
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bioinspired total syntheses of terpenoids
Cedric L.
Hugelshofer
and
Thomas
Magauer
*
Department of Chemistry and Pharmacy, Ludwig-Maximilians-University Munich, Butenandstrasse 5-13, 81377 Munich, Germany. E-mail: thomas.magauer@lmu.de
First published on 29th November 2016
Abstract
Nature's highly efficient routes for constructing natural products have inspired chemists to mimic these processes in a laboratory setting. This Perspective presents some recent examples of conceptually different bioinspired total syntheses of complex terpenoids and thereby aims to highlight the vast benefits offered by bioinspired strategies.
 Cedric L. Hugelshofer (left) and Thomas Magauer (right) | Thomas Magauer was born in Linz, Upper Austria in 1983. In 2002, he moved to Vienna to study chemistry at the University of Vienna. In 2007, he joined the laboratories of Prof. Johann Mulzer and under his guidance he developed enantioselective syntheses of the complex polyketide kendomycin and echinopines A and B. After graduating in 2009, he moved to Harvard University, USA, to begin postdoctoral studies with Prof. Andrew G. Myers. At Harvard University he worked on carbohydrates, chiral silicon protecting groups and developed a synthesis of natural and unnatural antiproliferative trioxacarcins. In 2012, he started his independent research as a Liebig-junior research group leader at the LMU Munich. Since 2013, his group has been supported by the Emmy Noether Program of the German Research Foundation (DFG). In 2016, the group received a Starting Grant of the European Research Council (ERC). |
Introduction
In the field of organic chemistry, a bioinspired synthesis or reaction sequence represents a series of transformations that mimics a hypothetical or proven biosynthesis of a natural product.1 While the imitated biosynthetic proposal generally has a firm basis in known chemical pathways,2 the mimicking transformations may, in contrast, be achieved using reagents and conditions that are not representative of natural systems.3 In such cases, where the focus primarily lies on emulating the underlying concept of a biosynthesis, it is deemed preferable to use the term “bioinspired” rather than “biomimetic” synthesis.In nature, relatively small and simple building blocks serve as precursors for the synthesis of complex molecules. Moreover, cascade reactions, where products of one transformation serve as the starting material for a subsequent reaction sequence, are triggered for the rapid increase of structural complexity.4 Striving to match nature's ingenuity for achieving the total synthesis of natural products is often an intractable ambition in a chemical laboratory. However, it can be advantageous to design synthetic routes bearing biosynthetic considerations in mind, or to devise strategies that feature just an individual bioinspired step.5 The development of bioinspired syntheses based on a formulated biosynthetic proposal can help to unveil links between apparently related natural products, especially if these coexist in an organism. Moreover, the identification of a common biosynthetic precursor can enable a general entry to a whole family of natural products. By inducing a specific reaction on a putative biosynthetic intermediate, the transformation might serve as an approach to corroborate a biosynthetic proposal. Contrarily, if the reaction would not succeed, the suggested biosynthesis would remain unsupported and might need to be revised. In addition to providing an understanding of how nature generates molecular complexity, bioinspired syntheses also stimulate the development of new reactions or result in alternative methods for the preparation of a desired target.6
In this Perspective, we wish to present some recent compelling examples of conceptually different bioinspired total syntheses of terpenoids. Elegant routes to stunningly complex natural products were first rendered possible and a wealth of fascinating discoveries has been made, highlighting the numerous benefits of pursuing bioinspired synthetic strategies.
