
An overview of the synthesis of acyl hydrazides from aldehydes and reactions of the products thereof
André
Shamsabadi
and
Vijay
Chudasama
*
Department of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK. E-mail: v.chudasama@ucl.ac.uk; Tel: +44 (0)207 679 2077
First published on 19th October 2016
Abstract
Herein a review on the methods for the formation and reaction of acyl hydrazides will be given. There is particular focus on the synthesis of acyl hydrazides from aldehyde precursors with examination of the various approaches (e.g. metal-based (rhodium, copper) and non-metal-based (aerobically- and photoorganocatalytically-initiated)) that have been used to achieve this. Finally, strategies to utilise acyl hydrazides for the formation of an array of useful entities will be detailed.
 André Shamsabadi | André Shamsabadi was born in London, UK. He received his undergraduate education at University College London (UCL) during which he was awarded the Jackson-Lewis Scholarship (2015), C. K. Ingold Prize (2015), Franz Sondheimer Prize (2016) and was a Dean's List commendee. In 2016, he was awarded a UCL Graduate School Research Scholarship, which allowed him to continue in a postgraduate position at UCL under the guidance of Dr Vijay Chudasama. His research interests are based in radical organic synthesis and aerobic C–H activation. |
 Vijay Chudasama | Dr Vijay Chudasama obtained his MSci degree and PhD from University College London in 2008 and 2011, respectively. Following post-doctoral studies under the supervision of Prof. Stephen Caddick, Vijay obtained a Ramsay Memorial Fellowship. During this time, he was made Technical Director of a biotechnology spin-out (ThioLogics™). In April 2015, he was awarded a lectureship at UCL for him to focus on the research areas of aerobic C–H activation and various aspects of Chemical Biology. Vijay's research has recently been highlighted by Forbes, Scientific American, CNN News, Nature Chemistry and the Royal Society of Chemistry. |
Acyl hydrazides (Fig. 1, otherwise referred to as ‘hydrazine imides’) represent synthetically versatile scaffolds that have been successfully exploited for the construction of various useful moieties such as thioesters, esters, amides1 and ketones,2 as well as building blocks for the creation of bioactive molecules such as hydroxamic acids3 and macrocylic enamides.4 They have been prepared from many methods through the years with a substantial increase in the number of reported methodologies in the past decade. Whilst there are some exceptions,5 the majority of acyl hydrazides are prepared from reaction of an aldehyde and an azodicarboxylate, which will be the focus of the initial part of this review. The latter part of the review will detail how these acyl hydrazide motifs have been used in organic synthesis.
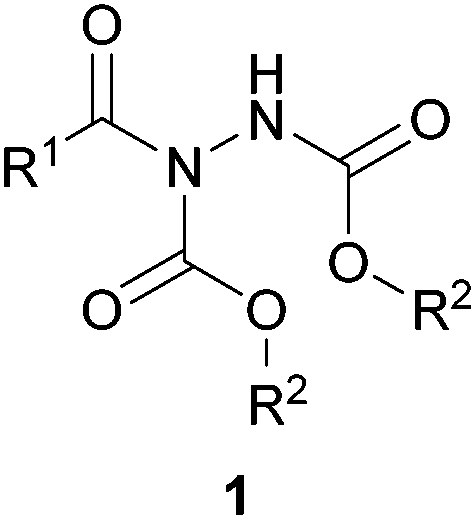 | ||
| Fig. 1 The general structure of an acyl hydrazide. | ||
Classical route to acyl hydrazides from aldehydes
The activation of C–H bonds is a popular and important area of organic chemistry,6–14 and it has been used to good effect in the synthesis of acyl hydrazides. Aldehydes are abundant, readily available entities that possess an innate and distinctive C–H bond, which makes them good candidates to participate in C–H activation reactions. Whilst a large number of C–H activation strategies, including those based on aldehydes, focus on pathways leading to C–C bond formation, C–N bond formation via this route is also an important category of reaction in organic synthesis. The formation of the acyl hydrazide functionality from aldehydes and azodicarboxylates via aldehydic C–H bond activation represents one such example in which a C–N bond is formed in a process known as hydroacylation (Scheme 1). The good efficiency of the reaction can be attributed to the strongly electron-deficient nature of the N–N double bond of azodicarboxylates; the low energy LUMO serve as an excellent radical/ionic-based15 nucleophile acceptor. | ||
| Scheme 1 Hydroacylation of azodicarboxylate 3 with aldehyde 2 to form azodicarboxylate 1. | ||
The reviewing of the formation of acyl hydrazides via hydroacylation will be separated into two sections: metal-catalysed and non-metal catalysed processes.
Metal-catalysed
Rhodium
The first metal-mediated aldehydic C–H activation reaction employing an azodicarboxylate as an electrophile was published by Lee and Otte in 2004.16 The groups initially had the intention to screen many transition metal catalysts in an attempt to promote a reaction between n-hexanal 2a and diisopropyl azodicarboxylate (DIAD) 3a (ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to make acyl hydrazide 1aa. However, in an initial control reaction, the reaction between n-hexanal and DIAD in THF, in the absence of catalyst, at 25 °C was examined. They obtained an approximate 1:1 ratio of acyl hydrazide 1aa and THF-adduct 4 (where C–H activation occurred at the ethereal α-position) with complete consumption of the azodicarboxylate after 3 days (Scheme 2).
:1) to make acyl hydrazide 1aa. However, in an initial control reaction, the reaction between n-hexanal and DIAD in THF, in the absence of catalyst, at 25 °C was examined. They obtained an approximate 1:1 ratio of acyl hydrazide 1aa and THF-adduct 4 (where C–H activation occurred at the ethereal α-position) with complete consumption of the azodicarboxylate after 3 days (Scheme 2).
 | ||
| Scheme 2 The hydroacylation reaction between n-hexanal 2a and DIAD 3a to form hydroacylation product 1aa and THF-adduct 4. | ||
Logically, in an attempt to eliminate formation of the THF-adduct, the reaction was carried in the absence of solvent. In this case, whilst the desired acyl hydrazide product was now formed exclusively the rate of reaction was very slow; complete conversion was only observed after 14 days. In an attempt to find a catalyst that would improve the rate whilst retaining the selectivity for acyl hydrazide 1aa, a wide variety of metal catalysts were screened in THF at 10 mol%. [Rh(OAc)2]2 was found to show the best catalytic activity with complete consumption of the azodicarboxylate observed after 15 minutes, however, it did provide a ca. 1:1 ratio of the acyl hydrazide 1aa and THF-adduct 4. The remainder of the screened catalysts, which included a wide variety of Pd, Ru and Rh species and Lewis acids, failed to show any significant activity above the background level (i.e. no catalyst) with the exception of [RuCl2(C6H6)2], which was still ca. 20% less efficient than [Rh(OAc)2]2.
A solvent screen with [Rh(OAc)2]2 as catalyst was then carried out to discover a reaction medium that would suppress the formation of the undesirable solvent-adduct by-product and promote formation of desired acyl hydrazide 1aa. Reactions carried out in hexane and toluene resulted in very slow reaction rates whilst use of other ethereal-based solvents such as Et2O and 1,4-dioxane resulted in the formation of their respective solvent-adducts compounds—albeit to a slightly smaller degree when compared to THF. Dichloromethane (DCM) proved to be especially detrimental to the reaction with no reaction observed at all, even after several days. Reaction in ethyl acetate (EtOAc), however, provided exclusively the acyl hydrazide product. Furthermore, [Rh(OAc)2]2 loadings as low as 1.5–2.0 mol% were tolerated before reaction yield was significantly affected in a negative manner. Under the optimised conditions (1.5–2.0 mol% of [Rh(OAc)2]2, 1.0 M, EtOAc, 25 °C), acyl hydrazide 1aa was obtained in 84% yield.
With conditions for the rhodium-catalysed procedure optimised (Scheme 3), the scope of the reaction with respect to aldehyde tolerance was next explored. Generally, the reaction appeared to perform quicker with smaller aldehydes (97% yield in 12 h when utilising propionaldehyde) with an increase in the number of carbon atoms roughly correlating with a slower rate of reaction being cited. Aldehydes with unsaturation at terminal or internal positions proceeded in slightly lower yields due to competing ene-type reaction pathways. With the exception of crotonaldehyde 2b (Scheme 4, 84% yield of acyl hydrazide 1ba), reactions featuring α,β-unsaturation or aromatic aldehydes were rather inefficient, with poor yields and long reaction times being observed; the presence of electron-donating groups (EDGs) on the aromatic moiety was particularly detrimental.
 | ||
| Scheme 3 Reaction was performed with 1.5–2.0 mol% [Rh(OAc)2]2 with a 1:1 ratio of aldehyde to azodicarboxylate in EtOAc (1.0 M) at 25 °C. | ||
 | ||
| Scheme 4 Reaction was performed with 1.5–2.0 mol% [Rh(OAc)2]2 with a 1:1 ratio of crotonaldehyde 2b to DIAD 3a in EtOAc (1.0 M) at 25 °C for 24 h. | ||
A radical mechanism was postulated for the addition of aldehyde to azodicarboxylate in the absence of catalyst and this was supported by the inhibition of the reaction in the presence of a radical scavenger. However, no mechanism was proposed for activation or for the formation of the product. Reaction with a catalyst loading of 10 mol% [Rh(OAc)2]2 was found to proceed up to 5 mol% of radical scavenger but with complete inhibition at 10 mol%. This led the authors to conclude that the rhodium-catalysed reaction may proceed via radical mechanism and that the role of rhodium catalyst was not completely clear at the time.
