
Chiroptical luminescent nanostructured cellulose films†
Erlantz
Lizundia
*ab,
Thanh-Dinh
Nguyen
b,
Jose L.
Vilas
a,
Wadood Y.
Hamad
c and
Mark J.
MacLachlan
 *b
*b
aMacromolecular Chemistry Research Group (LABQUIMAC), Department of Physical Chemistry, University of the Basque Country (UPV/EHU), Leioa 48940, Spain. E-mail: erlantz.liizundia@ehu.eus
bDepartment of Chemistry, University of British Columbia, 2036 Main Mall, Vancouver, British Columbia V6T 1Z1, Canada. E-mail: mmaclach@chem.ubc.ca
cFPInnovations, 2665 East Mall, Vancouver, British Columbia V6T 1Z4, Canada
First published on 21st December 2016
Abstract
In this work we report a straightforward and scalable method to fabricate luminescent and iridescent chiral nanomaterials with potential metal ion sensing capacity for environmental and biological applications. Nitrogen-doped carbon dots (N-CDs) were synthesized by a hydrothermal reaction of D-glucose and melamine. New free-standing chiral nematic composite materials with randomly distributed nanoparticles were prepared after N-CD co-assembled with CNCs via evaporation-induced self-assembly (EISA). The left-handed twisted, layered structure of chiral CNC composites remains intact for all the studied compositions, providing both iridescent and photoluminescent properties to our films. Structural, thermal, morphological and photophysical properties of the films were further explored. Overall, our experimental findings reveal that hydrothermal synthesis of melamine and D-glucose is an effective approach to obtain luminescent materials that can be combined with CNCs to obtain photonic films that retain long-range chiral nematic organization. These new functional materials have potential application as luminescent sensors for metal ions or bioimaging purposes.
Introduction
Since its discovery by Revol and co-workers in the 1990s,1 the long-range chiral nematic ordering of cellulose nanocrystals (CNCs) has opened new horizons for the development of novel functional materials with optical responsiveness.2–6 CNCs present a spindle-shaped structure and are extracted via controlled hydrolytic cleavage of the most plentiful biopolymer on the biosphere, cellulose.7–9 CNCs assemble into a stable structure via evaporation-induced self-assembly (EISA), resulting in films with brilliant iridescent colours when the helical pitch is on the order of the wavelength of visible light.1 In this framework, the development of novel chiral nematic materials with stimuli-responsive properties represents a promising approach for the fabrication of next generation functional materials with potential application in sensors, lasers, optical amplifiers, bioimaging and catalysts.10It would be expected that new functionalities may arise when left-handed chiral nematic CNCs host stimuli-responsive species. To date, rare-earth compounds such as ZrO2:Eu3+, Y2O3:Eu3+ or YVO4:Eu3+,11–13 gold nanoclusters,5 silver nanowires and CdS quantum dots (QD) have been utilized to develop chiral nematic cellulose films with new photonic properties.14,15 Although QDs present a tuneable and narrow emission spectrum that may be especially useful for sensing and optical switching, they are potentially toxic and usually require complex functionalization steps to achieve a homogeneous dispersion within their host matrix.15 Previous studies have shown that one of the main challenges to developing highly optically active chiral nematic structures is the synthesis of nanoparticles with suitable surface functional groups that allow the CNCs to maintain their long-range order upon co-assembly via EISA. The maximum concentrations that do not disturb the nematic order have been around ∼1.5% for CdS quantum dots,15 or ∼1.2 wt% for gold nanoclusters.5 It is therefore reasonable to seek inexpensive and straightforward methods for fabricating hydrophilic, optically active species from renewable resources to allow higher loadings.
Carbon dots (CDs), metal-free photoluminescent nanomaterials, have recently received enormous attention because of their low cost, stable photoluminescence (PL) and inherent physicochemical properties.16 To date, they have been used as efficient catalysts for the photodegradation of methyl blue,17 for multicolor patterning,18 for lighting systems,19 and for bioimaging.13 One of the important features of CDs is that their PL properties strongly depend on the initial materials used for their fabrication in contrast to that usually found in other semiconducting nanoparticles.20 These raw materials include, but are not limited to, soy milk,21 orange juice,22 ethanolamine, and ground coffee.23,24
CDs are usually obtained by solid-state synthesis at high temperatures, which raises the production costs.13 Therefore, it is desirable to develop a straightforward approach for their controlled fabrication. Soft solution-processing methods at low temperatures enable bottom-up design of novel nanomaterials with tailored functionalities.25 Hydrothermal synthesis is especially attractive for obtaining chemical reactions under “green” conditions and has proven effective for the fabrication of different nanomaterials.26 These CDs usually present many surface-functional groups on their surface (e.g., carbonyl, hydroxyl, carboxylate),27 making them highly compatible with both industrially relevant solvents (water, ethanol and acetone) and with hydrophilic nanomaterials such as CNCs.28 Furthermore, the introduction of heteroatoms within the nanoparticle core might endow CDs with improved photophysical performance. Accordingly, nitrogen doping has been proven to increase the quantum yield of CDs via the introduction of polyaromatic structures.29,30
Although remarkable progress has been made in the development of nanopaper based on carbon nanotubes,31–34 no reports could be found in the literature dealing with the incorporation of spherical carbonaceous nanoparticles with photoluminescent properties. Herein, we report a green, simple, scalable and fast hydrothermal approach to fabricate N-CDs. These nanoparticles retain the chiral nematic long-range ordering when they are co-assembled with CNCs over a wide range of compositions. The photophysical properties of the resulting free-standing and iridescent films were thoroughly explored and their potential use for luminescent sensing is highlighted. The synthetic approaches reported here provide new pathways for the development of luminescent nanoparticles with chemosensing capacity. As well, the materials have a unique combination of luminescent CDs with iridescent chiroptical properties that suggest advanced photonics applications.
Experimental
Chemicals
Analytical grade melamine (C3H6N6, ≥99%, ACROS Organics), D-glucose (C6H12O6, ≥98%, Sigma-Aldrich), zinc nitrate hexahydrate (Zn(NO3)2·6H2O, Sigma-Aldrich), and iron(III) chloride hexahydrate (FeCl3·6H2O, Sigma-Aldrich) were used as received without further purification. Cellulose nanocrystals (CNCs) were prepared as previously described from sulfuric acid hydrolysis of fully-bleached, commercial kraft softwood pulp.35,36 TEM of the CNCs indicated that they are 240 ± 62 nm in length and ∼10 nm in width (CNC length-distribution is shown in Fig. S5, ESI†).Preparation of N-CDs
In a typical procedure, 0.1 g of melamine and 0.4 g of D-glucose monosaccharide powders were added to 10 mL of deionized water and the resulting mixture was magnetically stirred at 80 °C for 1 h to ensure the complete dissolution of each component. Then, the solution was put into a 20 mL Teflon-lined autoclave, sealed and maintained at 160 °C for times ranging from 4 to 15 h. The autoclave was then allowed to cool to room temperature naturally over ∼2 h. The resulting product was composed of a carbonaceous char together with a fine brownish dispersion. This dispersion was sequentially filtered using a 0.45 μm pore-size PVDF filter membrane (Millex®-HV). For further purification, the product was dialyzed against distilled water through a dialysis membrane for 6 days (water was changed every 24 h). N-CDs remain inside the membrane, while low molecular weight ions and molecules diffuse through the membrane. The concentration of the N-CDs in the filtered, supernatant suspension was determined by allowing 3 g of the dispersion to evaporate for one week at 60 °C (the concentration was then calculated by dividing the mass of completely dried N-CDs by the mass of the initial aqueous dispersion).Preparation of N-CD/CNC solid photoluminescent films
The required amount of aqueous N-CDs dispersion (0.5 wt%) was added dropwise to 5 mL of CNC suspensions (pH 2.5, 4.2 wt%) to yield composites having 0.1, 0.2, 0.5, 1 and 2 wt% N-CDs. Resulting solutions were magnetically stirred for 1 h at room temperature and they were cast in 60 mm diameter polystyrene Petri dishes. The mixture was allowed to dry for 96 h. Neat CNC films were fabricated for comparison purposes. Films of ∼40 μm in thickness and 180 mg in weight were obtained.Characterization
Transmission electron microscope (TEM) images of nitrogen-doped C-dots (N-CDs) were obtained on a Hitachi H7600 electron microscope. Infrared spectra were obtained on a Nicolet 6700 FT-IR equipped with a smart orbit diamond attenuated total reflectance (ATR) attachment. Powder X-ray diffraction (PXRD) patterns of the samples were recorded on a D8 Advance X-ray diffractometer (λ = 1.540 Å). The chemical composition of N-CDs was investigated by X-ray photoelectron spectroscopy (XPS, Omicron & Leybold MAX200) using an Al Kα source. Elemental analyses (C/H/N) were performed at the UBC Microanalytical Services Laboratory, while elemental mapping images were obtained on a JEOL JSM-7001F scanning electron microscope (SEM) equipped with an energy-dispersive X-ray spectrometer (EDS) operated at an acceleration voltage of 10 kV. Carbon-13 nuclear magnetic resonance (13C NMR) spectra were collected on a Bruker AV400 spectrometer.Photoluminescence (PL) spectra were obtained on a PTI Photon Technology International fluorimeter using a 75 W arc lamp as a source with an excitation wavelength of 365 nm. For characterization of the raw N-CDs, 4 mL of aqueous dispersion was introduced in a 61FLUV10 quartz-to-Pyrex graded sealed fluorescence tube cuvette (cross section = 10 × 10 mm2, height = 45 mm). To verify that the photoluminescence is due to fluorescence and not to phosphorescence, the cuvette was degassed by bubbling argon through the sample for 15 min and then the cuvette was sealed. Obtained PL spectra before and after degassing were compared. Sensing experiments were carried out by adding small amounts of zinc nitrate hexahydrate or iron(III) chloride hexahydrate to aqueous N-CDs dispersions and by subsequently measuring the resulting PL spectra. N-CD/CNC solid films were carefully mounted on a quartz slide with double-sided tape to ensure that the film was centered on the emission beam and they were set at a 45 degree angle of the emission beam reflection.
Polarized optical microscopy (POM) with crossed polarizers was performed on an Olympus BX41 microscope. Scanning electron microscope (SEM) images of the samples were obtained on a Hitachi S4700 electron microscope. Samples were prepared by breaking films into small pieces and attaching them to aluminum stubs using double-sided adhesive tape and sputter coating with gold–palladium (5 nm). Two-photon laser-scanning microscopy (TPLSM) was performed on a Zen LSM 510 Laser Module. Small pieces of the films were mounted on quartz slides with cover glasses. The samples were excited on the cover glass surface under the excitation of a 730 nm laser and then scanned within several minutes. Images were acquired with the confocal detector unit with the pinhole fully open.
Thermogravimetric analysis (TGA) of the films (∼5 mg) was conducted at a heating rate of 10 °C min−1 under N2 atmosphere to 850 °C using a PerkinElmer Pyris 6 thermogravimetric analyzer. UV-Vis spectroscopy was conducted on a Cary 5000 spectrophotometer. Circular dichroism spectroscopy was recorded on a J-710 spectropolarimeter (JASCO) by attaching the films on quartz slides perpendicular to the beam path.
Results and discussion
Hydrothermal synthesis of luminescent N-CDs
Hydrothermal fabrication of carbon dots using different saccharides is a versatile approach to obtain novel luminescent materials.37,38 The hydrothermal reaction of saccharides yields carbonaceous nanomaterials,39 which could potentially be further functionalized in order to increase the photoluminescent properties of the resulting nanoparticles. In this context, melamine was used as a direct monomer source for the fabrication of N-CDs by a bottom-up approach. Since the –CHO aldehyde groups present in saccharides interact with the –NH2 amino moieties of melamine even at low temperature with no need for a catalyst,28 it was expected that the hydrothermal reaction of melamine and a saccharide (as a carbon source) would lead to novel N-containing carbon nanomaterials.Here we investigate the hydrothermal treatment of melamine and a natural saccharide, D-glucose (C6H12O6) at temperatures ranging from 140 to 180 °C. It was found that mainly carbonized materials were obtained at synthesis temperatures of about 180 °C, while temperatures of ∼140 °C and lower failed to give any N-CDs. Thus, a temperature of 160 °C was selected as the most suitable for the development of N-CDs. Fig. 1a displays the process through which N-CDs have been obtained. Two different components, a fine brownish stable dispersion and a black solid material, were obtained after the hydrothermal reaction of melamine and D-glucose at 160 °C. According to CNH elemental analysis (EA), the carbonaceous char is composed of 18.1% N, 50.3% C, and 4.6% H (and 27% O by difference), and represents ∼32 wt% of the whole reacted material. This black precipitate likely forms from the hydrothermally-induced carbonization of the carbohydrate together with melamine and has previously been observed during the formation of carbonaceous nanomaterials from N-containing biomass.40 The nitrogen content of this char is larger than the usual values reported to date and arises from the much larger nitrogen content of the reagents. In particular, melamine, our starting material, is 67% nitrogen by mass, which is substantially larger than the nitrogen content of other precursors of N-CDs; glucosamine and chitosan contain 6.5% and 9.4% N, respectively.40,41
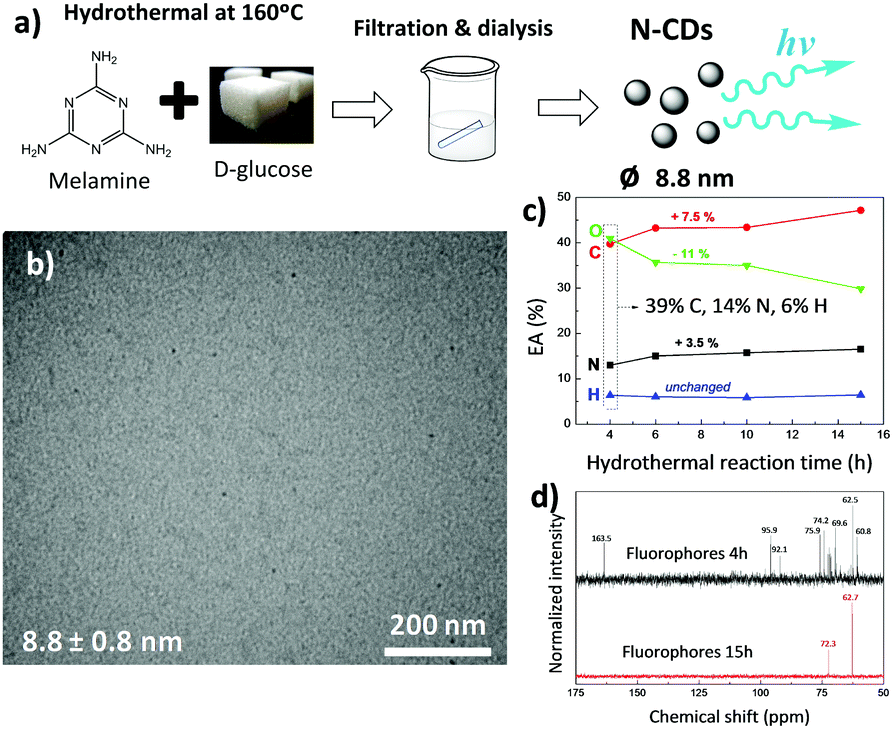 | ||
| Fig. 1 (a) Scheme showing the preparation of N-CDs from the hydrothermal reaction of melamine and D-glucose at 160 °C for 4 h; (b) representative TEM image of N-CDs having a diameter of 8.8 ± 0.8 nm; (c) C/H/N/O ratio obtained by elemental analysis (CHN) of diffused molecules (fluorophores) through the dialysis membrane for different hydrothermal reaction times; and (d) their corresponding 13C NMR spectra. | ||
After filtration of the brownish dispersion through a 0.45 μm pore-size polyvinylidene fluoride (PVDF) filter membrane, the fine brownish stable dispersion was further dialyzed against distilled water through a dialysis membrane for 6 days (reaction yield of about 1 wt%; Fig. S1a, ESI†). Transmission electron microscopy (TEM) images (Fig. 1b) show uniform, well-dispersed spherical nanoparticles having a mean diameter of 8.8 nm (statistics based on count of 50 particles), which falls within the typical size range for N-CDs formed after the hydrothermal or solvothermal treatment from different precursors.38
The material removed by dialysis represents 67 wt% of the whole reacted material and shows an increased luminescence in comparison with N-CDs (Fig. S1b, ESI†). The morphological features of these diffused molecules were analyzed by TEM (Fig. S2a, ESI†), but no nanoparticles were found. CNH elemental analysis of the dialyzed molecules shown in Fig. 1c reveals that upon reaction, oxygen is consumed at the expense of carbon and nitrogen (the amount of O decreases from 41 to 30 wt%, whereas the amount of C and N increases from 39 to 46.5 wt% and from 14 to 17.5 wt%, respectively), while 13C NMR spectra shown in Fig. 1d denotes a decreasing amount of compounds as reaction proceeds.
To explore the formation mechanism of N-CDs, carbon dots prepared after 10 h of hydrothermal reaction were analyzed by TEM (Fig. S2b, ESI†). We found that the size of N-CDs obtained after longer reaction times increased to 22.9 nm. Altogether, these data confirm that N-CDs are developed from carbonaceous precursor nuclei formed by the dehydration and fragmentation of melamine and D-glucose into soluble products that further polymerize/condense. These carbonaceous nanoparticles increase in size during the hydrothermal reaction by diffusion/linkage of fluorophores on the surface of nanoparticles.37
Fourier transform infrared spectroscopy (FTIR) was used to examine the chemical features of synthesized materials. As shown in Fig. 2a, both N-CDs and the organic fluorophores removed by dialysis present similar spectra with a broad absorption band at 3550–3000 cm−1 due to O–H/N–H stretching, together with narrower N–H scissoring and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and N–O stretching centred at 2930, 1562 and 1369 cm−1, respectively.42 Substances diffused through the dialysis membrane present CN and C–N stretching bands centred at 1630 and 1452 cm−1, which are typically found in heterocycles.42,43 N-CDs present two additional bands at 1618 and 1025 cm−1, which could be ascribed to the CC stretch of sp2 hybridized carbon and –COO− groups, respectively.44,45 This may be explained by the fact that the intermediates arising from melamine and D-glucose form a core composed of sp2 hybridized carbon atoms, with a surface covered by hydrophilic hydroxyl/carboxyl groups, as previously found in carbon dots derived from bovine serum albumin proteins.45 It may be expected that these surface groups would enable the dispersion of N-CDs within polar solvents and would act as a self-passivation layer for increasing their luminescent properties.46
C and N–O stretching centred at 2930, 1562 and 1369 cm−1, respectively.42 Substances diffused through the dialysis membrane present CN and C–N stretching bands centred at 1630 and 1452 cm−1, which are typically found in heterocycles.42,43 N-CDs present two additional bands at 1618 and 1025 cm−1, which could be ascribed to the CC stretch of sp2 hybridized carbon and –COO− groups, respectively.44,45 This may be explained by the fact that the intermediates arising from melamine and D-glucose form a core composed of sp2 hybridized carbon atoms, with a surface covered by hydrophilic hydroxyl/carboxyl groups, as previously found in carbon dots derived from bovine serum albumin proteins.45 It may be expected that these surface groups would enable the dispersion of N-CDs within polar solvents and would act as a self-passivation layer for increasing their luminescent properties.46
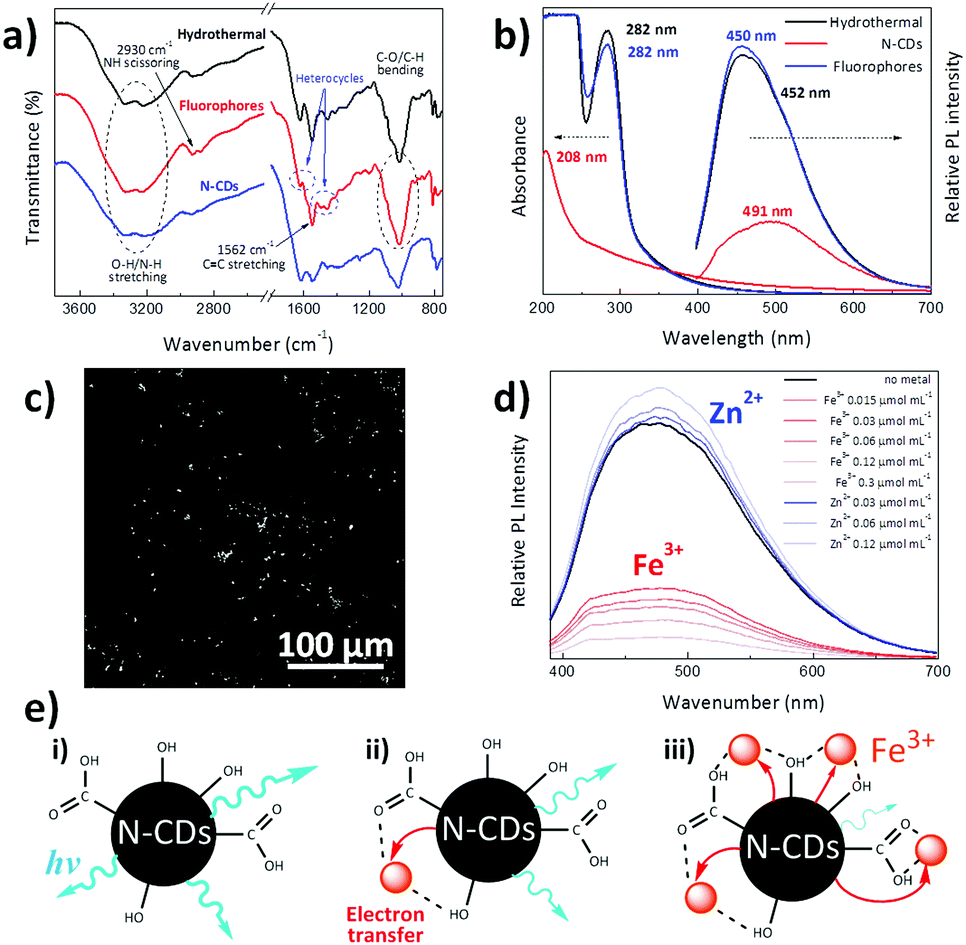 | ||
| Fig. 2 (a) FTIR spectra; (b) UV-Vis absorption and photoluminescence (PL) spectra of the whole hydrothermal reaction product, fluorophores and N-CDs after their separation by dialysis. (c) N-CDs observed under two-photon laser scanning microscopy (λex: 730 nm); (d) PL spectra of water-dispersed N-CDs under 365 nm incident light containing different concentrations of Fe3+ and Zn2+ ions. (e) A schematic of the proposed PL quenching mechanism of N-CDs in presence of low (ii) and high (iii) amounts of Fe3+. Note that the section (i) corresponds to the PL emission of water-dispersed N-CDs. | ||
The X-ray diffraction pattern (Fig. S3a, ESI†) of N-CDs reveals an overall amorphous structure as represented by a wide diffraction peak corresponding to the (002) diffraction facets of graphite centered at 22.4° 2θ, which, according to Bragg's law, corresponds to a d002 interlayer spacing of 0.39 nm. This interlayer spacing is 15% larger than that of pure graphite owing to the incorporation of heteroatoms on the basal plane which disturb the graphitic structure.13,47 Further evidence for the surface functional groups of N-CDs was obtained by X-ray photoelectron spectroscopy (XPS). Three main signals at 284, 399.5 and 532 eV assigned to C1s, N1s and O1s, respectively, are observed (Fig. S3b, ESI†), confirming the formation of nitrogen-doped carbon structures (a surface N atomic percentage of 2.2% is obtained).43 A dominant graphitic C1s peak centered at 284 eV is observed. This peak presents a broad asymmetric tail towards higher binding energy, denoting the presence of graphitic sp2 carbon atoms. Moreover, polyaromatic structures having C–N and CN groups are detected on the N1s spectrum as previously reported by Ding et al.42 As highlighted in Fig. S3c (ESI†), elemental mapping shows that C (yellow), N (pink) and O (cyan) atoms are uniformly distributed within the carbon dots. The amount of N within N-CDs measured by energy dispersive X-ray spectroscopy (EDS) is about 12.8 wt%, which agrees well with data obtained from EA.
As determined by EA, N-CDs have a carbon content of 45.7 wt%, 12.9 wt% of nitrogen and 2.2 wt% of hydrogen, the remaining 39.2 wt% being oxygen. The large amount of O within the N-CDs is remarkable in comparison with previously reported works (22% for N-CDs obtained by the pyrolysis of konjac flour,13 19.7% for N and S-doped carbon dots obtained using α-lipoic acid as the carbon source,42 19.4% for N-doped carbon nanodots fabricated after the calcination of waste chicken eggshell).48 It may be expected that this large amount of oxygen would help to achieve a good dispersion of N-CDs in both polar solvents and polar polymeric matrices. It is interesting to note that the surface N concentration is notably smaller than the total amount of N obtained by both EDS and EA, indicating that nitrogen is mainly present within the carbon dot core.29,30
Fig. 2b shows the UV-Vis absorption and photoluminescence (PL) spectra of the dispersion resulting from hydrothermal reaction for 4 h and the separated individual constituents, N-CDs and dialyzed organic fluorophores. The absorption peak located at ∼282 nm is attributed to the n–π* transition of CN and NO bonds present in the diffused molecules,49 while the broad absorption spectrum of N-CDs is characteristic of their band structure and complex energy levels,50 with its maximum (208 nm) ascribed to the π–π* electronic transitions of CC.49 On the other hand, the PL spectra indicate a stronger luminescence intensity of organic fluorophores than N-CDs, a fact that has already been found in carbogenic nanoparticles obtained from the pyrolysis of citric acid and ethanolamine.23 Fig. S4a (ESI†) presents the PL spectra of N-CDs obtained after hydrothermal reaction of melamine and glucose by themselves. Although no luminescence was found for hydrothermally-treated melamine and glucose alone, the N-CDs obtained after the hydrothermal reaction of melamine and glucose together yield an intense luminescence peak centred at 491 nm. This fact indicates that the hydrothermal reaction of melamine or glucose themselves does not lead to sufficient polymerization reactions to form luminescent particles. For instance, luminescent CDs have been obtained by hydrothermal reaction of glucose with KH2PO4 salt at 200 °C.24 To further verify that the photoluminescence is due to fluorescence and not to phosphorescence, the photoluminescence of the N-CDs aqueous dispersion was measured when the solution was saturated with air and after it had been degassed by bubbling argon through the sample for 15 min; virtually no differences in the photoluminescence spectra were found (Fig. S4b, ESI†). Several works have reported that CDs have short PL lifetimes ranging from 2.95 to 10.6 ns because of the radiative recombination of the excitons that give rise to fluorescence.46,51 More precisely, CDs prepared via a facile hydrothermal method and having surface carboxyl and hydroxyl groups present average photoluminescence lifetimes of about 6 ns with a double-exponential function, suggesting that there are two dominating states that contribute to overall PL, the energy gap and surface state transitions.52
Furthermore, as depicted in Fig. S4c (ESI†), N-CDs exhibit excitation-dependent emission behaviour, where the emission peak continuously red shifts with longer excitation wavelengths. Indeed, when the excitation wavelength (λex) is changed from 330 to 430 nm, the luminescence peak is shifted from 479 (indigo-blue) to 524 nm (green) with a notable intensity change in the emitted light. There is likely a distribution of particles in the dispersion whose absorption and luminescence varies across the spectrum. As well, the aggregation state of the N-CDs is anticipated to give excitation-dependent emissive properties.53 In the same way, Fig. S4d (ESI†) reveals that N-CDs synthesized from glucose and melamine show a concentration-dependent emission, as previously reported for other N-CD systems.13,42 The extended aggregation of N-CDs forms larger nanoparticles that decrease their band gap, leading to red emission shifting upon high N-CDs loading.53 Indeed, aggregation results in a shift on the emitted light from 466 to 591 nm when the concentration increases from 0.01 mg mL−1 to 0.5 mg mL−1.47
As a proof of concept, we evaluated the prospective applicability of these nanoparticles for bioimaging purposes (Fig. 2c). A drop of an aqueous N-CD dispersion (0.1 mg mL−1) was spread over a quartz substrate and was allowed to evaporate at room temperature to monitor unmodified N-CDs by two-photon laser-scanning microscopy (TPLSM). It is seen that N-CDs display a sharp contrast when excited under 730 nm light, making synthesized N-CDs useful for highly sensitive fluorescent probes for multiphoton bioimaging.54,55 Compared to one-photon confocal microscopy, this multiphoton imaging technique has the advantage of being less harmful to the living tissues since photons with lower energy (far red) are required to induce a luminescence response.56
Sensing of metal ions has received substantial attention as one of the main possible application areas of N-CDs, especially zinc and iron metals for the biological and environmental fields. While zinc is used by dozens of proteins to stabilize structures, iron is the fourth most abundant element in the Earth's crust by mass and is essential for oxygen transport, metabolism and many catalytic functions.18 Photoluminescence measurements at an excitation wavelength of 365 nm were carried out to explore the chemosensing capacity of aqueous suspensions of N-CDs to these metals via the addition of small amounts of Fe3+ and Zn2+ ions (Fig. 2d). The luminescence of N-CDs increases upon Zn2+ ion addition, while iron quenches their light emission. A similar PL increase in the presence of Zn2+ has been also reported to occur in nitrogen and phosphorus doped carbon nanodots synthesized via hydrothermal reaction of glucose, ammonia and phosphoric acid.51 We believe that coordination of the lone pairs on nitrogen atoms in the N-CDs eliminates non-radiative decay pathways typically enabled by lone pairs on N, thus leading to an enhancement of luminescence; this fluorescence enhancement is commonly observed in molecular zinc sensors.57 Conversely, the luminescence quenching is depicted in Fig. 2e, where it is shown that surface hydroxyl and carboxyl groups on the N-CDs interact with Fe3+ ions. This interaction allows a rapid non-radiative electron transfer from the excited N-CDs to the d-orbitals of trivalent Fe3+ ions, quenching the luminescence of N-CDs. As this electron transfer process increases with the presence of Fe3+, the greater the presence of such ions the lower would be the luminescence of N-CDs (Fig. 2e).18,58,59
Incorporation of N-CDs into chiral nematic CNC films
As shown in Fig. 3a, chiral nematic N-CD/CNC films were fabricated by the co-assembly of CNCs (240 ± 62 nm long and ∼10 nm wide; Fig. S5, ESI†) with N-CDs via EISA at room temperature with varying proportions. New strongly iridescent and crack-free, free-standing composite materials were formed (see Fig. S6, ESI† for further details). Visual observation shows that the structural coloration is notably modified upon the addition of different amounts of N-CDs, especially for the composite having 2 wt% N-CDs.60 Polarized optical microscopy (POM) images of neat CNCs and N-CD/CNC composites having 0.2 and 2 wt% N-CDs (Fig. 3b) reveal a striped texture originating from the strongly birefringent chiral nematic structure.61 The features observed by POM have spacing of several microns and clearly do not represent the helical pitch observed either by SEM or from the reflectance spectra. The structure of N-CD/CNC films was further investigated by means of scanning electron microscopy (SEM) (Fig. S7, ESI†). Incorporation of N-CDs does not substantially modify the structure of the neat CNC film. The usual long-range chiral nematic structure of pristine CNCs, where spindle-like features rotate in a counter-clockwise direction, is kept over the entire studied composition range. The repeating distance between twisted layers is found to be several hundred nanometers and progressively decreases upon addition of N-CDs. In view of these results, the change in ionic strength from adding N-CDs does not disrupt the formation of a chiral nematic liquid crystal by the CNCs.62 No signs of N-CDs, either individually distributed or in their aggregated state, could be observed due to their small particle size, confirming that the N-CDs are not present in large aggregates, but rather are distributed. An advantage of the EISA over hard-templating methods is that the co-assembly of CNCs together with N-CDs yields uniform structures because the individual composite constituents remain homogeneously distributed in solution at the molecular level.61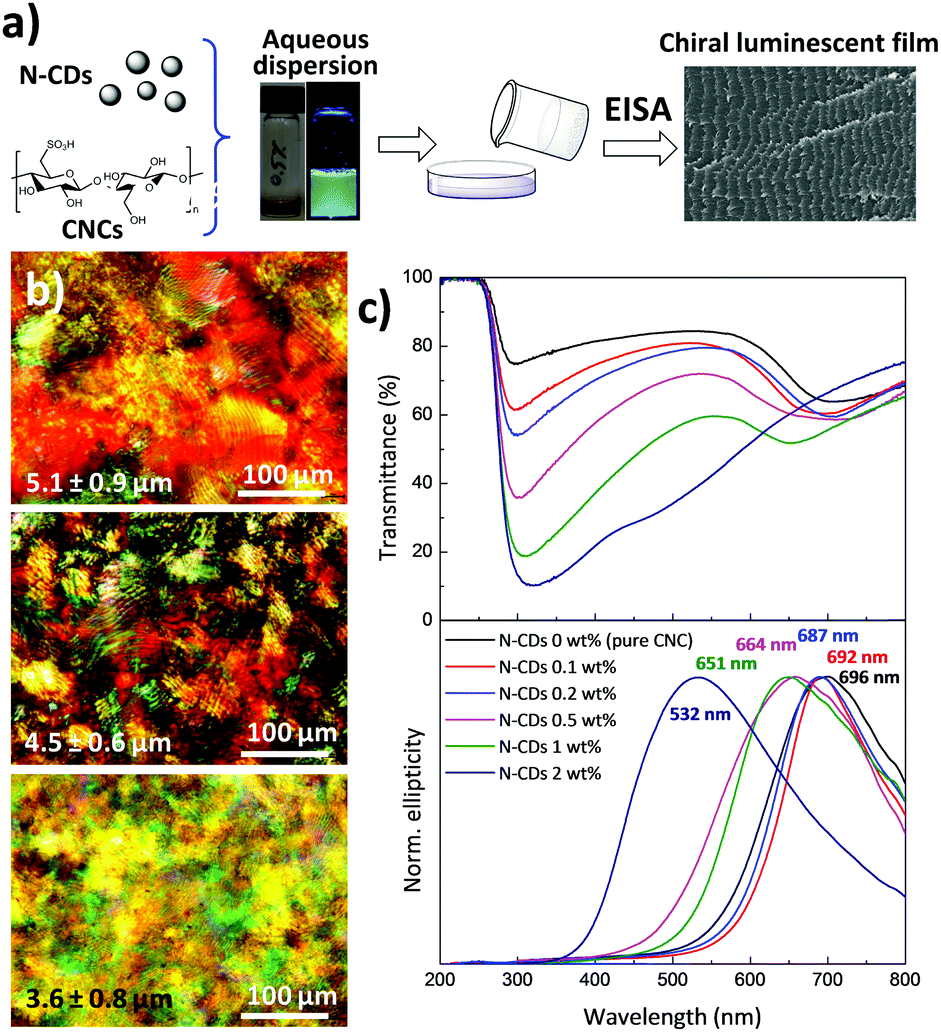 | ||
| Fig. 3 (a) Scheme showing the fabrication of luminescent chiral nematic N-CD/CNC films. (b) Polarized optical microscopy (POM) images and (c) UV-Vis (top) and circular dichroism (bottom) spectra of N-CD/CNC composite films. The spacing of the features observed in the fingerprint textures is indicated in each POM image. | ||
As determined by UV-Vis spectroscopy (Fig. 3c top), the chiroptical properties of the films are substantially modified after the addition of N-CDs. Films with concentrations up to 1 wt% show two well-resolved peaks, while N-CD/CNC 2 wt% presents a single very broad band with a maximum at 319 nm as a result of the overlapping of the CNC reflectance peak occurring at high wavelengths and the absorption at lower wavelengths arising from N-CDs.5 More precisely, the reflectance peak assigned to the CNCs is blue-shifted from 708 nm for neat CNCs to 463 nm for CNCs containing 2 wt% N-CDs, indicating a decrease in the helical pitch.63 The new intense absorption appearing at 288–335 nm may be attributed to the π–π* and n–π* transitions of CC and CO bonds of N-CDs, respectively, and increases its absorption wavelength with the presence of luminescent nanoparticles.38 Circular dichroism spectra of N-CD/CNC composites (Fig. 3c bottom) display intense signals with positive ellipticity for all the studied compositions. Because of the decrease of the helical pitch with the presence of N-CDs, the maximum circular dichroism signal shifts from 696 nm for neat CNCs to 532 nm for the CNC film containing 2 wt% N-CDs. The sharp circular dichroism bands obtained for all the compositions indicate the presence of homogeneous planar structures rather than chiral nematic polydomains,64 which matches well with reported POM and SEM images. Additionally, the full width at half maximum (FWHM) of circular dichroism signals increases from 206 to 246 nm for the neat CNC and N-CD/CNC 2 wt% film, respectively, suggesting a less ordered structure of the composites. Both reflected wavelength in UV-Vis and the positive circular dichroism peak wavelength closely match for all the compositions, implying that the left-handed chiral nematic order is maintained after incorporation of N-CDs.65,66 These effects are in good agreement with reported SEM images in Fig. S7 (ESI†). When these N-CDs are in suspension with CNCs, they are able to increase the ionic strength of the dispersion, which upon EISA leads to smaller helical pitches and therefore a blue shift in the spectral position of the CD signal, as previously shown in CNC dispersions having different NaCl concentrations.67 The small size of N-CDs allows them to be selectively introduced within the CNCs without perturbing their long-range ordering; in other words, the fact that N-CDs are introduced in between CNC twisted layers does not itself lead to an increase in the helical pitch (and thus a red-shift of CD signal).61 Overall, results shown in Fig. 3 demonstrate that N-CDs interact with their chiral nematic CNC matrix to give chiroptical nanostructured cellulose films. Preserving the chiral nematic structure of CNCs at high nanoparticle loadings could present an interesting step towards the development of multifunctional films because more stimulus responsive species would provide a larger signal upon exposure to analytes. It is noteworthy that the maximum amount of luminescent N-CDs that could be added without disturbing the chiral nematic structure of CNCs is ∼2 wt%, which is larger than the previously reported concentration of CdS quantum dots/silica (∼1.5%) without disrupting the chiral nematic order.15 We believe that the N-CDs have surface structures that are more chemically compatible with the CNCs, and do not disrupt the chiral nematic order.
Infrared spectra of the N-CD/CNC films (Fig. S8a, ESI†) show the characteristic IR bands corresponding to cellulose, a broad band in the 3650–3200 cm−1 (O–H stretching) and sharper bands at 2902 cm−1 (asymmetric and symmetric stretching of C–H groups), 1160 cm−1 (C–O–C bending) and 897 cm−1 (C–O–C asymmetric stretching of the β-glycosidic linkage).68 It can be seen that N-CD/CNC 2 wt% composite shows two new bands at 1618 and 1025 cm−1 corresponding to N-CDs (Fig. 2a), confirming the presence of N-CDs within the composite films. TGA results shown in Fig. S8b (ESI†) reveal a continuous increase in the thermal stability of composite films upon N-CD addition. In fact, the onset of thermodegradation (taken as the temperature at which the first 10% of the mass is lost) takes place 22 °C higher for the composite containing 2 wt% of N-CDs in comparison with pristine CNCs, implying that the depolymerization, dehydration and decomposition of cellulose glycosyl units may be delayed due to nitrogen release from the N-containing carbon dots upon heating. To date, scarce reports exist in which the thermal stability of chiral CNC films is significantly improved upon the addition of nanoparticles. For instance, lanthanide complexes do not change the thermal stability of TEMPO mediated oxidized nanofibrillated cellulose and it has been reported that several metal nanoparticles accelerate the decomposition of chiral CNC films at high temperatures.42,69
Photoluminescent properties of chiral nematic N-CD/CNC composite films
The N-CD dispersion within the host CNC matrix was evaluated by two-photon laser scanning microscopy (TPLSM). Fig. 4a displays a TPLSM micrograph of the N-CD/CNC 2 wt% film where the characteristic chiral nematic architecture was already observed (brown lines highlight these structures) together with randomly distributed bright spots (brown circles), corresponding to N-CDs. N-CDs are selectively located in between the fingerprint textured regions of CNCs. We speculate that, as shown in the high-magnification SEM image (Fig. 4a), N-CD/CNC films are able to keep their long range chiral nematic structure because N-CDs are excluded from the well-ordered sections.70 In other words, the presence of N-CDs does not inhibit the self-assembly process of CNCs, enabling the formation of repeating left-handed twisted layered structures at a N-CD concentration as high as 2 wt%. Photoluminescence properties of films having different N-CD concentrations are shown in Fig. 4b. Whereas neat CNC films show virtually no luminescence, N-CD/CNC composites present a blue emission when exposed to 365 nm UV light. The emission wavelength of composite films is red shifted from 498 to 511 nm upon N-CD addition. This is in agreement with the data shown in Fig. S4d (ESI†), where water-dispersed N-CDs show a concentration-dependent emission with a 25 nm red shift of the emitted light when the concentration increases from 0.01 to 0.5 mg mL−1. Moreover, it could be observed that the relative PL intensity is not proportional to the amount of incorporated N-CDs, indicating a substantial quenching effect. This aggregation-caused quenching (ACQ) of emitted light is already a well known phenomena reported more than sixty years ago and could be explained in terms of N-CDs aggregating upon loading.71 At low concentrations, i.e. <1 wt%, N-CDs remain well dispersed and all the nanoparticles emit blue light (∼498 nm) when photoexcited at 365 nm, while at higher concentrations the overall photoluminescence is decreased via non-radiative decay processes.72,73 Therefore, only the isolated N-CDs could emit photons, significantly decreasing the overall photoluminescence of the composites. In view of these results, it could be expected that the stabilization of the N-CDs before their co-assembly with CNCs would address this ACQ effect. As previously shown an amphiphilic stabilizer such as polyacrylic acid (PAA) can be used for that purpose.15 Since it was observed by the naked eye that N-CDs remain highly stable when they are found together with organic fluorophores (before dialysis), for the sake of comparison, CNCs were also co-assembled with the dispersion obtained after immediately after the hydrothermal reaction consisting of N-CDs and diffused organic fluorophores, and their PL properties were explored. Fig. S9 (ESI†) reveals that the PL intensity is about proportional to the amount of incorporated luminescent material, indicating that no substantial quenching effects occur as experimental conditions were kept unchanged. A future study may require the stabilization of N-CDs with organic molecules to avoid their ACQ within polymeric matrices.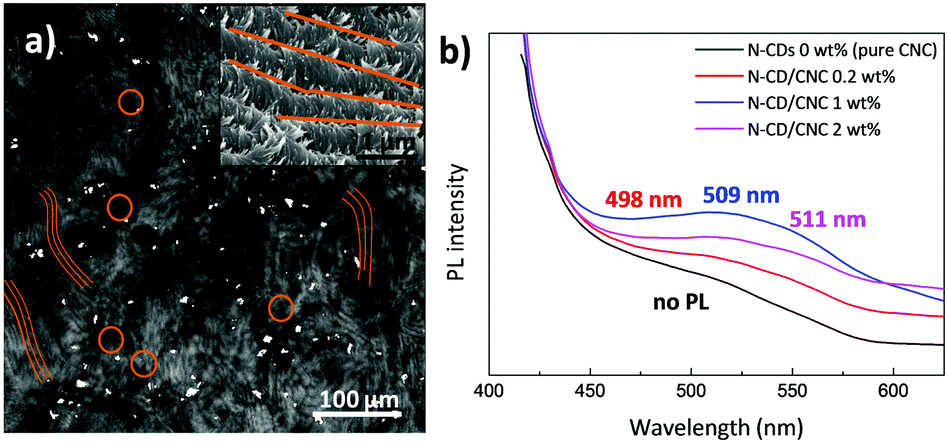 | ||
| Fig. 4 (a) Two-photon laser-scanning micrograph (485 × 485 μm2) of the chiral nematic N-CD/CNC 2 wt% composite having the characteristic fingerprint texture (its corresponding high-magnification SEM image is shown as an inset) and (b) PL spectroscopy of N-CD/CNC films. | ||
Conclusions
We demonstrate, for the first time, the preparation of free-standing, iridescent and luminescent chiral nematic N-CD/CNC composite materials after N-CD co-assembly with CNCs via evaporation-induced self-assembly (EISA). Our new method to fabricate these luminescent nanoparticles is simple, safe, and uses readily-available precursors. During the hydrothermal reaction of D-glucose and melamine, the organic fluorophores are consumed to yield N-CDs with increasing size from 8.8 to 22.9 nm. The addition of N-CDs to the CNC matrix increases the ionic strength of aqueous CNC dispersions, decreasing the helical pitch of the resulting films. All compositions display intense signals with positive ellipticity by circular dichroism spectroscopy, indicating the presence of homogeneous planar structures. Moreover, composite films show an increased thermal stability and intense iridescence under natural light together with blue emission when exposed to 365 nm UV light. As a result of the selective localization of N-CDs within the well-ordered CNC matrix, the left-handed twisted layered structure of chiral films remains intact with the addition of 2 wt% of N-CDs. In the form of water-dispersed nanoparticles or as free-standing films, these novel sustainable functional materials have potential utility as luminescent chemosensors for detecting metal ions or bioimaging purposes.Acknowledgements
E. L. thanks the University of the Basque Country (UPV/EHU) for a postdoctoral fellowship. The authors acknowledge financial support from the Basque Country Government (Ayudas para apoyar las actividades de los grupos de investigación del sistema universitario vasco, IT718-13), NSERC (Discovery Grant) and are thankful to FPInnovations and CelluForce Inc. for providing CNCs.Notes and references
- J. F. Revol, H. Bradford, J. Giasson, R. H. Marchessault and D. G. Gray, Int. J. Biol. Macromol., 1992, 14, 170–172 CrossRef CAS PubMed.
- B. Wang and A. Walther, ACS Nano, 2015, 9, 10637–10646 CrossRef CAS PubMed.
- I. Usov, G. Nyström, J. Adamcik, S. Handschin, C. Schütz, A. Fall, L. Bergström and R. Mezzenga, Nat. Commun., 2015, 6, 7564 CrossRef PubMed.
- C. Schütz, M. Agthe, A. B. Fall, K. Gordeyeva, V. Guccini, M. Salajková, T. S. Plivelic, J. P. F. Lagerwall, G. Salazar-Alvarez and L. Bergström, Langmuir, 2015, 31, 6507–6513 CrossRef PubMed.
- M. Schlesinger, M. Giese, L. K. Blusch, W. Y. Hamad and M. J. MacLachlan, Chem. Commun., 2015, 51, 530–533 RSC.
- M. Giese, L. K. Blusch, M. K. Khan, W. Y. Hamad and M. J. MacLachlan, Angew. Chem., Int. Ed., 2014, 53, 8880–8884 CrossRef CAS PubMed.
- R. H. Marchessault, F. F. Morehead and N. M. Walter, Nature, 1959, 184, 632–633 CrossRef CAS.
- B. G. Rånby, Acta Chem. Scand., 1949, 3, 649–650 CrossRef.
- A. Dufresne, Mater. Today, 2013, 16, 220–227 CrossRef CAS.
- A. Bobrovsky, K. Mochalov, V. Oleinikov, A. Sukhanova, A. Prudnikau, M. Artemyev, V. Shibaev and I. Nabiev, Adv. Mater., 2012, 24, 6216–6222 CrossRef CAS PubMed.
- G. Chu, W. Xu, D. Qu, Y. Wang, H. Song and Y. Xu, J. Mater. Chem. C, 2014, 2, 9189–9195 RSC.
- G. Chu, J. Feng, Y. Wang, X. Zhang, Y. Xu and H. Zhang, Dalton Trans., 2014, 43, 15321–15327 RSC.
- X. Teng, C. Ma, C. Ge, M. Yan, J. Yang, Y. Zhang, P. C. Morais and H. Bi, J. Mater. Chem. B, 2014, 2, 4631–4639 RSC.
- G. Chu, X. Wang, T. Chen, J. Gao, F. Gai, Y. Wang and Y. Xu, ACS Appl. Mater. Interfaces, 2015, 7, 11863–11870 CAS.
- T. D. Nguyen, W. Y. Hamad and M. J. MacLachlan, Adv. Funct. Mater., 2014, 24, 777–783 CrossRef CAS.
- S. N. Baker and G. A. Baker, Angew. Chem., Int. Ed., 2010, 49, 6726–6744 CrossRef CAS PubMed.
- H. Li, X. He, Z. Kang, H. Huang, Y. Liu, J. Liu, S. Lian, C. H. A. Tsang, X. Yang and S. T. Lee, Angew. Chem., Int. Ed., 2010, 49, 4430–4434 CrossRef CAS PubMed.
- S. Zhu, Q. Meng, L. Wang, J. Zhang, Y. Song, H. Jin, K. Zhang, H. Sun, H. Wang and B. Yang, Angew. Chem., Int. Ed., 2013, 52, 3953–3957 CrossRef CAS PubMed.
- W. Kwon, S. Do, J. Lee, S. Hwang, J. K. Kim and S. W. Rhee, Chem. Mater., 2013, 25, 1893–1899 CrossRef CAS.
- I. L. Medintz, H. T. Uyeda, E. R. Goldman and H. Mattoussi, Nat. Mater., 2005, 4, 435–446 CrossRef CAS PubMed.
- C. Zhu, J. Zhai and S. Dong, Chem. Commun., 2012, 48, 9367–9369 RSC.
- S. Sahu, B. Behera, T. K. Maiti and S. Mohapatra, Chem. Commun., 2012, 48, 8835–8837 RSC.
- M. J. Krysmann, A. Kelarakis, P. Dallas and E. P. Giannelis, J. Am. Chem. Soc., 2012, 134, 747–750 CrossRef CAS PubMed.
- P. C. Hsu and H. T. Chang, Chem. Commun., 2012, 48, 3984–3986 RSC.
- L. Li, G. Wu, G. Yang, J. Peng, J. Zhao and J. J. Zhu, Nanoscale, 2013, 5, 4015–4039 RSC.
- J. Jiang, K. Zhao, X. Xiao and L. Zhang, J. Am. Chem. Soc., 2012, 134, 4473–4476 CrossRef CAS PubMed.
- L. Bao, Z. L. Zhang, Z. Q. Tian, L. Zhang, C. Liu, Y. Lin, B. Qi and D. W. Pang, Adv. Mater., 2011, 23, 5801–5806 CrossRef CAS PubMed.
- X. Dong, Y. Su, H. Geng, Z. Li, C. Yang, X. Li and Y. Zhang, J. Mater. Chem. C, 2014, 2, 7477–7481 RSC.
- Z. Qian, J. Ma, X. Shan, H. Feng, L. Shao and J. Chen, Chem. – Eur. J., 2014, 20, 2254–2263 CrossRef CAS PubMed.
- D. Qu, M. Zheng, L. Zhang, H. Zhao, Z. Xie, X. Jing, R. E. Haddad, H. Fan and Z. Sun, Sci. Rep., 2014, 4, 5294 CAS.
- M. Salajkova, L. Valentini, Q. Zhou and L. A. Berglund, Compos. Sci. Technol., 2013, 87, 103–110 CrossRef CAS.
- M. M. Hamedi, A. Hajian, A. B. Fall, K. Hkansson, M. Salajkova, F. Lundell, L. Wgberg and L. A. Berglund, ACS Nano, 2014, 8, 2467–2476 CrossRef CAS PubMed.
- H. Koga, M. Nogi, N. Komoda, T. T. Nge, T. Sugahara and K. Suganuma, NPG Asia Mater., 2014, 6, e93 CrossRef CAS.
- L. Hu, G. Zheng, J. Yao, N. Liu, B. Weil, M. Eskilsson, E. Karabulut, Z. Ruan, S. Fan, J. T. Bloking, M. D. McGehee, L. Wågberg and Y. Cui, Energy Environ. Sci., 2013, 6, 513–518 CAS.
- K. E. Shopsowitz, H. Qi, W. Y. Hamad and M. J. MacLachlan, Nature, 2010, 468, 422–425 CrossRef CAS PubMed.
- K. E. Shopsowitz, W. Y. Hamad and M. J. MacLachlan, J. Am. Chem. Soc., 2012, 134, 867–870 CrossRef CAS PubMed.
- M. Sevilla and A. B. Fuertes, Carbon, 2009, 47, 2281–2289 CrossRef CAS.
- Y. Wang and A. Hu, J. Mater. Chem. C, 2014, 2, 6921–6939 RSC.
- Z. C. Yang, M. Wang, A. M. Yong, S. Y. Wong, X. H. Zhang, H. Tan, A. Y. Chang, X. Li and J. Wang, Chem. Commun., 2011, 47, 11615–11617 RSC.
- L. Zhao, L. Z. Fan, M. Q. Zhou, H. Guan, S. Qiao, M. Antonietti and M. M. Titirici, Adv. Mater., 2010, 22, 5202–5206 CrossRef CAS PubMed.
- L. Zhao, N. Baccile, S. Gross, Y. Zhang, W. Wei, Y. Sun, M. Antonietti and M. M. Titirici, Carbon, 2010, 48, 3778–3787 CrossRef CAS.
- H. Ding, J. S. Wei and H. M. Xiong, Nanoscale, 2014, 6, 13817–13823 RSC.
- T. Lai, E. Zheng, L. Chen, X. Wang, L. Kong, C. You, Y. Ruan and X. Weng, Nanoscale, 2013, 5, 8015–8021 RSC.
- L. Wu, H. Feng, M. Liu, K. Zhang and J. Li, Nanoscale, 2013, 5, 10839–10843 RSC.
- Q. Yang, L. Wei, X. Zheng and L. Xiao, Sci. Rep., 2015, 5, 17727 CrossRef CAS PubMed.
- Z. Yang, M. Xu, Y. Liu, F. He, F. Gao, Y. Su, H. Wei and Y. Zhang, Nanoscale, 2014, 6, 1890–1895 RSC.
- S. Chen, J.-W. Liu, M.-L. Chen, X.-W. Chen and J.-H. Wang, Chem. Commun., 2012, 48, 7637–7639 RSC.
- Y. Ke, B. Garg and Y. Ling, RSC Adv., 2014, 4, 58329–58336 RSC.
- Z. Luo, Y. Lu, L. A. Somers and A. T. C. Johnson, J. Am. Chem. Soc., 2009, 131, 898–899 CrossRef CAS PubMed.
- H. Li, Z. Kang, Y. Liu and S.-T. Lee, J. Mater. Chem., 2012, 22, 24230–24253 RSC.
- B. Shi, Y. Su, L. Zhang, M. Huang, R. Liu and S. Zhao, ACS Appl. Mater. Interfaces, 2016, 8, 10717–10725 CAS.
- X. Li, S. Zhang, S. A. Kulinich, Y. Liu and H. Zeng, Sci. Rep., 2014, 4, 4976 CAS.
- A. Sharma, T. Gadly, A. Gupta, A. Ballal, S. K. Ghosh and M. Kumbhakar, J. Phys. Chem. Lett., 2016, 7, 3695–3702 CrossRef CAS PubMed.
- S. M. Borisov and O. S. Wolfbeis, Chem. Rev., 2008, 108, 423–461 CrossRef CAS PubMed.
- D. W. Domaille, E. L. Que and C. J. Chang, Nat. Chem. Biol., 2008, 4, 168–175 CrossRef CAS PubMed.
- P. T. So, C. Y. Dong, B. R. Masters and K. M. Berland, Annu. Rev. Biomed. Eng., 2000, 2, 399–429 CrossRef CAS PubMed.
- P. Jiang and Z. Guo, Coord. Chem. Rev., 2004, 248, 205–229 CrossRef CAS.
- S. Liu, J. Tian, L. Wang, Y. Zhang, X. Qin, Y. Luo, A. M. Asiri, A. O. Al-Youbi and X. Sun, Adv. Mater., 2012, 24, 2037–2041 CrossRef CAS PubMed.
- Q. Mei, C. Jiang, G. Guan, K. Zhang, B. Liu, R. Liu and Z. Zhang, Chem. Commun., 2012, 48, 7468–7470 RSC.
- Q. Chen, P. Liu, C. Sheng, L. Zhou, Y. Duan and J. Zhang, RSC Adv., 2014, 4, 39301–39304 RSC.
- J. Xu, T. D. Nguyen, K. Xie, W. Y. Hamad and M. J. MacLachlan, Nanoscale, 2015, 7, 13215–13223 RSC.
- J. Pan, W. Hamad and S. K. Straus, Macromolecules, 2010, 43, 3851–3858 CrossRef CAS.
- J. A. Kelly, K. E. Shopsowitz, J. M. Ahn, W. Y. Hamad and M. J. MacLachlan, Langmuir, 2012, 28, 17256–17262 CrossRef CAS PubMed.
- Z. Yue and J. M. G. Cowie, Macromolecules, 2002, 35, 6572–6577 CrossRef CAS.
- R. Bardet, N. Belgacem and J. Bras, ACS Appl. Mater. Interfaces, 2015, 7, 4010–4018 CAS.
- S. Beck, J. Bouchard and R. Berry, Biomacromolecules, 2011, 12, 167–172 CrossRef CAS PubMed.
- A. Querejeta-Fernández, B. Kopera, K. S. Prado, A. Klinkova, M. Methot, G. Chauve, J. Bouchard, A. S. Helmy and E. Kumacheva, ACS Nano, 2015, 9, 10377–10385 CrossRef PubMed.
- E. Lizundia, J. L. Vilas and L. M. León, Carbohydr. Polym., 2015, 123, 256–265 CrossRef CAS PubMed.
- M. Miao, J. Zhao, X. Feng, Y. Cao, S. Cao, Y. Zhao, X. Ge, L. Sun, L. Shi and J. Fang, J. Mater. Chem. C, 2015, 3, 2511–2517 RSC.
- Q. Chen, P. Liu, F. Nan, L. Zhou and J. Zhang, Biomacromolecules, 2014, 15, 4343–4350 CrossRef CAS PubMed.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC.
- W. Kwon, G. Lee, S. Do, T. Joo and S. W. Rhee, Small, 2014, 10, 506–513 CrossRef CAS PubMed.
- S. Qu, X. Wang, Q. Lu, X. Liu and L. Wang, Angew. Chem., Int. Ed., 2012, 51, 12215–12218 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qm00225k |
| This journal is © the Partner Organisations 2017 |