Biosynthetic links
A fascinating study on the complex sesterterpenoid bolivianine (7) provided insight into its biosynthetic origin.7 Liu and co-workers proposed that the daunting heptacyclic skeleton of bolvianine (7), which contains nine stereogenic centers, might be biosynthetically derived from a Diels–Alder reaction cascade between onoseriolide (2) and β-E-ocimene (4) (Scheme 1).8 This biosynthetic proposal was particularly motivated by the discovery that terpenoids 2 and 4 are also found in Hedyosmum angustifolium, the small tree from which 7 had been isolated. Inspired by this hypothesis, a concise synthesis of onoseriolide (2) was first developed starting from verbenone (1). After oxidation of 2 to the corresponding aldehyde 3, it was discovered that heating a mixture of the latter product and β-E-ocimene (4) led to the envisioned intermolecular/intramolecular Diels–Alder cascade and furnished bolivianine (7) as the only isolable isomer. Impressively, three rings, four C–C bonds and five stereogenic centers were generated in this cascade transformation. While the first intermolecular cycloaddition reaction (5 → 6) required elevated temperatures to proceed, investigations on a related system showed that the intramolecular hetero-Diels–Alder reaction (6 → 7) spontaneously took place under ambient conditions. Further studies ruled out the possibility of an alternative cascade mechanism that was initiated by an intermolecular hetero-Diels–Alder reaction.8b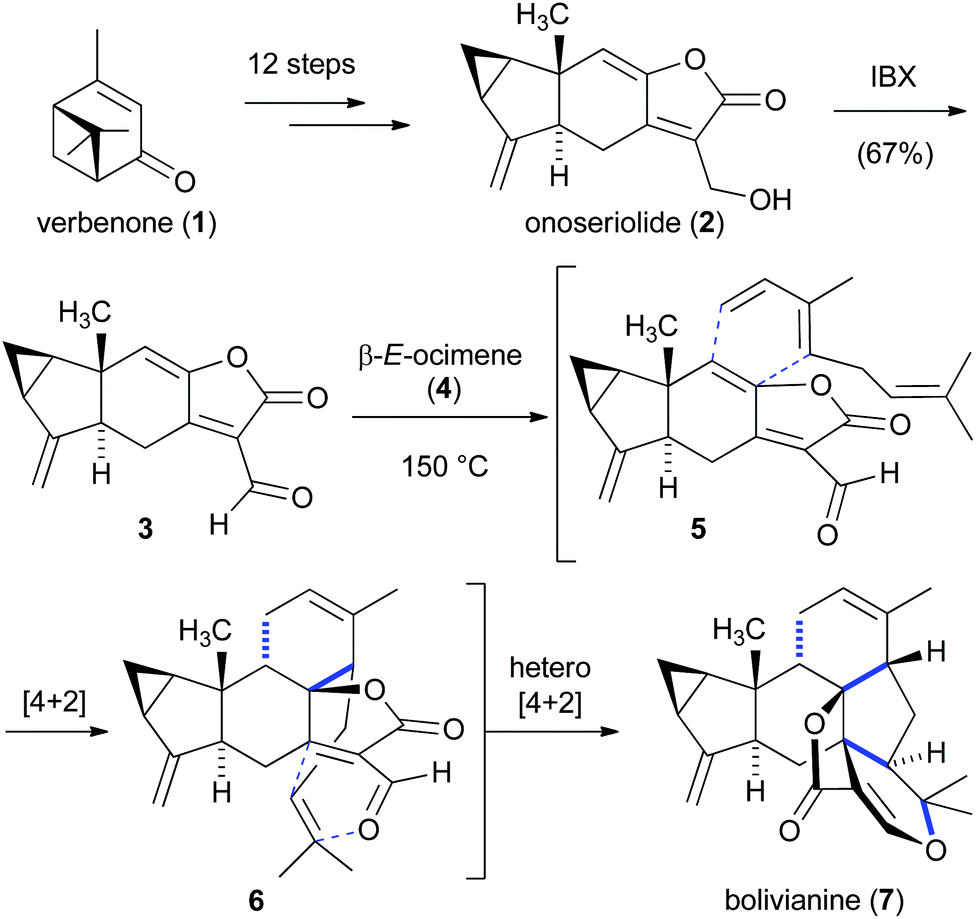 | ||
| Scheme 1 Bioinspired intermolecular/intramolecular Diels–Alder cascade for the synthesis of bolivianine (7). | ||
