DOI:
10.1039/C6OB02322C
(Communication)
Org. Biomol. Chem., 2017,
15, 65-68
Enantiospecific total syntheses of meroterpenoids (−)-F1839-I and (−)-corallidictyals B and D†
Received
25th October 2016
, Accepted 10th November 2016
First published on 10th November 2016
Abstract
Enantiospecific total syntheses of spiromeroterpenoid natural products (−)-F1839-I and (−)-corallidictyals B and D were achieved using the environmentally benign and highly atom economical Lewis acid catalysed Friedel–Crafts reaction and a highly regio- and stereoselective spirocyclic C–O bond formation reaction.
Introduction
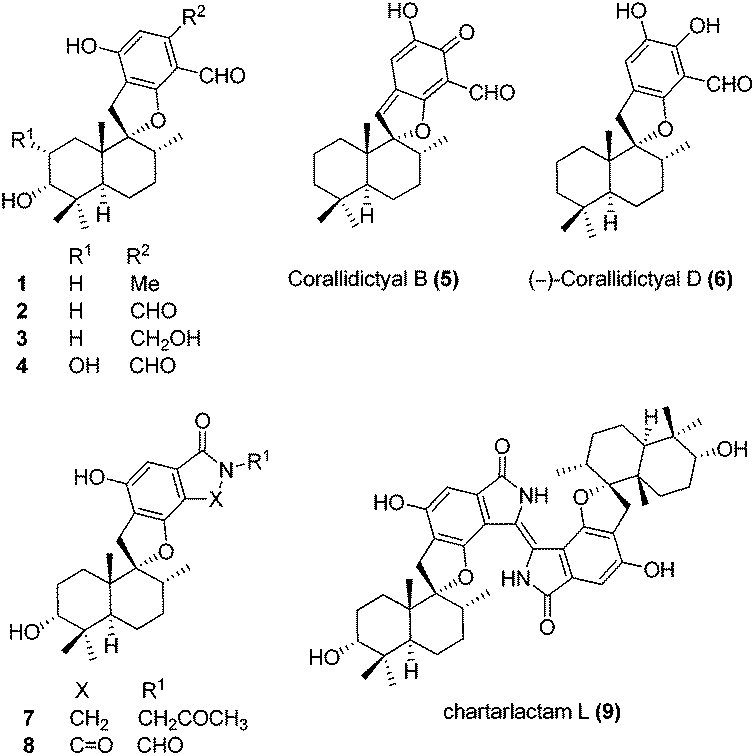
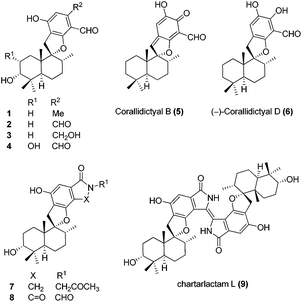
Meroterpenoids are hybrid natural products of both terpenoid and polyketide origin found widely in plants, fungi and bacteria. These secondary metabolites exhibit various biological activities, some of which are promising leads to clinical drugs. Meroterpenoid F1839-I (1) containing a spirocyclic structure was isolated by Endo and co-workers in 1994 from a fungal strain Stachybotrys sp., along with other structurally related natural products.1 F1839-I (1) is an inhibitor of pancreatic cholesterol esterase. A few examples of the related natural products with promising biological activity are, antiviral stachybotrydial (2),2 the myoinositol monophosphatase (IMPase) inhibitor L-671,776 (3)3 and the complement inhibitor K-76 (4).4 Spirosesquiterpene aldehyde corallidictyal B (5) was isolated by Chan et al.5 in 1994, from the marine sponge Aka coralliphagum, while corallidictyal D (6) was isolated from the same species by Köck and co-workers6 in 2007. Both natural products show protein kinase C inhibitory activity. In 2013, Dehai Li and co-workers7 isolated stachybotrin D (7), from the sponge-derived fungus S. chartarum MXH-X73, a novel non-nucleoside reverse transcriptase inhibitor of both wild-type HIV-1 and five NNRTI-resistant strains. Recently, Wenhan Lin and co-workers8 reported the isolation of chartarlactam A (8) and isoindolone-drimane dimer chartarlactam L (9), with a new skeleton, along with other related natural products showing antihyperlipidemic activity. Due to unique structural features and important biological activities, this class of natural products has attracted attention of synthetic chemists worldwide. To date, there is no report on the total synthesis of F1839-I reported in the literature. The first total synthesis of corallidictyal D (9) was achieved by Alvarez-Manzaneda and co-workers9a using spiroannulation of o-allyl phenols, developed in their laboratory. Recently, George et al.9b synthesised corallidictyal A–D, using Ag(I)-mediated oxidation to make corallidictyal B, and also reported a synthesis and structure revision of siphonodictyal B, proposing that the corallidictyals are artefacts of the isolation of siphonodictyal B.9c Most of the earlier approaches have relied upon the addition of a suitably protected arene nucleophile generated using pyrophoric t-BuLi, to a cyclic terpenoid moiety (Fig. 1).10
 |
| | Fig. 1 Representive examples of tetracyclic merosesquiterpenes. | |
Results and discussion
It is widely recognized that the Friedel–Crafts reaction is one of the most useful reactions for the formation of a C–C bond between an aromatic ring and an alkyl moiety. The potency of Friedel–Crafts alkylation is validated especially in the synthesis of structurally complex natural products. Despite the great importance of the Friedel–Crafts reaction for natural product synthesis, it has major drawbacks. Since stoichiometric or superstoichiometric amounts of Lewis or Brønsted acids and toxic alkyl halides have to be utilized, it leads to the generation of a lot of waste material. So to make this reaction economically benign, the development of the Friedel–Crafts reaction using a catalytic amount of a metal or an acid catalyst is highly desirable. Apart from this, the use of less toxic, alkylating reagents such as alcohols in place of alkyl halides would be a major improvement as water would be the only side product generated. In this regard, a lot of research has been done and many catalytic Friedel–Crafts reactions have been developed over the last few decades. In this manuscript, we report enantiospecific total syntheses of biologically intriguing and pharmacologically important meroterpenoid natural products F1839-I and corallidictyals B and D using the environmentally and economically benign, catalytic Friedel–Crafts alkylation reaction and a diastereoselective C–O bond formation reaction as key steps.
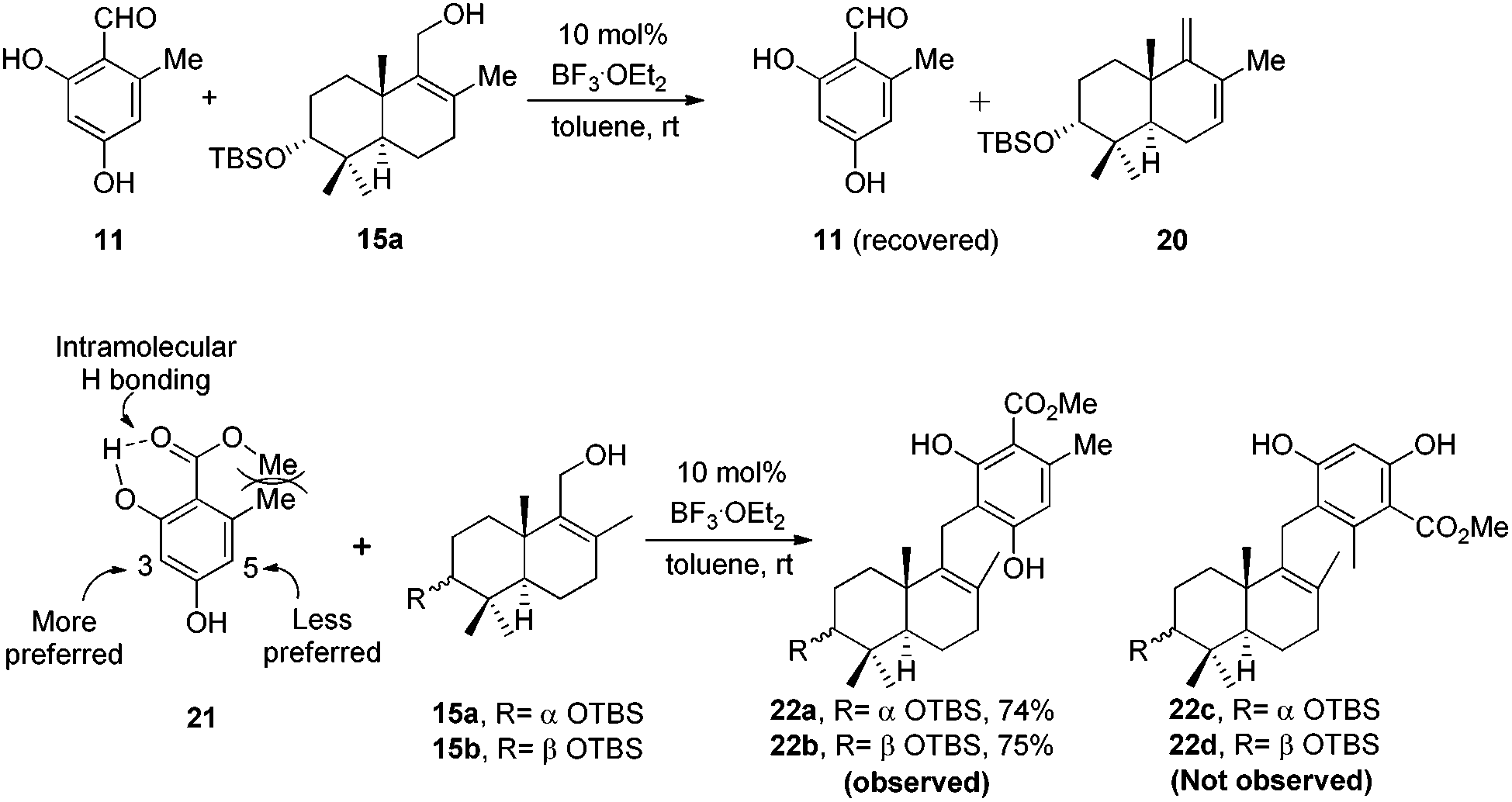
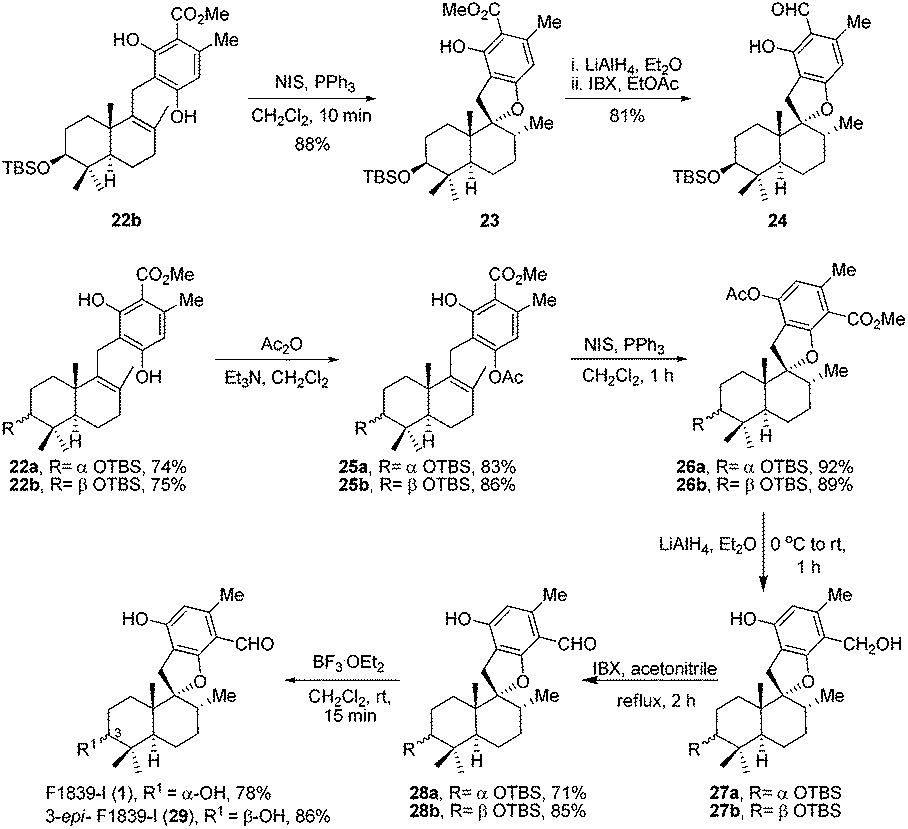
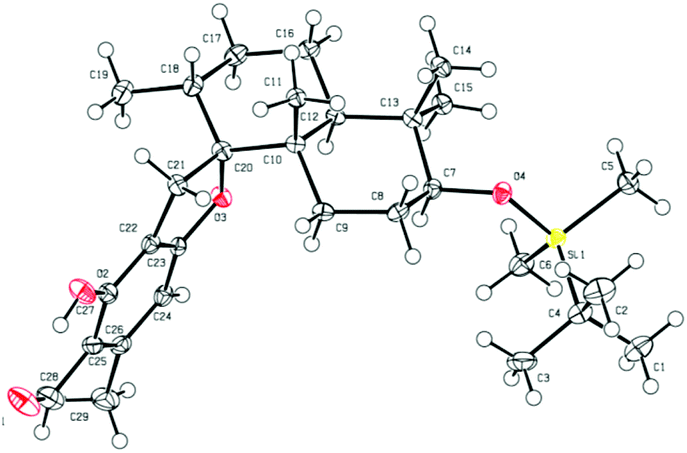
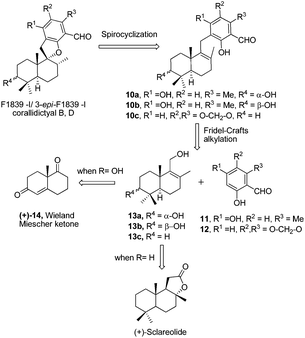
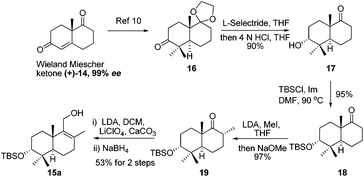
Retrosynthetic analysis of F1839-I, corallidictyals B and D is depicted in Scheme 1. It was envisioned that, F1839-I and corallidictyals B and D could be synthesized from compound 10a/b/c by diastereoselective spirocyclization. The key intermediates 10a/b/c, in turn could be prepared by the Friedel–Crafts reaction between commercially available resorcinol derivatives 11/12 and allylic alcohols 13a/b/c. The allylic alcohol 13a (when R4 = α-OH) required for the synthesis of F1839-I could be prepared from Wieland–Miescher ketone 14, whereas alcohol 13c (when R4 = H) required for the synthesis of corallidictyals B and D could be prepared from sclareolide. To begin with, the required alcohol 15a, for the synthesis of F1839-I (1), was prepared from the known trans decalone 16 in 7 steps. The required ketone 16 was prepared in 3 steps from Wieland–Miescher ketone (+)-14 (with 99% ee) by a known literature procedure.11 The ketone functionality of 16 was selectively reduced to an axial alcohol by L-selectride in THF, followed by deprotection of the ketal using 4 N HCl to afford compound 17 in 90% yield, which was converted to the corresponding TBS-ether 18 in 95% yield. Compound 18 on treatment with LDA followed by quenching of the generated enolate by methyl iodide furnished the alkylation product as a mixture of diastereomers. The crude mixture of diastereomers was epimerized to the single isomer 19 having a methyl group in the equatorial position using sodium methoxide in methanol. Ketone 19 was converted to the desired alcohol 15a in two steps using dichloromethyllithium, which was generated in situ from dichloromethane and LDA at −95 °C followed by reflux in THF. The solvents were removed and the residue was further treated with HMPA, lithium perchlorate, and CaCO3 at 130 °C, to furnish the corresponding aldehyde. It was reduced without further purification to the allylic alcohol 15a by NaBH4 in 53% yield (for 2 steps). Alcohol 15b required for the synthesis of (−)-3-epi-F1839-I was prepared from 1,3-cyclohexanedione using a known literature reported procedure.12 After having appropriately substituted alcohol 15a in hand, it was subjected to the Friedel–Crafts reaction. Unfortunately the Friedel–Crafts reaction of the allylic alcohol 15a and the benzaldehyde derivative 11 in the presence of 10 mol% BF3·OEt2 in toluene or CH2Cl2, failed to generate the expected coupling product; instead a benzaldehyde derivative was recovered, while the allyl alcohol 15a generated the elimination product 20 (Scheme 2). It was contemplated that the presence of a formyl group on the aromatic ring must have caused the deactivation of the resorcinol derivative and hence lowering its reactivity for Friedel–Crafts alkylation. To tune the reactivity of the aromatic part with terpenoid, the formyl group in 11 was replaced by an ester group (as an ester is comparatively less electron withdrawing than an aldehyde). Surprisingly, this change resulted in smooth Friedel–Crafts alkylation between ester 21 and alcohol 15a/15b to afford single regioisomers 22a and 22b in 74% and 75% yields, respectively. Although there are two positions (C-3 and C-5) available in structure 21 (Scheme 3), Friedel–Crafts alkylation took place preferentially at the C-3 position, probably due to an intramolecular hydrogen bonding between the ester and the adjacent phenolic OH group, hence C-3 attack is more preferred due to less steric crowding, compared to the more sterically crowded C-5 position. The next task was regio-, chemo- and diastereoselective C–O bond formation to generate the spirocyclic skeleton of the natural product. Compound 22b under Alvarez-Manzaneda conditions (10 mol% PPh3 and NIS)9a furnished the spirodihydrobenzofuran derivative 23 in 88% yield. Compound 23 upon LiAlH4 reduction, followed by IBX oxidation furnished aldehyde 24 in 81% yield. The structure of 24 (confirmed by X-ray crystallography13) proves that spirocyclization occurred with the undesired phenolic OH group, indicating the higher reactivity of the free phenolic OH group compared to the chelated phenolic OH group with the ester group. The structure of 24 also proves the regioselectivity of the Friedel–Crafts reaction between arene 21 and allyl alcohols 15a/b. Next the selective protection of the hydroxyl group was considered to avoid the formation of a mixture of regioisomers. Hydrogen bonding between hydroxyl and ester groups on the aromatic ring was exploited for selective acetylation of other hydroxyl groups by the treatment of compounds 22a and 22b with acetic anhydride in the presence of triethyl amine in CH2Cl2 at 0 °C for 8 h to generate acetates 25a and 25b in 83% and 86% yields, respectively. These compounds under Alvarez-Manzaneda conditions (10 mol% PPh3 and NIS),9a furnished the required spirodihydrobenzofuran derivatives 26a and 26b in 92% and 89% yields, respectively, as a single diastereomer (confirmed by NMR spectroscopy of the crude material). The acetate and ester groups in 26a and 26b were reduced by LiAlH4 to afford compounds 27a and 27b. Reduction of the ester was performed with great precautions as slight variation in temperature or reaction time leads to direct conversion of the ester group to the methyl group. Compounds 27a and 27b were quite unstable and hence oxidised to aldehydes without further purification using IBX in acetonitrile to furnish the required aldehydes 28a and 28b in 71% and 85% yields, respectively. Although our initial attempts to deprotect the TBS group, using TBAF and HF·pyridine were unsuccessful, finally we got success with BF3·OEt2, which furnished (−)-F1839-I (1) and (−)-3-epi-F1839-I (29) in 78% and 86% yields, respectively, thus completing the first total synthesis of (−)-F1839-I (1) in 13 longest linear steps with 13.7% overall yield from ketone 16 (Scheme 4). The spectral data (IR, 1H, 13C, and HRMS) were in complete agreement with those reported in the literature1 (Fig. 2).
 |
| | Scheme 1 Retrosynthetic analysis. | |
 |
| | Scheme 2 Synthesis of allyl alcohol 15a. | |
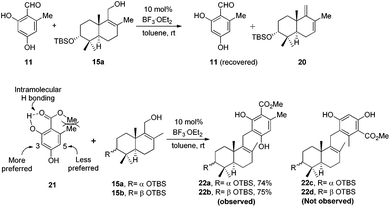 |
| | Scheme 3 Friedel–Crafts alkylation between 21 and 15a/b. | |
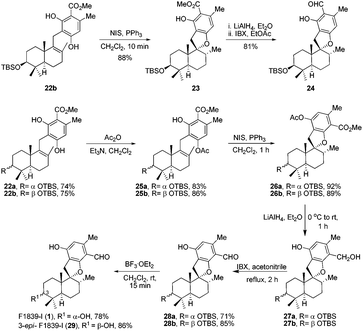 |
| | Scheme 4 Synthesis of F1839-I (1) and 3-epi-F1839-I (29). | |
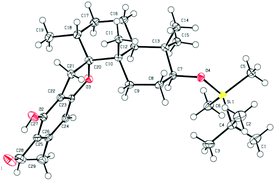 |
| | Fig. 2 ORTEP diagram for 24. | |
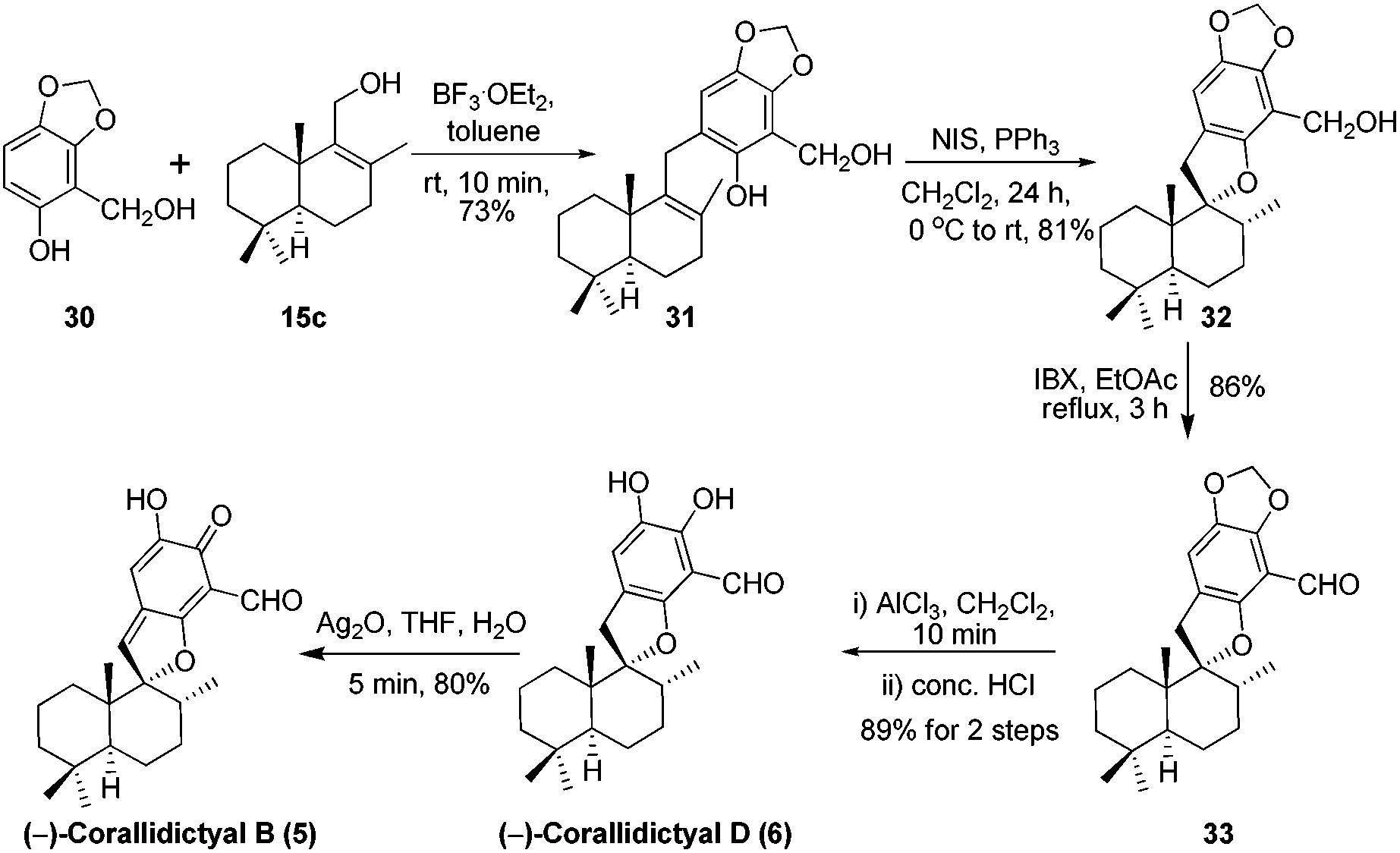
Next we targeted the syntheses of corallidictyals B (5) and D (6). To begin with, the required alcohol 15c for the synthesis of corallidictyals B and D, was prepared from sclareolide in six steps using a known literature procedure.14 Next the commercially available sesamol derivative 30 and alcohol 15c when treated with 10 mol% BF3·OEt2 in toluene, furnished compound 31 in 73% yield. Compound 31 under Alvarez-Manzaneda conditions (10 mol% PPh3 and NIS)9a yielded spirodihydrobenzofuran 32 in 81% yield. Oxidation of alcohol 32 using IBX in ethyl acetate under reflux conditions, followed by acetal deprotection using AlCl3 furnished corallidictyal D (6) in 89% yield. Dearomatization of corallidictyal D (6) by Ag2O in THF–H2O afforded corallidictyal B (5) in 81% yield. The spectral data (IR, 1H, 13C, and HRMS) of corallidictyal D (6) and corallidictyal B (5) were in complete agreement with those reported in the literature.5,6 Thus, the enantiospecific first total synthesis of corallidictyal B was achieved from sclareolide in 14.9% overall yield in 11 longest linear steps from sclareolide (Scheme 5).
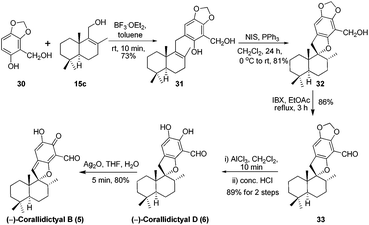 |
| | Scheme 5 Total synthesis of corallidictyals B (5) and D (6). | |
Conclusions
Total syntheses of (−)-F1839-I and (−)-corallidictyals B and D were achieved in 13, 11 and 10 steps with overall yields of 13.7%, 14.9% and 18.6%, respectively. Key transformations include the catalytic Friedel–Crafts reaction between an electron rich arene and a primary allylic alcohol to form the complete carbon framework of meroterpenoid natural products. It is noteworthy that the synthetic strategy described here is a step economy process and no extra protecting group manipulations were required in the total synthesis of spiromeroterpenoid natural products.
Acknowledgements
We thank Mr Mayank Gupta and Dr Kapil Tomar for their help in solving the crystal structure. B. D. D. thanks CSIR, New Delhi and S. A. thanks UGC, New Delhi for the award of a research fellowship. Financial support from IIT Kanpur is gratefully acknowledged.
Notes and references
- K. Sakai, K. Watanabe, K. Masuda, M. Tsuji, K. Hasumi and A. Endo, J. Antibiot., 1995, 48, 447 Search PubMed.
-
(a) W. A. Ayer and S. Miao, Can. J. Chem., 1993, 71, 487–493 CrossRef CAS;
(b) T.-W. Lin, W.-W. Chang, C.-C. Chen and Y.-C. Tsai, Biochem. Biophys. Res. Commun., 2005, 331, 953–957 Search PubMed.
- Y. K. T. Lam, C. F. Wichmann, M. S. Meinz, L. Guariglia, R. A. Giacobbe, S. Mochales, L. Kong, S. S. Honeycutt, D. Zink, G. F. Bills, L. Huang, R. W. Burg, R. L. Monaghan, R. Jackson, G. Reid, J. J. Maguire, A. T. McKnight and C. I. Ragan, J. Antibiot., 1992, 45, 1397–1403 Search PubMed.
- W. Miyazaki, H. Tamaoka, M. Shinohara, H. Kaise, T. Izawa, Y. Nakano, T. Kinoshita, K. Hong and K. Inoue, Microbiol. Immunol., 1980, 24, 1091–1098 Search PubMed.
- J. A. Chan, A. J. Freyer, B. K. Carte, M. E. Hemling, G. A. Hofmann, M. R. Mattern, M. A. Mentzer and J. W. Westley, J. Nat. Prod., 1994, 57, 1543–1548 Search PubMed.
- A. Grube, M. Assmann, E. Lichte, F. Sasse, J. R. Pawlik and M. Köck, J. Nat. Prod., 2007, 70, 504–509 Search PubMed.
- X. Ma, L. Li, T. Zhu, M. Ba, G. Li, Q. Gu, Y. Guo and D. Li, J. Nat. Prod., 2013, 76, 2298–2306 CrossRef CAS PubMed.
- Y. Li, C. Wu, D. Liu, P. Proksch, P. Guo and W. Lin, J. Nat. Prod., 2014, 77, 138–147 CrossRef CAS PubMed.
-
(a) M. J. Cano, H. Bouanou, R. Tapia, E. Alvarez, R. Alvarez-Manzaneda, R. Chahboun and E. Alvarez-Manzaneda, J. Org. Chem., 2013, 78, 9196 CrossRef CAS PubMed;
(b) A. W. Markwell-Heys and J. H. George, Org. Biomol. Chem., 2016, 14, 5546–5549 Search PubMed;
(c) A. W. Markwell-Heys, K. K. W. Kuan and J. H. George, Org. Lett., 2015, 17, 4228–4231 Search PubMed.
-
(a) E. J. Corey and J. Das, J. Am. Chem. Soc., 1982, 104, 5551–5553 Search PubMed;
(b) J. E. McMurry and M. D. Erion, J. Am. Chem. Soc., 1985, 107, 2712–2720 Search PubMed;
(c) A. S. Kende, W.-P. Deng, M. Zhong and X.-C. Guo, Org. Lett., 2003, 5, 1785–1788 Search PubMed.
- K. Yasui, K. Kawada, K. Kagawa, K. Tokura, K. Kitadokoro and Y. Ikenishi, Chem. Pharm. Bull., 1993, 41, 1698 Search PubMed.
- M. E. Jung and B. A. Duclos, Tetrahedron, 2006, 62, 9321 Search PubMed.
- CCDC 1515143 contains the supplementary crystallographic data for compound 24.
- J. H. George, J. E. Baldwin and R. M. Adlington, Org. Lett., 2010, 12, 2394 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1515143. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ob02322c |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here.