DOI:
10.1039/C6OB01665K
(Review Article)
Org. Biomol. Chem., 2017,
15, 34-50
Recent advances in the chemistry of metallated azetidines
Received
2nd August 2016
, Accepted 13th October 2016
First published on 13th October 2016
Abstract
The almost unexplored four-membered heterocycles azetidines, represent a particularly interesting class of molecules, among the family of saturated nitrogen heterocycles. Although often challenging to synthesize, substituted azetidines strongly attract chemists because of their importance in catalysis, stereoselective synthesis and medicinal chemistry. This review aims to give a brief summary of modern developments in direct metal-based functionalization of the azetidine ring, focusing on the regio- and stereoselectivity of these reactions, as well as on some useful synthetic applications. It will be highlighted, in particular, how an interplay of factors such as structure, substitutions at both nitrogen and carbon atoms and coordinative phenomena deeply influence the reactivity of the corresponding metallated species, paving the way for easy planning a site-selective functionalization of azetidines.
 Daniele Antermite | Daniele Antermite obtained his MS degree in Chemistry and Pharmaceutical Technology at the University of Bari (Italy) in April 2016. In 2014, he was Erasmus trainee at the Karlsruhe Institute of Technology (Germany) in the group of Dr N. Jung and Prof. S. Bräse. In 2015, as Erasmus student, carried out his master thesis, focused on organometallic (lithium and magnesium) chemistry, at the University of Vienna (Austria) under the joint supervision of Prof. R. Luisi, Dr L. Degennaro and Dr V. Pace. In October 2016, was hired as PhD student at Imperial College of London in the group of Dr J. A. Bull, here he will be working on palladium-catalysed directed C(sp3)–H arylation of saturated heterocycles. |
 Leonardo Degennaro | Leonardo Degennaro was born in 1972 in Bitonto. He obtained the master degree in Chemistry and Pharmaceutical Technology in 1999 and the PhD in Applied Chemical and Enzymatic Synthesis in 2003. In 2002 he was “visiting scholar” at the University of Groningen under the supervision of Prof. B. L. Feringa. In 2006 he was appointed assistant professor in Organic Chemistry at the Department of Pharmacy of the University of Bari. In 2011 he has been “visiting assistant professor” at the University of Kyoto working in the group of Prof. J.-I. Yoshida. The research activity is aimed at developing new stereocontrolled synthesis by using small heterocycles and organometallic species, and microreactor technology. |
 Renzo Luisi | Renzo Luisi is an Associate Professor of Organic Chemistry at the University of Bari “A. Moro” – Italy. He graduated summa cum laude in Chemistry and Pharmaceutical Technology at the University of Bari (Italy) in 1996 and got the PhD in Chemical Sciences in 2000. He was appointed Assistant Professor at the University of Bari in 2001, and Associate Professor of Organic Chemistry in 2005. The main research interests rely on organometallic chemistry (mainly lithium and boron chemistry), synthesis and reactivity of small heterocyles, asymmetric synthesis, dynamic NMR spectroscopy and flow chemistry. He published about 100 papers in peer-reviewed international journals, book chapters, reviews and hold several international collaborations. |
Introduction
Saturated nitrogen-containing heterocycles represent one of the most privileged structural motifs among natural products, marketed drugs and bioactive molecules and their synthesis and elaboration continue to constitute a major area of synthetic chemical research.1 In recent years, interest in the effective access to substituted heterocycle derivatives has become even more relevant due to the increasing demand for enhanced degree of saturation and three-dimensionality in drug candidates.2,3 Enhanced 3D shapes (directly connected with the increased fraction of sp3 carbon atoms) have been proposed to lead to more beneficial physicochemical, pharmacokinetic and safety properties, such as reduced lipophilicity and improved solubility, permeability and off-target promiscuity.2c,d