Similarly guided by the logic of identifying biosynthetic relationships between terpenoids co-occurring in a plant, we recently reported a strategy that culminated in the asymmetric total synthesis of eighteen members of the leucosceptroid family of natural products.9 Our synthetic route involved the convergent union of the two building blocks 8 and 9, and the development of a novel dilactol aldol-type condensation to forge the tricyclic skeleton of 10. This condensation provided access to gram quantities of a highly functionalized leucosceptroid core structure,10 which paved the way for the preparation of leucosceptroid A (10) (Scheme 2). After having conducted initial studies aimed at revealing biogenetic relationships between individual leucosceptroid members using a model system,11 we investigated the bioinspired photo-oxidation of leucosceptroid A (10). It was discovered that exposure of 10 to singlet oxygen cleanly furnished endo-peroxide 11, which upon sequential reduction and aldol reaction of a γ-keto aldehyde intermediate provided leucosceptroid C (12). Alternatively, an efficient Kornblum–DeLaMare-type rearrangement12 of 11 gave rise to leucosceptroid P (13).13
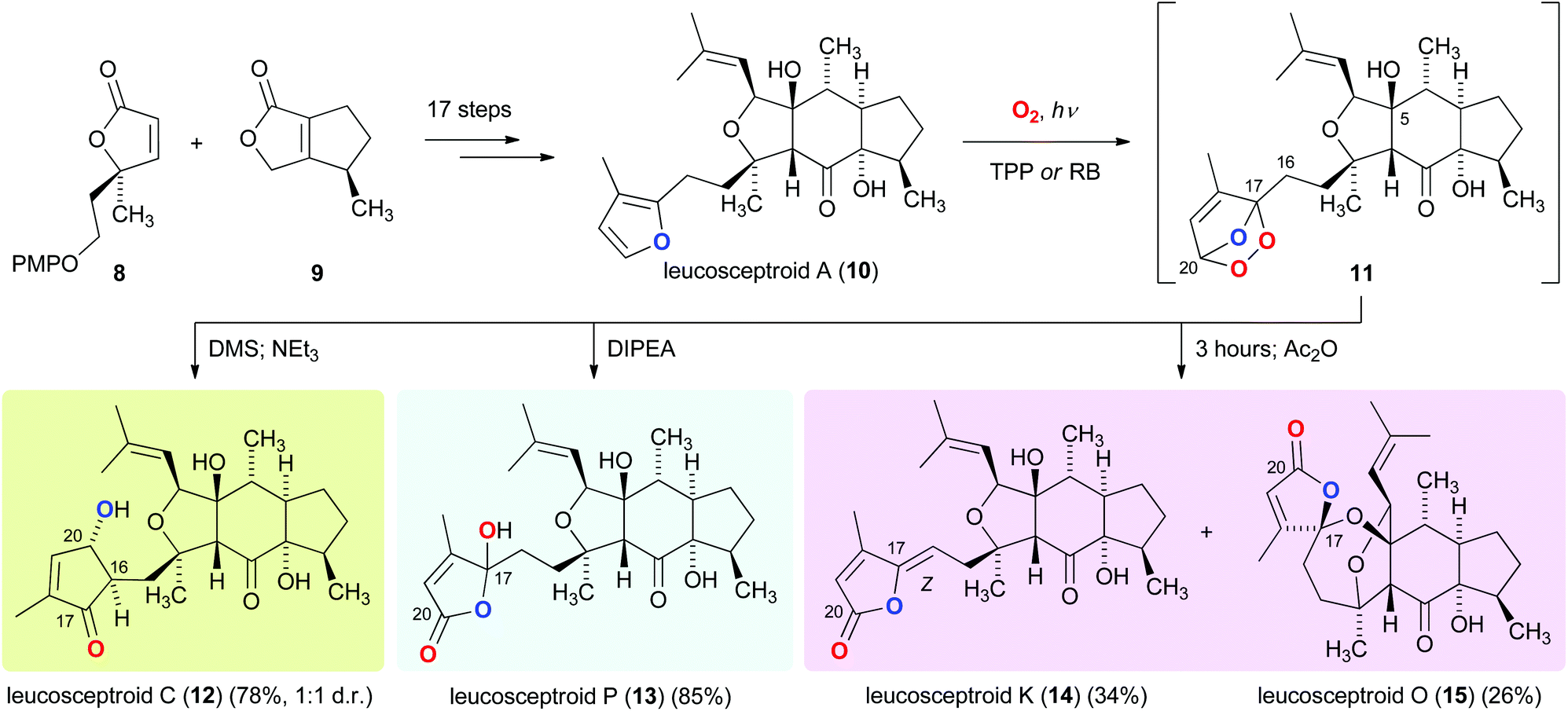 | ||
| Scheme 2 Photo-oxidation of leucosceptroid A (10) for the bioinspired total synthesis of leucosceptroids C (12), P (13), K (14), and O (15). | ||
Moreover, we discovered that generation of endo-peroxide 11, followed by purposeful idleness led to arguably the most exciting transformation. Upon standing in solution at 23 °C, 11 slowly underwent competing elimination and spirocyclization to afford, after dehydration, leucosceptroids K (14) and O (15). The result of this bioinspired transformation proved highly instructive regarding two different aspects. Firstly, by concurring with the circumstance that only the Z isomer of leucosceptroid K (14) has been isolated to date. We hypothesized that a crucial hydrogen bonding interaction between OH-5 and the endo-peroxide moiety in 11 results in a selective elimination reaction to form 