2009, 131, 7558–7559 CrossRef CAS PubMed;
(f) D. E. Olson and J. Du Bois, J. Am. Chem. Soc., 2008, 130, 11248–11249 CrossRef CAS PubMed.
- For selected reports on Rh-catalyzed cascade cyclopropanation/skeletal rearrangement reactions, see:
(a) C.-E. Kim, S. Park, D. Eom, B. Seo and P. H. Lee, Org. Lett., 2014, 16, 1900–1903 CrossRef CAS PubMed;
(b) S. Rajasekar and P. Anbarasan, J. Org. Chem., 2014, 79, 8428–8434 CrossRef CAS PubMed;
(c) R.-Q. Ran, J. He, S.-D. Xiu, K.-B. Wang and C.-Y. Li, Org. Lett., 2014, 16, 3704–3707 CrossRef CAS PubMed;
(d) S. W. Kwok, L. Zhang, N. P. Grimster and V. V. Fokin, Angew. Chem., Int. Ed., 2014, 53, 3452–3456 CrossRef CAS PubMed;
(e) E. E. Schultz, V. N. G. Lindsay and R. Sarpong, Angew. Chem., Int. Ed., 2014, 53, 9904–9908 CrossRef CAS PubMed;
(f) Y. Tian, Y. Wang, H. Shang, X. Xu and Y. Tang, Org. Biomol. Chem., 2015, 13, 612–619 RSC;
(g) Y.-S. Zhang, X.-Y. Tang and M. Shi, Org. Chem. Front., 2015, 2, 1516–1520 RSC;
(h) T. Miura, T. Nakamuro, C.-J. Liang and M. Murakami, J. Am. Chem. Soc., 2014, 136, 15905–15908 CrossRef CAS PubMed.
- For selected reports on Rh-catalyzed ylide formations, see:
(a) H. J. Jeon, D. J. Jung, J. H. Kim, Y. Kim, J. Bouffard and S. Lee, J. Org. Chem., 2014, 79, 9865–9871 CrossRef CAS PubMed;
(b) X. Lei, L. Li, Y.-P. He and Y. Tang, Org. Lett., 2015, 17, 5224–5227 CrossRef CAS PubMed;
(c) D. J. Lee, J. Shin and E. J. Yoo, Chem. Commun., 2014, 50, 6620–6622 RSC;
(d) A. Boyer, Org. Lett., 2014, 16, 1660–1663 CrossRef CAS PubMed;
(e) Y.-S. Zhang, X.-Y. Tang and M. Shi, Chem. Commun., 2014, 50, 15971–15974 RSC;
(f) J. He, Y. Shi, W. Cheng, Z. Man, D. Yang and C.-Y. Li, Angew. Chem., Int. Ed., 2016, 55, 4557–4561 CrossRef CAS PubMed.
-
(a) K. P. Kornecki and J. F. Berry, Chem. – Eur. J., 2011, 17, 5827–5832 CrossRef CAS PubMed;
(b) K. P. Kornecki and J. F. Berry, Eur. J. Inorg. Chem., 2012, 562–568 CrossRef CAS;
(c) K. P. Kornecki and J. F. Berry, Chem. Commun., 2012, 48, 12097–12099 RSC.
- X. Zhang, Z. Ke, N. J. DeYonker, H. Xu, Z.-F. Li, X. Xu, X. Zhang, C.-Y. Su, D. L. Phillips and C. Zhao, J. Org. Chem., 2013, 78, 12460–12468 CrossRef CAS PubMed.
- K. Chen, Z.-Z. Zhu, J.-X. Liu, X.-Y. Tang, Y. Wei and M. Shi, Chem. Commun., 2016, 52, 350–353 RSC.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
-
(a) A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS;
(b) J. P. Perdew, K. Burke and Y. Wang, Phys. Rev. B: Condens. Matter, 1996, 54, 16533–16539 CrossRef CAS.
-
(a) U. Steinbrenner, A. Bergner, M. Dolg and H. Stoll, Mol. Phys., 1994, 82, 3–11 CrossRef CAS;
(b) A. Henglein, J. Phys. Chem., 1993, 97, 5457–5471 CrossRef CAS;
(c) M. Kaupp, P. v. R. Schleyer, H. Stoll and H. Preuss, J. Chem. Phys., 1991, 94, 1360–1366 CrossRef CAS.
- W. H. Lam, K. C. Lam, Z. Lin, S. Shimada, R. N. Perutz and T. B. Marder, Dalton Trans., 2004, 1556–1562 RSC.
-
(a) X. Lin, C. Zhao, C.-M. Che, Z. Ke and D. L. Phillips, Chem. – Asian J., 2007, 2, 1101–1108 CrossRef CAS PubMed;
(b) X. Lin, Y. Xi and J. Sun, Comput. Theor. Chem., 2012, 999, 74–82 CrossRef CAS;
(c) X. Lin, J. Sun, Y. Xi and B. Pang, Comput. Theor. Chem., 2011, 963, 284–289 CrossRef CAS;
(d) R. Lorpitthaya, Z.-Z. Xie, K. B. Sophy, J.-L. Kuo and X.-W. Liu, Chem. – Eur. J., 2010, 16, 588–594 CrossRef CAS PubMed;
(e) R. Lorpitthaya, Z.-Z. Xie, J.-L. Kuo and X.-W. Liu, Chem. – Eur. J., 2008, 14, 1561–1570 CrossRef CAS PubMed.