Copper
With a view to improve on the poor yields obtained for hydroacylation of azodicarboxylates with aromatic aldehydes when using [Rh(OAc)2]2,16 Peng et al. explored the use of an alternative metal; cheap and environmentally benign copper.17 A reaction using 2 equivalents of benzaldehyde to DIAD was used as a model for optimisation, where various copper species at 20 mol% were trialled; copper(II) acetate monohydrate proved to be the leading candidate.After settling on the catalyst, the influence of the reaction medium was next explored. Although no discernible pattern between solvent properties (e.g. polarity, viscosity) and reaction efficiency were identified, solvent was shown to heavily influence the reaction (Table 1). Despite the lack of a trend within the individual series, as with the work by Lee and Otte,16 the best yield was observed with ethyl acetate being employed as solvent and dichloromethane was once again shown to be amongst the worst performing solvents (Table 1).
|
|
||
|---|---|---|
| Entry | Solvent | Yield/% |
| Reaction conditions: DIAD (0.5 mmol), benzaldehyde (1 mmol), Cu(OAc)2·H2O (20 mol%), solvent (5 mL), RT, 48 h. | ||
| 1 | EtOAc | 98 |
| 2 | MeCN | 90 |
| 3 | MeOH | 72 |
| 4 | THF | 60 |
| 5 | CH2Cl2 | 52 |

With respect to catalyst loading, whilst amounts above 20 mol% did not improve yield, the rate of reaction rate was slightly faster (36 h compared to 48 h). Interestingly, although catalyst loadings below 20 mol% led to suboptimal reaction yields, the yields were still very high (84% yield at 10 mol%) and, perhaps as expected, the absence of catalyst resulted in a highly inefficient reaction.
Finally, lowering the ratio of benzaldehyde to DIAD was explored: from 2:1 to 1.2:1. Interestingly, whilst lowering the equivalence of benzaldehyde resulted in a decrease in yield, at the 1.2:1 cut-off in their study a high yield of 82% was still observed. It is slightly disappointing that the reaction was not trialled with a 1:1 ratio of starting materials to make a fair comparison with the article by Lee and Otte,16 and that all subsequent exemplification took place with an excess of aldehyde (2 equivalents) as the aldehyde component is generally perceived as the more valuable entity of the two starting materials.
The optimised conditions (Scheme 5) did provide good to excellent yields on application of both aromatic and aliphatic aldehydes (60–99%). There also seemed to be no reduction in yield when employing larger aldehydes as was the case when using the rhodium acetate catalyst.16 The use of copper(II) acetate monohydrate as the coupling catalyst17 generally provided superior yields when compared to applying rhodium(II) acetate,16 especially in the utilisation of electron-rich aromatic aldehydes. For example, application of 4-methoxybenzaldehyde provided a higher reaction yield of its corresponding acyl hydrazide (86% compared to 46%) and a marked improvement in the use of 4-dimethylaminobenzaldehyde (60% compared to 26%) was also observed. Heterocyclic aldehydes were also tolerated and gave good to excellent yields of hydroacylation product, thus broadening the scope of the original reaction discovery further (Scheme 6). Interestingly, unlike the work by Lee and Otte,16 no competing ene-type reactions were observed on application of copper (e.g. for reaction with an aromatic aldehyde possessing a para-dimethylamino group). However, these conclusions should all be caveated by the fact that a vast excess of aldehyde was used and thus that if such an excess of aldehyde was used in the presence of [Rh(OAc)2]2 similar yields/conclusions may have been observed/drawn.
 | ||
| Scheme 5 Reaction was performed with 20 mol% Cu(OAc)2 with a 2:1 ratio of aldehyde to DIAD 3a in EtOAc (5 mL) at room temperature. | ||
 | ||
| Scheme 6 Reaction was performed with 20 mol% Cu(OAc)2 with a 2:1 ratio of aldehyde 2d to DIAD 3a in EtOAc (5 mL) at room temperature. | ||
Nothing definitive was determined about the mechanism of the transformation using copper but a radical mechanism via a chain reaction process was proposed. The radical mechanism hypothesis was supported by observation of complete inhibition of reaction when it was carried out with 4 mol% of hydroquinone, a radical scavenger.
Copper oxide nanoparticles
In 2012, Mandal et al. reported the use of copper(II) oxide nanoparticles as effective catalysts in the hydroacylation reaction of aldehydes and azodicarboxylates.18 The group aimed to improve on the issues with past coupling methods such as long reaction times and poor to no reusability of the active catalyst. The reactions used by Mandal et al. marked the first time a heterogeneous catalyst was effectively used to achieve hydroacylation via azodicarboxylate functionalisation. Initially, a solvent screen with silica-supported copper(II) oxide nanoparticles was conducted in a few common organic solvents with 1.2 equivalents of aldehyde to azodicarboxylate. The screening process showed that acetonitrile was the optimal reaction solvent, providing the highest yield at 60 °C (Table 2). Slightly puzzlingly, however, ethyl acetate was not trialled in the solvent screen despite it being the best solvent in the aforementioned metal-catalysed coupling reactions.16,17|
|
||
|---|---|---|
| Entry | Solvent | Yield/% |
| Reaction conditions: DIAD (1 mmol), benzaldehyde (1.2 mmol), CuO-np/SiO2 (10 mol%), solvent (5 mL), 60 °C, 24 h. | ||
| 1 | MeCN | 88 |
| 2 | MeOH | 73 |
| 3 | 1,4-Dioxane | 69 |
| 4 | THF | 62 |
| 5 | Toluene | 56 |

Under the optimal solvent and temperature conditions, a few other potential reaction catalysts were trialled including the use of copper(II) oxide nanoparticles without silica support (Table 3). It was shown that the silica support network resulted in a more efficient reaction and that, under the conditions studied, silica-supported copper(II) oxide nanoparticles provided superior yields to the copper(II) acetate previously utilised by Peng et al.,17 as well as to trichlorides of ruthenium and rhodium. Finally, it was shown that lowering the catalyst loadings led to a reduction in reaction efficiency and that a 10 mol% loading was necessary for good yield to be achieved.
|
|
||
|---|---|---|
| Entry | Catalyst | Yield/% |
| Reaction conditions: DIAD (1 mmol), benzaldehyde (1.2 mmol), CuO-np/SiO2 (10 mol%), MeCN (5 mL), 60 °C, 24 h. | ||
| 1 | CuO-np/SiO2 | 88 |
| 2 | Cu(OAc)2 | 73 |
| 3 | CuO-np | 69 |
| 4 | RuCl3 | 62 |
| 5 | RhCl3 | 56 |
| 6 | Without catalyst | No reaction |

With optimised conditions in hand, reactions with various aldehydes were trialled and it was observed that aromatic aldehydes provided comparable yields (60–95%) of their corresponding acyl hydrazides when compared to the methodology developed by Peng et al. (i.e. copper(II) acetate catalysis).17 Electron-rich aromatic aldehydes proved to be better reaction partners than electron-deficient aromatic aldehydes (e.g. use of 4-methoxybenzaldehyde achieved an 86% yield of its resultant acyl hydrazide in 24 h whilst 4-nitrobenzaldehyde afforded a 67% yield in 96 h). Reaction with aliphatic aldehydes provided excellent yields of hydroacylation products (90–93%) in relatively short reaction times (15–18 h) and, consistent with previous studies,16,17 proved to be more efficient than reaction with aromatic aldehydes.
When compared to the copper(II) acetate monohydrate catalyst utilised by Peng et al.,17 the use of copper(II) oxide nanoparticles appears to be an attractive alternative due to requiring a lower equivalence of aldehyde and a lower catalyst loading. Furthermore, the silica-supported CuO catalyst is easily recovered by filtration and can be recycled. Mandal et al. showed that even after the fifth cycle of catalysing the optimised reaction of benzaldehyde with DIAD, the change in yield was minimal (83% from 85%).18 On average, the use of supported copper(II) oxide nanoparticles also allowed shorter reaction times. However, the copper(II) acetate monohydrate process described by Peng et al. operates at room temperature (Table 4); this temperature factor may be critical when employing aldehydes that are prone to facile decarbonylation, polymerisation or are volatile.
| Reaction conditions | Cu(OAc)2·H2O | CuO-np/SiO2 |
|---|---|---|
| Solvent | EtOAc | MeCN |
| Temperature /°C | RT | 60 |
| Catalyst loading/mol% | 20 | 10 |
| Aldehyde equivalence | 2 | 1.2 |
Zinc
Following from the work with copper(II) acetate monohydrate,17 Qin et al. studied the use of an alternative metal catalyst (i.e. zinc, also inexpensive and environmentally benign) in an attempt to further improve efficiency when using aromatic aldehydes in the hydroacylation of azodicarboxylates.19 Taking inspiration from contemporary reactions that employ zinc acetate as a catalyst, e.g. the hydrosilylation of aldehydes,20 Qin et al. set about trialling zinc(II) acetate dihydrate as a catalyst for hydroacylation. With the primary target of improving the yields and reaction times when utilising aromatic aldehydes, the reaction of benzaldehyde and DIAD (2:1 ratio) was chosen as the model reaction to screen solvents, catalyst loading and aldehyde equivalency. The reaction efficiency was found to be highly dependent on solvent (Table 5) with acetonitrile proving to be the optimal organic solvent. Interestingly, in this case, ethyl acetate, which previously outperformed acetonitrile when using a rhodium or copper acetate catalyst, had a limiting effect on yield.16,17
|
|
||
|---|---|---|
| Entry | Solvent | Yield/% |
| Reaction conditions: DIAD (0.5 mmol), benzaldehyde (1 mmol), Zn(OAc)2·2H2O (20 mol%), solvent (3 mL), RT, 48 h. | ||
| 1 | MeCN | 80 |
| 2 | 1,4-Dioxane | 61 |
| 3 | DMF | 56 |
| 4 | EtOAc | 54 |
| 5 | DCM | 52 |
| 6 | MeOH | 51 |
| 7 | THF | 42 |
| 8 | EtOH | 38 |

Similar to the circumstance when using copper(II) acetate monohydrate as the catalyst, the use of catalyst loadings greater than 20 mol% did not result in an improvement in yield and lowering the loading below this resulted in a decrease in efficiency. Reaction yield also decreased when operating with aldehyde equivalency of below 2 (1.5:1 and 1.2:1 resulted in 54% and 30% yield respectively). However, the drop off in yield when operating even slightly below optimal conditions was sharp, unlike when using copper(II) acetate monohydrate.17
With optimised conditions determined, the scope of the reaction was explored. Disappointingly, the reaction times and yields, in all circumstances, were poorer for both aromatic and aliphatic when compared to analogous reactions catalysed by Cu(OAc)2·H2O. Nonetheless, it can be concluded that zinc(II) acetate dihydrate is an alternative to copper(II) acetate monohydrate, albeit with no clearly discernible advantages at this stage.
Tungsten
In 2013, a group led by Ryu and Fagnoni showed that the hydroacylation of azodicarboxylates by aldehydes can be achieved under irradiation using tetrabutylammonium decatungstate (TBADT) as the active catalyst.21 The reaction was initially used to activate C–H bonds in cycloalkyl or heterocyclic substrates, where an acyl radical intermediate could be formed through the use of carbon monoxide under high pressure. However, the reaction conditions were also later found to be applicable to direct aldehydic C–H bond activation (Scheme 7), a far more efficient process to carbonylating alkyl intermediates. | ||
| Scheme 7 The hydroacylation reaction performed under irradiation between aldehyde and DIAD 3a utilising TBADT as an active catalyst. | ||
It should be noted that due to the use of irradiation in the reaction protocol, ethereal solvents (owing to the formation of solvent-adduct side products) and unsaturated aldehydes (owing to activating hetero-Diels–Alder pathways) were considered unsuitable for this process. Two different irradiation sources were utilised by the authors: a 300 W Xe lamp and a SolarBox (Xe lamp, 500 W m−2). These methods were utilised interchangeably throughout the study with the SolarBox generally providing higher yields (Table 6, Scheme 8).
 | ||
| Scheme 8
a Reaction was performed with 2 mol% TBADT with a 1:1 ratio of cyclohexanecarboxaldehyde 2f to DIAD 3a in MeCN (5 mL) at room temperature for 20 h. b Reaction was performed with 2 mol% TBADT with a 1:1.2 ratio of cyclohexanecarboxaldehyde 2f to DIAD 3a in MeCN (5 mL) at room temperature for 2 h. | ||
|
|
||
|---|---|---|
| Entry | Reaction conditions | Yield/% |
| Reaction conditions: DIAD (0.5 mmol), heptaldehyde (1–1.2 mmol), TBADT (2 mol%), MeCN (5 mL), hν, 2 h. | ||
| 1 | SolarBox (Xe lamp, 500 W m−2), TBADT (2 mol%) | 76 |
| 2 | 300 W Xe lamp, TBADT (2 mol%) | 64 |
| 3 | 300 W Xe lamp, without catalyst | 4 |

The methodology has a number of desirable characteristics such as employment of the aldehyde (which is generally seen as the more valuable reaction precursor) as the limiting reagent as well as boasting the fastest reaction rate via a metal-mediated pathway (2 h). Although, moderate to good yields were achieved for both aliphatic and aromatic aldehydes (ca. 60–70%), these were generally lower than those achieved in the aforementioned metal-mediated processes.17,18 Other limitations were also present, such as not being able to utilise unsaturated aldehydes and/or aldehyde that are susceptible to decarbonylation (e.g. phenylactaldehyde). Despite the limitations associated with the methodology, in 2015, a group led by Ravelli and Protti in collaboration with Fagnoni showed that the photoreaction can be used in a continuous-flow process, which could serve as a very effective tool for large-scale production of acyl hydrazide compounds.22
Cobalt
In 2014, Xu et al. reported the co-administration of a Lewis acidic cobalt species and a Brønsted acid to achieve hydroacylation of azodicarboxylates.23 Having identified the reaction of 4-methoxybenzaldehyde 2g with azodicarboxylate as a particular problem reaction with previous methods,16,24 the reaction utilising this aldehyde and diethyl azodicarboxylate (DEAD) 3b was used as the model for optimisation studies. Initially, various cobalt salts were screened, with CoCO3 providing the highest yield when used in conjunction with TFA as the Lewis acid additive (Table 7). Here, similarly to the work reported by Mandal et al.,18 1.2 equivalents of aldehyde relative to azodicarboxylate was used.|
|
||
|---|---|---|
| Entry | Lewis acid | Yield/% |
| Reaction conditions: DEAD (0.2 mmol), 4-methoxybenzaldehyde (0.24 mmol), Lewis acid (20 mol%), TFA (5 mol%), MeCN (0.2 mL), 12 h. | ||
| 1 | CoCO3 | 91 |
| 2 | Co(OAc)2 | 74 |
| 3 | CoCl2 | 56 |
| 4 | Co(acac)2 | 26 |

In further optimisation studies, use of various solvents revealed that the reaction was not compatible with ethereal-based reaction mediums with complex mixtures being obtained when employing THF or Et2O (Table 8). Again rather surprisingly, EtOAc was not trialled even though it was previously shown to be an optimal solvent in rhodium and copper acetate catalysed reactions.16,17 In terms of catalyst loading, there was no reduction in yield upon decreasing the catalyst loading from 20 mol% to 10 mol%, however, further reductions compromised reaction yield greatly.
|
|
||
|---|---|---|
| Entry | Solvent | Yield/% |
| Reaction conditions: DEAD (0.2 mmol), 4-methoxybenzaldehyde (0.24 mmol), CoCO3 (20 mol%), TFA (5 mol%), solvent (0.2 mL), RT, 12 h. | ||
| 1 | CH2Cl2 | 91 |
| 2 | MeCN | 88 |
| 3 | THF | Complex mixture |
| 4 | Et2O | Complex mixture |
| 5 | MeOH | <5 |
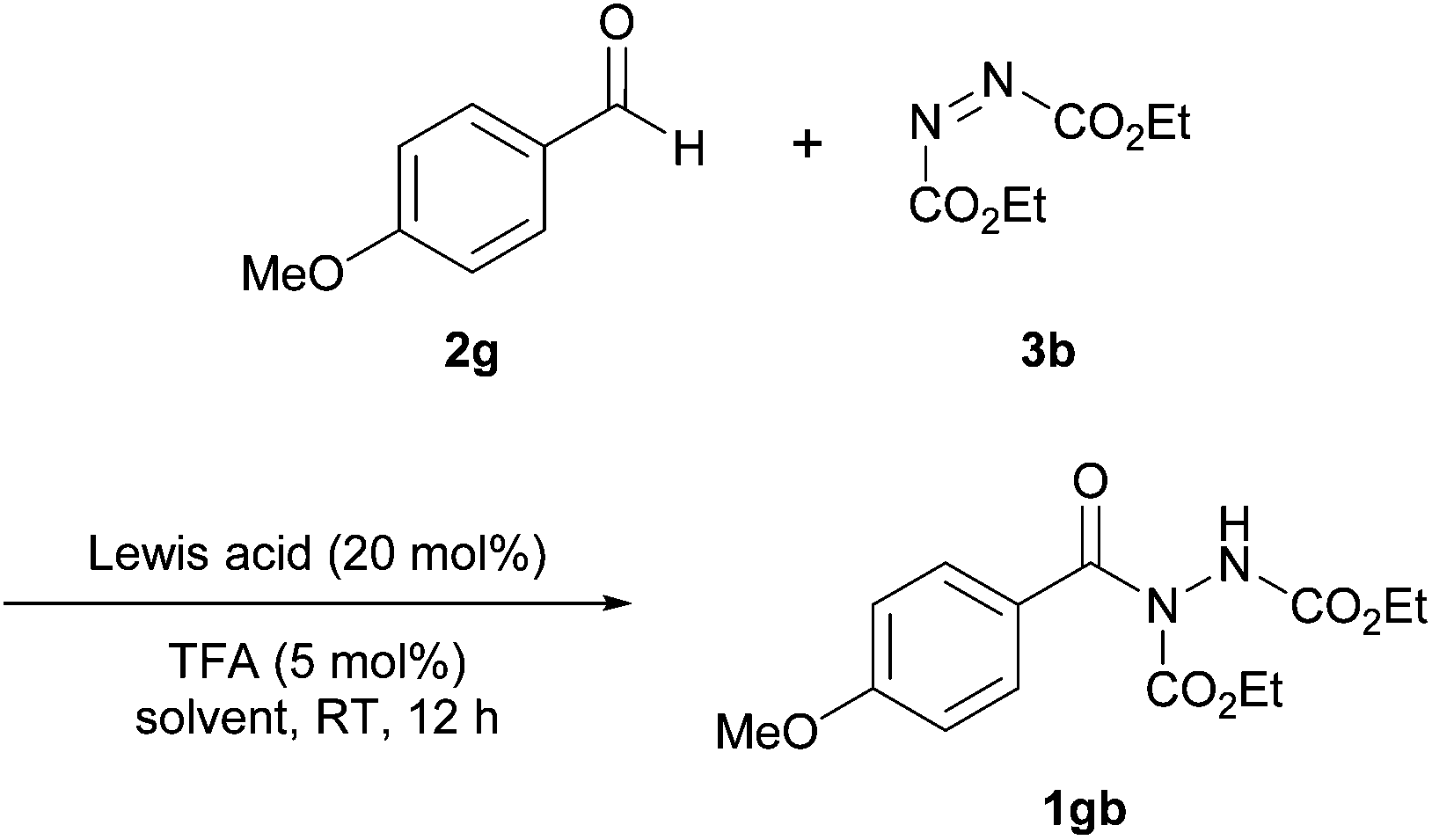
Finally, and perhaps as expected, employment of a Brønsted acid was found to be necessary in order to achieve good to excellent yield. The reaction of 4-methoxybenzaldehyde and DEAD was also shown to be highly inefficient in the presence of no or small amounts of TFA; 1 mol% and 0 mol% both afforded <10% of acyl hydrazide 1gb.
With optimised conditions in hand, a broad range of aldehydes were trialled under the reaction conditions. Aliphatic aldehydes of varying degrees of branching and chain length gave excellent yields of 94–96% across the seven trialled aldehydes. The reaction methodology seemed to be limited by steric hindrance factors as opposed to electronic when employing aromatic aldehydes. ortho-Substituted aromatic aldehydes gave lower yields (77–80%) when compared to meta- or para-substituted aromatic aldehydes (92–94%) regardless of the electron donating or withdrawing ability of the substituents. The reaction was however less tolerant of unsaturation in the aldehyde moiety with only a 52% yield achieved with cinnamaldehyde.
Mechanistic studies were also carried out with radical scavenger 2,2,6,6-(tetramethylpiperidin-1-yl)oxy (TEMPO). Using either a catalytic or stoichiometric amount of TEMPO resulted in the complete suppression of the formation of the desired product. To probe this further, electron paramagnetic resonance (EPR) experiments were performed on a mixture of cobalt(II) carbonate, 2-methylbutyraldehyde, DEAD and TFA. Where a strong signal was observed, Xu proposed that this was due to the presence of a nitrogen-centred radical. No signal was detected with the omission of either CoCO3 or TFA. A radical mechanism was thus proposed (Scheme 9). The cobalt species was thought to facilitate the formation of an acyl radical 5 from the initial aldehyde 2 prior to addition of the acyl radical to the azodicarboxylate 3b to form a nitrogen-centred radical 6xb—this was proposed to be the radical species observed by EPR. This radical species was then thought to abstract a hydrogen atom from another molecule of aldehyde to give the acyl hydrazide product 1xb and regenerate the acyl radical 5.
 | ||
| Scheme 9 Suggested chain radical mechanism for the formation of acyl hydrazide 1xb utilising a cobalt catalyst. | ||
Cobalt-impregnated magnetite
Pérez and Ramón investigated the use of a cobalt catalyst immobilised on a magnetite surface for use in azodicarboxylate hydroacylation.25 The methodology marked the second time that a heterogeneous catalyst was used to catalyse the reaction of aldehydes and azodicarboxylates, i.e. following on from the work by Mandal et al.18Initially, it was in fact an iridium catalyst impregnated on magnetite that was selected to catalyse a reaction between benzaldehyde and DIAD (1.2 equivalents of aldehyde to azodicarboxylate were employed), which served as a model reaction for scrutinising reaction conditions. It was shown that reaction yield increased with increasing reaction temperature up to 60 °C; further increase in temperature led to a sharp decrease in reaction efficiency (Table 9). The optimal temperature of 60 °C is analogous to that used for the methodology utilising CuO nanoparticles as the heterogeneous catalyst.18
|
|
||
|---|---|---|
| Entry | Temperature/°C | Yield/% |
| Reaction conditions: DIAD (1 mmol), benzaldehyde (1.2 mmol), IrO2·Fe3O4 (0.13 mol%), MeCN (1 mL), 72 h. | ||
| 1 | 25 | 2 |
| 2 | 40 | 18 |
| 3 | 50 | 32 |
| 4 | 60 | 65 |
| 5 | 70 | 15 |
| 6 | 100 | 9 |

When screening solvents (Table 10), it was observed that the greatest yield of acyl hydrazide 1ca was achieved when using trichloroethylene as the reaction medium (80%); this also provided the descried product in the quickest reaction time (24 h). It should also be noted, however, that the reaction seemed to proceed well in water (70%).

Finally, an extensive number of metal oxides and loading levels were trialled as the active catalyst. High yields were achieved when using either CoO or NiO (60–90%), with the highest yield being observed with 1.42 mol% of CoO impregnated on magnetite. An additional experiment conducted under an argon atmosphere led to a slight increase in reaction yield (93% against 90%). As anticipated, reaction without the use of a catalyst gave a poor yield of acyl hydrazide 1ca (42%).
Exploring the scope, the reaction was observed to proceed exceptionally well with most aliphatic aldehydes. Reaction yields were however compromised when using tertiary or α,β-unsaturated aliphatic aldehydes (74–75%). The reaction also worked excellently with halo-substituted aromatic aldehydes (87–95%) but the introduction of EDGs on the aryl group resulted in lower yields.
Non-metal catalysed
Aerobic activated
In 2009, the use of room temperature ionic liquids (RTILs) to achieve the reaction of an aldehyde and an azodicarboxylate via hydroacylation was proposed by a group led by Ni and Headley.24 Not only was this seen as an alternative to using metal catalysts, which can be expensive and toxic, but also as a potential solution for the suboptimal yields achieved through the use of aromatic aldehydes in most metal-mediated hydroacylation processes due to the milder reaction conditions. In addition, RTILs are easily recyclable and therefore can be seen as a more sustainable alternative.In this procedure, the reaction of propionaldehyde and DIAD was selected as the model reaction for optimisation studies. RTILs with 1-butyl-3-methylimidazolium ([BMIM] 7) as the cationic moiety were explored (Fig. 2). When trialling various anionic moieties, bis(trifluoromethanesulfonyl)imide), [NTf2]− exhibited the highest reaction efficiency (90% yield in 23 h); compared to using BF4− or PF6− anions (Table 11).
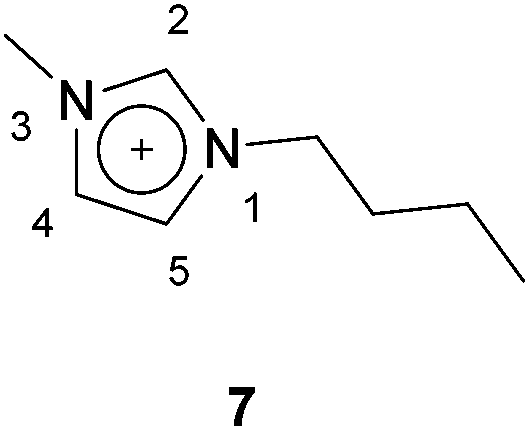 | ||
| Fig. 2 The chemical structure of the 1-butyl-3-methylimidazolium ([BMIM]) cation. | ||
|
|
|||
|---|---|---|---|
| Entry | Solvent | Reaction time/h | Yield/% |
| Reaction conditions: DIAD (0.5 mmol), propionaldehyde (1 mmol), ionic liquid (0.5 mL), RT. | |||
| 1 | [BMIM][NTf2] | 23 | 90 |
| 2 | [BMIM][BF4] | 124 | 40 |
| 3 | [BMIM][PF6] | 124 | <10 |
| 4 | [BMIM][(C2F5SO2)2N] | 18 | 92 |

When rationalising why the different anions would play a significant role in the reaction, Ni and Headley proposed that the larger [NTf2]− anion would result in a larger cation/anion separation allowing for a more effective interaction between the negative charge of the carbonyl oxygen and the cationic portion of the ionic liquid. This interaction was suggested to assist in activation of the C–H bond in aldehydes. To investigate this statement further, a reaction was carried out using [BMIM][(C2F5SO2)2N] as solvent. This ionic liquid has a similar molecular structure to [BMIM][NTf2] but the anion is slightly larger in size and therefore a greater cation/anion separation was expected. As a consequence, the reaction would be expected to proceed more efficiently and, fitting with hypothesis, this is what was observed (Table 11).
Further investigations were also carried out to determine if the acidity of the proton at the C-2 position of [BMIM] aided in the stabilisation of a key intermediate to assist C–H activation of the aldehyde in their proposed pathway. To investigate this claim, an alternative ionic liquid which was identical in structural composition with the exception of a methyl group replacing the H atom at the C-2 position, was utilised as the reaction medium. Employing this ionic liquid resulted in the reaction proceeding in a comparable manner, indicating that the C-2 proton of the imidazolium cation does not influence the reaction rate or yield to any substantial degree.
In terms of optimisation, systematically decreasing the equivalence of aldehyde to azodicarboxylate resulted in longer reaction times and lower reaction yields when going below a ratio of 1.5:1 (Table 12). It was also found that increasing the reaction temperature to 40 °C greatly improved reaction rate (reaction completed in 3 h compared to 23 h) with a concomitant slight improvement in reaction yield.
|
|
|||
|---|---|---|---|
| Entry | Aldehyde equivalence | Reaction time/h | Yield/% |
| Reaction conditions: DEAD (0.5 mmol), propionaldehyde, ionic liquid (0.5 mL), RT. | |||
| 1 | 2 | 23 | 90 |
| 2 | 1.5 | 32 | 92 |
| 3 | 1 | 96 | 70 |

When probing the scope of the hydroacylation reaction using [BMIM][NTf2] as the medium under the optimised conditions, it was found that different substituents on aliphatic aldehydes greatly influenced reaction times and yields. Saturated aldehydes with either linear or branched substituents afforded the desired hydroacylation products in fast reaction times and in excellent yields (∼99%). In terms of aliphatic aldehydes, it was only the presence of a phenyl group at the γ-position that resulted in a notable lower reaction yield (88%) and a slower rate of reaction (48 h). Following a discovery by Chudasama et al.,26 it was shown that this is likely due to a competing intramolecular 1,5-H-atom abstraction pathway (Scheme 10). Yields were heavily compromised when utilising unsaturated aldehydes with olefin functionality at either an internal or terminal position (40–43% in 48 h). The reason for this was the signification formation of a bisazodicarboxylate due to competing ene-type reactions; the same problem observed by Lee's group when using a rhodium acetate catalyst.16 Employment of aromatic aldehydes led to much longer reaction times (>120 h). Aromatic aldehydes bearing H or Cl atoms were well tolerated but those with strong EWGs or EDGs (e.g. methoxy and nitro groups) performed very poorly in the reaction conditions (<10% yield in 168 h).
 | ||
| Scheme 10 Suggested alternative intramolecular H-atom abstraction pathway to account for the lower yields using aldehyde 2i. | ||
In contrast to previous research in the area, the authors tested the tolerance of different azodicarboxylates. Azodicarboxylates displaying differing ester groups (aliphatic and aromatic) were trialled. Interestingly, the azodicarboxylate substrate behaved in a similar way to how the aldehyde substrate did with aliphatic substituents (e.g. isopropyl, ethyl) resulting in efficient reactions in a relatively fast manner and aromatic substituents (benzoyl) generally leading to poorer yields with slower rates.
A radical mechanism was assumed by Ni and Headley, and following research provided by Chudasama et al. (vide infra),27 it is likely to be initiated via aerobic activation of the aldehyde following exposure of the starting materials to air (Scheme 11). Caddick and co-workers proposed that an intermediate acyl radical 5 is generated through interaction of aldehyde 2 with dioxygen. In the absence of an alternative pathway, the acyl radical will further react with dioxygen and additional instances of aldehyde 2 to form its corresponding carboxylic acid 13 in the aldehyde auto-oxidation process. The presence of the azodicarboxylate 3, however, provides a radical trapping reagent which results in the formation of a nitrogen-centred radical species 6. This species can react with another instance of starting aldehyde to initiate a chain process and form acyl hydrazide 1 (Scheme 12), i.e. in an analogous manner to that proposed by Xu et al.23
 | ||
| Scheme 11 Aldehyde auto-oxidation process. | ||
 | ||
| Scheme 12 Aerobically initiated radical hydroacylation reaction between aldehyde 2 and azodicarboxylate 3. | ||
In 2010, Ni and Headley28 demonstrated that the hydroacylation of azodicarboxylate by aldehyde could be carried out applying water in place of an RTIL as the reaction solvent. The use of water as the reaction solvent is extremely beneficial from a green chemistry standpoint and the absence of a catalyst provides numerous benefits as common issues such as cost, purification and homogeneity can be avoided. With the organic substrates being immiscible in water, the reaction is said to happen ‘on water’, although this was only proposed at a later date.29 Similarly to their previous experiments utilising the ionic liquid, the reaction between 2 equivalents of propionaldehyde 2h to DIAD 3a was used as a model to probe for whether water was the best solvent; this was observed to be the case.
When exploring the scope of the reaction, yields were generally comparable to those achieved using ionic liquid as the reaction medium,24 but this protocol had the additional benefits of taking place at room temperature and using water as the solvent (beneficial in terms of cost, safety, environment), albeit at the slight compromise of having slightly longer reaction times. With optimised conditions in hand, excellent yields were achieved with aliphatic aldehydes (92–98% utilising saturated aldehydes) whilst only moderate yields were achieved with aromatic aldehydes (59–80%). The reaction also tolerated EWGs and EDGs on the aromatic ring to much a greater extent compared to using ionic liquids as the solvent. Again, unsaturation in the aldehyde motif led to formation of ene-type by-products, thus heavily compromising the effectiveness of the reaction.
Later in 2010, Chudasama et al. conducted a more thorough set of experiments of achieving the hydroacylation reaction ‘on water’ (Scheme 13) but with the goal of utilising the aldehyde as the limiting reagent; an important characteristic of a reaction to be adapted for use with valuable aldehydes. It was research conducted by the group that suggested that the hydroacylation reaction in the absence of any apparent catalyst is likely to occur through trapping of an acyl radical intermediate generated by the aldehyde auto-oxidation process.26a
 | ||
| Scheme 13 Reaction was performed ‘on water’ with a 1:1.2 ratio of aldehyde to azodicarboxylate at 21 °C. | ||
The reaction of aldehydes with DIAD and DEAD using air for activation and water as solvent was achieved in moderate to excellent yields (55–91%) but with aliphatic aldehydes displaying a large substrate scope (e.g. ester, epoxide, acetal and alkynyl functionalities). Benzaldehyde and 4-flurorbenzaldehyde achieved respectable yields (75–79%), however, electron donating groups were not so well tolerated; the introduction of a methoxy group at the para-position resulted in 44% yield. Most significantly, however, reaction rates and to some extent yields were correlated to the rate at which aldehydes auto-oxidised in air rather than to the structure of the aldehyde, which provided better understanding.
The substrate scope explored also included the trialling of enantiopure aldehydes in the reaction conditions, which was thought to be highly significant and to tie into the popular of using organocatalysis to make enantiopure aldehydes.30,31 Aldehydes bearing α-enantioenriched chiral centres were treated with DIAD and this resulted in acyl hydrazides with essentially complete retention of stereochemistry (98–99% ree); this marked the first example of retention of enantiomeric excess of an aldehyde in the field hydroacylation (Scheme 14). The rationale for this behaviour was attributed to the intermediate acyl radicals being σ-type radicals and the reaction conditions not allowing acid- or base-promoted racemisation to take place.
 | ||
| Scheme 14 Reaction was performed ‘on water’ with a 1:1.2 ratio of aldehyde 2j to DIAD 3a at 21 °C. | ||
Pyridine catalysed
In 2015, Muthusubramanian and co-workers reported a pyridine-based metal-free methodology to achieve the hydroacylation of azodicarboxylates with aldehydes with drastically reduced reaction times.32 An initial reaction utilising 1.2 equivalents of benzaldehyde to DIAD in DMF, under reflux conditions, using a pyridine catalyst achieved a good yield of acyl hydrazide (65%) with the main limitation in yield being due to suboptimal conversion. To improve conversion, the reaction was repeated under microwave irradiation at 140 °C. To their delight the reaction was now complete in an improved yield (85%) and within only 5 minutes.Encouraged by their preliminary experiments, the authors trialled various nitrogen based catalysts for the hydroacylation of DIAD by benzaldehyde (Table 13). However, an improvement yield was not observed, and the use of different reaction solvents also failed to improve yield beyond that achieved in DMF (Table 14). Notably, however, moderate yields were achieved when using water as solvent.
|
|
||
|---|---|---|
| Entry | Catalyst | Yield/% |
| Reaction conditions: DIAD (1 mmol), benzaldehyde (1.2 mmol), catalyst (0.1 mmol), DMF (1 mL), microwave irradiation, 10 min. | ||
| 1 | Pyridine | 85 |
| 2 | Isoquinoline | 71 |
| 3 | Quinoline | 63 |
| 4 | Diethylamine | Trace |
| 5 | DABCO | Trace |
| 6 | Triethylamine | 25 |
| 7 | Piperidine | Trace |
| 8 | — | 25 |

|
|
||
|---|---|---|
| Entry | Solvent | Yield/% |
| Reaction conditions: DIAD (1 mmol), benzaldehyde (1.2 mmol), pyridine (0.1 mmol), solvent (1 mL), microwave irradiation, 10 min. | ||
| 1 | DMF | 85 |
| 2 | Water | 60 |
| 3 | MeOH | 53 |
| 4 | Toluene | 27 |
| 5 | 1,4-Dioxane | 23 |
| 6 | THF | 20 |
| 7 | DCE | Trace |

Under the optimised conditions, hydroacylation of DIAD with a wide range of aromatic aldehydes in only 10 minutes resulted in good yield to excellent yields (76–89%). However, it should be noted that the exemplification was limited to a small number of alkyl- and halo-substituted aromatic moieties. The only aliphatic aldehyde trialled was propionaldehyde, which resulted in a 76% yield of the corresponding acyl hydrazide. It was also somewhat disappointing that no unsaturated aldehydes were trialled as this had proven to be a particular limitation in previous azodicarboxylate hydroacylation reactions.16,24,28
A mechanism based on a zwitterionic intermediate 15 following nucleophilic attack of pyridine 14 onto the azodicarboxylate functionality was tentatively proposed (Scheme 15). The anion on the zwitterionic intermediate 16 was thought to react with an aldehyde to form an unstable 4-membered ring species 17 that collapsed to form the acyl hydrazide product 1xa.
 | ||
| Scheme 15 Tentative mechanism for the formation of acyl hydrazide 1xavia a pyridine-catalysed reaction. | ||
Photooragnocatalytic
In 2014, Kokotos et al. proposed a new method of achieving the hydroacylation of azodicarboxylates with aldehydes in a short reaction time by using activated ketones as photoorganocatalysts.33 At this point, only the work by Ryu and Fagnoni21 had achieved the hydroacylation process in a photocatalytic fashion through the use of tetrabutylammonium decatungstate, where the employment of an expensive Xe lamp (300 W) resulted in an issue with reaction scope (methodology incompatible with unsaturated substrates or ethereal solvents).The methodology applied by Kokotos et al., in contrast, involved using 1.5 equivalents of aldehyde to azodicarboxylate whilst employing ordinary household lamps (2 × 15 W). A model reaction of 1.5 equivalents of heptaldehyde with respect to DIAD was used for reaction optimisation, initially in terms of the photoorganocatalyst being employed (Scheme 16). To their delight, employment of photoorganocatalysts phenylglyoxylic acid 22 and ethyl phenylglyoxylate 19 resulted in a quantitative yield of acyl hydrazide 1ea. It was also observed that a decrease in the activation of the ketone, e.g. substitution of the phenyl group in phenylglyoxic acid for an ethyl group, resulted in a significant decrease in reaction efficiency (26% compared to 100%).
 | ||
| Scheme 16 Reaction was performed under light irradiation (2 × 15 W household lamps) with 10 mol% of photoorganocatalyst and a 1.5:1 ratio of heptaldehyde 2e to DIAD 3a in pet. ether (40–60 °C, 0.5 mL) at room temperature for 2 h. | ||
With the ideal photoinitiator (phenylglyoxylic acid 22) determined, the reaction was scrutinised with respect to solvent. Petroleum ether (40–60 °C) provided the best results with near quantitative formation of acyl hydrazide 1ea (Table 15) being achieved; it should also be noted that an excellent yield was recorded ‘on water’ and good yields were obtained across the series of solvents generally.
|
|
||
|---|---|---|
| Entry | Solvent | Yield/% |
| Reaction conditions: DIAD (1 mmol), heptaldehyde (1.5 mmol), (0.1 mmol), solvent (0.5 mL), phenylglyoxylic acid (10 mol%), RT, 2 h, light irradiation. | ||
| 1 | Pet. ether (40–60 °C) | 99 |
| 2 | Water | 92 |
| 3 | CH2Cl2 | 82 |
| 4 | Hexane | 78 |
| 5 | MeCN | 76 |

Using petroleum ether (40–60 °C) as the reaction solvent, conditions of employing equimolar quantities of aldehyde and azodicarboxylate led to a decrease in yield (76% compared to >99%). Application of 1.5 equivalents of the azodicarboxylate gave an excellent yield of acyl hydrazide 1ea (96%). Although this yield is slightly lower when compared to using 1.5 equivalents of aldehyde (>99%), it does suggest that the reaction could potentially be used for efficient hydroacylation with aldehyde employed as limiting reagent.
Examining the scope of the reaction showed it to work very efficiently with aliphatic aldehydes (67–99%) excluding those displaying α,β-unsaturation (52–69%), which also required a much longer reaction time (24–48 h). Aromatic aldehydes, as observed with many other methods, were generally less well tolerated under the conditions. A notable exception is where benzaldehyde was used—here an exceptional yield of its corresponding acyl hydrazide (99%) was obtained.
Utilising acyl hydrazides
The acyl hydrazide moiety displays a number of interesting chemical properties (Fig. 3, e.g. acidic proton, relatively weak N–N bond, acyl group, carbamate functionality, often a stable crystalline solid), which can be exploited to achieve transformation into important and desirable chemical functionalities; these will discussed in turn. | ||
| Fig. 3 Structural and chemical characteristics of acyl hydrazides. | ||
Functionalisation at N–H bond
In 2004, Kim and Lee4 employed a rhodium acetate catalyst to form acyl hydrazides and then either allylated or alkylated at the acidic N–H site by using α-bromoallyl or α-bromoacetate derivatives as electrophiles under basic conditions (Scheme 17). The group then took advantage of the stereodynamics of the substituted acyl hydriazide (restricted N–N bond rotation with CO–N–N–CO dihedral angle of approximately 90°) to achieve a ring-closing metathesis by employment of Grubbs’ second generation catalyst (Scheme 18). Following a number of further steps, in the total synthesis of macrocycle 30, the relatively weak N–N bond is broken through the use of ammoniacal sodium metal (Scheme 19). The specific properties of the acyl hydrazide motif were vital to achieve desired goal in this research. | ||
| Scheme 17 Reaction conditions (step 1): azodicarboxylate (1 eq.), aldehyde (1 eq.), [Rh(OAc)2]2 (2 mol%), RT, 24 h. Reaction conditions (step 2): acyl hydrazide (1 eq.) was isolated and dissolved in DMF (2 mL), Cs2CO3 (1.2 eq.), allyl bromide (2 eq.), RT, 2 h. | ||
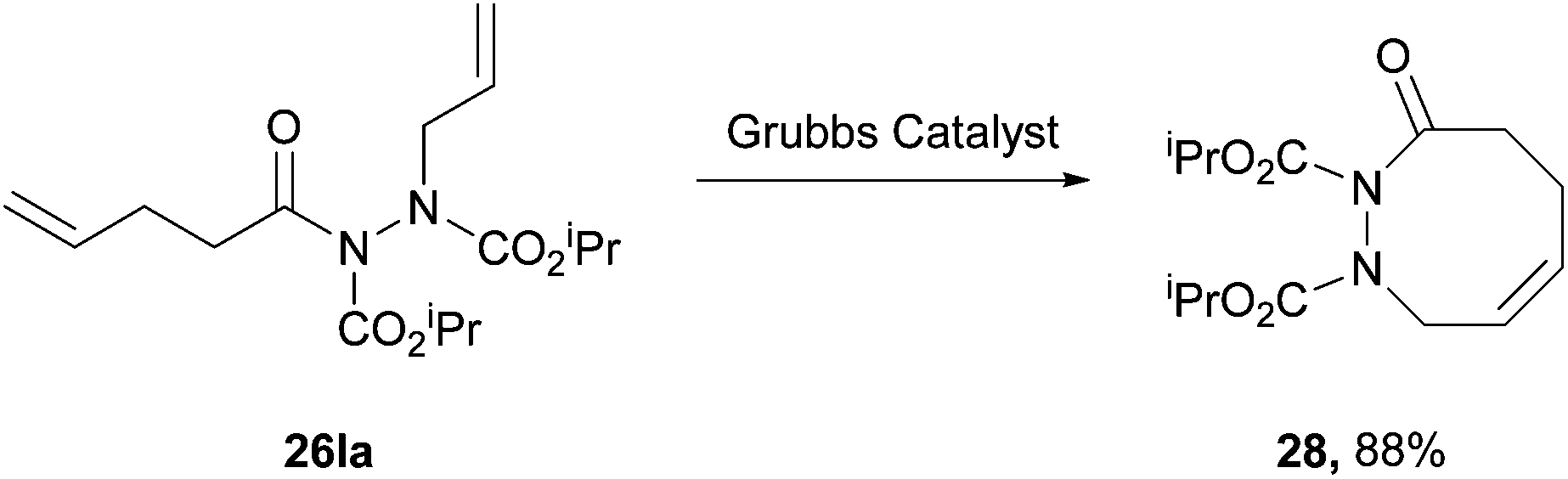 | ||
| Scheme 18 Reaction conditions: diene 26la in CH2Cl2 (0.002 M), Grubbs second generation catalyst (5 mol% in 1 mL CH2Cl2), nitrogen atmosphere, reflux, 2 h. | ||
 | ||
| Scheme 19 Reaction conditions: bicycle 29 (0.39 mmol), THF (1.5 mL), NH3 (10 mL), Na metal, −78 °C, 2.5 h. | ||
Very early research into the reactivity of acyl hydrazides by Kalinin and co-workers34 also showed that a silicon group can be added to the acidic site. Here, 1.5 equivalents of bis(trimethylsilyl)acetamide (BSA) was used as the silylating agent to achieve an 85% yield of the silylated acyl hydrazide 32 (Scheme 20). The authors noted that hydrazine derivatives featuring a trimethylsilyl group on the nitrogen can act as convenient models in the study of conformational transitions in hydrazines.
 | ||
| Scheme 20 Reaction conditions: acyl hydrazide 3mc (0.10 mol), CH2Cl2 (5 mL), BSA 31 (0.15 mol), 60–80 °C, 8 h. | ||
Acyl hydrazides as acyl donors
Another branch of chemical transformations that have been carried out with acyl hydrazides is the effective nucleophilic substitution at the acyl site. As of now, these transformations have been successfully used in the formation of various valuable chemical entities, e.g. asymmetric ketones,2 esters,1 thioesters,1 amides,1 and hydroxamic acids.3 | ||
| Scheme 21 Formation of metal complex 35 upon treatment of Weinreb amide 33 with organometallic reagent 34. | ||
To their delight, the formation of ketones from aryl acyl hydrazides proved effective with the utilisation of either 2.5 equivalents of an aliphatic Grignard reagent at −78 °C, or 2.5 equivalents of an aromatic Grignard reagent (this required warming up to 0 °C from −78 °C owing to the decreased nucleophilicity of the reagent). Good to excellent yields of 68–92% were achieved upon addition of Grignard reagents to aryl acyl hydrazides displaying various functionalities. In the exceptional cases where the use of an aliphatic Grignard reagent gave inefficient reaction, i.e. when employing acyl hydrazides displaying nitro, cyano, ortho-fluoro or ortho-methyl functionalities, aromatic Grignard reagents performed very well due to their tuned down reactivity (Scheme 22).
 | ||
| Scheme 22 Reaction conditions: acyl hydrazide 1xa (0.5 mmol), phenylmagnesium bromide (1.25 mmol), THF (10 mL), −78 to 0 °C, 1 h. | ||
 | ||
| Scheme 23 Reaction conditions: (i) DIAD (0.6 mmol), aldehyde (0.5 mmol), DMF (0.5 mL), 21 °C, 24 h; (ii) alcohol (0.55 mmol), Cs2CO3 (0.50 mmol), 16 h. | ||
 | ||
| Scheme 24 Reaction conditions: (i) DIAD (0.6 mmol), aldehyde (0.5 mmol), DMF (0.5 mL), 21 °C, 24 h; (ii) thiol (0.55 mmol), Cs2CO3 (0.50 mmol), 16 h. | ||
 | ||
| Scheme 25 Reaction conditions: (i) DIAD (0.6 mmol), aldehyde (0.5 mmol), DMF (0.5 mL), 21 °C, 24 h; (ii) amine (0.55 mmol), 16 h. | ||
A similar one-pot approach was utilised by Papadopoulos and Kokotos3 in the formation of hydroxamic acids where the in situ generation of the acyl hydrazide functionality allowed a nucleophilic addition–elimination step of hydroxylamine at the acyl position. The method of choice for the formation of the acyl hydrazide was the author's previously described photoorganocatalytic process utilising phenylglyoxylic acid 22 as photoinitiator (Scheme 26).33 The group also demonstrated the effectiveness of the reaction in the final step of the total synthesis of the drug vorinostat 44n, where a 47% transformational yield was achieved upon application of a valuable aldehyde 2n (Scheme 27).
 | ||
| Scheme 26 Reaction conditions (step 1): DIAD (0.50 mmol), aldehyde (0.75 mmol), phenylglyoxylic acid (0.05 mmol), pet. ether (40–60 °C, 1 mL), light irradiation, 90 min to 48 h. Reaction conditions (step 2): hydroxylamine hydrochloride (1.50 mmol), triethylamine (1.50 mmol), CH2Cl2 (4 mL), RT, 20 h. | ||
 | ||
| Scheme 27 Reaction conditions: (i) DIAD (0.50 mmol), aldehyde (0.75 mmol), phenylglyoxylic acid (0.05 mmol), pet. ether (40–60 °C, 1 mL), light irradiation, 90 min to 48 h; (ii) hydroxylamine hydrochloride (1.50 mmol), triethylamine (1.50 mmol), CH2Cl2 (4 mL), RT, 20 h. | ||
 | ||
| Fig. 4 The chemical structure of the 3-alkyl-5-aryl-1,3,4-oxadiazol-2(3H)-one heterocycle. | ||
 | ||
| Scheme 28 Proposed mechanisms for the formation of oxadiazolones. | ||
The reaction can tolerate variation of the alkyl group at the 3-position by changing the azodicarboxylate used to make the Mitsunobu reagent 46, and to the aryl moiety at the 5-position via selection of a specific carboxylic acid. Interestingly, a clear trend relating the size of the alkyl group on the azodicarboxylate and the efficiency of the cyclisation was observed. In an optimisation study utilising 1.2 equivalents of the Mitsunobu reagent relative to carboxylic acid (Scheme 29), primary alkyl azodicarboxylates (dimethyl azodicarboxylate (DMAD), DEAD or dibenzyl azodicarboxylate, (DBAD)) provided superior yields of the corresponding oxadiazolone product (52–66%) when compared to utilising Mitsunobu reagents exhibiting a secondary (isopropyl group in DIAD) or tertiary (tert-butyl group in di-t-butyl azodicarboxylate (DtBAD)) alkyl group (0–10%).
 | ||
| Scheme 29 Proposed mechanisms for the formation of oxadiazolones. | ||
The authors then carried out tests to determine whether the cyclisation step was occurring thermally and/or as a consequence of components in the reaction mixture. It was found that heating of purified acyl hydrazide intermediate 1cb with or without solvent did not result in the formation of the heterocyclic product. Perhaps as expected, heating the acyl hydrazide with 1 equivalent of the Mitsunobu reagent for 5 h did however afford the desired product (54% yield). Interestingly, utilisation of a catalytic quantity of the Mitsunobu reagent (0.05 eq.) still achieved a similar result over 19 h (49%). They therefore assumed that the presence of the Mitsunobu reagent or perhaps its by-products (triphenylphosphine oxide or dialkyl 1,2-hydrazinedicarboxylate) where important in facilitating the cyclisation step. Further experiments were then carried out where heating the acyl hydrazide 1cb in the presence of 1 equivalent of triphenylphosphine oxide for 5 h afforded only an 11% yield of the heterocycle, whereas subjecting the acyl hydrazide to heat in the presence of 0.2 equivalents of dialkyl 1,2-hydrazinedicarboxylate for 24 h did not lead to any formation of the desired product. The authors then decided to test the hypothesis that the cyclisation mechanism is facilitated by heating acyl hydrazide in the presence of base (Scheme 29). It was found that the addition of either sodium ethoxide (EtONa), sodium hydride (NaH) or DBU achieved comparable yields of the desired product (42–64% when utilised in a catalytic amount over 2 h) when compared to the one-pot formation of oxadiazolones (Scheme 28).
Conclusions
Acyl hydrazides are proving to be increasingly effective and versatile entities that have been exploited for conversion into various desirable functionalities. The efficient formation of acyl hydrazides is therefore becoming an important consideration, with (as described) many distinct methods for their preparation being furnished in recent years. More or less each and every methodology, be it metal-mediated or aerobically-initiated, has shown it has a role to play depending on what the scientist requires from their synthesis (e.g. it may be short reaction time, limited to use of certain solvents, require photochemical activation, need heterogeneous catalysis, etc.). These methods are not only providing new ways of making acyl hydrazides but are also leading to novel methods in which they can be exploited, e.g. one-pot conversions that take advantage of the reactivity of acyl hydrazides in situ.Acknowledgements
We gratefully acknowledge Prof. Alwyn Davies for useful discussions.Notes and references
- A. Maruani, M. T. W. Lee, G. Watkins, A. R. Akhbar, H. Baggs, A. Shamsabadi, D. A. Richards and V. Chudasama, RSC Adv., 2016, 6, 3372–3376 RSC.
- A. R. Akhbar, V. Chudasama, R. J. Fitzmaurice, L. Powell and S. Caddick, Chem. Commun., 2014, 50, 743–746 RSC.
- G. N. Papadopoulos and C. G. Kokotos, Chem. – Eur. J., 2016, 22, 6964–6967 CrossRef CAS PubMed.
- Y. J. Kim and D. Lee, Org. Lett., 2004, 6, 4351–4353 CrossRef CAS PubMed.
- (a) O. Sugimoto, T. Arakaki, H. Kamio and K. Tanji, Chem. Commun., 2014, 50, 7314 RSC; (b) D. H. R. Barton, N. Ozbalik and B. Vacher, Tetrahedron, 1988, 44, 7385–7392 CrossRef CAS; (c) D. L. Hughes and R. A. Reamer, J. Org. Chem., 1996, 61, 2967–2971 CrossRef CAS PubMed; (d) W.-C. Yang, S.-S. Weng, A. Ramasamy, G. Rajeshwaren, Y.-Y. Liaob and C.-T. Chen, Org. Biomol. Chem., 2015, 13, 2385–2392 RSC.
- S. De Sarkar, W. Liu, S. I. Kozhushkov and L. Ackermann, Adv. Synth. Catal., 2014, 356, 1461–1479 CrossRef CAS.
- P. Gandeepan and C. H. Cheng, Chem. – Asian J., 2015, 10, 824–838 CrossRef CAS PubMed.
- S. A. Girard, T. Knauber and C. J. Li, Angew. Chem., Int. Ed., 2014, 53, 74–100 CrossRef CAS PubMed.
- K. Godula and D. Sames, Science, 2006, 312, 67–73 CrossRef CAS PubMed.
- J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507–514 CrossRef CAS PubMed.
- F. Liron, J. Oble, M. M. Lorion and G. Poli, Eur. J. Org. Chem., 2014, 5863–5883 CrossRef CAS.
- J. Schranck, A. Tlili and M. Beller, Angew. Chem., Int. Ed., 2014, 53, 9426–9428 CrossRef CAS PubMed.
- Y. Segawa, T. Maekawa and K. Itami, Angew. Chem., Int. Ed., 2015, 54, 66–81 CrossRef CAS PubMed.
- R. A. Sheldon, Green Chem., 2008, 10, 359–360 RSC.
- V. Nair, R. S. Menon, A. R. Sreekanth, N. Abhilash and A. T. Biju, Acc. Chem. Res., 2006, 39, 520–530 CrossRef CAS PubMed.
- D. Lee and R. D. Otte, J. Org. Chem., 2004, 69, 3569–3571 CrossRef CAS PubMed.
- Y. Qin, Q. Peng, J. Song and D. Zhou, Tetrahedron Lett., 2011, 52, 5880–5883 CrossRef CAS.
- S. M. Inamdar, V. K. More and S. K. Mandal, Chem. Lett., 2012, 41, 1484–1486 CrossRef CAS.
- Y. Qin, D. Zhou and M. Li, Lett. Org. Chem., 2012, 9, 267–272 CrossRef CAS.
- T. Inagaki, Y. Yamada, T. P. Le, A. Furuta, J. I. Ito and H. Nishiyama, Synlett, 2009, 253–256 CAS.
- I. Ryu, A. Tani, T. Fukuyama, D. Ravelli, S. Montanaro and M. Fagnoni, Org. Lett., 2013, 15, 2554–2557 CrossRef CAS PubMed.
- F. Bonassi, D. Ravelli, S. Protti and M. Fagnoni, Adv. Synth. Catal., 2015, 357, 3687–3695 CrossRef CAS.
- H.-B. Zhang, Y. Wang, Y. Gu and P.-F. Xu, RSC Adv., 2014, 4, 27796–27799 RSC.
- B. Ni, Q. Zhang, S. Garre and A. D. Headley, Adv. Synth. Catal., 2009, 351, 875–880 CrossRef CAS.
- J. M. Pérez and D. J. Ramón, Adv. Synth. Catal., 2014, 356, 3039–3047 CrossRef.
- (a) V. Chudasama, A. R. Akhbar, K. A. Bahou, R. J. Fitzmaurice and S. Caddick, Org. Biomol. Chem., 2013, 11, 7301–7317 RSC; (b) V. Chudasama, J. M. Ahern, R. J. Fitzmaurice and S. Caddick, Tetrahedron Lett., 2011, 52, 1067–1069 CrossRef CAS.
- V. Chudasama, J. M. Ahern, D. V. Dhokia, R. J. Fitzmaurice and S. Caddick, Chem. Commun., 2011, 47, 3269–3271 RSC.
- Q. Zhang, E. Parker, A. D. Headley and B. Ni, Synlett, 2010, 2453–2456 CAS.
- V. Chudasama, R. J. Fitzmaurice, J. M. Ahern and S. Caddick, Chem. Commun., 2010, 46, 133–135 RSC.
- A. B. Northrup and D. W. C. MacMillan, J. Am. Chem. Soc., 2002, 124, 6798–6799 CrossRef CAS PubMed.
- S. P. Brown, M. P. Brochu, C. J. Sinz and D. W. C. MacMillan, J. Am. Chem. Soc., 2003, 125, 10808–10809 CrossRef CAS PubMed.
- A. Mariappan, K. Rajaguru, S. Muthusubramanian and N. Bhuvanesh, Tetrahedron Lett., 2015, 56, 338–341 CrossRef CAS.
- G. N. Papadopoulos, D. Limnios and C. G. Kokotos, Chem. – Eur. J., 2014, 20, 13811–13814 CrossRef CAS PubMed.
- A. V. Kalinin, É. T. Apasov, S. V. Bugaeva, S. L. Ioffe and V. A. Tartakovskii, Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.), 1983, 32, 1282–1284 CrossRef.
- R. K. Dieter, Tetrahedron, 1999, 55, 4177–4236 CrossRef CAS.
- S. Balasubramaniam and I. S. Aidhen, Synthesis, 2008, 3707–3738 CAS.
- S. Nahm and S. M. Weinreb, Tetrahedron Lett., 1981, 22, 3815–3818 CrossRef CAS.
- J. L. Romine, S. W. Martin, N. A. Meanwell, V. K. Gribkoff, C. G. Boissard, S. I. Dworetzky, J. Natale, S. Moon, A. Ortiz, S. Yeleswaram, L. Pajor, Q. Gao and J. E. Starrett, J. Med. Chem., 2007, 50, 528–542 CrossRef CAS PubMed.
- F. Mazouz, S. Gueddari, C. Burstein, D. Mansuy and R. Milcent, J. Med. Chem., 1993, 36, 1157–1167 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |