
Fluorometabolite biosynthesis: isotopically labelled glycerol incorporations into the antibiotic nucleocidin in Streptomyces calvus†
Axel
Bartholomé
a,
Jeffrey E.
Janso
b,
Usa
Reilly
b and
David
O'Hagan
*a
aUniversity of St Andrews, School of Chemistry and Centre for Biomolecular Sciences, North Haugh, St Andrews, Fife KY16 9ST, UK. E-mail: do1@st-andrews.ac.uk; Fax: +44 (0)13344 63800; Tel: +44 (0)1334 467171
bWorldwide Research and Development, Medicinal Sciences, Pfizer, 445 Eastern Point Rd, Groton, CT 06340, USA
First published on 9th November 2016
Abstract
Deuterium and carbon-13 labelled glycerols have been fed to Streptomyces calvus fermentations and isotope incorporation into the fluorine containing antibiotic nucleocidin have been evaluated by 19F-NMR. A single deuterium atom was incorporated from [2H5]- and (R)-[2H2]-glycerol into C-5′ of the antibiotic, suggesting that an oxidation occurs at this carbon after ribose ring assembly from glycerol (pentose phosphate pathway), during nucleocidin biosynthesis.
Fluorine containing natural products are exceedingly rare in nature.1 The toxin fluoroacetate (FAc) 1 is the most widely distributed natural product of this class where it has been identified in many plants2 and a few bacteria.3,4 The mode of fluoroacetate biosynthesis in plants has not been established, however its biosynthesis in bacteria is known.5Streptomyces cattleya is a bacterium that co-produces FAc 1 and 4-fluorothreonine (4-FT) 2.3 Studies carried out with S. cattleya determined that the fluorination enzyme (fluorinase) catalyses a nucleophilic substitution reaction between fluoride ion and S-adenosyl-L-methionine (Ado-Met) 3 to generate 5′-fluorodeoxyadenosine (FDA) 4.6 This is the only biosynthetic fluorination enzyme known. It has subsequently been identified by gene mining in several other bacteria.7 FDA 4 is then processed to FAc 1 and 4-FT 2. A combination of isotopic labelling studies, including isotopically labelled glycerol 5, and an understanding of the individual steps in the biosynthesis of 1 and 2 have determined that the C5′ fluoromethyl and C4′ carbon of FDA 4 are converted to FAc 1 and C3 and C4 of 4-FT 2, the circled regions in Scheme 1.8 It has also been established that both hydrogens of the fluoromethyl group of FDA 4 are retained in the conversion to the fluoromethyl groups of 1 and 2.9
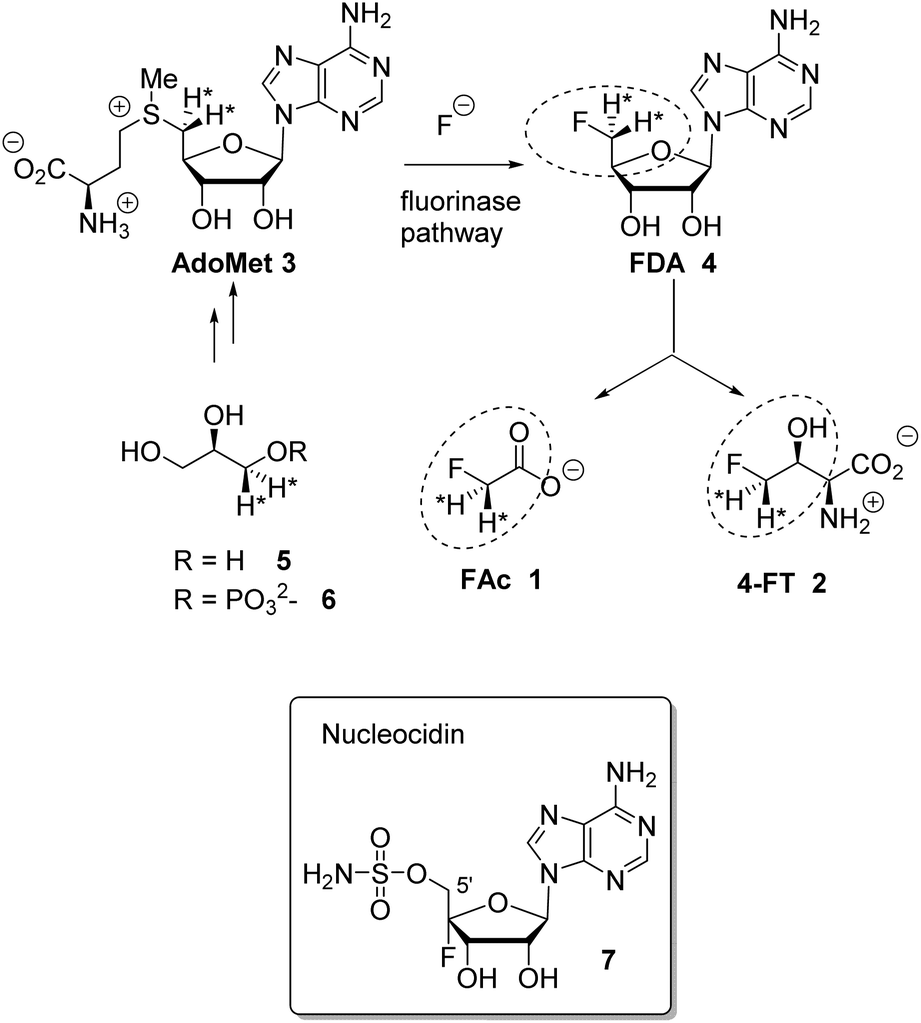 | ||
| Scheme 1 Fluorinase reaction of AdoMet 3 to give FDA 4 and then the relationship between FDA and FAc 1 and 4-FT 2. Incorporation of (R)-[2H2]-glycerol determined that both fluoromethyl hydrogens at C5′ of FDA 4 are retained in the fluorometabolites.9 Nucleocidin 7 is a metabolite of S. calvus. | ||
The antibiotic nucleocidin 7 produced by the actinomycete soil bacterium, Streptomyces calvus is another example of the rare fluorometabolites.10 Its biosynthesis is unknown; however, the presence of the fluorine atom at the C4′ position of the ribose ring11 in 7 is inconsistent with the involvement of the fluorinase found in S. cattleya. Full genome sequencing of the nucleocidin producer S. calvus ATCC 13382 has not identified a fluorinase gene,12 thus S. calvus may employ a very different process for enzymatic C–F bond formation relative to bacterial FAc 1 production.
Ever since its isolation, from S. calvus in 1956,10 nucleocidin 7 production has proved problematic, with consistently low titres and unpredictable titres. Publically deposited strains appear to have lost the ability to produce nucleocidin.12–14 Some progress has been made to understand the inconsistent production, and it has been observed that a mutation of the bldA gene, encoding a Leu-tRNA, is apparent in the publically available strains. Correction of this mutation was recently shown12 to re-establish production in S. calvus. In another study13 mutation of the rpoB gene (rifamycin resistance) led to an apparent increase in nucleocidin production. In this study we use an in-house strain of S. calvus T-3018 held by Pfizer. This strain does not have a bldA mutation, although production is still fickle with very low titres. Nucleocidin production can however be monitored, even at low production levels, directly from extracts by 19F{1H}-NMR. As a first step towards elucidating the biosynthetic pathway to fluorometabolite 7 we now report the incorporation of various isotopically labelled glycerols 5a–d.
For this study we have supplemented fermentation cultures of S. calvus with glycerols [2-13C]-5a, [2H5]-5b, (R)-[2H2]-5c and (S)-[2H2]-5d. Despite the low titres, we envisaged that the incorporation of deuterium or carbon-13 isotopes close to the fluorine atom of the antibiotic could be reported visually by 19F{1H}-NMR due to heavy atom induced chemical shifts of the fluorine resonance, and/or 13C–19F-spin–spin coupling in the case of carbon-13 incorporation.
A first experiment explored pulse feeding of glycerol [2-13C]-5a to a final concentration of 8 mM, in a shake flask fermentation of S. calvus. Cultures were worked up after 20 days as previously described.13 Nucleocidin is excreted from the bacterial cells and thus the cells were spun down in a centrifuge and the supernatant was extracted into butanol. The extract was concentrated and was then analysed by 19F{1H}-NMR. The resultant spectrum is shown in Fig. 1a. It is clear that the fluorine signal for nucleocidin has accompanying satellites due to 1JCF (232 Hz) coupling, consistent with a population (∼25%) of nucleocidin 7 molecules enriched with carbon-13 at C4′. The satellite signals are off-centre relative to the natural abundance 19F-NMR signal due to a heavy atom α-shift.8 This incorporation is entirely consistent with pentose phosphate pathway involvement in ribose biosynthesis where carbons C3–C5 of D-ribose derive from the exogenously added glycerol, feeding into the C3 metabolite pool via sn-glycerol-3-phosphate 6 (Scheme 2).15
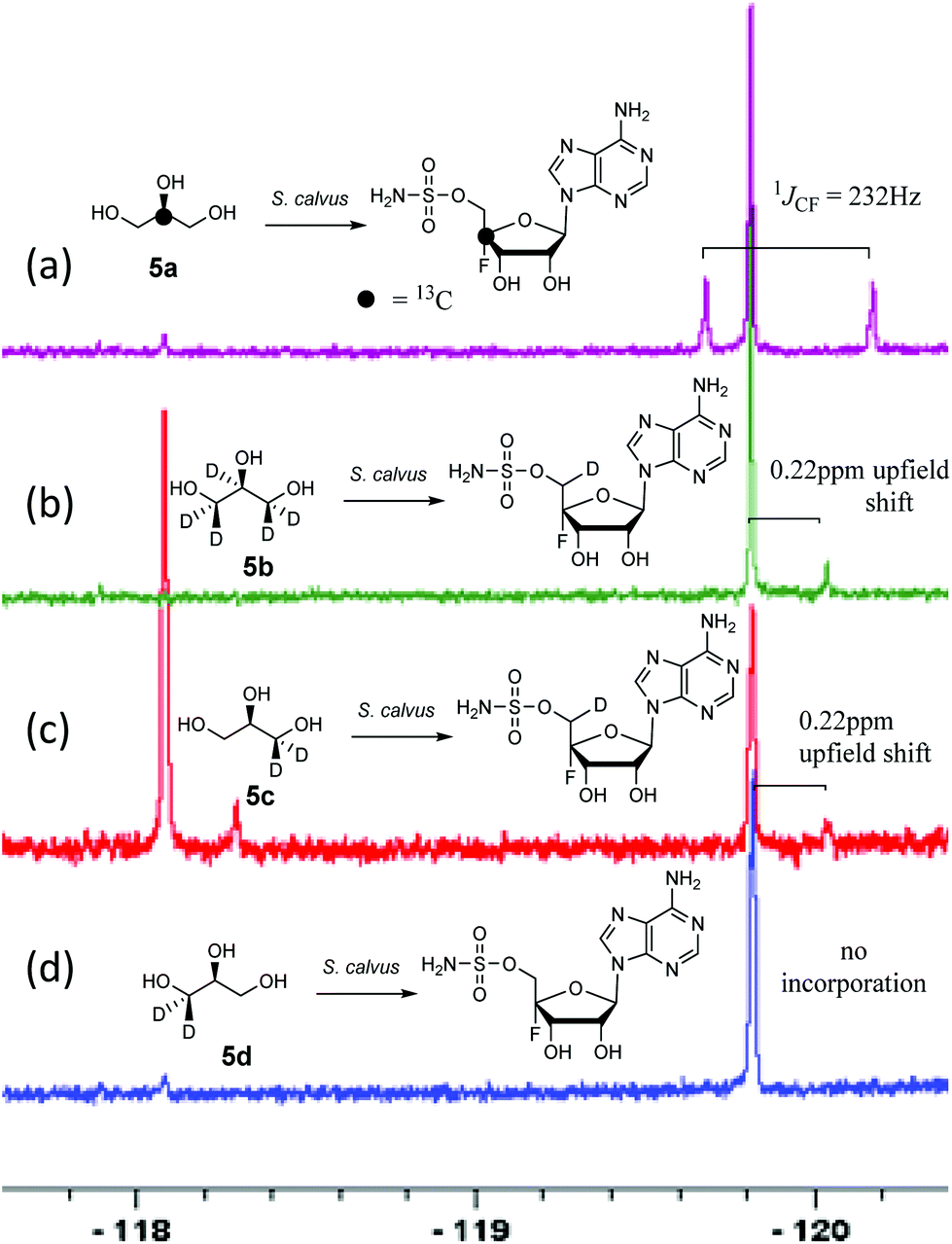 | ||
| Fig. 1 19F{1H}NMR of nucleocidin 7 (−119.8 ppm) after glycerol 5 feeding experiments to S. calvus fermentations. An unknown metabolite (−118.1 ppm) is also present in some spectra. (a) [2-13C]-glycerol 5a; (b) [2H5]-glycerol 5b; (c) (R)-[2H2]-glycerol 5c; (d) (S)-[2H2]-glycerol 5d. A pictorial summary of the isotopic labelling outcome is given in each case. | ||
 | ||
| Scheme 2 The pentose phosphate pathway showing the fate of glycerol through to ribose-5-phosphate. Glycerol 5e has a virtual labelling pattern which is a composite of the experimentally administered glycerols 5a–d, and also highlights the stereochemical fate of the (pro-R) hydroxymethyl carbon (■). This pathway predicts incorporation of a C3 glycerol unit (bold C–C bonds) into C-3–C-5 of ribose-5-phosphate, with the retention of two deuteriums at C-5 and the loss of deuteriums at C-3 and C-4. | ||
Addition of perdeuterated glycerol 5b (final conc. 8 mM) to a S. calvus fermentation led to the incorporation of deuterium (∼5%) into nucleocidin 7 as determined by an upfield shift in the resultant {1H}19F-NMR spectrum as shown in Fig. 1b. The magnitude of this upfield shift at 0.22 ppm is consistent with a single vicinal deuterium incorporated β- to the fluorine atom. There is a F–C–C–D angular dependence on the magnitude of the shift ranging from 0.15 ppm (0°) to 0.35 ppm (180°) as determined in classic studies by Lambert and Greifenstein.16 The observed value is intermediate between these extremes and consistent with a single vicinal C–D (γ-shift) bond approximately at 90° to the C–F bond. This experiment does not however distinguish the location of the incorporation between C3′ or C5′ of the ribose ring of 7. To achieve such a distinction, comparative incorporation experiments were explored with the enantiomeric glycerols (R)-[2H2]-5c and (S)-[2H2]-5d. It is already established that the pro-R arm of glycerol delivers C5′ of ribose through the pentose phosphate pathway, and that the pro-S arm delivers C3′ of ribose.15,17 Glycerols 5c and 5d were prepared synthetically with enantiomeric purities >95% ee, as previously described.9 The resultant 19F{1H}-NMR spectra from each of these glycerol feeding experiments are shown in Fig. 1c and d. It is clear that only glycerol (R)-[2H2]-5c results in the incorporation (∼8%) of deuterium into nucleocidin 7, and again only a single deuterium atom survives from the double labelled precursor. It follows from the known stereochemistry of glycerol processing through the pentose phosphate pathway, that this deuterium is located at C5′.15,17
In this experiment a second fluorometabolite was observed in the 19F{1H}-NMR spectrum with a signal at −118.1 ppm. This metabolite is detected periodically in nucleocidin extracts although its occurrence and level is unpredictable in our experience. Such a fluoro metabolite was also observed in the recent study where nucleocidin production was elicited by reversing the bldA mutation.12 Its structure is unknown although given the similar level of deuterium incorporation from glycerol 5c, it appears to be metabolically related to nucleocidin either as a biosynthetic precursor, or as a metabolite.
For isomer (S)-[2H2]-5d the absence of any deuterium at C3′ in 7 is consistent with sn-glycerol-3-phosphate 6 being processed to ribulose-5-phosphate via xylulose-5-phosphate, and the action of D-ribulose-5-phosphate 3-epimerase, with exchange of the surviving deuterium with bulk solvent.18 The predicted labelling pattern of ribose-5-phosphate from glycerol is illustrated in Scheme 2.
The outcome where both 5b and 5c glycerols contribute only one deuterium to C5′ of nucleocidin can be contrasted with the biosynthesis of FAc, 1 and 4-FT, 2 in S. cattleya discussed above and shown in Scheme 1. In those cases two deuterium atoms were incorporated into the fluorometabolites, and by implication into the C5′ ribose carbon of FDA 4.9a For nucleocidin only one deuterium is incorporated.
In summary, this study provides the first biosynthetic data on nucleocidin 7 assembly from isotope labelling studies. It is shown that glycerol is incorporated into the ribose ring moiety of nucleocidin, consistent with expectation via the pentose phosphate pathway. However, the presence of a single deuterium only at the C5′ position of nucleocidin 7 after feeding experiments with glycerols 5b and 5c suggests that a hydrogen is lost from this carbon after ribose ring assembly but prior to, or concominant with, fluorine introduction. This observation places a constraint on working hypotheses addressing nucleocidin biosynthesis.
Acknowledgements
We are grateful to Dr Alessandra Eustáquio for helping to establish this research project.References
- D. O'Hagan and H. Deng, Chem. Rev., 2015, 115, 634–649 CrossRef PubMed.
- (a) T. McEwan, Nature, 1964, 202, 827 CrossRef; (b) P. B. Oelrichs and T. McEwan, Nature, 1961, 190, 586–587 CrossRef.
- M. Sanada, T. Miyano, S. Iwadare, J. M. Williamson, B. H. Arison, J. L. Smith, A. W. Douglas, J. M. Liesch and E. Inamine, J. Antibiot., 1986, 39, 259–265 CrossRef CAS PubMed.
- (a) S. Huang, L. Ma, M. H. Tong, Y. Yu, D. O'Hagan and H. Deng, Org. Biol. Chem., 2014, 12, 4828–4831 RSC; (b) L. Ma, A. Bartholomé, M. H. Tong, Z. Qin, Y. Yu, T. Shepherd, K. Kyeremeh, H. Deng and D. O'Hagan, Chem. Sci., 2015, 6, 1414–1419 RSC.
- H. Deng, S. M. Cross, R. P. McGlinchey, J. T. G. Hamilton and D. O'Hagan, Chem. Biol., 2008, 15, 1268–1276 CrossRef CAS PubMed; X. M. Zhi, S. Hackl, M. N. Thaker, L. Kalan, C. Weber, D. S. Urgast, E. M. Krupp, A. Brewer, S. Vanner, A. Szawiola, G. Yim, J. Feldmann, A. Bechthold, S. Vanner and D. L. Zechel, ChemBioChem, 2015, 16, 2498–2506 CrossRef PubMed.
- (a) D. O. Hagan, C. Schaffrath, S. L. Cobb, J. T. G. Hamilton and C. D. Murphy, Nature, 2002, 416, 279 CrossRef PubMed; (b) C. Dong, F. L. Huang, H. Deng, C. Schaffrath, J. B. Spencer, D. O'Hagan and J. H. Naismith, Nature, 2004, 427, 561–565 CrossRef CAS PubMed.
- H. Deng, L. Ma, N. Bandaranayaka, Z. Qin, G. Mann, K. Kyeremeh, Y. Yu, T. Shepherd, J. H. Naismith and D. O'Hagan, ChemBioChem, 2014, 15, 364–368 CrossRef CAS PubMed.
- J. T. G. Hamilton, C. D. Murphy, M. R. Amin, D. O'Hagan and D. B. Harper, J. Chem. Soc., Perkin Trans. 1, 1998, 759–767 RSC.
- (a) J. Nieschalk, J. T. G. Hamilton, C. D. Murphy, D. B. Harper and D. O'Hagan, Chem. Commun., 1997, 799–800 RSC; (b) R. E. Hill, A. Iwanow, B. G. Sayer, W. Wysocka and I. D. Spencer, J. Biol. Chem., 1987, 262, 7463–7471 CAS.
- S. O. Thomas, V. L. Singleton, J. A. Lowry, R. W. Sharpe, L. M. Pruess, J. N. Porter, J. H. Mowat and N. Bohonos, Antibiot. Annu., 1956, 1956–1957, 716–721 Search PubMed.
- G. O. Morton, J. E. Lancaster, G. E. Van Lear, W. Fulmor and W. E. Meyer, J. Am. Chem. Soc., 1969, 91, 1535–1537 CrossRef CAS PubMed.
- X. M. Zhu, S. Hackl, M. N. Thaker, L. Kalan, C. Weber, D. S. Urgast, E. M. Krupp, A. Brewer, S. Vanner, A. Szawiola, G. Yim, J. Feldmann, A. Bechtold, G. D. Wright and D. L. Zechel, ChemBioChem, 2015, 16, 2498–2506 CrossRef CAS PubMed.
- K. Fukuda, T. Tamura, Y. Segawa, Y. Mutaguchi and K. Inagaki, Actinomycetologica, 2009, 23, 51–55 CrossRef CAS.
- A. R. Maguire, W. D. Meng, S. M. Roberts and A. J. Willets, J. Chem. Soc., Perkin Trans. 1, 1993, 1795–1808 RSC.
- D. C. Crans and G. M. Whitesides, J. Am. Chem. Soc., 1985, 107, 7008–7018 CrossRef.
- (a) J. B. Lambert and L. G. Greifenstein, J. Am. Chem. Soc., 1974, 96, 5120–5124 CrossRef CAS; (b) J. B. Lambert and L. G. Greifenstein, J. Am. Chem. Soc., 1973, 95, 6150–6152 CrossRef CAS.
- W. Van Winden, P. Verheijen and S. Heijnen, Metab. Eng., 2001, 3, 151–162 CrossRef CAS PubMed.
- (a) Y.-R. Chen, F. W. Larimer, E. H. Serpersu and F. C. Hartman, J. Biol. Chem., 1999, 22, 2132–2136 CrossRef; (b) M. W. McDonough and W. A. Wood, J. Biol. Chem., 1961, 236, 1220–1224 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ob02291j |
| This journal is © The Royal Society of Chemistry 2017 |