- The energy profile for the reaction of 1a was also investigated at the B3PW91-D3/BS1//BPW91/BS1 level, including dispersion corrections. For details, see the ESI.†.
-
(a) R. Seeger and J. A. Pople, J. Chem. Phys., 1977, 66, 3045–3050 CrossRef CAS;
(b) R. Bauernschmitt and R. Ahlrichs, J. Chem. Phys., 1996, 104, 9047–9052 CrossRef CAS.
-
(a) K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS;
(b) H. P. Hratchian and H. B. Schlegel, J. Chem. Phys., 2004, 120, 9918–9924 CrossRef CAS PubMed;
(c) H. P. Hratchian and H. B. Schlegel, J. Chem. Theory Comput., 2005, 1, 61–69 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
-
(a) X. Hong, B. M. Trost and K. N. Houk, J. Am. Chem. Soc., 2013, 135, 6588–6600 CrossRef CAS PubMed;
(b) X. Xu, P. Liu, A. Lesser, L. E. Sirois, P. A. Wender and K. N. Houk, J. Am. Chem. Soc., 2012, 134, 11012–11025 CrossRef CAS PubMed;
(c) T. J. L. Mustard, P. A. Wender and P. H.-Y. Cheong, ACS Catal., 2015, 5, 1758–1763 CrossRef CAS PubMed;
(d) P. Liu, L. E. Sirois, P. H.-Y. Cheong, Z.-X. Yu, I. V. Hartung, H. Rieck, P. A. Wender and K. N. Houk, J. Am. Chem. Soc., 2010, 132, 10127–10135 CrossRef CAS PubMed;
(e) T. Sperger, I. A. Sanhueza, I. Kalvet and F. Schoenebeck, Chem. Rev., 2015, 115, 9532–9586 CrossRef CAS PubMed;
(f) Y. Park, S. Ahn, D. Kang and M.-H. Baik, Acc. Chem. Res., 2016, 49, 1263–1270 CrossRef CAS PubMed;
(g) R. N. Straker, Q. Peng, A. Mekareeya, R. S. Paton and E. A. Anderson, Nat. Commun., 2016, 7, 10109 CrossRef CAS PubMed.
- Attempts to investigate the open-shell singlet pathways have also been made, however, the intermediates and transition states on open-shell singlet pathways always collapsed to a close-shell singlet state.
-
(a) Q. Zhang, C. Wu, L. Zhou and J. Li, Organometallics, 2013, 32, 415–426 CrossRef CAS;
(b) J. G. Harrison, O. Gutierrez, N. Jana, T. G. Driver and D. J. Tantillo, J. Am. Chem. Soc., 2016, 138, 487–490 CrossRef CAS PubMed.
- For recent examples of metal-based carbene or nitrene radicals, see:
(a) T. Bach, B. Schlummer and K. Harms, Chem. Commun., 2000, 287–288 RSC;
(b) T. Bach, B. Schlummer and K. Harms, Chem. – Eur. J., 2001, 7, 2581–2594 CrossRef CAS PubMed;
(c) M. P. Doyle, Angew. Chem., Int. Ed., 2009, 48, 850–852 CrossRef CAS PubMed;
(d) H. Lu, W. I. Dzik, X. Xu, L. Wojtas, B. de Bruin and X. P. Zhang, J. Am. Chem. Soc., 2011, 133, 8518–8521 CrossRef CAS PubMed;
(e) V. Lyaskovskyy, A. I. O. Suarez, H. Lu, H. Jiang, X. P. Zhang and B. de Bruin, J. Am. Chem. Soc., 2011, 133, 12264–12273 CrossRef CAS PubMed;
(f) L.-M. Jin, X. Xu, H. Lu, X. Cui, L. Wojtas and X. P. Zhang, Angew. Chem., Int. Ed., 2013, 52, 5309–5313 CrossRef CAS PubMed;
(g) L.-M. Jin, H. Lu, Y. Cui, C. L. Lizardi, T. N. Arzua, L. Wojtas, X. Cui and X. P. Zhang, Chem. Sci., 2014, 5, 2422–2427 RSC;
(h) J. Zhang, J. Jiang, D. Xu, Q. Luo, H. Wang, J. Chen, H. Li, Y. Wang and X. Wan, Angew. Chem., Int. Ed., 2015, 54, 1231–1235 CrossRef CAS PubMed.
- The corresponding intermediate 1IN7 with the singlet state is unstable, and cannot be located.
- The corresponding intermediate 1IN11 with the singlet state is also unstable, and cannot be located.
Footnote |
| † Electronic supplementary information (ESI) available: Electronic energies, enthalpies, free energies, and solvation free energies (in Hartrees) and archive entries including Cartesian coordinates for all of the relevant species depicted in Schemes 3–9. See DOI: 10.1039/c6qo00713a |
|
| This journal is © the Partner Organisations 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Xu-Bo
Hu
b,
Kai
Chen
b and
Min
Shi
ab
*a,
Xu-Bo
Hu
b,
Kai
Chen
b and
Min
Shi
ab