14. Secondly, identification of leucosceptroid A (10) as the likely biosynthetic precursor of leucosceptroid O (15) was found to be an invaluable discovery. We had initially suggested that the spirocyclic motif in 15 might also be derived from leucosceptroids P (13) or K (14) by virtue of intramolecular spiroketalization, a transformation that we had failed to emulate in the laboratory.13
Bioinspired oxidations
An enthralling example of a bioinspired oxidation also involving singlet oxygen as a powerful reagent14 was recently reported in the total synthesis of cardamom peroxide (20).15 This natural product features an oxidized, dimeric monoterpene framework and incorporates a bridging seven-membered endo-peroxide. While the biosynthetic origin of 20 was unknown at the outset of the studies, Maimone and Hu hypothesized that two monoterpene units and three units of molecular oxygen might be the fundamental building blocks (Scheme 3).16 Moreover, the endo-peroxide motif was considered to arise from an unusual 7-endo cyclization of a peroxy radical (as shown in 19).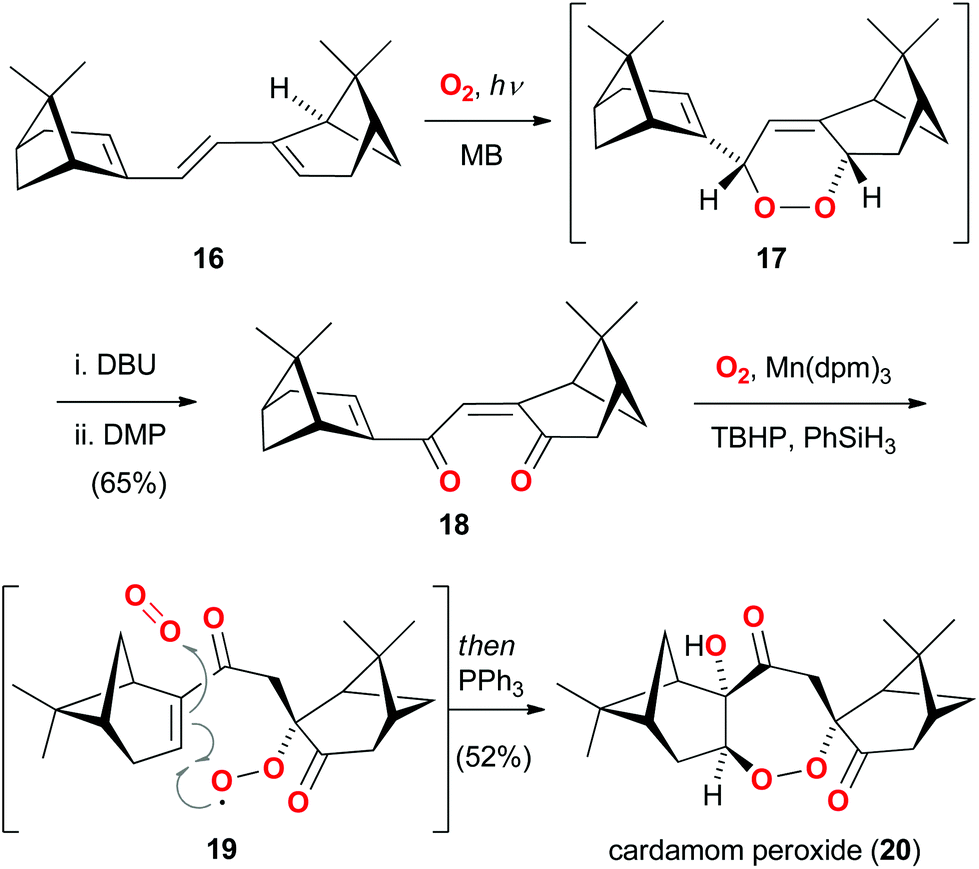 | ||
| Scheme 3 Maimone's concise total synthesis of cardamom peroxide (20) based on an oxygen stitching strategy. | ||
In their strategy, myrtenal was first dimerized to triene 16, which underwent smooth [4 + 2] cycloaddition with singlet oxygen. The intermediate endo-peroxide 17 was then converted to dienone 18via sequential Kornblum–DeLaMare rearrangement and oxidation. Exposure of 18 to molecular oxygen and a manganese catalyst led to an impressive tandem hydroperoxidation process, which presumably involved the postulated intermediate 19. A final reduction with triphenylphosphine completed the four-step synthesis of cardamom peroxide (20) and corroborated the biosynthetic proposal involving a 7-endo cyclization of a peroxy radical.
