DOI:
10.1039/C6QM00235H
(Research Article)
Mater. Chem. Front., 2017,
1, 967-972
Unsymmetrical α-diimine palladium catalysts and their properties in olefin (co)polymerization†
Received
26th September 2016
, Accepted 6th December 2016
First published on 14th December 2016
Abstract
Previously, it was demonstrated that nickel complexes bearing unsymmetrical α-diimine ligands generated polymers with a broad molecular weight distribution in ethylene polymerization. In this contribution, the influence of some unsymmetrical α-diimine ligands on the catalytic properties of their palladium complexes was investigated in ethylene polymerization, ethylene–methyl acrylate copolymerization and 1-hexene polymerization. Quite interestingly, the unsymmetrical α-diimine ligand structures did not lead to a broad molecular weight distribution in these polymerization and copolymerization reactions. In addition, the influence of different substituents (OMe, Me, Cl and NO2) and different backbone structures was studied. The palladium complexes bearing electron-donating groups showed higher thermal stability and afforded polymers with a much higher molecular weight. The palladium complex bearing naphthalene backbone showed a much lower catalytic activity, generating polymers or copolymers with a much lower molecular weight. The mechanical and surface properties of the resulting polymer and copolymer were also investigated.
Introduction
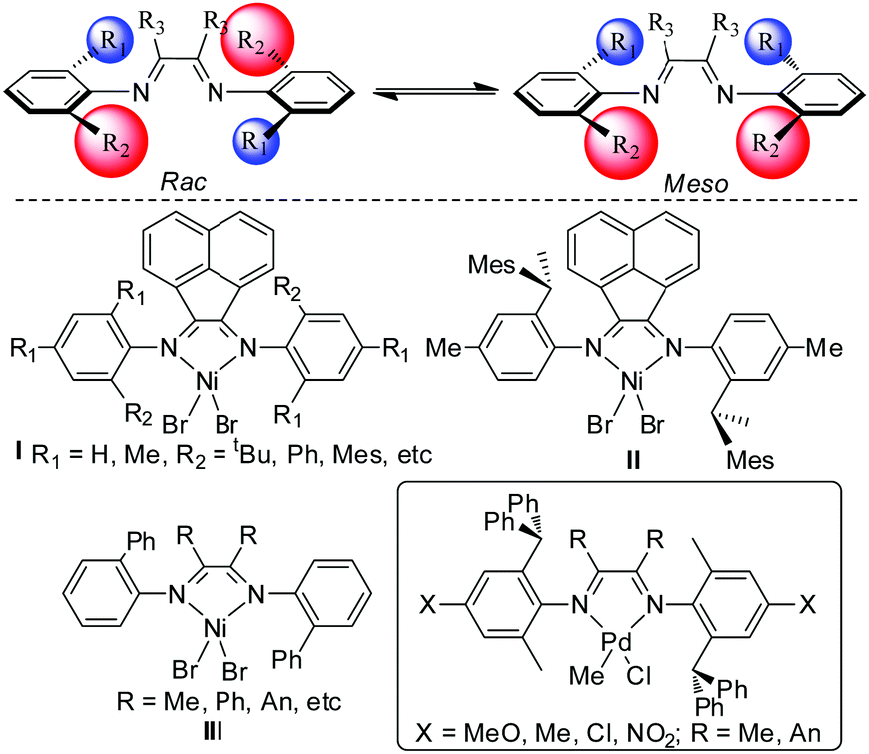
The α-diimine Ni(II) and Pd(II) catalyst system is one of the most extensively studied olefin polymerization catalyst systems.1 Despite some recent research efforts in salicylaldimine Ni(II) catalysts2 and phosphine-sulfonate Pd(II) catalysts,3 the α-diimine system remains to be of great interest in this field.4 These complexes possess many unique properties. For example, the chain walking feature enables the synthesis of a highly branched polymer microstructure using ethylene as the only feedstock. The α-diimine Ni(II) complexes are highly active in ethylene polymerization, with activities comparable with many early transition metal catalysts. The α-diimine Pd(II) complexes can copolymerize olefin with various polar vinyl monomers including acrylates, vinyl ketones, vinyl ethers, etc.1 α-diimine is surprisingly versatile and a huge amount of different α-diimine ligands have been designed and synthesized. Among them, unsymmetrical α-diimine ligand structures are very fascinating.5 The interest in exploring unsymmetrical α-diimine ligands partly originates from the possibility of realizing stereo-controlled α-olefin polymerization.
The interconversion between the rac and meso forms of unsymmetrical α-diimine ligands depends mainly on the steric bulkiness of the substituents. When the substituents are large enough, the interconversion becomes sufficiently slow at ambient temperatures. As a result, the two forms can be separated using column chromatography. Pellecchia et al. isolated the rac and the meso forms of some α-diimine Ni(II) catalysts (Scheme 1, I) and studied their performance in propylene polymerization.6 Interestingly, the two forms showed more than 5 times difference in activity and ca. 1.5 times difference in polymer molecular weight. The stereo-regularity of the resulting polypropylene was also totally different. Later on, Coates et al. prepared the pure rac form of the α-diimine Ni(II) catalyst (Scheme 1, II) and studied its properties in stereo-controlled propylene polymerization.7 Wu's group reported some unsymmetrical α-diimine ligands and the corresponding Ni(II) complexes comprising mixtures of the rac and meso forms (Scheme 1, I and III).8
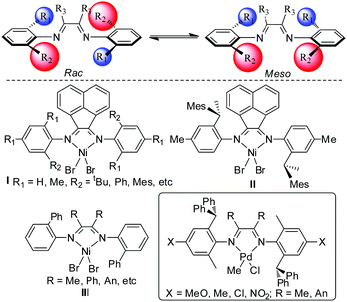 |
| | Scheme 1 Interconversion between the rac and meso forms for unsymmetrical α-diimine ligands and some previously reported unsymmetrical α-diimine ligands. | |
In ethylene polymerization, these Ni(II) complexes led to the formation of bimodal polyethylene with a very broad molecular weight distribution. High molecular weight polyethylene possesses high toughness, slow crystallization rate and high melt viscosity while low molecular weight polyethylene possesses high stiffness, fast crystallization rate and low melt viscosity. The generation of tailor polyolefins with a broad molecular weight distribution is highly desired in industry due to their unique properties for processing and applications.9 As such, the generation of such kinds of polyolefin materials in one pot is greatly advantageous.
Based on the above studies, it can be concluded that the rac and meso forms of the α-diimine Ni(II) catalysts showed very different reactivities in olefin polymerization. In contrast to the many studies of unsymmetrical α-diimine Ni(II) catalysts, there has been very little information on the properties of unsymmetrical α-diimine Pd(II) catalysts. In this contribution, we prepared some unsymmetrical α-diimine ligands and the corresponding Pd(II) catalysts. In addition, some electron donating and withdrawing substituents were installed in the ligand structure to explore the influence of the electronic effect on the polymerization properties.10 The performance of these Pd(II) complexes in ethylene polymerization, ethylene–methyl acrylate (E–MA) copolymerization and 1-hexene polymerization was investigated.
Results and discussion
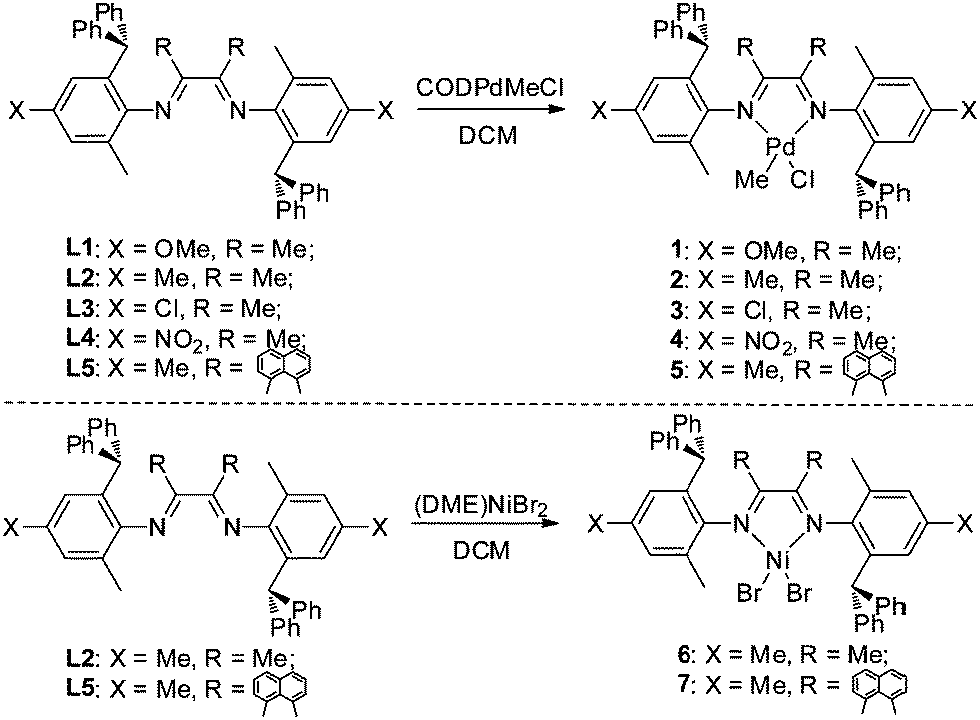
Synthesis and characterization of α-diimine ligands (L1–L5) and the metal complexes (1–7)
The α-diimine ligands (L1–L5) were easily prepared from the condensation reactions of anilines with ketones at 45–80% yields (Scheme 2). These ligands were characterized by 1H, 13C NMR and mass spectrometry. Subsequently, the reaction with 1 eq. of (COD)PdMeCl (COD = 1,5-cyclooctadiene) afforded the desired Pd complexes (1–5) in 35–76% yields. The Pd complexes were characterized by 1H, 13C NMR, mass spectrometry and elemental analysis. The Ni complexes (6 and 7) were prepared for comparison by the reaction of 1 eq. of (DME)NiBr2 (DME = ethylene glycol dimethyl ether) with L2 and L5.
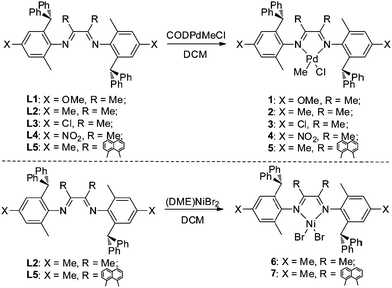 |
| | Scheme 2 Synthesis of the unsymmetrical α-diimine metal complexes. | |
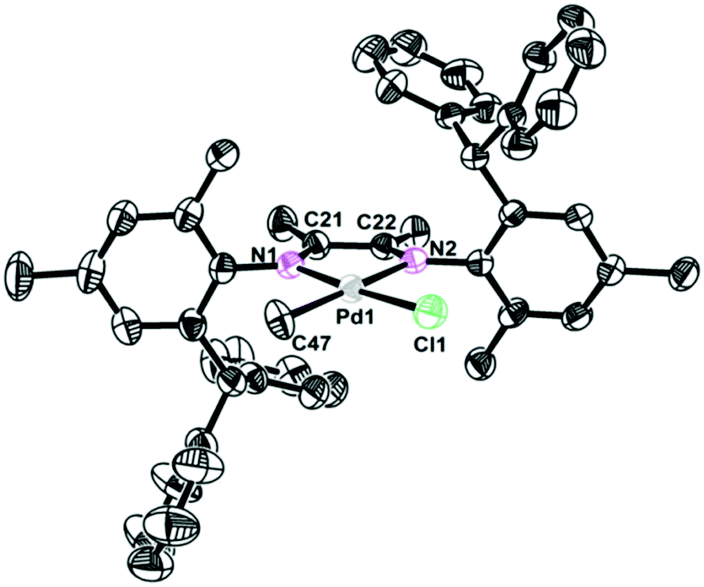
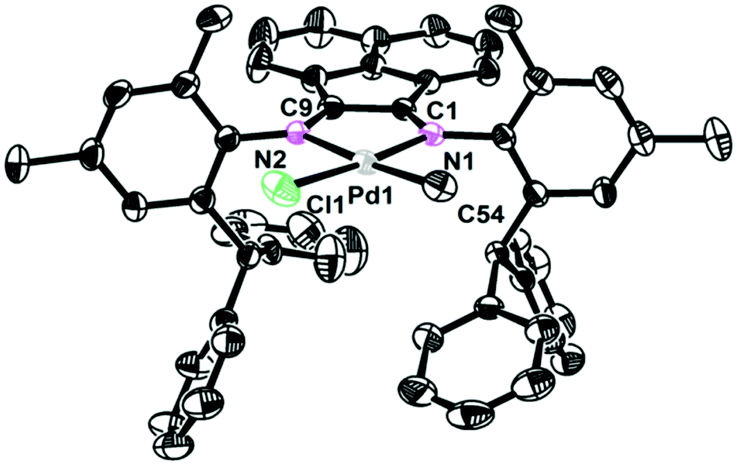
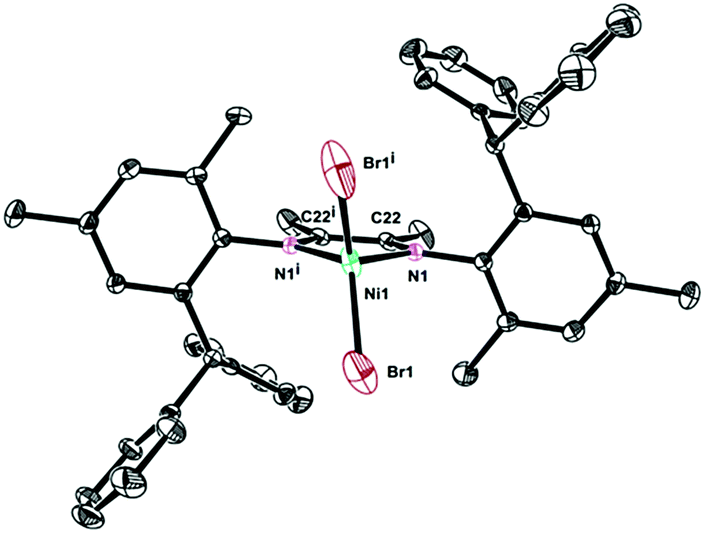
The single crystals of complexes 2, 5 and 6 were grown by layering hexane onto the CH2Cl2 solution at room temperature. The physical appearance of the crystals is uniform. The molecular structures of complexes 2, 5 and 6 were determined by X-ray diffraction analysis (Fig. 1–3). As expected, a square planar geometry was observed at the Pd center and a distorted tetrahedron geometry was observed for the Ni center. The bond angles and bond distances are typical for previously reported α-diimine Pd(II) complexes. Interestingly, the rac form was observed for complexes 2 and 6, while the meso form was observed for complex 5. Despite the similar structures of complexes 2 and 6, they showed very different behaviors in ethylene polymerization.
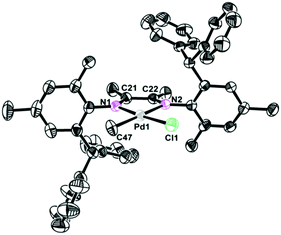 |
| | Fig. 1 Molecular structure of 2. Hydrogen atoms have been omitted for clarity. Atoms are drawn at 30% probability level. Selected bond distances (Å) and angles (deg): Pd1–N1 2.071(6), Pd1–C47 2.101(7), Pd1–N2 2.149(6), Pd1–Cl1 2.287(2) N1–Pd1–C47 97.6(3), N1–Pd1–N2 77.0(2), C47–Pd1–Cl1 87.4(2), N2–Pd1–Cl1 98.04(17). | |
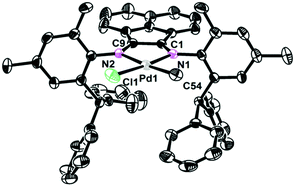 |
| | Fig. 2 Molecular structure of 5. Hydrogen atoms have been omitted for clarity. Atoms are drawn at 30% probability level. Selected bond distances (Å) and angles (deg): Pd1–C54 2.045(4), Pd1–N1 2.054(3), Pd1–N2 2.181(3), Pd1–Cl1 2.2835(13), C54–Pd1–N1 95.10(17), N1–Pd1–N2 79.40(13), C54–Pd1–Cl1 89.31(15), N2–Pd1–Cl1 96.11(10). | |
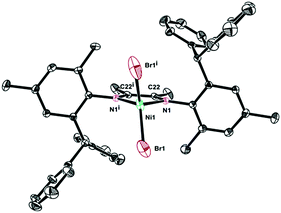 |
| | Fig. 3 Molecular structure of 6. Hydrogen atoms have been omitted for clarity. Atoms are drawn at 25% probability level. Selected bond distances (Å) and angles (deg): Ni1–Br1 1.988(5), Ni1–N1 2.2956(18), N1i–Ni1–N1 81.5(3), Br1–Ni1–Br1i 124.59(16), N1i–Ni1–Br1 116.22(13), N1–Ni1–Br1 105.24(14). | |
In the 1H and 13C NMR spectra of the Pd complexes 1–5, only one set of chemical resonances was observed for each case. In particular, for complex 1, only one Pd–Me signal and one set of chemical resonances was observed in the 1H NMR spectrum at 60 °C, −40 °C and −80 °C. This suggests that the exchange between the rac and meso forms for the Pd complexes is fast on the NMR time scale. It is reasonable to assume that the fast exchange would be maintained in the cationic (α-diimine)PdR+ active species during polymerization. This is probably a key reason that very narrow PDIs were observed for the Pd complexes in ethylene polymerization and copolymerization reactions. In contrast, the exchange is relatively slow in the Ni complexes, leading to broad polymer PDIs.
Ethylene polymerization studies
An in situ activation process using 1.2 eq. of tetrakis(3,5-bis(trifluoromethyl)phenyl)borate (NaBAF) was employed in the polymerization and copolymerization studies. These Pd complexes are all highly active in ethylene polymerization (Table 1). The catalytic activity increased when the polymerization temperature was increased from 20 to 40 °C. When the temperature was further increased to 60 °C, the activity decreased dramatically (Table 1, entries 3–5). Complex 3 bearing a Cl substituent showed the highest activity among these complexes, with an activity of up to 17.0 × 104 g mol−1 h−1 (Table 1, entries 6 and 7). Comparing with complex 2, complex 5 with the naphthalene backbone showed a much lower catalytic activity and a much lower polyethylene molecular weight (Table 1, entries 10 and 11). This might originate from the lower steric bulkiness of ligand L5 compared with L2 and correspondingly the lower thermal stability of complex 5. The acenaphthyl backbone is planar and could pull the aryl substituents backwards. In contrast, the methyl backbone is three-dimensional and could push the aryl substituents forward. Such kinds of steric effects have been reported previously in α-diimine nickel and palladium systems (the methyl backbone led to the formation of a polymer with a much higher molecular weight compared with the acenaphthyl backbone).10b,11
Table 1 Ethylene homopolymerization with the Pd and Ni complexes at 9 atma
| Ent. |
Cat. |
[cat] (μmol) |
T (°C) |
t (h) |
Yield (g) |
Act.b |
M
n
(104) |
PDIc |
B
|
|
Polymerization conditions: entries 1–11: 43 mL of toluene, 2 mL of CH2Cl2, 1.2 eq. NaBAF; entries 12 and 13: 100 mL of toluene, 2 mL of dichloromethane, 600 eq. MAO, 9 atm.
Activity is in unit of 104 g mol−1 h−1.
Determined by GPC using universal calibration.
B = branches per 1000 carbons, branching numbers were determined using 1H NMR spectroscopy.
|
| 1 |
1
|
10 |
20 |
3 |
0.4 |
1.33 |
34.3 |
1.06 |
67 |
| 2 |
1
|
10 |
40 |
3 |
1.27 |
4.23 |
22.5 |
1.54 |
66 |
| 3 |
2
|
10 |
20 |
3 |
1.4 |
4.67 |
31.6 |
1.07 |
65 |
| 4 |
2
|
10 |
40 |
3 |
2.72 |
9.07 |
20.3 |
1.43 |
68 |
| 5 |
2
|
10 |
60 |
3 |
1.35 |
4.50 |
6.7 |
1.68 |
67 |
| 6 |
3
|
10 |
20 |
3 |
1.5 |
5.00 |
23.5 |
1.32 |
67 |
| 7 |
3
|
10 |
40 |
3 |
5.1 |
17.0 |
16.4 |
2.07 |
68 |
| 8 |
4
|
10 |
20 |
3 |
1.2 |
4.00 |
17.2 |
1.37 |
68 |
| 9 |
4
|
10 |
40 |
3 |
0.88 |
2.93 |
16.0 |
1.78 |
73 |
| 10 |
5
|
10 |
20 |
3 |
0.7 |
2.33 |
2.7 |
1.51 |
65 |
| 11 |
5
|
10 |
40 |
3 |
1.15 |
3.83 |
2.3 |
2.33 |
62 |
| 12 |
6
|
3 |
30 |
0.5 |
2.88 |
192 |
5.3 |
4.98 |
50 |
| 13 |
7
|
3 |
30 |
0.5 |
2.49 |
166 |
4.1 |
6.41 |
48 |
Generally, the catalysts bearing electron-donating substituents (complexes 1 and 2) generated polyethylene with a higher molecular weight than the catalysts bearing electron-withdrawing substituents (complexes 3 and 4). Complexes 1, 2 and 3 could maintain high activity after 24 h at room temperature (Fig. S1, ESI†). Specifically for complexes 1 and 2, the living polymerization feature was observed at 20° (PDI very close to 1, Table 1, entries 1 and 3). The polymer branching density is largely independent of the α-diimine ligand structure and the polymerization conditions, which agrees well with previously reported α-diimine palladium complexes.1 Most interestingly, very narrow PDIs (less than 2) were observed for all the cases. This suggests that the rac and the meso forms of the palladium complexes possess very similar properties in ethylene polymerization, which is totally different from previously reported α-diimine nickel complexes. The Ni complexes 6 and 7 studied in this work also led to the formation of polyethylene with very broad PDIs (Table 1, entries 12 and 13, Fig. S2, ESI†), which agrees very well with previous reports.
E–MA copolymerization studies
Under the same activation conditions, these palladium complexes except complex 4 were able to incorporate the MA monomer during ethylene polymerization (Table 2). When the MA monomer was introduced, both the catalytic activity and the polymer molecular weight were greatly reduced. MA incorporation ratios of 3–4% were observed for complexes 1, 2, 3 and 5 (Table 2, entries 2, 4, 6 and 10). The NO2 substituent in complex 4 makes the Pd center more electrophilic and more prone to MA poisoning, which is probably the main reason for the absence of MA incorporation (Table 2, entry 8). This electronic effect also agrees well with previous studies on α-diimine catalyst systems.7 Similar to ethylene homopolymerization, the branching densities of the copolymer were relatively independent of the electronic properties of the ligands.
Table 2 Ethylene homopolymerization and ethylene/MA copolymerization with the palladium complexes at 1 atma
| Ent. |
Cat. |
[MA] (M) |
T (°C) |
X
MA
(%) |
Yield (g) |
Act.c |
M
n
(104) |
PDId |
B
|
|
Conditions: total volume of toluene and MA: 43 mL, 2 mL of CH2Cl2, 10 μmol catalyst, 1.2 eq. NaBAF, 12 h.
Determined by 1H NMR spectroscopy.
Activity is in unit of 103 g mol−1 h−1.
Determined by GPC using universal calibration.
B = branches per 1000 carbons, branching numbers were determined using 1H NMR spectroscopy, the branches ending with functional groups are added to the total branches.
|
| 1 |
1
|
0 |
25 |
— |
4.50 |
142 |
14.2 |
2.16 |
63 |
| 2 |
1
|
1 |
25 |
4.3 |
0.11 |
0.92 |
5.4 |
1.40 |
76 |
| 3 |
2
|
0 |
25 |
— |
3.62 |
134 |
13.4 |
2.39 |
66 |
| 4 |
2
|
1 |
25 |
4.2 |
0.09 |
0.75 |
3.5 |
1.30 |
84 |
| 5 |
3
|
0 |
25 |
— |
3.41 |
125 |
12.5 |
2.55 |
69 |
| 6 |
3
|
1 |
25 |
3.2 |
0.044 |
0.37 |
1.8 |
2.05 |
88 |
| 7 |
4
|
0 |
25 |
— |
0.81 |
93 |
9.3 |
1.84 |
70 |
| 8 |
4
|
1 |
25 |
0 |
0.018 |
0.15 |
1.0 |
1.41 |
77 |
| 9 |
5
|
0 |
25 |
— |
1.02 |
17 |
1.7 |
1.83 |
64 |
| 10 |
5
|
1 |
25 |
4.8 |
0.054 |
0.45 |
2.4 |
1.74 |
83 |
1-Hexene polymerization studies
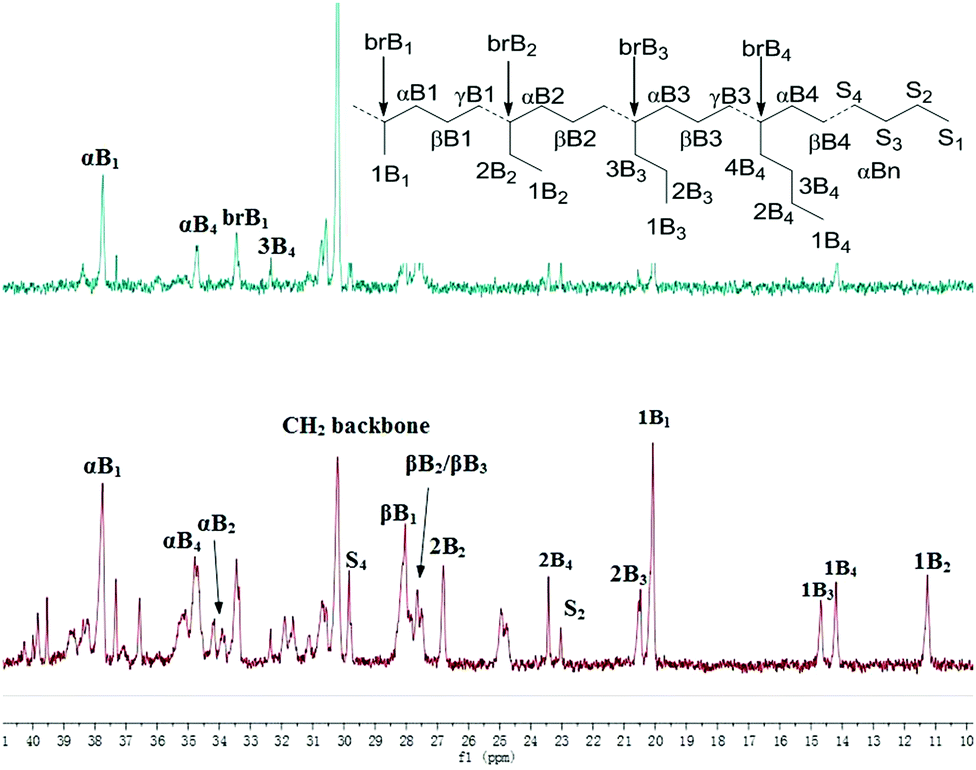
In 1-hexene polymerization, similar activities were observed for complexes 1, 2 and 3 (Table 3, entries 1–3). Complexes 4 and 5 showed much lower activities (Table 3, entries 4 and 5), which may be due to their lower thermal stability and steric effect in the case of 5. Similar to ethylene homopolymerization, the complexes bearing electron donating substituents led to the formation of polymers with a higher molecular weight. In addition, similar branching densities were observed among these Pd complexes. Interestingly, completely different microstructures were observed for poly(1-hexene) generated from catalysts 1 and 4, which is probably due to the ligand electronic effects. Only methyl and butyl branches were observed for catalyst 1, while a significant amount of ethyl and propyl branches were observed for catalyst 4 (Fig. 4).
Table 3 1-Hexene homopolymerization with the palladium complexesa
| Ent. |
Cat. |
[cat] (μmol) |
T (°C) |
t (h) |
Yield (g) |
Act.b |
Mnc (104) |
PDIc |
B
|
|
Polymerization conditions: 5 mL of toluene, 5 mL of hexane, 2 mL of dichloromethane, 1.2 eq. NaBAF.
Activity is in unit of 104 g mol−1 h−1.
Determined by GPC using universal calibration.
B = branches per 1000 carbons, branching numbers were determined using 1H NMR spectroscopy.
|
| 1 |
1
|
10 |
25 |
3 |
1.21 |
4.03 |
4.9 |
1.31 |
89 |
| 2 |
2
|
10 |
25 |
3 |
1.20 |
4.00 |
5.0 |
1.36 |
88 |
| 3 |
3
|
10 |
25 |
3 |
1.08 |
3.60 |
2.8 |
1.90 |
89 |
| 4 |
4
|
10 |
25 |
3 |
0.26 |
0.87 |
1.8 |
1.69 |
93 |
| 5 |
5
|
10 |
25 |
3 |
0.56 |
1.87 |
0.8 |
1.43 |
79 |
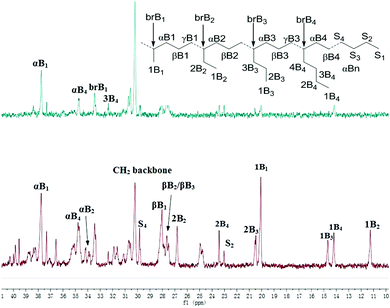 |
| | Fig. 4
13C NMR spectrum of the poly(1-hexene) generated catalysts 1 and 4 (Table 3, entries 1 and 4). | |
Properties of polyethylene and the E–MA copolymer
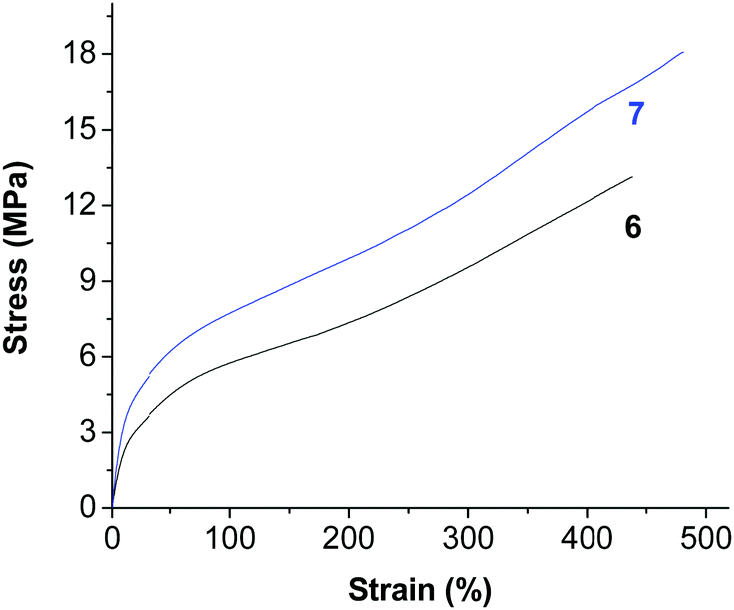
The polyethylene generated using the nickel complexes 6 and 7 showed relatively high melting points (109.6 °C for 6 and 96.9 °C for 7, Fig. S40 and S41, ESI†). In addition, these polymer materials showed very nice mechanical properties (Fig. 5, Fig. S3–S5, ESI†). Very high tensile strengths (of up to 18 MPa) and very good elastic properties (a strain at break close to 500%) were observed for these materials. The higher molecular weight and the broad molecular weight distribution may both contribute to the good performance.
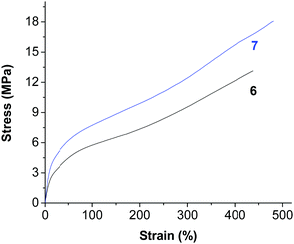 |
| | Fig. 5 Stress versus strain for polyethylene generated using complexes 6 and 7 (samples from Table 1, entries 12 and 13). | |
The incorporation of the MA unit in the polyolefin materials could dramatically improve their surface properties (related to the paintability, printability etc.). At a 3.2% MA incorporation ratio, the water contact angle can be reduced to 81° from 100° of pure polyethylene (Fig. 6). The angle could be further reduced to 74° when the MA incorporation was increased to 4.3%. When the MA unit was hydrolysed to the COOH group, the water contact angle was reduced to 41°. This clearly demonstrates the significant influence of even a small amount of polar functionalities on the properties of the polyolefin materials.
 |
| | Fig. 6 Water contact angle of polyethylene (sample from Table 2, entry 1), E–MA copolymer (samples from Table 2, entries 6 and 2) and the hydrolysed copolymer. | |
Conclusions
A series of α-diimine palladium complexes bearing unsymmetrical ligand structures were prepared and characterized. Different substituents (OMe, Me, Cl and NO2) and backbone structure were installed to study their influence on the properties of the metal complexes. The properties of these palladium complexes were investigated in ethylene polymerization, ethylene/methyl acrylate copolymerization and 1-hexene polymerization reactions. The most interesting and surprising finding of these studies is that these unsymmetrical α-diimine palladium complexes generated monodispersed polymers and copolymers with narrow PDIs. This is totally different from the Ni complex bearing these unsymmetrical ligands as well as previously reported unsymmetrical α-diimine Ni complexes, which led to the formation of polymers with very broad or bimodal GPC curves.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC, 21960071, 51522306), the Anhui Provincial Natural Science Foundation (1608085MB29), the Fundamental Research Funds for the Central Universities (WK3450000001), and the Recruitment Program of Global Experts. We thank Dr S. M. Zhou (HFNL, USTC) for the determination of the crystal structures.
Notes and references
-
(a) A. Nakamura, S. Ito and K. Nozaki, Chem. Rev., 2009, 109, 5215 CrossRef CAS PubMed;
(b) E. Y. X. Chen, Chem. Rev., 2009, 109, 5157 CrossRef CAS PubMed;
(c) Z. Guan and C. S. Popeney, Top. Organomet. Chem., 2009, 26, 179 CrossRef CAS;
(d) D. H. Camacho and Z. Guan, Chem. Commun., 2010, 46, 7879 RSC;
(e) Z. Dong and Z. Ye, Polym. Chem., 2012, 3, 286 RSC;
(f) Z. Ye, L. Xu, Z. Dong and P. Xiang, Chem. Commun., 2013, 49, 6235 RSC;
(g) D. Takeuchi, Polym. J., 2012, 44, 919 CrossRef CAS;
(h) Y. Chen, L. Wang, H. Yu, Y. Zhao, R. Sun, G. Jing, J. Huang, H. Khalid, N. M. Abbasi and M. Akram, Prog. Polym. Sci., 2015, 45, 23–43 CrossRef CAS;
(i) L. H. Guo and C. L. Chen, Sci. China: Chem., 2015, 58, 1663 CrossRef CAS;
(j) L. H. Guo, S. Y. Dai, X. L. Sui and C. L. Chen, ACS Catal., 2016, 6, 428 CrossRef CAS.
-
(a) T. R. Younkin, E. F. Connor, J. I. Henderson, S. K. Friedrich, R. H. Grubbs and D. A. Bansleben, Science, 2000, 287, 460 CrossRef CAS PubMed;
(b) H. L. Mu, L. Pan, D. P. Song and Y. S. Li, Chem. Rev., 2015, 115, 12091 CrossRef CAS PubMed;
(c) H. Makio, H. Terao, A. Iwashita and T. Fujita, Chem. Rev., 2011, 111, 2363 CrossRef CAS PubMed;
(d) M. Delferro, J. P. McInnis and T. J. Marks, Organometallics, 2010, 29, 5040 CrossRef CAS;
(e) M. P. Weberski, C. L. Chen, M. Delferro, C. Zuccaccia, A. Macchioni and T. J. Marks, Organometallics, 2012, 31, 3773 CrossRef CAS;
(f) M. P. Weberski, C. L. Chen, M. Delferro and T. J. Marks, Chem. – Eur. J., 2012, 18, 10715 CrossRef CAS PubMed;
(g) A. Osichow, I. Göttker-Schnetmann and S. Mecking, Organometallics, 2013, 32, 5239 CrossRef CAS;
(h) C. J. Stephenson, J. P. McInnis, C. L. Chen, M. P. Weberski, A. Motta, M. Delferro and T. J. Marks, ACS Catal., 2014, 4, 999 CrossRef CAS;
(i) T. Wiedemann, G. Voit, A. Tchernook, P. Roesle, I. Göttker-Schnetmann and S. Mecking, J. Am. Chem. Soc., 2014, 136, 2078 CrossRef CAS PubMed;
(j) F. Ölscher, I. Göttker-Schnetmann, V. Monteil and S. Mecking, J. Am. Chem. Soc., 2015, 137, 14819 CrossRef PubMed;
(k) J. C. Wang, E. D. Yao, Z. T. Chen and Y. G. Ma, Macromolecules, 2015, 48, 5504 CrossRef CAS;
(l) X. H. Hu, S. Y. Dai and C. L. Chen, Dalton Trans., 2016, 45, 1496 RSC.
-
(a) A. Nakamura, T. M. J. Anselment, J. Claverie, B. Goodall, R. F. Jordan, S. Mecking, B. Rieger, A. Sen, P. W. N. M. Van Leeuwen and K. Nozaki, Acc. Chem. Res., 2013, 46, 1438 CrossRef CAS PubMed;
(b) B. P. Carrow and K. Nozaki, Macromolecules, 2014, 47, 2541 CrossRef CAS;
(c) N. D. Contrella, J. R. Sampson and R. F. Jordan, Organometallics, 2014, 33, 3546 CrossRef CAS;
(d) Y. Ota, S. Ito, J. Kuroda, Y. Okumura and K. Nozaki, J. Am. Chem. Soc., 2014, 136, 11898 CrossRef CAS PubMed;
(e) R. Nakano and K. Nozaki, J. Am. Chem. Soc., 2015, 137, 10934 CrossRef CAS PubMed;
(f) Z. B. Jian, B. C. Moritz and S. Mecking, J. Am. Chem. Soc., 2015, 137, 2836 CrossRef CAS PubMed;
(g) Z. B. Jian and S. Mecking, Angew. Chem., Int. Ed., 2015, 54, 15845 CrossRef CAS PubMed;
(h) Y. L. Zhang, Y. C. Cao, X. B. Leng, C. Chen and Z. Huang, Organometallics, 2014, 33, 3738 CrossRef CAS;
(i) X. L. Sui, S. Y. Dai and C. L. Chen, ACS Catal., 2015, 5, 5932 CrossRef CAS;
(j) M. Chen, B. P. Yang and C. L. Chen, Angew. Chem., Int. Ed., 2015, 54, 15520 CrossRef CAS PubMed;
(k) M. Chen, B. P. Yang and C. L. Chen, Synlett, 2016, 1297 CrossRef;
(l) Z. X. Wu, M. Chen and C. L. Chen, Organometallics, 2016, 35, 1472 CrossRef CAS.
-
(a) Z. Guan, P. M. Cotts, E. F. McCord and S. J. McLain, Science, 1999, 283, 2059 CrossRef CAS PubMed;
(b) D. H. Leung, J. W. Ziller and Z. Guan, J. Am. Chem. Soc., 2008, 130, 7538 CrossRef CAS PubMed;
(c) C. L. Chen, S. Luo and R. F. Jordan, J. Am. Chem. Soc., 2008, 130, 12892 CrossRef CAS PubMed;
(d) F. S. Liu, H. B. Hu, Y. Xu, L. H. Guo, S. B. Zai, K. M. Song, H. Y. Gao, L. Zhang, F. M. Zhu and Q. Wu, Macromolecules, 2009, 42, 7789 CrossRef CAS;
(e) M. M. Wegner, A. K. Ott and B. Rieger, Macromolecules, 2010, 43, 3624 CrossRef CAS;
(f) C. L. Chen and R. F. Jordan, J. Am. Chem. Soc., 2010, 132, 10254 CrossRef CAS PubMed;
(g) C. L. Chen, S. Luo and R. F. Jordan, J. Am. Chem. Soc., 2010, 132, 5273 CrossRef CAS PubMed;
(h) H. Liu, W. Z. Zhao, X. Hao, C. Redshaw, W. Huang and W. H. Sun, Organometallics, 2011, 30, 2418 CrossRef CAS;
(i) D. Zhang, E. T. Nadres, M. Brookhart and O. Daugulis, Organometallics, 2013, 32, 5136 CrossRef CAS;
(j) J. L. Rhinehart, L. A. Brown and B. K. Long, J. Am. Chem. Soc., 2013, 135, 16316 CrossRef CAS PubMed;
(k) J. L. Rhinehart, N. E. Mitchell and B. K. Long, ACS Catal., 2014, 4, 2501 CrossRef CAS;
(l) T. Vaidya, K. Klimovica, A. M. LaPointe, I. Keresztes, E. B. Lobkovsky, O. Daugulis and G. W. Coates, J. Am. Chem. Soc., 2014, 136, 7213 CrossRef CAS PubMed;
(m) K. E. Allen, J. Campos, O. Daugulis and M. Brookhart, ACS Catal., 2015, 5, 456 CrossRef CAS;
(n) S. Takano, D. Takeuchi, K. Osakada, N. Akamatsu and A. Shishido, Angew. Chem., Int. Ed., 2014, 53, 9246 CrossRef CAS PubMed;
(o) L. Zhu, Z. S. Fu, H. J. Pan, W. Feng, C. L. Chen and Z. Q. Fan, Dalton Trans., 2014, 43, 2900 RSC;
(p) R. K. Wang, X. L. Sui, W. M. Pang and C. L. Chen, ChemCatChem, 2016, 8, 434 CrossRef CAS;
(q) W. P. Zou and C. L. Chen, Organometallics, 2016, 35, 1794 CrossRef CAS;
(r) R. K. Wang, M. H. Zhao and C. L. Chen, Polym. Chem., 2016, 7, 3933 RSC;
(s) S. Y. Dai and C. L. Chen, Angew. Chem., Int. Ed., 2016, 55, 13281 CrossRef CAS PubMed.
-
(a) M. Jeon and S. Y. Kim, Polym. J., 2008, 40, 409 CrossRef CAS;
(b) A. Meduri, T. Montini, F. Ragaini, P. Fornasiero, E. Zangrando and B. Milani, ChemCatChem, 2013, 5, 1170–1183 CrossRef CAS;
(c) V. Rosar, A. Meduri, T. Montini, F. Fini, C. Carfagna, P. Fornasiero, G. Balducci, E. Zangrando and B. Milani, ChemCatChem, 2014, 6, 2403–2418 CrossRef CAS.
- D. Pappalardo, M. Mazzeo, S. Antinucci and C. Pellecchia, Macromolecules, 2000, 33, 9483 CrossRef CAS.
-
(a) J. M. Rose, A. E. Cherian and G. W. Coates, J. Am. Chem. Soc., 2006, 128, 4186 CrossRef CAS PubMed;
(b) A. E. Cherian, J. M. Rose, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2005, 127, 13770 CrossRef CAS PubMed.
-
(a) H. Zou, F. M. Zhu, Q. Wu, J. Y. Ai and S. A. Lin, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 1325 CrossRef CAS;
(b) H. Zou, S. Hu, H. H. Huang, F. M. Zhu and Q. Wu, Eur. Polym. J., 2007, 43, 3882 CrossRef CAS;
(c) H. Y. Gao, F. S. Liu, H. B. Hu, F. M. Zhu and Q. Wu, Chin. J. Polym. Sci., 2013, 31, 563 CrossRef CAS.
- M. Sturzel, S. Mihan and R. Mulhaupt, Chem. Rev., 2016, 116, 1398 CrossRef PubMed.
-
(a) C. S. Popeney and Z. Guan, Organometallics, 2005, 24, 1145 CrossRef CAS;
(b) C. S Popeney and Z. Guan, Macromolecules, 2010, 43, 4091 CrossRef;
(c) C. S. Popeney, C. M. Levins and Z. Guan, Organometallics, 2011, 30, 2432 CrossRef CAS;
(d) S. Y. Dai, X. L. Sui and C. L. Chen, Angew. Chem., Int. Ed., 2015, 54, 9948 CrossRef CAS PubMed;
(e) L. H. Guo, X. L. Sui, S. Y. Dai and C. L. Chen, Polymers, 2016, 8, 37 CrossRef.
- D. P. Gates, S. A. Svejda, E. Onate, C. M. Killian, L. K. Johnson, P. S. White and M. Brookhart, Macromolecules, 2000, 33, 2320 CrossRef CAS.
|
| This journal is © the Partner Organisations 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a
*a