
Direct conversion of coordination compounds into Ni2P nanoparticles entrapped in 3D mesoporous graphene for an efficient hydrogen evolution reaction†
Sungeun
Jeoung
a,
Bora
Seo
a,
Jeong Min
Hwang
a,
Sang Hoon
Joo
ab and
Hoi Ri
Moon
*a
aDepartment of Chemistry, School of Natural Science, Ulsan National Institute of Science and Technology (UNIST), UNIST-gil 50, Ulsan 44919, Republic of Korea. E-mail: hoirimoon@unist.ac.kr; Fax: +82-52-217-2019; Tel: +82-52-217-2928
bDepartment of Chemical Engineering, School of Energy and Chemical Engineering, Ulsan National Institute of Science and Technology (UNIST), UNIST-gil 50, Ulsan 44919, Republic of Korea. E-mail: shjoo@unist.ac.kr
First published on 9th December 2016
Abstract
This paper reports a simple preparation route to a composite of small Ni2P nanoparticles (NPs) entrapped in 3D mesoporous graphene by the thermal conversion of a coordination compound followed by phosphidation. Recently, transition metal phosphides (TMPs) have gained increasing attention owing to their promising potential as non-precious metal catalysts in the hydrogen evolution reaction (HER). In order to enhance the catalytic activity of TMPs, researchers have sought to synthesize small TMP NPs to increase the catalytically active surface area. Although surfactant-mediated syntheses can produce small TMP NPs, a cumbersome surfactant removal step is necessary to generate catalytically active clean surfaces. Interfacing TMP NPs with carbon nanomaterials is another promising approach to boost the catalytic performance by providing high electrical conductivity and durability. However, the synthesis of composites of TMP NPs and carbon demands multiple synthetic steps, including the preparation of TMP NPs, synthesis of carbon nanomaterials, and dispersion of TMP NPs onto the carbon support. The essence of our approach toward the 3D graphene encapsulating Ni2P NPs (Ni2P@mesoG) lies in the utilization of the conversion phenomenon of [Ni2(EDTA)] (EDTA = ethylenediaminetetraacetate). The thermolysis of [Ni2(EDTA)] at 600 °C produces a composite of single-crystalline 5 nm-sized Ni NPs individually entrapped in 3D mesoG (Ni@mesoG), and the following phosphidation completely converts the Ni NPs to single-crystalline Ni2P NPs in mesoG (Ni2P@mesoG) without agglomeration. This solvent-free thermal conversion route to the Ni2P@mesoG composite is simple and scalable. Notably, graphitic shell layers in Ni2P@mesoG stabilize small Ni2P NPs possessing a large active surface area, and facilitate the electron transfer due to the intimate contact between them. Consequently, the use of Ni2P@mosoG exhibits superior electrocatalytic HER activity and durability in both strong acidic and basic media.
Introduction
Hydrogen is an appealing energy carrier as a future replacement for fossil fuels.1,2 However, hydrogen production is currently dominated by the steam reforming of hydrocarbons and the partial oxidation of methane, both of which remain highly dependent on fossil fuels.3,4 To realize a hydrogen economy, hydrogen production via clean and carbon-neutral methods is necessary. Water electrolysis or splitting is considered to be especially promising for hydrogen production because it offers environmental benignity and high product purity.5–8 High-performance catalysts are integral to water electrolysis; thus far, platinum is the most efficient catalyst for the hydrogen evolution reaction (HER). However, the high cost and scarcity of this precious metal have motivated the development of alternatives such as transition metal oxides,9,10 chalcogenides,11–17 and phosphides.18–29 Metal phosphides in particular have gained increasing attention for their high HER activity. Typical preparation methods for transition metal phosphides (TMPs) include the direct reduction of phosphate in H2,30 solid-state reactions using red or white phosphorus,31,32 decomposition of high-boiling-point tri-n-octylphosphine (TOP)33,34 or P(SiMe3)3,35,36 and solvothermal or hydrothermal synthesis.37 However, the above methods typically yield very large TMP particulates. Although surfactant-mediated syntheses can produce small TMP nanoparticles (NPs), a cumbersome surfactant removal step is necessary to generate catalytically active clean surfaces. Hence, the surfactant-free synthesis of metal phosphide NPs of less than 10 nm in diameter is still a challenge.For electrocatalytic applications, interfacing of the active species with carbon nanomaterials is highly useful; the high electrical conductivities and large surface areas of carbons can boost the overall catalytic performance.38–40 Conventionally, composites of TMP NPs and carbon are prepared via multiple synthetic steps, including the preparation of TMP NPs, synthesis of carbon nanomaterials, and dispersion of TMP NPs onto the carbon support.
Herein, we report a simple preparation route to 3D few-layer graphene encapsulating small (ca. 5 nm) Ni2P NPs (Ni2P@mesoG), and characterize the use of this composite as an efficient electrocatalyst for the HER. The Ni2P@mesoG composite can be prepared simply by the thermal decomposition of the [Ni2(EDTA)] (EDTA = ethylenediaminetetraacetate) complex, followed by phosphidation (Scheme 1). The design of the Ni2P@mesoG electrocatalyst allows for the generation of small NPs, formation of intimate contact between the carbon support and active NPs, and protection of the NPs by stable graphitic shells in highly corrosive environments.41,42
 | ||
| Scheme 1 Schematic illustration of the Ni2P NPs entrapped in 3D mesoporous graphene, Ni2P@mesoG. | ||
Experimental
Materials and characterization methods
All chemicals and solvents were of reagent grade and were used without further purification. Raman spectroscopy measurements were performed using a micro-Raman system (WITec) with an excitation energy of 2.41 eV (532 nm). Elemental analyses were performed at the UNIST Central Research Facilities Center (UCRF) at the Ulsan National Institute of Science and Technology (UNIST). X-ray powder diffraction (XRPD) data were recorded on a Bruker D2 phaser diffractometer at 30 kV and 10 mA for Cu Kα (λ = 1.54050 Å), with a step size of 0.02° in 2θ. N2 sorption isotherms of samples were obtained using a BELSORP-max at 77 K. Prior to the adsorption measurements, the samples were evacuated (p < 10−5 mbar) at 100 °C for 2 h. The specific surface area was determined in the relative pressure range from 0.05 to 0.3 of the Brunauer–Emmett–Teller (BET) plot, and the total pore volume was calculated from the amount adsorbed at a relative pressure of about 0.98–0.99. X-ray photoelectron spectroscopy (XPS) was performed on a K-alpha from Thermo Fisher. Transmission electron microscopy (TEM) images, high-resolution TEM (HR-TEM) images, and energy dispersive X-ray spectrometry (EDS) mapping images were obtained using a JEOL JEM-2100F microscope.Preparation of the materials
Electrochemical measurements
All electrochemical measurements were performed on an IviumStat electrochemical analyzer at room temperature under atmospheric pressure using a three-electrode electrochemical cell. A graphite rod and Ag/AgCl (in saturated KCl solution) were used as the counter electrode and the reference electrode, respectively. All potentials in this report were converted to the reversible hydrogen electrode (RHE) scale. For electrochemical measurements, a rotating ring-disk electrode (RRDE) containing glassy carbon (GC) as the central disk (4 mm diameter, 0.126 cm2 area) was used as the working electrode. The RRDE was polished with a 1.0 μm alumina suspension, and then with a 0.3 μm suspension to generate a mirror finish before use. Catalyst inks were prepared by mixing 30 mg of the catalyst with 60 μL of Nafion (5 wt% in isopropanol, Sigma-Aldrich) in a solution of 60 μL DI water, 600 μL of EtOH (99.9%), and the mixture was sonicated for 30 min to produce a homogeneous slurry. Afterwards, 3 μL of the catalyst ink was dropped onto the GC disk and dried at 60 °C for 5 min. The resulting catalyst loading on GC was 1 mg cm−2. Before electrochemical measurements, the electrolyte was bubbled with N2 for 20 min. The electrochemical impedance spectroscopy (EIS) was measured in a frequency range of 10 kHz to 1 Hz at an overpotential of 100 mV. The polarization curves in this paper were plotted after compensating for the ohmic drops with Rs (series resistance) values obtained from the EIS measurements. The polarization curves for the hydrogen evolution reaction (HER) were recorded by sweeping the potential from 0.1 V to −0.4 V (vs. RHE) at a scan rate of 2 mV s−1 with a rotation speed of 1500 rpm. Chronopotentiometry (CP) was conducted by exerting a static current density of −10 mA cm−2 for 24 h.Results and discussion
Previously, we reported the synthesis of 3D mesoG via the thermal conversion of a Ni2+ coordination compound, [Ni2(EDTA)].43 The heat treatment of [Ni2(EDTA)] at 1000 °C under an inert atmosphere produced severely agglomerated Ni metals on the carbon matrix, and the following etching process yielded 3D interconnected hollow N-doped graphene shells consisting of 3–4 layers with small, uniform mesopores of ca. 4 nm, referred to as mesoG (Fig. S1, ESI†). In contrast, a mild thermolysis at 600 °C produced a composite of Ni NPs encapsulated in 3D mesoG (Ni@mesoG), evidenced by TEM. As shown in Fig. 1a, the Ni NPs are well-distributed throughout the mesoG matrix with an average diameter of 5.0 ± 0.4 nm. A HR-TEM micrograph shows that each Ni NP inside the carbon shell is single-crystalline, exhibiting lattice fringes separated by 0.20 nm. This distance is coincident with the (111) interplanar d-spacing of cubic nickel (JCPDS file no. 71-4655). The XRPD pattern of Ni@mesoG (Fig. 1c) is also indexed as a single-phase cubic Ni0 structure, and the crystallite sizes of the Ni NPs determined from the XRPD data are about 5.5 nm, which agrees well with the observations from the TEM images. The Ni NPs individually entrapped in the mesoG phase were converted to Ni2P NPs upon solid-state phosphidation with NaH2PO2 at 400 °C, producing Ni2P NPs entrapped in a graphitic carbon shell (Ni2P@mesoG). The TEM image (Fig. 1b) shows that the 3D graphene morphology is well-retained and the size of Ni2P NPs is 6.0 ± 1.4 nm without agglomeration after phosphidation. The small increment in the NP size could result from the increase in cell volume by 154% during the phase conversion from Ni to Ni2P. Furthermore, the HR-TEM image of the NPs in the carbon matrix revealed that a single-crystalline Ni NP is converted to a single-crystalline Ni2P NP, with a (111) interplanar d-spacing of 0.22 nm that corresponds to hexagonal nickel phosphide (Ni2P, JCPDS file no. 89-2742). The complete transformation of Ni@mesoG to Ni2P@mesoG is verified by XRPD patterns (Fig. 1c). The EDS mapping images (Fig. S2, ESI†) further confirm that both Ni and P elements are uniformly distributed over the mesoG matrix, along with the N element originating from the EDTA ligands.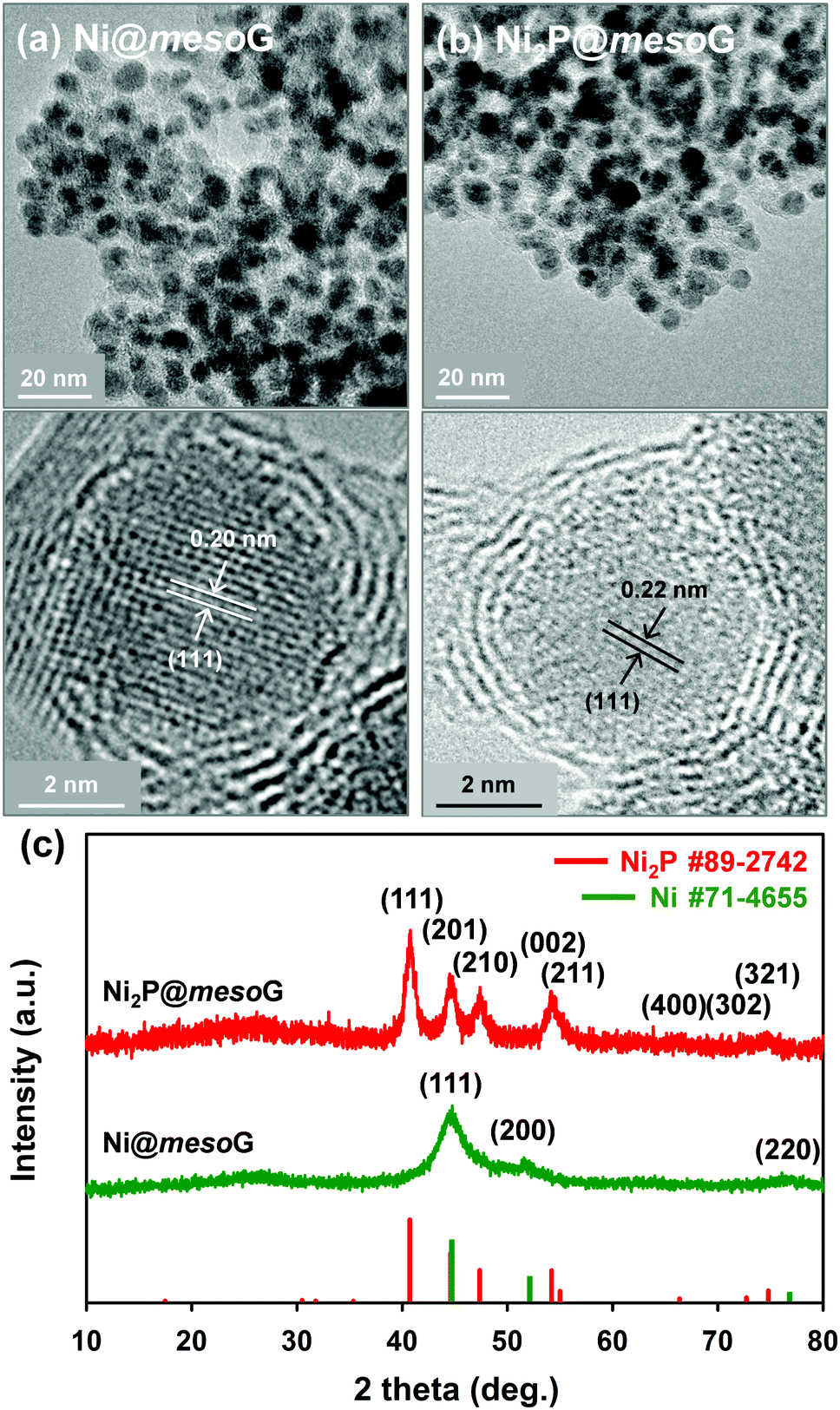 | ||
| Fig. 1 (a) TEM and HR-TEM images of Ni@mesoG, (b) TEM and HR-TEM images of Ni2P@mesoG, and (c) XRPD patterns of Ni@mesoG (bottom) and Ni2P@mesoG (top). | ||
To explore the structural and electronic properties of Ni@mesoG and Ni2P@mesoG, Raman spectroscopy was utilized (Fig. 2a). The D band is related to disordered carbon, while the G band is related to graphitic sp2 carbon. The relative intensity ratio of the D and G bands (ID/IG) is generally proportional to the number of defects in a graphitic carbon. D and G bands were observed at 1349 and 1591 cm−1 for Ni@mesoG, and 1346 and 1599 cm−1 for Ni2P@mesoG, respectively. Both composites showed the same ID/IG value of 0.98, indicating that the graphitic nature is well-maintained after thermal phosphidation. The chemical states of Ni, P, and C in Ni2P@mesoG were substantiated by XPS. The Ni 2p3/2 XPS spectrum (Fig. 2b) indicates the formation of Ni–P bonds (853.0 eV) and nickel phosphate (856.6 and 861.0 eV) on the surface of Ni2P with exposure to air. The peaks at 130.0 eV for Ni–P bonds and 134.3 eV for phosphate in the spectrum of P 2p3/2 (Fig. 2c) also shows good agreement with the Ni 2p3/2 spectra. The C 1s XPS spectra (Fig. 2d) exhibit peaks at 284.3, 284.9, and 287.8 eV, attributed to sp2 graphitic carbon, sp3 diamond-like carbon, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, respectively. In particular, the peak for sp2 carbon is dominant, indicating that the mesoG surface is highly graphitic. Ni@mesoG and Ni2P@mesoG exhibited typical type IV isotherms with an H3-type hysteresis loop over a relative pressure range of 0.42 < P/P0 < 0.99, characteristic of mesoporous materials (Fig. S3, ESI†). The porosity of Ni@mesoG provided a high BET surface area of 208 m2 g−1 and a large total pore volume of 0.49 cm3 g−1, and this was translated to Ni2P@mesoG with 125 m2 g−1 of surface area and 0.36 cm3 g−1 of total pore volume.
O, respectively. In particular, the peak for sp2 carbon is dominant, indicating that the mesoG surface is highly graphitic. Ni@mesoG and Ni2P@mesoG exhibited typical type IV isotherms with an H3-type hysteresis loop over a relative pressure range of 0.42 < P/P0 < 0.99, characteristic of mesoporous materials (Fig. S3, ESI†). The porosity of Ni@mesoG provided a high BET surface area of 208 m2 g−1 and a large total pore volume of 0.49 cm3 g−1, and this was translated to Ni2P@mesoG with 125 m2 g−1 of surface area and 0.36 cm3 g−1 of total pore volume.
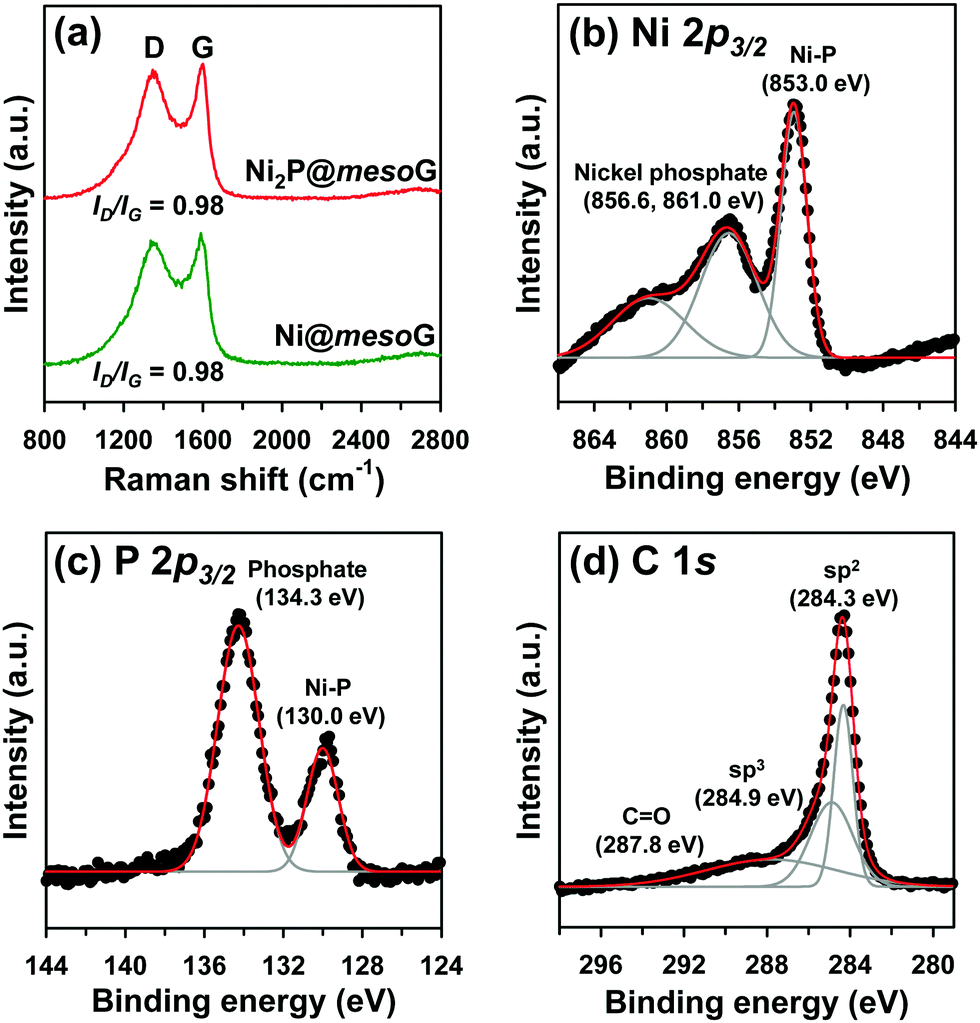 | ||
| Fig. 2 (a) Raman spectra of Ni@mesoG and Ni2P@mesoG, (b) Ni 2p3/2 XPS spectra of Ni2P@mesoG, (c) P 2p3/2 XPS spectra of Ni2P@mesoG, and (d) C 1s XPS spectra of Ni2P@mesoG. | ||
We also prepared Ni2P@mesoG composites at different phosphidation temperatures of 300, 500, and 600 °C (Fig. S4–S9, ESI†). The XRPD patterns (Fig. S4, ESI†) showed that the composite phosphidated at 300 °C contained Ni0 as well as Ni2P phases, indicating insufficient phosphidation. Higher-temperature (500 °C and 600 °C) phosphidation generated pure Ni2P, but some Ni2P NPs emerged from the graphene shell and agglomerated to form larger particles in these samples (Fig. S5, ESI†). In addition, nitrogen adsorption experiments (Fig. S6, ESI†) revealed that decreased BET surface areas and pore volumes with increases in the phosphidation temperature (175 m2 g−1 and 0.46 cm3 g−1 for 300 °C, 125 m2 g−1 and 0.36 cm3 g−1 for 400 °C, 111 m2 g−1 and 0.32 cm3 g−1 for 500 °C, and 47.2 m2 g−1 and 0.18 cm3 g−1 for 600 °C). Based on these results, the Ni2P@mesoG composite phosphidating at 400 °C was chosen to explore the electrocatalytic performance for the HER.
The electrocatalytic HER activity of the Ni2P@mesoG was investigated in both acidic and alkaline media using a three-electrode setup (see details of electrochemical measurements in the ESI†). All polarization curves here were obtained after compensating for the ohmic drop (iR) loss arising from the solution resistance. To examine the encapsulation effect of the graphitic carbon layers, Ni2P NPs grown on 3D graphene shells (Ni2P/mesoG) were prepared and characterized as HER catalysts for comparison. The structure and morphology of the Ni2P/mesoG composite were characterized by XRPD and TEM (Fig. S10, ESI†). Unlike Ni2P@mesoG, in which the Ni2P NPs were encapsulated by graphitic carbon shells, the Ni2P NPs grown outside the carbon layers were severely agglomerated, producing large particles. For comparison, the HER performances of mesoG and 20% Pt/C (HiSPEC 3000, Johnson-Matthey) were also tested. The catalyst loading for each material was 1 mg cm−2 except for the Pt/C (14 μgPt cm−2).
Fig. 3a shows the HER polarization curves in an acidic medium (0.5 M H2SO4). As expected, the 20% Pt/C catalysts exhibit the best HER performance with negligible overpotential among the compared samples, while mesoG shows only marginal HER activity. Importantly, the Ni2P@mesoG exhibits the highest HER performance among the mesoG-based samples, as evidenced by the high current density with a low overpotential (Fig. 3a). The overpotential to drive a current density of −10 mA cm−2 is 98 mV for Ni2P@mesoG. By increasing the catalyst loading to 3 mg cm−2, Ni2P@mesoG requires only 79 mV of overpotential to achieve a current density of −10 mA cm−2 (Fig. S10a, ESI†). In contrast, Ni2P/mesoG requires a relatively larger overpotential (139 mV) to drive the same current density (Fig. 3a). The reaction kinetics is examined with the Tafel slopes, extracted from the linear portion of the Tafel plots that are derived from the polarization curves (Fig. 3b). The Tafel slopes of Ni2P@mesoG and Ni2P/mesoG are 56 and 60 mV dec−1, respectively. The relatively smaller Tafel slope of Ni2P@mesoG indicates more favourable reaction kinetics during the HER in acidic media. The Tafel slope of 56 mV dec−1 for Ni2P@mesoG may suggest that the HER over this catalyst occurs via the Volmer–Heyrovsky HER mechanism in acidic media.44 The charge transfer kinetics during HER is assessed using EIS (Fig. 3c). The dramatically smaller semicircle in the Nyquist plot of Ni2P@mesoG compared to that in the plot of Ni2P/mesoG in acidic media clearly suggests a lower charge transfer resistance in Ni2P@mesoG. We extensively compared the HER performance of Ni2P@mesoG with previous nickel phosphide based HER catalysts in terms of overpotential and Tafel slopes (Table S1, ESI†). The comparison makes the Ni2P@mesoG as one of the high-performing HER catalysts. We note that as HER activities are highly dependent on experimental conditions such as catalyst loading, more valid comparison may be made with the data measured under similar experimental conditions. The superior HER activity in Ni2P@mesoG is attributed to the synergistic contribution of the following factors. The formation of small (ca. 5 nm) Ni2P NPs could provide a large active surface area. Moreover, the intimate contact between the Ni2P NPs and carbon shells also facilitates electron transfer during the HER.38
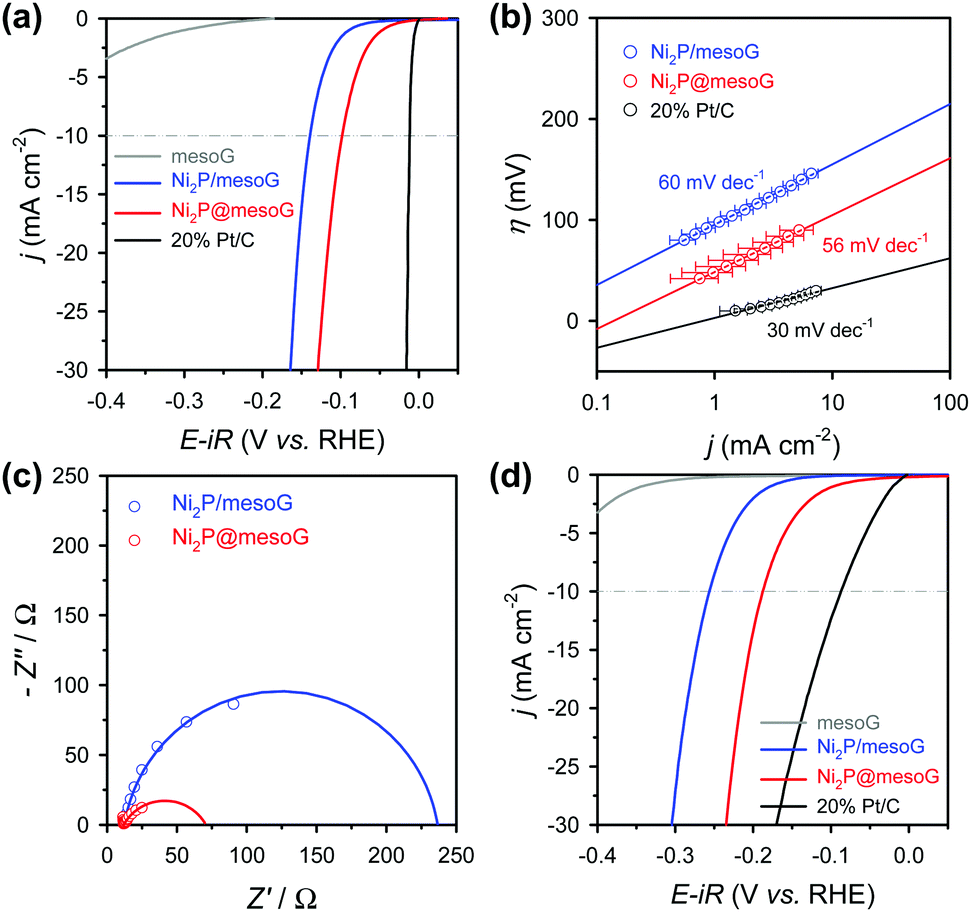 | ||
| Fig. 3 Electrocatalytic HER performances. (a) HER polarization curves in 0.5 M H2SO4. (b) Corresponding Tafel plots. The polarization curves and Tafel plots are depicted with and without the iR correction, respectively. (c) Nyquist plots for the impedance spectra obtained at η = 100 mV (vs. RHE) in 0.5 M H2SO4. Empty circles and solid lines represent numerical raw data and fitting results, respectively. (d) HER polarization curves in 1 M KOH. | ||
Excellent HER performance is also demonstrated by Ni2P@mesoG in an alkaline medium (Fig. 3d). The HER activity measured in the basic medium of 1 M KOH reveals that, excepting the Pt/C catalyst, the Ni2P@mesoG shows the best HER performance among the compared samples in terms of overpotential and Tafel slopes. The overpotentials required to drive a current density of −10 mA cm−2 are 188 mV and 256 mV for Ni2P@mesoG and Ni2P/mesoG, respectively. The overpotential for Ni2P@mesoG required to achieve −10 mA cm−2 could be decreased to 112 mV with a higher catalyst loading (3 mg cm−2) (Fig. S11b, ESI†). The lower charge transfer resistance in Ni2P@mesoG relative to that in Ni2P/mesoG was also observed from the Nyquist plots of the catalysts in the alkaline medium (Fig. S12a, ESI†). The Tafel slopes of Ni2P@mesoG and Ni2P/mesoG were 99 and 100 mV dec−1, respectively (Fig. S12b, ESI†).
Electrochemical durability is critical for the practical applicability of a material. CP was conducted by maintaining a current density of −10 mA cm−2 over 24 h in acidic media (Fig. 4a). The Ni2P@mesoG catalyst exhibits high durability, with little decay in the HER activity and good morphology maintenance over 24 h of electrolysis. TEM images obtained after the CP measurements reveal that the Ni2P remains encapsulated by graphitic carbon layers with a small particle size (Fig. 4b). The stable nature of Ni2P@mesoG indicates the physically protective role of the graphitic carbon layers.
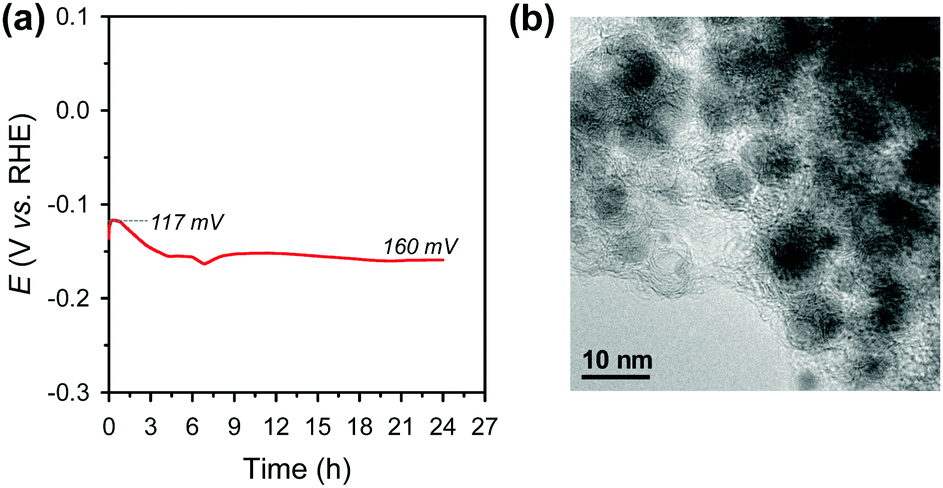 | ||
| Fig. 4 (a) Chronopotentiometry (CP) curve measured at a current density of −10 mA cm−2 in 0.5 M H2SO4. Data are displayed without iR-correction. (b) TEM image after the CP measurements. | ||
Conclusions
In conclusion, we have reported a simple preparation route to the Ni2P@mesoG composite via the thermal decomposition of a coordination complex and subsequent phosphidation. Importantly, in Ni2P@mesoG, the graphene layers act both as a protective and an enhancing matrix for catalytically active Ni2P NPs, which leads to high electrocatalytic activity and durability for HER in both strong acidic and basic media. This simple preparation route should be widely applicable to synthesize the composite materials of metal pnictide or metal chalcogenide NPs entrapped in graphitic carbon matrices for highly efficient electrocatalysts.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) Grant funded by the Korean Government (MSIP) (NRF-2016R1A5A1009405 and 2015M1A2A2056560). B. S. acknowledges the Global PhD Fellowship (2013H1A2A1032647).Notes and references
- M. S. Dresselhaus and I. L. Thomas, Nature, 2001, 414, 332 CrossRef CAS PubMed.
- S. Chu and A. Majundar, Nature, 2012, 488, 294 CrossRef CAS PubMed.
- J. A. Turner, Science, 2004, 305, 972 CrossRef CAS PubMed.
- D. R. Palo, Chem. Rev., 2007, 107, 3992 CrossRef CAS PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446 CrossRef CAS PubMed.
- K. S. Joya, Y. F. Joya, K. Ocakoglu and R. v. d. Krol, Angew. Chem., Int. Ed., 2013, 52, 10426 CrossRef CAS PubMed.
- Y. Zhao, X. Jia, G. Chen, L. Shang, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung, D. O’Hare and T. Zhang, J. Am. Chem. Soc., 2016, 138, 6517 CrossRef CAS PubMed.
- X. Jia, Y. Zhao, G. Chen, L. Shang, R. Shi, X. Kang, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Energy Mater., 2016, 6, 1502585 CrossRef.
- M. Gong, W. Zhou, M.-C. Tsai, J. Zhou, M. Guan, M.-C. Lin, B. Zhang, Y. Hu, D.-Y. Wang, J. Yang, S. J. Pennycook, B.-J. Hwang and H. Dai, Nat. Commun., 2014, 5, 4695 CrossRef CAS PubMed.
- Y. H. Li, P. F. Liu, L. F. Pan, H. F. Wang, Z. Z. Yang, L. R. Zheng, P. Hu, H. J. Zhao, L. Gu and H. G. Yang, Nat. Commun., 2015, 6, 8064 CrossRef CAS PubMed.
- T. F. Jaramillo, K. P. Jørgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100 CrossRef CAS PubMed.
- Y. Li, H. Wang, L. Xie, Y. Liang, G. Hong and H. Dai, J. Am. Chem. Soc., 2011, 133, 7296 CrossRef CAS PubMed.
- J. Kibsgaard, Z. Chen, B. N. Reinecke and T. F. Jaramillo, Nat. Mater., 2012, 11, 963 CrossRef CAS PubMed.
- D. Kong, J. J. Cha, H. Wang, H. R. Lee and Y. Cui, Energy Environ. Sci., 2013, 6, 3553 CAS.
- B. Seo, G. Y. Jung, Y. J. Sa, H. Y. Jeong, J. Y. Cheon, J. H. Lee, H. Y. Kim, J. C. Kim, H. S. Shin, S. K. Kwak and S. H. Joo, ACS Nano, 2015, 9, 3728 CrossRef CAS PubMed.
- B. Seo, H. Y. Jeong, S. Y. Hong, A. Zak and S. H. Joo, Chem. Commun., 2015, 51, 8334 RSC.
- D. Yoon, B. Seo, J. Lee, K. S. Nam, B. Kim, S. Park, H. Baik, S. H. Joo and K. Lee, Energy Environ. Sci., 2016, 9, 850 CAS.
- E. J. Popczun, J. R. McKone, C. G. Read, A. J. Biacchi, A. M. Wiltrout, N. S. Lewis and R. E. Schaak, J. Am. Chem. Soc., 2013, 135, 9267 CrossRef CAS PubMed.
- J. Tian, Q. Liu, A. M. Asiri and X. Sun, J. Am. Chem. Soc., 2014, 136, 7587 CrossRef CAS PubMed.
- Y. Liang, Q. Liu, A. M. Asiri, X. Sun and Y. Luo, ACS Catal., 2014, 4, 4065 CrossRef CAS.
- Z. Pu, Q. Liu, A. M. Asiri and X. Sun, Nanoscale, 2014, 6, 11031 RSC.
- P. Jiang, Q. Liu and X. Sun, Nanoscale, 2014, 6, 13440 RSC.
- Z. Pu, Q. Liu, A. M. Asiri and X. Sun, ACS Appl. Mater. Interfaces, 2014, 6, 21874 CAS.
- Z. Xing, Q. Liu, A. M. Asiri and X. Sun, ACS Catal., 2015, 5, 145 CrossRef CAS.
- P. Xiao, W. Chen and X. Wang, Adv. Energy Mater., 2015, 5, 1500985 CrossRef.
- X. Wang, Y. V. Kolen’ko, X.-Q. Bao, K. Kovnir and L. Liu, Angew. Chem., Int. Ed., 2015, 54, 8188 CrossRef CAS PubMed.
- A. Han, S. Jin, H. Chen, H. Ji, Z. Sun and P. Du, J. Mater. Chem. A, 2015, 3, 1941 CAS.
- B. Seo, D. S. Baek, Y. J. Sa and S. H. Joo, CrystEngComm, 2016, 18, 6083 RSC.
- C. Tang, L. Gan, R. Zhang, W. Lu, X. Jiang, A. M. Asiri, X. Sun, J. Wang and L. Chen, Nano Lett., 2016, 16, 6617 CrossRef CAS PubMed.
- X. Wang, P. Clark and S. T. Oyama, J. Catal., 2002, 208, 321 CrossRef CAS.
- S. Carenco, X. F. Le Goff, J. Shi, L. Roiban, O. Ersen, C. Boissière, C. Sanchez and N. Mézailles, Chem. Mater., 2011, 23, 2270 CrossRef CAS.
- Y. Ni, L. Jin and J. Hong, Nanoscale, 2011, 3, 196 RSC.
- J. Park, B. Koo, K. Y. Yoon, Y. Hwang, M. Kang, J.-G. Park and T. Hyeon, J. Am. Chem. Soc., 2005, 127, 8433 CrossRef CAS PubMed.
- E. Muthuswamy, G. H. L. Savithra and S. L. Brock, ACS Nano, 2011, 5, 2402 CrossRef CAS PubMed.
- L. Li and P. Reiss, J. Am. Chem. Soc., 2008, 130, 11588 CrossRef CAS PubMed.
- W.-S. Ojo, S. Xu, F. Delpech, C. Nayral and B. Chaudret, Angew. Chem., Int. Ed., 2012, 51, 738 CrossRef CAS PubMed.
- Y. Deng, Y. Zhou, Y. Yao and J. Wang, New J. Chem., 2013, 37, 4083 RSC.
- Y. Liang, Y. Li, H. Wang and H. Dai, J. Am. Chem. Soc., 2013, 135, 2013 CrossRef CAS PubMed.
- X. Huang, C. Tan, Z. Yin and H. Zhang, Adv. Mater., 2014, 26, 2185 CrossRef CAS PubMed.
- L. Jiao, Y.-X. Zhou and H.-L. Jiang, Chem. Sci., 2016, 7, 1690 RSC.
- M. Tavakkoli, T. Kallio, O. Reynaud, A. G. Nasibulin, C. Johans, J. Sainio, H. Jiang, E. I. Kauppinen and K. Laasonen, Angew. Chem., Int. Ed., 2015, 54, 4535 CrossRef CAS PubMed.
- J. Dang, P. Ren, D. Deng and X. Bao, Angew. Chem., Int. Ed., 2015, 54, 2100 CrossRef PubMed.
- K. J. Lee, Y. J. Sa, H. Y. Jeong, C. W. Bielawski, S. H. Joo and H. R. Moon, Chem. Commun., 2015, 51, 6773 RSC.
- W. C. Sheng, H. A. Gasteiger and Y. Shao-Horn, J. Electrochem. Soc., 2010, 157, B1529 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic data, XRPD pattern, TGA, and nitrogen sorption data. See DOI: 10.1039/c6qm00269b |
| This journal is © the Partner Organisations 2017 |