A conceptually quite different approach for the total synthesis of natural products was adopted by Baran, who hypothesized that a strategy, which holistically mimics nature's two-phase terpenoid synthesis would prove advantageous.17 Following the first (cyclase) phase, where the carbon framework is constructed from linear hydrocarbon building blocks, the second (oxidase) phase would involve divergent oxidation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–H bonds to generate structural diversity. Inspired by nature's efficiency in creating a myriad of complex terpenoids, Baran and co-workers initiated a research program aimed at replicating the biosynthesis of taxanes.18 In 2012, the artificial first phase synthesis of the taxane family was outlined, and taxadienone (23) was introduced as the laboratory's synthetic cyclase phase endpoint (Scheme 4).19 Since this prelude to an ultimate total synthesis of Taxol, considerable advances have been reported regarding translation of the oxidase phase and in the ascent of the ‘oxidase phase pyramid’.20
C and C–H bonds to generate structural diversity. Inspired by nature's efficiency in creating a myriad of complex terpenoids, Baran and co-workers initiated a research program aimed at replicating the biosynthesis of taxanes.18 In 2012, the artificial first phase synthesis of the taxane family was outlined, and taxadienone (23) was introduced as the laboratory's synthetic cyclase phase endpoint (Scheme 4).19 Since this prelude to an ultimate total synthesis of Taxol, considerable advances have been reported regarding translation of the oxidase phase and in the ascent of the ‘oxidase phase pyramid’.20
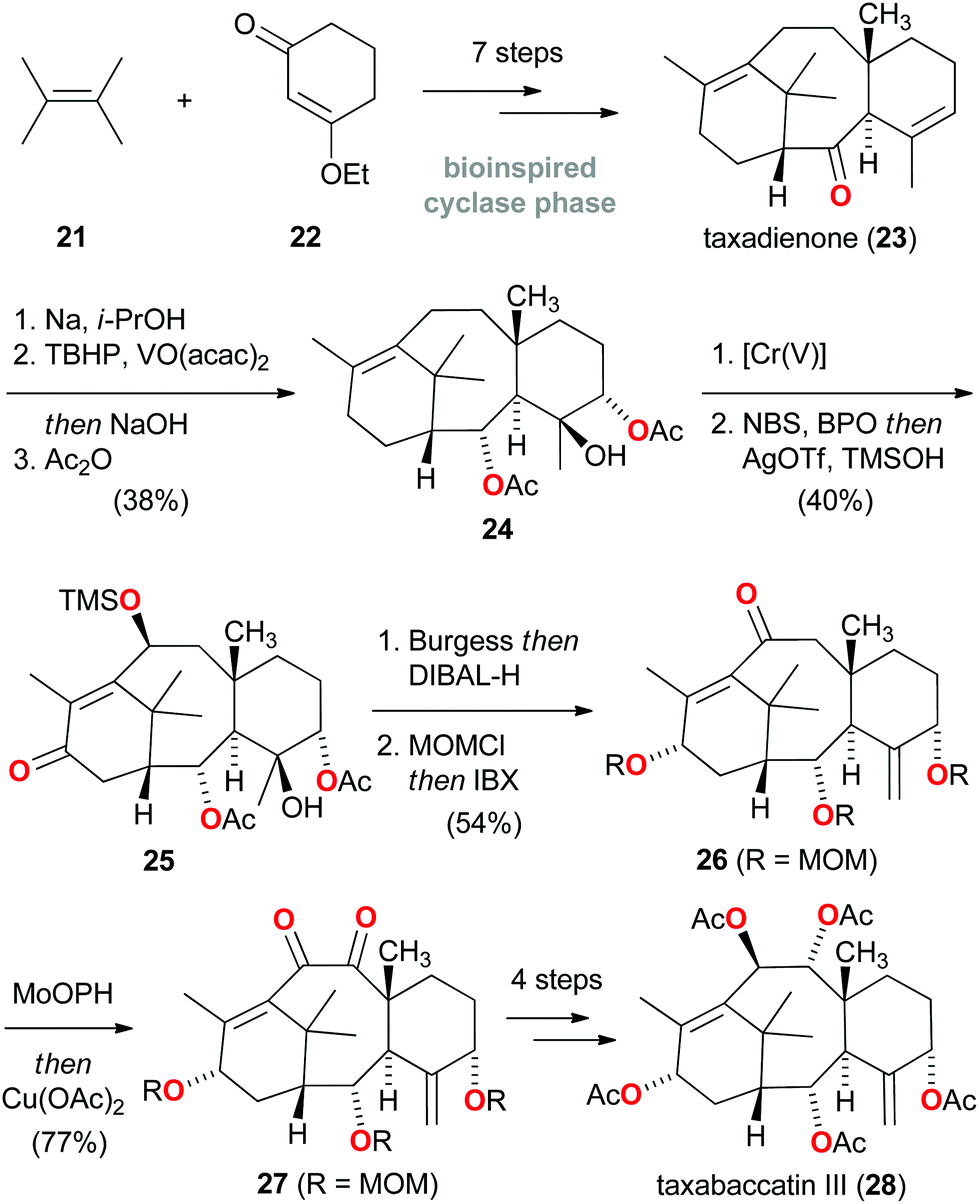 | ||
| Scheme 4 Baran's bioinspired two-phase terpenoid total synthesis of taxabaccatin III (28). | ||
In this regard, the successful synthesis of ‘level 7’ taxabaccatin III (28), featuring five oxidized carbon atoms and two degrees of unsaturation, was recently reported starting from taxadienone (23).21 As outlined in Scheme 4, C–O bonds were sequentially introduced into the carbon framework using a carefully choreographed sequence of reactions involving epoxidation (23 → 24), a novel chromium(V)-mediated allylic oxidation and radical-based C–H functionalization (24 → 25), and α-hydroxylation (26 → 27). These sequential, regio- and stereoselective C–H oxidations highlight that a creative bioinspired synthesis may constitute a huge driving force for the development of novel methods and can stimulate considerable advances in a field of chemistry.22 While further ascension of the ‘oxidase phase pyramid’ to eventually reach Taxol certainly remains a significant challenge, this total synthesis of taxabaccatin III (28) constitutes a successful proof of concept of the bioinspired two-phase terpenoid synthesis principle.
Pursuing a biosynthetic hypothesis
Recent work by our group focused on developing a total synthesis of the diterpenoid dictyoxetane (35), which contains an oxetane embedded in a unique dioxatricyclic ring system.23 Following several unsuccessful approaches to construct the highly substituted oxetane,24 our synthetic journey led us to pursue a bioinspired strategy. Guided by a biosynthetic hypothesis, which proposed three sequential exo-tet cyclizations for the formation of the dioxatricyclic substructure of 35,25 we prepared epoxide 30 from tricycle 29 (Scheme 5). Pleasingly, this benzyl-protected analog of the suggested biosynthetic intermediate could be converted to epoxide 31via the postulated 3-exo cyclization. However, all subsequent attempts to induce the 5-exo cyclization to form trans-tetrahydrofuran 32 failed under a wide range of conditions. Having become aware that the unfavourable conformation of the seven-membered ring in 31 prevented the envisioned 5-exo etherification, we devised a crucial modification of the originally proposed biosynthesis, wherein oxetane formation would occur prior to that of the strained trans-tetrahydrofuran ring.26 Towards this end, a highly selective photo-oxidation of the silyl ether derived from tricycle 29 furnished allylic alcohol 33. Formation of the allylic mesylate followed by 4-exo cyclization then efficiently afforded oxetane 34. Concurring with our modified hypothesis, treatment of 34 with N-iodosuccinimide led to smooth 5-exo cyclization, completing the construction of the synthetically challenging dioxatricyclic substructure, and giving rise to dictyoxetane (35) after hydrogenolysis.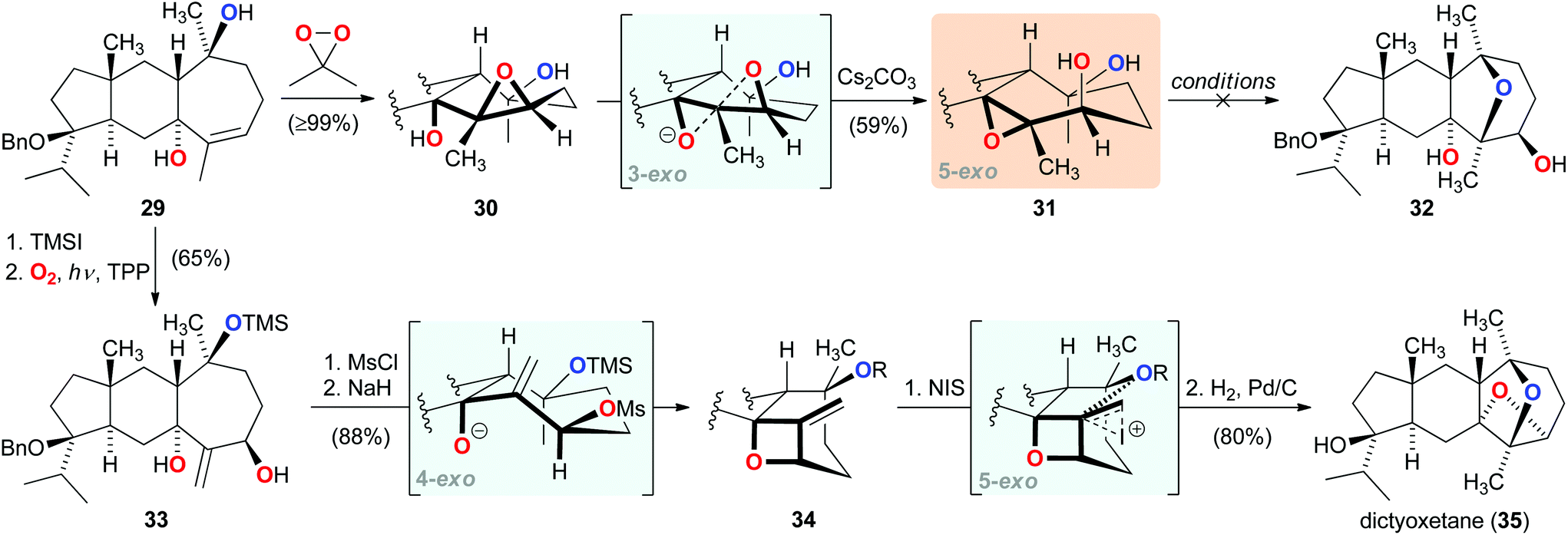 | ||
| Scheme 5 Bioinspired consecutive 4-exo-tet and 5-exo-tet cyclizations in the total synthesis of dictyoxetane (35). | ||
Conclusion and outlook
This Perspective has highlighted how several complex terpenoids were constructed by emulating processes taking place in nature, and disclosed benefits of applying the concept of bioinspired synthesis. Bioinspired total synthesis complements non-bioinspired approaches, and has resulted in a wealth of stunning discoveries, which arguably would not have been revealed otherwise.Besides unravelling biosynthetic links between natural products and disclosing how complex terpenoids are formed in nature, it was shown that a bioinspired approach may also stimulate innovation and the development of new reactions. As underpinned by our studies on dictyoxetane (35), it should be remembered that a total synthesis based on a biosynthetic proposal does not represent definitive proof of a corresponding biogenetic pathway, nor does a failed attempt to translate a hypothesis imply it is necessarily incorrect. However, our studies exemplify that bioinspired chemistry may favourably build upon hypotheses and ultimately result in a successful synthetic strategy.
Bioinspired synthesis has become one of the most efficient approaches to make natural products since its birth 100 years ago. What has to be done in the 21st century? Expanding the chemist's toolbox (reagents and catalysts) and increasing the repertoire of chemical transformations (reactions) are prerequisites to successfully mimic not only individual steps but whole sequences of a biosynthesis. This will be also necessary to further improve the overall efficiency and practicability of current bioinspired syntheses in order to facilitate the implementation into industrial processes and ultimately outcompete nature. By having presented the attractiveness and tremendous benefits of pursuing bioinspired strategies, we hope this article will stimulate and inspire further research groups to bear biosynthetic considerations in mind while devising synthetic routes.
Acknowledgements
We gratefully acknowledge financial support from the Funds of the Chemical Industry (Sachkostenzuschuss and Dozentenpreis to T. M. and Kekulé Mobility Fellowship to C. L. H.) and the German Research Foundation (Emmy Noether Fellowship, SFB 749 and SFB 152 to T. M.). We thank Dr Julius R. Reyes (LMU Munich) for helpful discussions during the preparation of this manuscript.Notes and references
- M. C. de la Torre and M. A. Sierra, Angew. Chem., Int. Ed., 2004, 43, 160 CrossRef CAS PubMed.
- E. Gravel and E. Poupon, Eur. J. Org. Chem., 2008, 27 CrossRef.
- M. Razzak and J. K. de Brabander, Nat. Chem. Biol., 2011, 7, 865 CrossRef CAS PubMed.
- (a) K. C. Nicolau, D. J. Edmonds and P. G. Bulger, Angew. Chem., Int. Ed., 2006, 45, 7134 CrossRef PubMed; (b) R. Ardkhean, D. F. J. Caputo, S. M. Morrow, H. Shi, Y. Xiong and E. A. Anderson, Chem. Soc. Rev., 2016, 45, 1557 RSC.
- P. G. Bulger, S. K. Bagal and R. Marquez, Nat. Prod. Rep., 2008, 25, 254 RSC.
- A. M. Armaly, Y. C. DePorre, E. J. Groso, P. S. Riehl and C. S. Schindler, Chem. Rev., 2015, 115, 9232 CrossRef CAS PubMed.
- L. Acebey, M. Sauvain, S. Beck, C. Moulis, A. Gimenez and V. Jullian, Org. Lett., 2007, 9, 4693 CrossRef CAS PubMed.
- (a) C. Yuan, B. Du, L. Yang and B. Liu, J. Am. Chem. Soc., 2013, 135, 9291 CrossRef CAS PubMed; (b) B. Du, C. Yuan, T. Yu, L. Yang, Y. Yang, B. Liu and S. Qin, Chem. – Eur. J., 2014, 20, 2613 CrossRef CAS PubMed.
- (a) S.-H. Luo, Q. Luo, X.-M. Niu, M.-J. Xie, X. Zhao, B. Schneider, J. Gershenzon and S.-H. Li, Angew. Chem., Int. Ed., 2010, 49, 4471 CrossRef CAS PubMed; (b) S.-H. Luo, L.-H. Weng, M.-J. Xie, X.-N. Li, J. Hua, X. Zhao and S.-H. Li, Org. Lett., 2011, 13, 1864 CrossRef CAS PubMed; (c) S.-H. Luo, J. Hua, C.-H. Li, S.-X. Jing, Y. Liu, X.-N. Li, X. Zhao and S.-H. Li, Org. Lett., 2012, 14, 5768 CrossRef CAS PubMed; (d) S.-H. Luo, J. Hua, X.-M. Niu, Y. Liu, C.-H. Li, Y.-Y. Zhou, S.-X. Jing, X. Zhao and S.-H. Li, Phytochemistry, 2013, 86, 29 CrossRef CAS PubMed; (e) S.-H. Luo, J. Hua, C.-H. Li, Y. Liu, X.-N. Li, X. Zhao and S.-H. Li, Tetrahedron Lett., 2013, 54, 235 CrossRef CAS; (f) X. Huang, L. Song, J. Xu, G. Zhu and B. Liu, Angew. Chem., Int. Ed., 2013, 52, 952 CrossRef CAS PubMed; (g) S. Guo, J. Liu and D. Ma, Angew. Chem., Int. Ed., 2015, 54, 1298 CrossRef CAS PubMed.
- (a) C. L. Hugelshofer and T. Magauer, Angew. Chem., Int. Ed., 2014, 53, 11351 CrossRef CAS PubMed; (b) C. L. Hugelshofer and T. Magauer, Synlett, 2015, 572 CAS.
- S.-H. Luo, C. L. Hugelshofer, J. Hua, S.-X. Jing, C.-H. Li, Y. Liu, X.-N. Li, X. Zhao, T. Magauer and S.-H. Li, Org. Lett., 2014, 16, 6416 CrossRef CAS PubMed.
- N. Kornblum and H. E. DeLaMare, J. Am. Chem. Soc., 1951, 73, 880 CrossRef CAS.
- C. L. Hugelshofer and T. Magauer, J. Am. Chem. Soc., 2015, 137, 3807 CrossRef CAS PubMed.
- (a) I. Margaros, T. Montagnon, M. Tofi, E. Pavlakos and G. Vassilikogiannakis, Tetrahedron, 2006, 62, 5308 CrossRef CAS; (b) T. Montagnon, M. Tofi and G. Vassilikogiannakis, Acc. Chem. Res., 2008, 41, 1001 CrossRef CAS PubMed; (c) A. A. Ghogare and A. Greer, Chem. Rev., 2016, 116, 9994 CrossRef CAS PubMed.
- S. Kamchonwongpaisan, C. Nilanonta, B. Tarnchompoo, C. Thebtaranonth, Y. Thebtaranonth, Y. Yuthavong, P. Kongsaeree and J. Clardy, Tetrahedron Lett., 1995, 36, 1821 CrossRef CAS.
- X. Hu and T. J. Maimone, J. Am. Chem. Soc., 2014, 136, 5287 CrossRef CAS PubMed.
- K. Chen and P. S. Baran, Nature, 2009, 459, 824 CrossRef CAS PubMed.
- Y. Ishihara and P. S. Baran, Synlett, 2010, 1733 CAS.
- A. Mendoza, Y. Ishihara and P. S. Baran, Nat. Chem., 2012, 4, 21 CrossRef CAS PubMed.
- N. C. Wilde, M. Isomura, A. Mendoza and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 4909 CrossRef CAS PubMed.
- C. Yuan, Y. Jin, N. C. Wild and P. S. Baran, Angew. Chem., Int. Ed., 2016, 55, 8280 CrossRef CAS PubMed.
- (a) M. S. Chen and M. C. White, Science, 2007, 318, 783 CrossRef CAS PubMed; (b) T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362 CrossRef CAS PubMed.
- K. C. Pullaiah, R. K. Surapaneni, C. B. Rao, K. F. Albizati, B. W. Sullivan, D. J. Faulkner, H. Cun-heng and J. Clardy, J. Org. Chem., 1985, 50, 3665 CrossRef CAS.
- C. L. Hugelshofer and T. Magauer, Chem. – Eur. J., 2016, 22, 15125 CrossRef CAS PubMed.
- J. Reinecke and H. M. R. Hoffmann, Chem. – Eur. J., 1995, 1, 368 CrossRef CAS.
- C. L. Hugelshofer and T. Magauer, J. Am. Chem. Soc., 2016, 138, 6420 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |