
Water-dispersible hyperbranched conjugated polymer nanoparticles with sulfonate terminal groups for amplified fluorescence sensing of trace TNT in aqueous solution†
Xiaofu
Wu
a,
Hao
Hang
b,
Hua
Li
b,
Yonghong
Chen
b,
Hui
Tong
 *a and
Lixiang
Wang
*a
*a and
Lixiang
Wang
*a
aState Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, P. R. China. E-mail: chemtonghui@ciac.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 18th May 2017
Abstract
Conjugated polymers (CPs) have been recognized as promising fluorescent sensing materials for 2,4,6-trinitrotoluene (TNT) vapor. However, their applications for TNT detection in aqueous solution are restricted because of their low sensitivity (detection limits >90 nM). Here we report highly sensitive detection of TNT by water-dispersible hyperbranched conjugated polymer nanoparticles (HCPN-S) with hydrophobic CP cores and hydrophilic sulfonate terminal groups. Based on efficient TNT encapsulation by numerous hydrophobic cavities inside the hyperbranched CP core, HCPN-S exhibits a quenching constant of 1.21 × 106 M−1, two to three orders of magnitude higher than those of CP analogues, allowing amplified detection of TNT with a detection limit of 0.8 ppb (3.7 nM), which is the best result for CP-based fluorescent sensors for TNT detection in water reported so far. Furthermore, HCPN-S can selectively detect TNT even in the presence of 2,4,6-trinitrophenol and other nitro compounds.
Introduction
2,4,6-Trinitrotoluene (TNT) is not only a well-known explosive, but also considered as an environmental contaminant. The existence of excess TNT in water can severely affect humans and other living organisms, and even lead to serious liver, eye, and neurological damage. The US Environmental Protection Agency (EPA) has set the maximum permissible level of 2 ppb (9.25 nM) for TNT in drinking water,1 whereas TNT concentrations in ground water near some military facilities or unexploded ordnance are even as high as 500 ppb.2 Various sophisticated spectroscopic technologies, such as ion mobility spectrometry,3 surface enhanced Raman spectrometry,4 and gas chromatography coupled with mass spectrometry,5 have been used for TNT detection. However, they usually suffer from several limitations, such as low sensitivity, poor selectivity, and being expensive and complicated, and thus are unsuitable for real-time field detection of TNT. Currently, amplified photoluminescence (PL) quenching of conjugated polymers (CPs) based on photoinduced electron transfer (PET) is considered to be one of the most sensitive explosive detection methods.6 Numerous CP materials have been designed for the sensitive detection of TNT.7–12 However, unlike the extensive fluorescent CP studies on TNT detection in vapor or organic solution phases, only a few CPs, CP-based hybrid materials, and CP nanoparticles have been developed for TNT detection in water,9–12 mainly because the hydrophobic nature of CPs results in poor water solubility and weak luminescence in aqueous medium.13,14 Up to now, most CP-based fluorescent sensors have allowed TNT detection down to a sub-micromolar level in the aqueous phase, which is still much higher than the maximum safety limit (9.25 nM) set by the EPA. Furthermore, selectivity is another critical issue for TNT detection in practical applications. In particular, 2,4,6-trinitrophenol (TNP), another commonly used nitro-explosive, always displayed stronger quenching ability than TNT in solution phases.9,15 Hence, it is still highly desirable to explore novel CP materials for sensitive and selective detection of trace TNT in an aqueous environment.CP nanoparticles (CPNs), primarily prepared by emulsion or reprecipitation of linear CPs,16 have emerged as an important class of fluorescent sensing materials due to their exceptional brightness, excellent photostability and good water-dispersibility, which are also attractive properties for explosive detection. However, reports of CPNs as fluorescent TNT sensors have only achieved limited success in terms of both sensitivity and selectivity.12,14,17 Unlike most CPNs based on linear CPs, we sought to construct hyperbranched conjugated polymer nanoparticles (HCPNs) as fluorescent sensors.18 Due to their highly three-dimensional (3D) branched topological structures and the numerous terminal groups in their periphery,19 hyperbranched polymers have been widely studied as hosts with good guest-encapsulation ability, which have potential applications in drug delivery, bioimaging, catalysis, etc.20 With this in mind, we envisioned exploiting the possibility to capture and sense trace TNT in aqueous solution with our HCPNs based on the encapsulation effect. However, these HCPNs suffer from poor water-dispersibility due to their intrinsic hydrophobic CP structures.
Here, we report the preparation of water-dispersible hyperbranched conjugated polymer nanoparticles (HCPN-S) by attaching hydrophilic sulfonate terminal groups on the hydrophobic hyperbranched conjugated polymer core. As illustrated in Scheme 1, hydrophilic sulfonate terminal groups will stabilize HCPN-S in water, while the hydrophobic cavities inside the hyperbranched core will encapsulate and enrich hydrophobic TNT from the aqueous environment, which may greatly facilitate the interaction between HCPN and TNT. Additionally, the use of negatively charged terminal units will help to prevent acidic TNP from quenching the fluorescence of HCPN-S and lead to selective detection of TNT without significant interference by TNP in water. As a result, HCPN-S shows amplified fluorescence quenching response and superior sensitivity for TNT at nanomolar concentrations in aqueous solution. In particular, highly selective detection of TNT in water is realized even in the presence of TNP and other nitro-explosives.
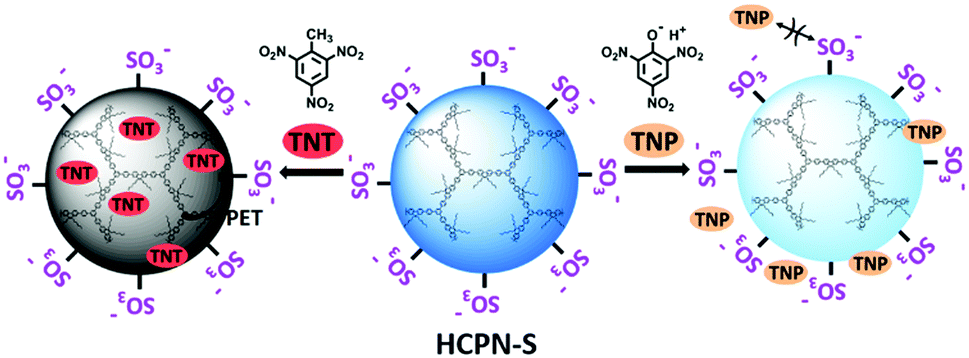 | ||
| Scheme 1 Sensitive and selective detection of TNT in water: the encapsulation of TNT molecules inside the hydrophobic cavities of hyperbranched cores to boost the sensitivity (left) and the electrostatic repulsion of TNP molecules with negatively charged terminal groups to enhance the selectivity (right). | ||
Results and discussion
To obtain water-dispersible HCPN-S, its precursor HCPN-OH with phenol terminal groups was synthesized by the Suzuki cross-coupling polymerization of 9,9-dihexyl-2,7-dibromofluorene and 1,3,5-tris-{4-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolane)phenyl}-benzene in a toluene-in-water miniemulsion, followed by the addition of 4-[4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl]-phenol and 4-bromophenol as end-capped agents (Scheme 2). Subsequently, HCPN-OH was treated with sodium tert-butoxide and 1,4-butanesultone to obtain HCPN-S. The proton signals at 2.3 and 3.9 ppm in the 1H NMR spectrum and the absorption peaks at 1000–1300 cm−1 in the FT-IR spectrum revealed the presence of –O(CH2)4SO3− terminal groups in HCPN-S (Fig. S1 and S2, ESI†), which was further supported by the appearance of the sulfur element in its elemental analysis (Table S1, ESI†). Their water contact angles also confirmed that the hydrophobic HCPN-OH (87.1 ± 2.5°) was converted into the more hydrophilic HCPN-S (41.7 ± 2.6°) (Fig. S3, ESI†). For comparison, we also prepared a linear conjugated polymer nanoparticle (LCPN) aqueous dispersion via miniemulsification of a linear CP analogue (LCP) with sodium dodecyl sulfate (SDS) as the surfactant (Scheme S1, ESI†).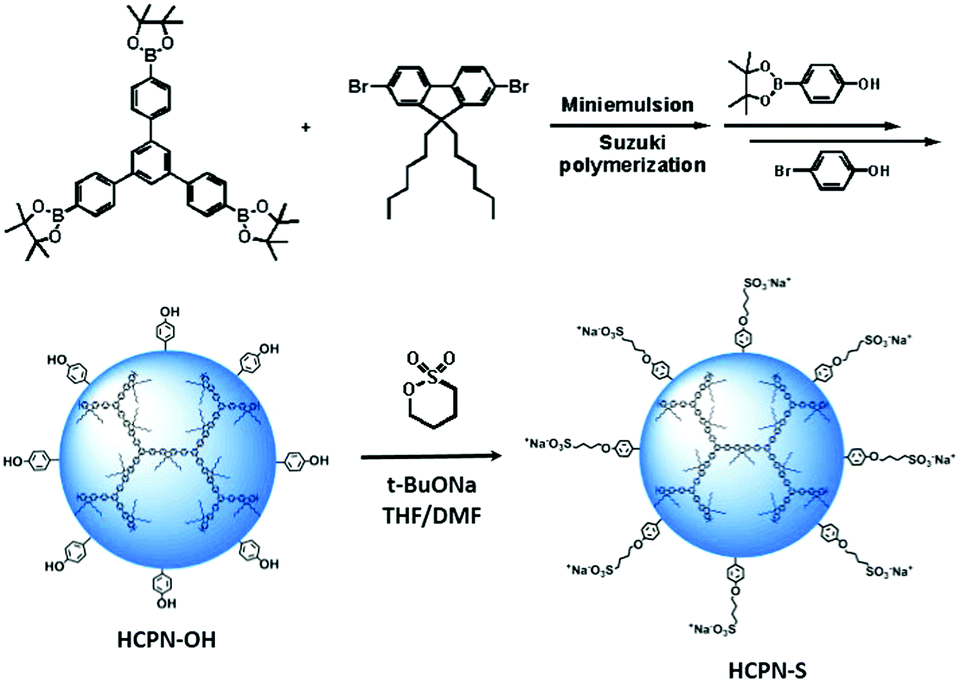 | ||
| Scheme 2 The preparation of HCPN-S and its precursor HCPN-OH. | ||
HCPN-S is dispersible in DMF, DMSO and H2O, but insoluble in other common organic solvents. Homogeneous and transparent dispersions without any insoluble solid could be obtained (Fig. S5, ESI†). Moreover, the dispersion was found to be stable over two weeks in both H2O and DMF. The particle morphology and size of both the HCPN-OH dispersion in THF and HCPN-S dispersion in H2O were assessed by transmission electron microscopy (TEM) (Fig. 1a and b). Both of them maintained the spherical morphology with an average diameter of around 25 nm (Fig. 1c and Fig. S6, ESI†). The LCPN dispersion also had a spherical morphology and a similar average particle size (21 nm) (Fig. S7, ESI†).
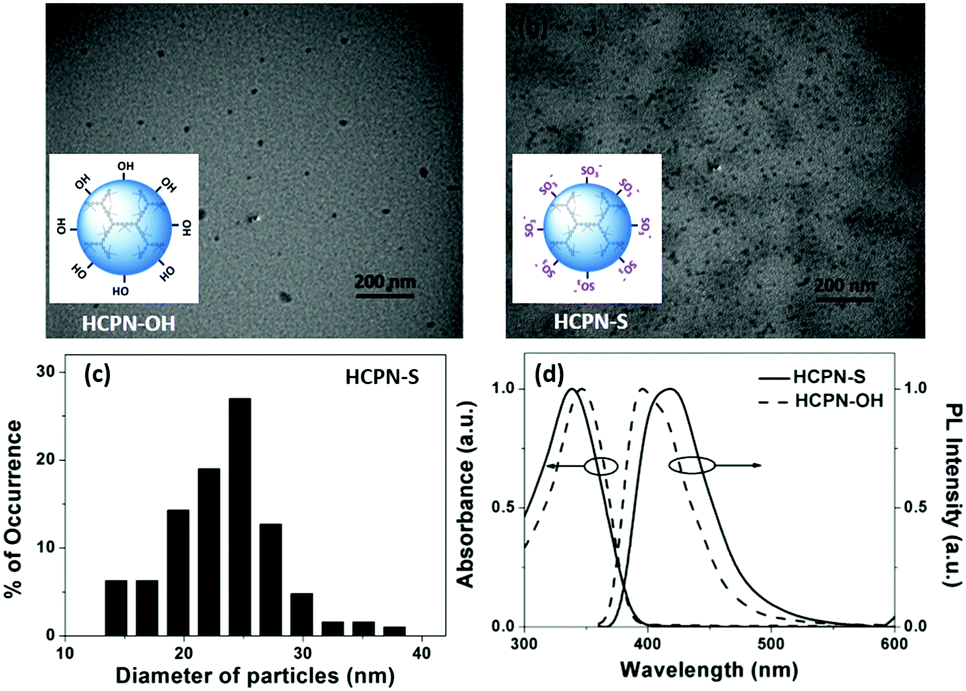 | ||
| Fig. 1 TEM images obtained by drop-casting. (a) THF dispersion of HCPN-OH and (b) water dispersion of HCPN-S onto copper grids. (c) Size distribution histogram using the TEM images of HCPN-S. (d) Absorption and emission spectra of dispersions of HCPN-S and HCPN-OH. | ||
The UV-vis absorption and fluorescence spectra of HCPN-OH and HCPN-S were also examined (Fig. 1d). Their absorption spectra were quite similar to the peaks at 345 nm and 340 nm, respectively. Both polymers exhibited intensive blue emission with quantum yields higher than 20% (Table S2, ESI†). The fluorescence maximum of HCPN-OH was at 396 nm, while HCPN-S exhibited a red-shifted PL peak at 419 nm. The LCPN dispersion also showed blue emission with absorption and emission peaks at 340 nm and 394 nm, respectively (Fig. S8, ESI†).
To demonstrate the sensing behavior of HCPN-S toward TNT in water, fluorometric titration was performed at first. When 30 μM TNT was added into the HCPN-S dispersion, the PL intensity of HCPN-S at 419 nm decreased dramatically by 27-fold (Fig. 2a). A good linear correlation was found over a large concentration range from 1.0 × 10−8 M to 1.5 × 10−5 M (R2 = 0.9989) (Fig. 2b). HCPN-S exhibited a Stern–Volmer quenching constant, Ksv, of 1.21 × 106 M−1 with a limit of detection (LOD) down to 3.7 nM (0.8 ppb) based on the standard method reported.8b To the best of our knowledge, HCPN-S was among the most sensitive CP-based fluorescent sensors for TNT detection in water (Table S3, ESI†). Similar Ksv values were only observed for the fluorescence quenching of conjugated polymer nanoaggregates by trinitrobenzene (TNB) at the micromolar level in THF/water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9).13b Upon comparing with HCPN-S, the quenching efficiency of HCPN-OH towards TNT was much lower (Fig. S9, ESI†). Like most CP-based TNT sensors,8,18 its Ksv was only 2.21 × 103 M−1, 3 orders of magnitude lower than that of HCPN-S. These results indicated that the introduction of hydrophilic terminal groups not only provided water dispersibility but also greatly amplified the sensitivity towards TNT. For comparison, the fluorescence quenching response of the LCPN dispersion and LCP to TNT was also investigated (Fig. 2b and Fig. S10, S11, ESI†). Similarly, the LCPN dispersion in water displayed a higher quenching efficiency (Ksv: 2.12 × 104 M−1) than LCP in THF (Ksv: 2.68 × 103 M−1). The higher sensitivity of HCPN-S and LCPN relative to HCPN-OH and LCP can be attributed to the capture of TNT molecules by a hydrophobic effect.14,21 However, it must be noted that although HCPN-OH and LCP in THF had similar Ksv values, the HCPN-S dispersion showed two orders of magnitude higher Ksv than LCPN (Fig. 2b). The LOD (3.0 μM) of LCPN was also far inferior to that of HCPN-S (3.7 nM). Considering the similar particle size and negatively charged anionic shell, their different sensing ability should originate from their different cores. As illustrated in Fig. 2c, the hyperbranched core of HCPN-S will entrap TNT molecules within the inner cavities, resulting in a large PET interface and an enhanced quenching effect. In contrast, the tangled and compact stacking polymer chains in the core of LCPN will prevent TNT from entering the inside of the particles, leading to a longer PET distance and a poor quenching effect. Therefore, the amplified fluorescence sensing of TNT by HCPN-S is mainly due to its hyperbranched core with numerous hydrophobic cavities that can encapsulate hydrophobic TNT into these void spaces.
:9).13b Upon comparing with HCPN-S, the quenching efficiency of HCPN-OH towards TNT was much lower (Fig. S9, ESI†). Like most CP-based TNT sensors,8,18 its Ksv was only 2.21 × 103 M−1, 3 orders of magnitude lower than that of HCPN-S. These results indicated that the introduction of hydrophilic terminal groups not only provided water dispersibility but also greatly amplified the sensitivity towards TNT. For comparison, the fluorescence quenching response of the LCPN dispersion and LCP to TNT was also investigated (Fig. 2b and Fig. S10, S11, ESI†). Similarly, the LCPN dispersion in water displayed a higher quenching efficiency (Ksv: 2.12 × 104 M−1) than LCP in THF (Ksv: 2.68 × 103 M−1). The higher sensitivity of HCPN-S and LCPN relative to HCPN-OH and LCP can be attributed to the capture of TNT molecules by a hydrophobic effect.14,21 However, it must be noted that although HCPN-OH and LCP in THF had similar Ksv values, the HCPN-S dispersion showed two orders of magnitude higher Ksv than LCPN (Fig. 2b). The LOD (3.0 μM) of LCPN was also far inferior to that of HCPN-S (3.7 nM). Considering the similar particle size and negatively charged anionic shell, their different sensing ability should originate from their different cores. As illustrated in Fig. 2c, the hyperbranched core of HCPN-S will entrap TNT molecules within the inner cavities, resulting in a large PET interface and an enhanced quenching effect. In contrast, the tangled and compact stacking polymer chains in the core of LCPN will prevent TNT from entering the inside of the particles, leading to a longer PET distance and a poor quenching effect. Therefore, the amplified fluorescence sensing of TNT by HCPN-S is mainly due to its hyperbranched core with numerous hydrophobic cavities that can encapsulate hydrophobic TNT into these void spaces.
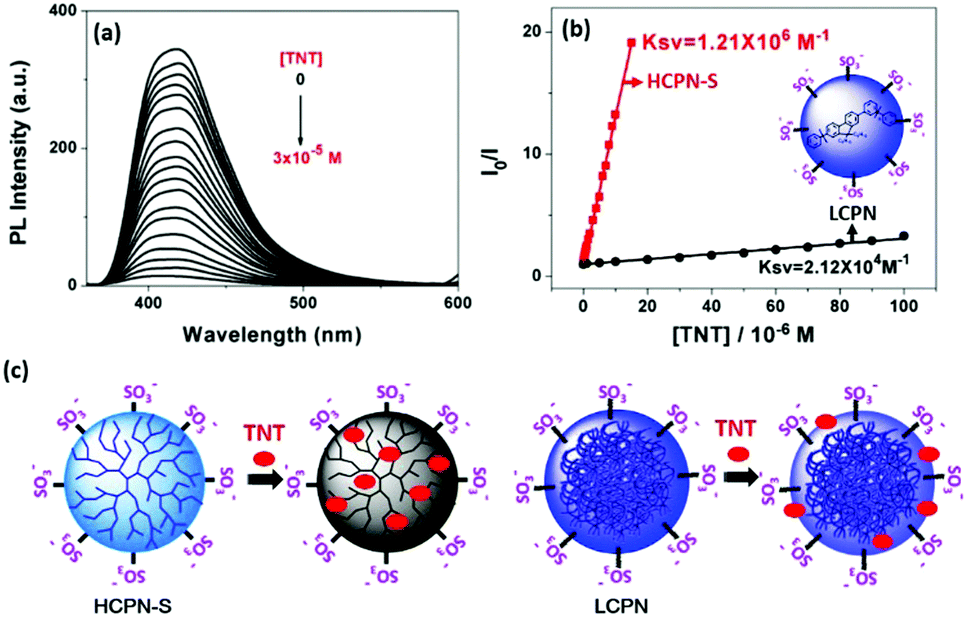 | ||
| Fig. 2 (a) Fluorescence response profiles of the HCPN-S dispersion upon addition of TNT. The excitation wavelength was 340 nm. (b) Stern–Volmer plots for the fluorescence quenching of HCPN-S at 419 nm and LCPN at 394 nm by TNT. (c) The schematic illustration of the proposed mechanism for different sensitivities between HCPN-S and LCPN. | ||
To further reveal the origin of the significantly enhanced sensing properties of HCPN-S, Coumarin-6 was chosen as a hydrophobic dye to demonstrate its guest-encapsulation ability. Coumarin-6, a bright green fluorescent hydrophobic dye in organic solvents, is almost insoluble and nonemissive in water. Upon addition of HCPN-S, the mixture showed the typical Coumarin-6 green emission at 500 nm upon excitation at 458 nm (the absorption peak of Coumarin-6), indicating that HCPN-S could effectively encapsulate Coumarin-6 and provide a hydrophobic microenvironment for it. Moreover, upon excitation at 340 nm (the absorption peak of HCPN-S), the emission of HCPN-S disappeared completely, accompanied by the dramatically enhanced emission of Coumarin-6, which was 12-fold stronger than that by exciting Coumarin-6 at 458 nm directly, revealing an efficient energy transfer process from HCPN-S to Coumarin-6 (Fig. S12, ESI†).22 The energy transfer efficiency was estimated to be 95%. Under a UV lamp, it was clearly seen by the naked eye that the nonemissive aqueous dispersion of Coumarin-6 and the blue emission of HCPN-S turned into the brightly green emission of Coumarin-6 in their mixture (Fig. 3a inset). Although the addition of LCPN could also provoke the increased PL of Coumarin-6 upon excitation at 340 nm (the absorption peak of the LCPN), the blue LCPN emission was still strong (Fig. 3b and Fig. S13, ESI†), indicating the incomplete energy transfer from LCPN to Coumarin-6. The energy transfer efficiency (47%) is only half that of HCPN-S, further proving the more efficient encapsulation of the hydrophobic guest by HCPN-S than LCPN.
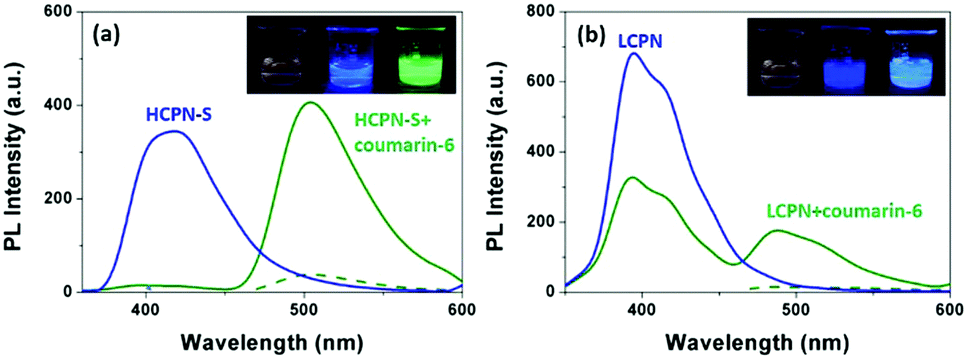 | ||
| Fig. 3 (a) Fluorescence spectra of HCPN-S (blue solid line) excited at 340 nm, and a Coumarin-6/HCPN-S mixture excited at 458 nm (green dot line) and 340 nm (green solid line). Inset: Images of Coumarin-6 (left), HCPN-S (middle) and the Coumarin-6/HCPN-S mixture (right). (b) Fluorescence spectra of LCPN (blue solid line) excited at 340 nm, and a Coumarin-6/LCPN mixture excited at 458 nm (green dot line) and 340 nm (green solid line). Inset: Images of Coumarin-6 (left), LCPN (middle) and the Coumarin-6/LCPN mixture (right). | ||
The response of the HCPN-S to TNT is not only sensitive but also selective. When different analytes, including TNT, TNP, 2,4-dinitrotoluene (DNT), nitrobenzene (NB), nitromethane (NM), 1,3,5-trinitro-1,3,5-triazinane (RDX), cyclotetramethylene tetranitramine (HMX), toluene (TOL), chlorobenzene (CB) and phenol (PhOH), were added into the HCPN-S aqueous dispersion, only TNT significantly attenuated its emission (Fig. 4a). Just 10 μM TNT could induce 12.7-fold PL quenching of HCPN-S, while TNP only induced about 1.7-fold PL quenching. Furthermore, the Ksv value for TNP is 8.39 × 104 M−1, which was more than 14-fold lower than that of TNT, indicating the good selectivity of HCPN-S (Fig. 4b). In the aqueous solution, acidic TNP can easily disassociate to form a negatively charged anion, which kept it away from HCPN-S with negatively charged sulfonate terminal groups because of an electrostatic repulsion effect, leading to a limited PET process and a lower quenching efficiency. Unlike HCPN-S, both LCPN and HCPN-OH were quenched by TNP more efficiently (Fig. S17 and S18, ESI†), as for most CP-based fluorescent sensors.8d,e,9 For LCPN, its Ksv value for TNP (7.13 × 104 M−1) was close to the result of HCPN-S. However, it was still higher than that of TNT (2.12 × 104 M−1), due to the poor TNT encapsulation of LCPN. Therefore, the hydrophilic sulfonate terminal groups and the hydrophobic hyperbranched core of HCPN-S cooperated to enhance not only the sensitivity but also the selectivity for TNT detection.
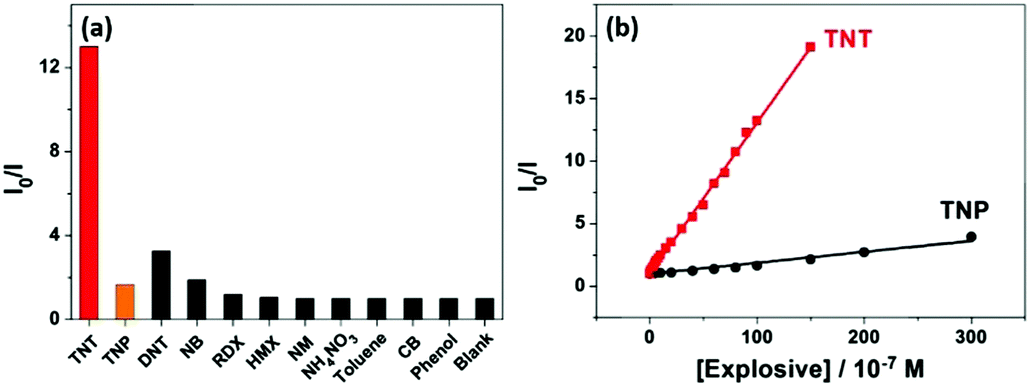 | ||
| Fig. 4 (a) Fluorescence response of HCPN-S upon addition of various analytes (concentrations: 10 μM for TNT, TNP, DNT and NB; 100 μM for other analytes). (b) Stern–Volmer plots for the fluorescence quenching of HCPN-S at 419 nm by TNT and TNP. | ||
Conclusions
In conclusion, a water-dispersible hyperbranched conjugated polymer nanoparticle (HCPN-S) dispersion was successfully prepared by Suzuki-coupling polymerization in an oil-in-water miniemulsion followed by post-functionalization to introduce hydrophilic sulfonate terminal units on the hydrophobic hyperbranched conjugated polymer core. Based on the combination of hydrophobic encapsulation and electrostatic repulsion interaction, highly sensitive and selective sensing of TNT can be achieved. HCPN-S was capable of sensing TNT in water with a detection limit at the nanomolar level (3.7 nM, 0.8 ppb), which was three orders of magnitude lower than that of the conjugated polymer nanoparticle based on a linear analogue. The quenching constant reached 1.21 × 106 M−1, which was among the best results for TNT sensing in water by fluorescent sensors based on conjugated polymers or conjugated polymer nanoparticles. Specifically, the selective recognition for TNT even in the presence of 2,4,6-trinitrophenol and other nitro compounds in water was also realized. Considering their structure versatility and design flexibility, we anticipate that our effort will benefit future research on developing fluorescent sensors based on hyperbranched conjugated polymer nanoparticles.Experimental
Materials
All the chemicals and reagents were used as received from commercial sources without further purification. Solvents for chemical synthesis were purified according to standard procedures. 1,3,5-Tris-{4-(4,4,5,5-tetramethyl-[1,3,2] dioxaborolane)phenyl}-benzene18d and the linear conjugated polymer, LCP,18a was synthesized as previously described.Measurement and characterization
1H and 13C NMR spectra were recorded on Bruker AV-300 with CDCl3 and DMF-d7 as solvents. IR spectra were obtained using a FT-IR Bruker Vertex 70 spectrometer at a nominal resolution of 2 cm−1. Elemental analysis was performed using a Bio-Rad elemental analysis system. Thermal gravimetric analysis (TGA) was performed at a heating rate of 10 °C min−1 under a flow of N2 with a PerkinElmer-TGA7 system. Transmission electron microscopy (TEM) imaging was performed using a Philips-FEI Tecnai F20 microscope (Philips, The Netherlands) at an accelerating voltage of 200 kV. The samples for TEM were prepared by drop-casting nanoparticle dispersions (0.01 mg mL−1) onto copper grids. UV-visible absorption measurements were carried out on a Perkin-Elmer Lambda 35 UV-vis spectrometer at a scan rate of 480 nm min−1. Fluorescence emission spectra were recorded on a Perkin-Elmer LS 50B luminescence spectrometer with Xenon discharge lamp excitation. The samples for absorption and fluorescence measurements were prepared as follows: the nanoparticles were dispersed in THF or H2O with a concentration of 5 mg L−1, and then the dispersions were ultrasonicated for 15 min. The fluorescence titration experiment was carried out by adding small aliquots of explosive solutions to 3.00 mL of the nanoparticle dispersions.The preparation of HCPN-OH
Under an argon atmosphere, 9,9-dihexyl-2,7-dibromofluorene (177 mg, 0.36 mmol), 1,3,5-tris-{4-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolane)phenyl}-benzene (164 mg, 0.24 mmol) and Pd(PPh3)4 (2 mg) were dissolved in degassed toluene (6 mL). The mixture solution was added to a stirred solution of cetyltrimethylammonium bromide (CTAB) (5.82 g, 16 mmol) in degassed deionized water (90 mL). Then the mixture was ultrasonicated in an ultrasonic bath at 50 °C for 20 min. A solution of 2 M aqueous K2CO3 (3.0 mL) was added under these conditions. After being ultrasonicated for another 10 min, the reaction emulsion was stirred in an oil bath at 85 °C for 24 h under an argon atmosphere. The end-capped agent, 4-[4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl]-phenol, was added. After being stirred for 4 h, 4-bromophenol was added. After being reacted for another 4 h, the resulting mixture was poured into saturated NaCl aqueous solution, and dichloromethane was added. The organic layer was separated, and most of the solvents were removed. The residue was precipitated in methanol. The solid was separated by centrifugation, and then placed into methanol again, ultrasonicated for 20 min and centrifugated. This procedure was repeated two times. The obtained solid was extracted by Soxhlet extraction with methanol and acetone for 1 day, respectively, and dried at 80 °C under vacuum for 24 h to afford the final nanoparticles (156 mg, 82%), which could be readily dispersed in many common organic solvents, such as THF, toluene, dichloromethane, etc.The preparation of HCPN-S
To a stirred hot solution of excessive sodium tert-butoxide in dry DMF (30 mL) at a temperature of 85 °C, a HCPN-OH (50 mg) dispersion in dry THF (30 mL) was added dropwise. The mixture was stirred for 2 h, and then a solution of 1,4-butanesultone (0.5 mL) in dry DMF (15 mL) was added dropwise. The mixture was stirred at 85 °C for 48 h. Methanol (20 mL) was added and stirred for 30 min. The final suspension was separated by centrifugation. The precipitated solid was placed into methanol and ultrasonicated for 20 min, and then separated by centrifugation. The solid was placed into THF and ultrasonicated for 20 min, and then separated by centrifugation. The obtained solid was dried at 80 °C under vacuum for 24 h to afford the final nanoparticles.The preparation of LCPN
LCPN was prepared according to the previous literature.23 The polymer LCP (25 mg) was dissolved in CHCl3 (2 mL) and added to 1 (wt)% SDS aqueous solution. After stirring for 1 h at room temperature and ultrasonicating for 5 min, the emulsion was stirred at 62 °C for 3 h in order to evaporate the CHCl3. The aqueous LCPN dispersion with a polymer concentration of 1 mg mL−1 was obtained and stored under seal. For the analysis and measurement, the dispersion of LCPN with a polymer concentration of 5 mg L−1 was obtained by diluting this stock solution.Acknowledgements
This paper was financially supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB12010200), the 973 Project (2009CB623601 and 2009CB930603), the Science Fund for Creative Research Groups (No. 20921061), and the National Natural Science Foundation of China (No. 21574131, 51173179, 21204080, 21074130, 21674111 and 21322403).Notes and references
- Agency for Toxic Substances and Disease Registry, Toxicological Profile for 2,4,6-Trinitrotoluene, U.S. Department of Health and Human Services, 1995.
- F. Fant, A. De Sloovere, K. Matthijsen, C. Marle, S. El Fantroussi and W. Verstraete, Environmental Pollution, Elsevier Science, Oxford, 2000, vol. 111, p. 503 Search PubMed.
- G. A. Eiceman and J. A. Stone, Anal. Chem., 2004, 76, 390A CAS.
- D. S. Moore, Rev. Sci. Instrum., 2004, 75, 2499 CrossRef CAS.
- N. L. Sanders, S. Kothari, G. Huang, G. Salazar and R. G. Cooks, Anal. Chem., 2010, 82, 5313 CrossRef CAS PubMed.
- (a) S. W. Thomas III, G. D. Joly and T. M. Swager, Chem. Rev., 2007, 107, 1339 CrossRef PubMed; (b) Y. Salinas, R. Martínez-Máñez, M. D. Marcos, F. Sancenón, A. M. Costero, M. Parraad and S. Gil, Chem. Soc. Rev., 2012, 41, 1261 RSC; (c) S. Rochat and T. M. Swager, ACS Appl. Mater. Interfaces, 2013, 5, 4488 CrossRef CAS PubMed; (d) X. Sun, Y. Wang and Y. Lei, Chem. Soc. Rev., 2015, 44, 8019 RSC; (e) J. Cao, J. X. Feng and S. Wu, Chin. J. Polym. Sci., 2014, 32, 371 CrossRef CAS.
- (a) J.-S. Yang and T. M. Swager, J. Am. Chem. Soc., 1998, 120, 5321 CrossRef CAS; (b) H. Nie, Y. Zhao, M. Zhang, Y. Ma, M. Baumgarten and K. Müllen, Chem. Commun., 2011, 47, 1234 RSC; (c) J. L. Novotney and W. R. Dichtel, ACS Macro Lett., 2013, 2, 423 CrossRef CAS; (d) Y. Wang, Y. Gao, L. Chen, Y. Fu, D. Zhu, Q. He, H. Cao, J. Cheng, R. Zhang, S. Zheng and S. Yan, RSC Adv., 2015, 5, 4853 RSC.
- (a) A. Narayanan, O. P. Varnavski, T. M. Swager and T. Goodson, J. Phys. Chem. C, 2008, 112, 881 CrossRef CAS; (b) L. Feng, H. Li, Y. Qu and C. Lu, Chem. Commun., 2012, 48, 4633 RSC; (c) T. L. Duniho, B. J. Laughlin, A. A. Buelt, W. F. Baker, C. A. Conrad and R. C. Smith, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1487 CrossRef CAS; (d) W. Shu, C. Guan, W. Guo, C. Wang and Y. Shen, J. Mater. Chem., 2012, 22, 3075 RSC; (e) L.-L. Zhou, M. Li, H.-Y. Lu and C.-F. Chen, Polym. Chem., 2016, 7, 310 RSC.
- (a) H. Sohn, R. M. Calhoun, M. J. Sailor and W. C. Trogler, Angew. Chem., Int. Ed., 2001, 40, 2104 CrossRef CAS; (b) B. Xu, X. Wu, H. Li, H. Tong and L. Wang, Macromolecules, 2011, 44, 5089 CrossRef CAS; (c) A. Räupke, A. Palma-Cando, E. Shkura, P. Teckhausen, A. Polywka, P. Görrn, U. Scherf and T. Riedl, Sci. Rep., 2016, 6, 29118 CrossRef PubMed.
- (a) G. He, N. Yan, J. Yang, H. Wang, L. Ding, S. Yin and Y. Fang, Macromolecules, 2011, 44, 4759 CrossRef CAS; (b) S. B. Jagtap, D. D. Potphode, T. K. Ghorpade, A. K. Palai, M. Patri and S. P. Mishra, Polymer, 2014, 55, 2792 CrossRef CAS.
- H. Zhang, L. Feng, B. Liu, C. Tong and C. Lü, Dyes Pigm., 2014, 101, 122 CrossRef CAS.
- (a) S. J. Toal, D. Magde and W. C. Trogler, Chem. Commun., 2005, 5465 RSC; (b) P. Marks, S. Cohen and M. Levine, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4150 CrossRef CAS.
- (a) J. Liu, Y. Zhong, J. W. Y. Lam, P. Lu, Y. Hong, Y. Yu, Y. Yue, M. Faisal, H. H. Y. Sung, I. D. Williams, K. S. Wong and B. Z. Tang, Macromolecules, 2010, 43, 4921 CrossRef CAS; (b) W. Dong, T. Fei, A. Palma-Cando and U. Scherf, Polym. Chem., 2014, 5, 4048 RSC; (c) S. Long, Y. Wan and A. D. Xia, Acta Chim. Sin., 2015, 73, 723 CrossRef CAS; (d) W. Dong, J. Pina, Y. Pan, E. Preis, J. S. S. d. Melo and U. Scherf, Polymer, 2015, 76, 173 CrossRef CAS; (e) A. Palma-Cando and U. Scherf, ACS Appl. Mater. Interfaces, 2015, 7, 11127 CrossRef CAS PubMed.
- (a) X. Wang, Y. Guo, D. Li, H. Chen and R.-C. Sun, Chem. Commun., 2012, 48, 5569 RSC; (b) N. Alizadeh, A. Akbarinejad and A. Ghoorchian, ACS Appl. Mater. Interfaces, 2016, 8, 24901 CrossRef CAS PubMed.
- (a) K. Shiraishi, T. Sanji and M. Tanaka, ACS Appl. Mater. Interfaces, 2009, 1, 1379 CrossRef CAS PubMed; (b) X. Sun, Y. Liu, G. Shaw, A. Carrier, S. Dey, J. Zhao and Y. Lei, ACS Appl. Mater. Interfaces, 2015, 7, 13189 CrossRef CAS PubMed.
- (a) J. Pecher and S. Mecking, Chem. Rev., 2010, 110, 6260 CrossRef CAS PubMed; (b) C. Wu and D. T. Chiu, Angew. Chem., Int. Ed., 2013, 52, 3086 CrossRef CAS PubMed; (c) K. Li and B. Liu, Chem. Soc. Rev., 2014, 43, 6570 RSC.
- (a) S. Bandyopadhyay, P. Pallavi, A. G. Anil and A. Patra, Polym. Chem., 2015, 6, 3775 RSC; (b) J. Huang, J. Gu, Z. Meng, X. Jia and K. Xi, Nanoscale, 2015, 7, 15413 RSC; (c) A. H. Malik, S. Hussain, A. Kalita and P. K. Iyer, ACS Appl. Mater. Interfaces, 2015, 7, 26968 CrossRef CAS PubMed.
- (a) H. Li, X. Wu, B. Xu, H. Tong and L. Wang, RSC Adv., 2013, 3, 8645 RSC; (b) X. Wu, H. Li, B. Xu, H. Tong and L. Wang, Polym. Chem., 2014, 5, 4521 RSC; (c) X. Wu, H. Li, Y. Xu, B. Xu, H. Tong and L. Wang, Nanoscale, 2014, 6, 2375 RSC; (d) X. Wu, H. Li, Y. Xu, H. Tong and L. Wang, Polym. Chem., 2015, 6, 2305 RSC; (e) Y. Xu, X. Wu, Y. Chen, H. Hang, H. Tong and L. Wang, Polym. Chem., 2016, 7, 4542 RSC.
- (a) T. V. Richter, C. Bühler and S. Ludwigs, J. Am. Chem. Soc., 2012, 134, 43 CrossRef CAS PubMed; (b) J. Wang, J. Mei, E. Zhao, Z. Song, A. Qin, J. Z. Sun and B. Z. Tang, Macromolecules, 2012, 45, 7692 CrossRef CAS; (c) F. Qiu, D. Wang, Q. Zhu, L. Zhu, G. Tong, Y. Lu, D. Yan and X. Zhu, Biomacromolecules, 2014, 15, 1355 CrossRef CAS PubMed.
- (a) S. E. Stiriba, H. Kautz and H. Frey, J. Am. Chem. Soc., 2002, 124, 9698 CrossRef CAS PubMed; (b) C. S. Popeney, M. C. Lukowiak, C. Böttcher, B. S. Welker, D. Mangoldt, G. Gunkel, Z. Guan and R. Haag, ACS Macro Lett., 2012, 1, 564 CrossRef CAS; (c) G. Gunkel-Grabole, M. L. S. Sigg, S. Lörcher, C. G. Palivan and W. P. Meier, Biomater. Sci., 2015, 3, 25 RSC; (d) D. Wang, T. Zhao, X. Zhu, D. Yan and W. Wang, Chem. Soc. Rev., 2015, 44, 4023 RSC; (e) C. Zhan, S. Li, J. Cuib and Y. Chen, Chin. J. Polym. Sci., 2015, 33, 920 CrossRef CAS.
- (a) P. Beyazkilic, A. Yildirim and M. Bayindir, Nanoscale, 2014, 6, 15203 RSC; (b) K. Boonkitpatarakul, Y. Yodt, N. Niamnontc and M. Sukwattanasinitt, RSC Adv., 2015, 5, 33306 RSC.
- A. Patra, J.-M. Koenen and U. Scherf, Chem. Commun., 2011, 47, 9612–9614 RSC.
- K. Landfester, R. Montenegro, U. Scherf, R. Guntner, U. Asawapirom, S. Patil, D. Neher and T. Kietzke, Adv. Mater., 2002, 14, 651 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7qm00173h |
| This journal is © the Partner Organisations 2017 |