Among the family of saturated N-bearing heterocycles, the almost unexplored azetidines represent a particularly interesting class of compounds, since they own a good compromise between a satisfactory stability and a strong molecular rigidity, allowing an efficient tuning of pharmacological properties displayed by molecules bearing this moiety.4 Historically, the chemistry of their higher and lower homologues have received much more attention from synthetic chemists with respect to this four-membered aza-heterocycle, as evidenced by several commercial applications.5 Despite this, the azetidine core is emerging as highly appealing within medicinal and agrochemical industries because of its occurrence in various biologically active natural compounds6 and drug candidates (Fig. 1).4b,7 Moreover, azetidines have also found some applications as ligands in metal-catalyzed transformation and as chiral auxiliaries.8 The most representative examples, between the numerous practical applications of the azetidine ring, are likely related to recently marketed drugs such as Azelnipidine (Calblock®)9 and Ximelagatran (Exanta®)10 or to a large variety of 3- and 2-substituted azetidines showing different pharmacological activities (Fig. 1).11 The main reason for the lagging of azetidine chemistry behind its larger and smaller family counterparts, should be recognized in the large lack of effective methodologies associated with the synthesis and functionalisation of this particular ring.5e,f Given these premises, the development of robust synthetic methods to access these building blocks is certainly of high value. Nevertheless, over the past few decades, a varieties of protocols for the preparation of azetidines have been reported.12 The most common strategies for the synthesis of 2-substituted azetidines involve intramolecular cyclizations of open-chain structures (Scheme 1, path a) or the reduction of readily available 2-azetidinones (Scheme 1, path b).4b,13 It is worth mentioning the possibility to perform also an asymmetric dehydrative cyclization of 1,3-amino alcohols using 1,1′-carbonyldiimidazole to synthesize 2,3-disubstituted azetidines.14 However, the well-known kinetically unfavorable cyclization, generally limits these kind of synthetic strategies, resulting often in only moderate yields. Moreover, many of these procedures require several steps and functional group transformations.15 An interesting three-step nucleophilic addition/ring contraction of α-bromopyrrolidinones has been recently reported by Blanc and coworkers to obtain the corresponding racemic α-carbonylated azeditines (Scheme 1, path c).16 On the other hand, protocols for the aziridine ring expansion using dimethylsulfoxonium methylide have also been documented (Scheme 1, path d).4a,b An unresolved limitation of most of these strategies is that the substituents are required to be present on the precursors, prior to the ring formation, thus not enabling straightforward structure–activity relationship (SAR) studies and compounds libraries synthesis of pharmaceutical interest. In this sense, Jamison and co-workers in 2015 described an advantageous two-step synthesis of optically active 2-alkyl azetidines (Scheme 1, path e).17 This latter strategy employs a highly regioselective nickel-catalyzed cross-coupling of enantiomerically pure N-tosyl-aziridines with aliphatic organozinc reagents, followed by an intramolecular cyclization. A key factor for the successful outcome of the reaction was the presence of a thiophenyl function tethered to the aziridine ring, which can be easily activated by methylation to trigger the subsequent cyclization.
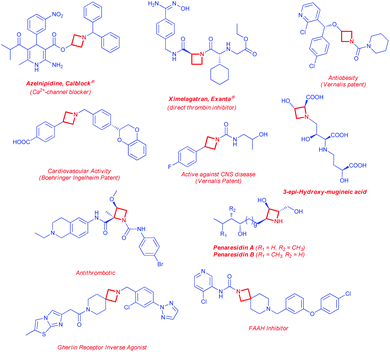 |
| | Fig. 1 Noteworthy examples of biologically active drug leads and natural products incorporating the azetidine nucleus. | |
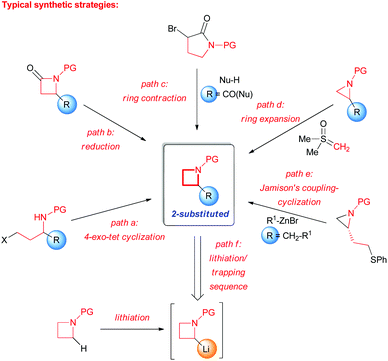 |
| | Scheme 1 Representative methods for the synthesis of 2-substituted azetidines. PG = protecting group. | |
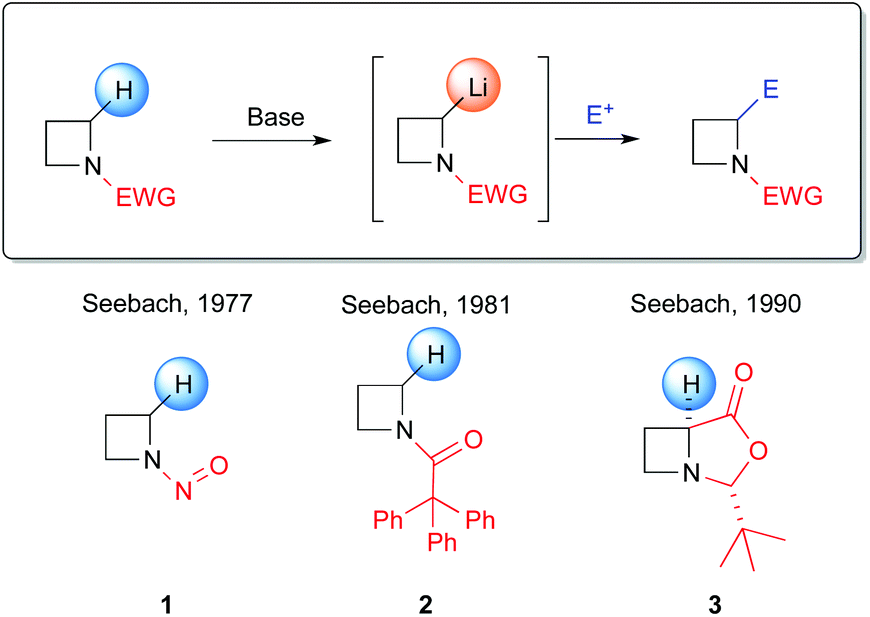
In this context, a highly appealing alternative strategy that traditionally has been largely employed for the assembly of α-branched amines is undoubtedly represented by the α-deprotonation of a N-protected (activated) amine by lithium bases, followed by the trapping with a suitable electrophile (Scheme 1, path f). However, the chemistry of lithiated azetidines has been considerably less explored, compared to metallated aziridines, pyrrolidines or piperidines. To date, very few examples of lithiation of four-membered azetidines have been reported. Pioneering works in this field were carried out by Seebach et al. between 1970s and 1980s, dealing with azetidines bearing electron-withdrawing groups (EWG), both on the nitrogen atom and on the C2, to activate the α-C–H bond by increasing its acidity (Scheme 2).18 However, several drawbacks are related to these protocols, including carcinogenicity associated to the lithiation of N-nitrosoazetidine 1 with LDA (THF, −78 °C)18a and poor prospects for later development of an asymmetric application due to the use of tBuLi to achieve the metallation of N-(triphenylacetyl)azetidine 2 (THF, −40 °C).18b,19 In addition, competitive ortho-lithiation at the triphenylacetyl protecting group, followed by a carbamoyl 1–3 shift was reported.18b Furthermore, no useful synthetic applications were reported at that time.
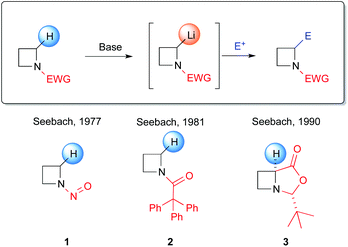 |
| | Scheme 2 First reported examples of α-lithiated azetidines. | |
Only recently, more systematic studies on the α-lithiation of N-protected azetidines have been performed, allowing a better understanding of their reactivity and furnishing mechanistic insights, that could help to fill the knowledge gap. In this review we will discuss recent advances on the reactivity of metallated azetidines, with a focus on their regio- and stereoselective metallation in general and lithiation in particular, also highlighting some useful synthetic applications. Special attention will be dedicated to the interplay of factors such as structure, substitutions at both nitrogen and carbon atoms and complexation phenomena that plays a pivotal role in controlling the reactivity of the corresponding metallated systems.
Lithiation of N-EWG azetidines
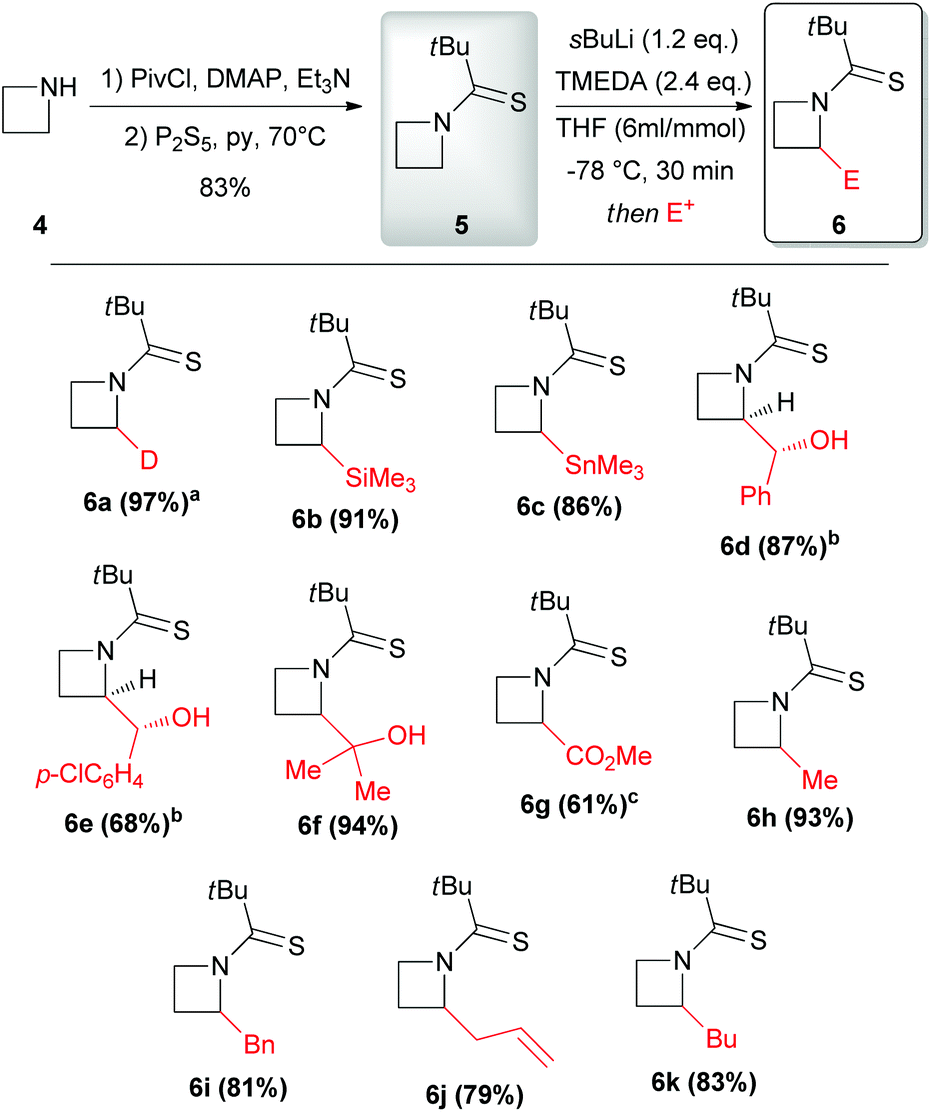
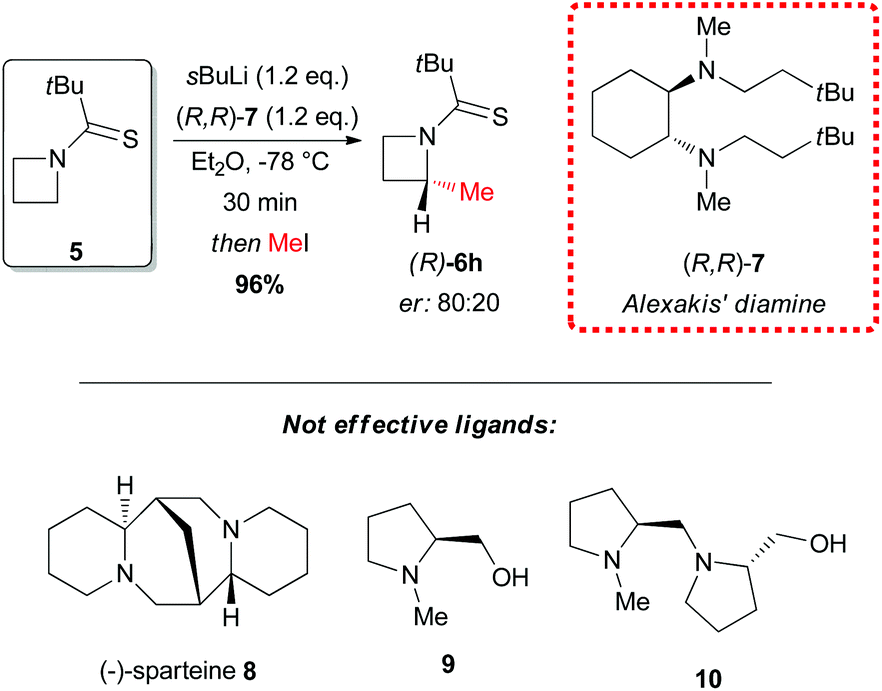
In 2010 Hodgson reported the first example of effective α-lithiation–trapping sequence at the 2-position of azetidines bearing the unusual Seebach's20N-thiopivaloyl activating group (Scheme 3).21 The starting thioamide 5 could be obtained by the simple N-protection of azetidine 4 with pivaloyl chloride, followed by treatment with P2S5. Subsequent lithiation with sBuLi/TMEDA in THF at −78 °C and trapping with several electrophiles after 30 min gave 2-substituted azetidines 6a–k in excellent yields (Scheme 3). This protocol tolerated a large variety of different electrophiles including silyl- and stannyl chlorides, carbonyl derivatives (including enolizable ones, such as acetone) and alkyl halides. The latter case is notable, since alkylation of dipole-stabilized organolithiums can be sometimes inefficient because of possible SET (single electron transfer) process.22 The reactions using prochiral aldehydes occurred with acceptable diastereoselectivity leading to the isolation of products 6d and 6e as main diasteromers.23 Different EWG on the nitrogen atom, such Boc, tBuSO, tBuSO2, PO(OEt)2, were found to be unsuitable for effective α-lithiation/trapping sequence on unsubstituted azetidines, in contrast to what observed in the case of the corresponding aziridines.24 Undoubtedly, the N-thiopivaloyl group played a main role for the successful outcome of this transformation, since when α-metallation of the corresponding pivalamide derivate was performed, only the attack of sBuLi at the carbonyl group was observed, while the use of LTMP returned only unreacted starting material. The rationale at the basis of this unique effectiveness (with respect to the classical activating groups) is unknown, but the authors suggested it could be found in the combination of α-position activation and a reduced or even absent tendency for the attack of the lithium base at the thiocarbonyl moiety. It should be stressed that this method allowed also an effective asymmetric variant, by replacing TMEDA with different chiral ligands.21 Lithiation of N-thiopivaloyl azetidine 5 using Alexakis’ trans-cyclohexane diamine (R,R)-7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 25 in Et2O gave, after trapping with MeI, α-methyl azetidine (R)-6h in excellent yield (96%) and 80:20 er (Scheme 4). However, the use of different chiral ligands, including (−)-sparteine 8 or proline-based ligands 9 and 10, as well as different solvents (ethereal and not), resulted in lower enantiomeric ratios. Remarkably, the sense of the stereoselectivity of the reaction was opposite to that previously obtained with N-Boc pyrrolidine and N-Boc piperidine, using sBuLi and (−)-sparteine 8 or (R,R)-7.26 Moreover, clean removal of the N-thiopivaloyl protecting group was achieved for azetidine 6i, by using MeLi; conversion in the corresponding pivalamide 12 could also be obtained by using CH3CO3H (Scheme 5).21
25 in Et2O gave, after trapping with MeI, α-methyl azetidine (R)-6h in excellent yield (96%) and 80:20 er (Scheme 4). However, the use of different chiral ligands, including (−)-sparteine 8 or proline-based ligands 9 and 10, as well as different solvents (ethereal and not), resulted in lower enantiomeric ratios. Remarkably, the sense of the stereoselectivity of the reaction was opposite to that previously obtained with N-Boc pyrrolidine and N-Boc piperidine, using sBuLi and (−)-sparteine 8 or (R,R)-7.26 Moreover, clean removal of the N-thiopivaloyl protecting group was achieved for azetidine 6i, by using MeLi; conversion in the corresponding pivalamide 12 could also be obtained by using CH3CO3H (Scheme 5).21
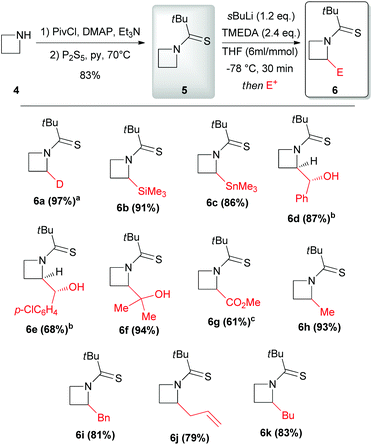 |
| | Scheme 3 Preparation, lithiation and electrophilic trapping of N-thiopivalamide 5. All presented yields are referred to the isolated product. a100% D, determined by GC-MS and 1H NMR. bMain diastereomer. cIsolated as the methyl ester following reaction with TMSCHN2. py = pyridine, DMAP = 4-dimethylaminopyridine, Piv = pivaloyl. | |
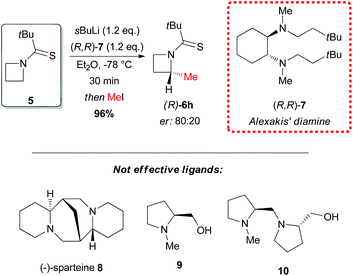 |
| | Scheme 4 Asymmetric lithiation–methylation sequence of N-thiopivaloyl azetidine 5. | |
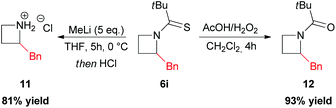 |
| | Scheme 5 Removal and transformation of the N-thiopivaloyl functionality. | |
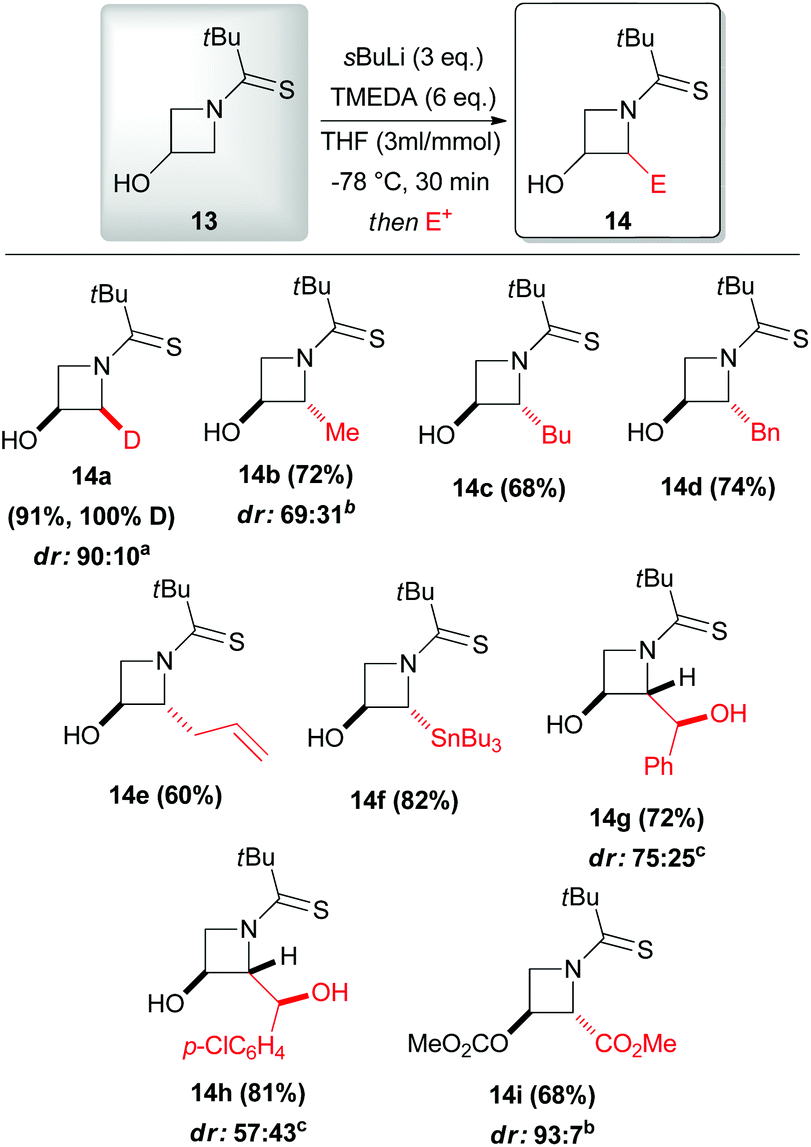
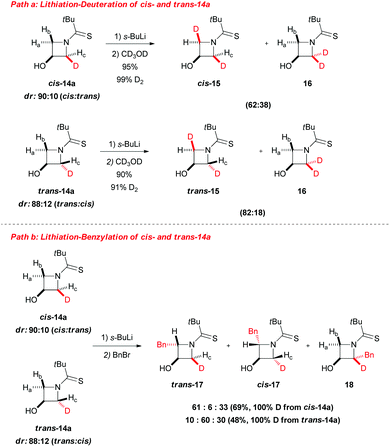
Some years later, the same author extended such a lithiation approach to N-thiopivaloylazetidin-3-ol 13, allowing access to a range of 2-substituted 3-hydroxyazetidines 14a–i with generally excellent trans-diastereoselectivity. Remarkably, only in the case of deuteration the cis-isomer was found to be the major product (Scheme 6).27 To the optimal outcome of the reaction were required 3 equivalents of sBuLi, because of the presence of the free hydroxyl substituent, and a higher concentration than in the case of unsubstituted N-thiopivaloyl azetidine 5. The best thermal condition was demonstrated to be −78 °C, since higher temperature led to reduced diasteromeric ratios and no significant improvement was observed at lower temperatures as well. Moreover, the use of TMEDA in THF was also found to be crucial for obtaining optimal yields. The reaction with Mander's reagent (methyl cyanoformate) provided C- and O-methoxycarbonylated azetidine 14i, which is highly attractive, due to the possibility of accessing azetidine-based amino acids (Scheme 6). Noteworthy, subsequent mechanistic investigation on cis-14a and trans-14a allowed to get more insights into the stereoselectivity of the lithiation–electrophilic trapping sequence (Scheme 7). Based on deuterium labeling studies, Hodgson suggested that the initial α-deprotonation is preferentially, but not exclusively, trans-stereoselective (Scheme 7, path a): indeed, removal of the proton trans to hydroxyl was hindered in the case of trans-14a due to the presence of the trans-deuterium, accounting for the unequal proportions of 2,2-dideuterated adducts 16 (primary kinetic isotope effect). Moreover, lithiation–benzylation experiments performed on cis-14a and trans-14a led to mixtures of three possible diastereomers (Scheme 7, path b). Intriguingly, only trans-2,2-derivate 18 was observed starting either from cis-14a and trans-14a. Thus, these results suggest that there is a strong preference for electrophilic trapping to occur trans- to the C-3 lithium alkoxide (aside from deuteration that occurred with a strong cis- bias, where probably the alkoxide may have an important role in directing the deuteration cis to itself),28 regardless of whether a cis or trans proton is initially removed at C-2. In other words, the stereochemical outcome of the lithiated azetidinol trapping depends on the electrophile used but is independent of which α-proton is initially abstracted. However, it should be highlighted that such a kind of speculation must be treated with caution at this early stage, because it is not possible to completely exclude an initial cis-deprotonation and because we cannot deduce any information on the stereochemical (in)stability of the lithiated intermediate.
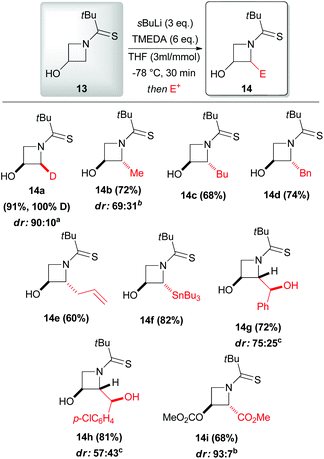 |
| | Scheme 6 Electrophilic incorporation into N-thiopivaloylazetidin-3-ol 13. All presented yields are referred to the isolated product. aRatio of cis/trans: major diastereoisomer shown. bRatio of trans/cis: major diastereoisomer shown. cMixture of epimers at side-chain carbinol: major diastereoisomer shown. | |
 |
| | Scheme 7 Stereochemical investigations on the lithiation and electrophilic trapping of Azetidinol 14a. | |
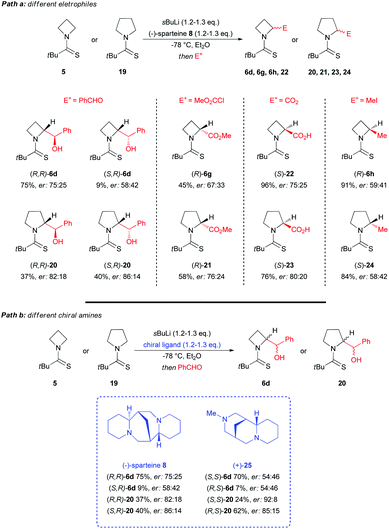
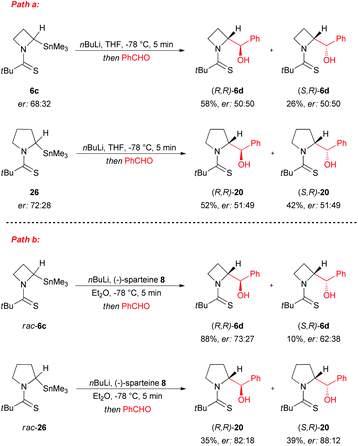
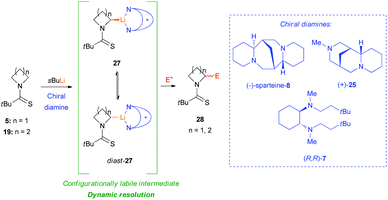
In this context, recently, O'Brien et al. clearly demonstrated the intrinsic configurational instability of lithiated N-thiopivaloyl azetidine intermediates at −78 °C in THF or Et2O, through seminal mechanistic studies.29 As a direct evidence of this, a wide variation of enantioselectivity and sense of induction was reported, in almost the same reaction conditions, for the lithiation–trapping of both N-thiopivaloyl azetidine 5 and N-thiopivaloyl pyrrolidine 19, when different electrophiles or different chiral diamines were employed (Scheme 8). As highlighted, the variability of the sense of asymmetric induction strictly depends on the electrophile used. Indeed, with reference to the configuration of the α-carbon, adducts formed using benzaldehyde ((R,R)-6d, (S,R)-6d, (R,R)-20 and (S,R)-20) or methyl chloroformate ((R)-6g and (R)-21) presented an opposite configuration to those obtained by trapping with CO2 ((S)-22 and (S)-23); whilst, with MeI, products were found to have opposite stereochemistry starting from azetidine or pyrrolidine ((R)-6h and (S)-24, respectively). In the same way, by using different chiral diamines, it resulted in a significant variation in the level of enantioselectivity starting from azetidine 5 or pyrrolidine 19. Moreover, when the same lithiated intermediates were generated via tin–lithium exchange starting from stannanes 6c (68:32 er) and 26 (72:28 er), by using n-BuLi in THF at −78 °C and benzaldehyde as electrophile, essentially racemic diastereomers (R,R)-6d, (S,R)-6d and (R,R)-20, (S,R)-20, were obtained respectively in the absence of any chiral ligand (Scheme 9, path a). On the other hand when the same reaction was performed on racemic stannanes 6c and 26, in the presence of (−)-sparteine-8, diasteromeric adducts (R,R)-6d, (S,R)-6d and (R,R)-20, (S,R)-20 were obtained with a degree of enantioselectivity comparable to that achieved with sBuLi/(−)-sparteine 8 from azetidine 5 and pyrrolidine 19 (Scheme 9, path b). Based on the observation that the organolithium species generated also from N-thiopivaloyl pyrrolidine 19 is not configurationally stable at −78 °C, these authors first concluded that the reason of the difference, in terms of stability, compared to corresponding N-Boc pyrrolidines and piperidines (which, on the other hand, are well known to generate configurationally stable organolithium species),26,30 should be the N-thiopivaloyl group itself, which probably leads to a weaker C–Li bond. In addition, by excluding the possibility of an asymmetric deprotonation to give a configurationally stable lithiated intermediate, it appears clear that the lithiation–trapping, in the presence of a chiral ligand, of N-thiopivaloyl azetidine 5 and pyrrolidine 19 must proceed through dynamic resolution of configurationally labile diasteromeric organolithium intermediates 27 and diast-27 (Scheme 10). It is also reasonable to address the variable enantioselectivity, observed with different electrophiles (under the same conditions), to different rates of trapping, driving to a different configuration of the major product.
 |
| | Scheme 8 Asymmetric lithiation–trapping of N-thiopivaloyl azetidine 5 and pyrrolidine 19 with different electrophiles and chiral diamines. Major enantiomers shown. | |
 |
| | Scheme 9 Investigation on the configurational stability of lithiated N-thiopivaloyl azetidine and pyrrolidine generated through tin–lithium exchange. | |
 |
| | Scheme 10 Dynamic resolution in stereoselective lithiation–trapping of N-thiopivaloyl aza-heterocycles 5 and 19. | |
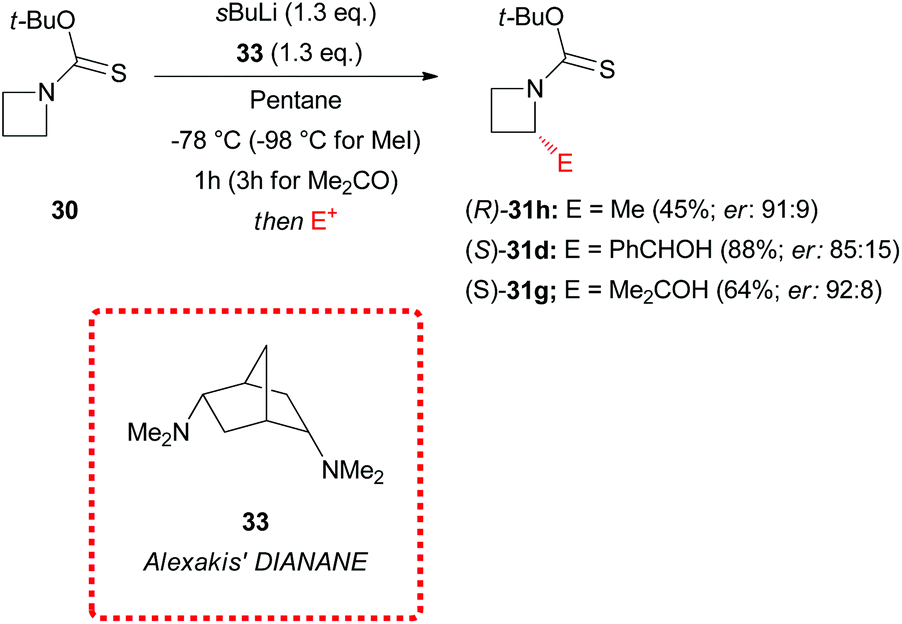
In 2015 Hodgson and coworkers described also the effectiveness of the tert-butoxythiocarbonyl group (Botc) in promoting azetidine α-lithiation, allowing at the same time mild conditions for deprotection.31N-Botc-azetidine 30 could be generated in very good yield (88%) by reaction of the readily available dithiocarbonic acid O-tert-butyl ester 29 with azetidine 4 (Scheme 11). Intriguing, no evidence of formation of the competitive dithiocarbamate, which was reported for reaction of tertiary alkyl xanthate esters with amines,32 was observed. Significantly, N-Botc-azetidine 30 could be efficiently lithiated under similar conditions to those previously described for N-thiopivaloyl-azetidine 5, and subsequently trapped with a wide range of electrophiles to give 2-substituted N-Boc-azetidines 31a–j (Scheme 11). Remarkably, silylation, stannylation, alkylation and reaction with carbonyl compounds occurred smoothly (with only a slight lowering of the yields observed for more electron-rich electrophiles such as p-anisaldehyde). As mentioned, N-Botc-azetidines underwent effective deprotection under mild acidic conditions (no ring opening was observed) as demonstrated for α-methyl N-Botc-azetidine 31h, which was quantitatively converted into the corresponding α-methyl-azetidine 32 by using TFA.
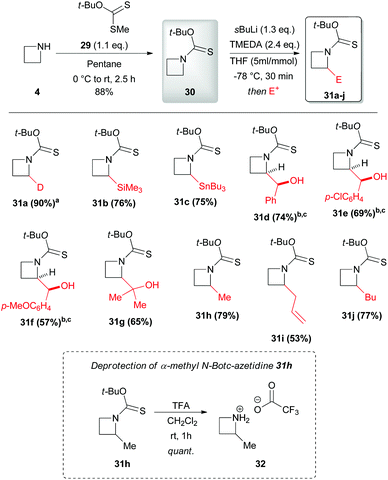 |
| | Scheme 11 Preparation, lithiation and electophilic trapping of N-Botc-azetidine 30. Deprotection of 31h with TFA. All presented yields are referred to the isolated product. a95% D, determined by mass spectrometry. bdr = 70:30, major diastereomer shown. c1.0 equivalent of sBuLi was used. | |
Alexakis’ DIANANE 33 (N,N,N′,N′-endo,endo-tetramethyl-2,5-diaminonorbornane)33 proved to be the optimal chiral ligand for enantioselective α-electrophilic substitution of N-Botc-azetidine 30, if compared to (−)-sparteine 8 and trans-cyclohexane diamine (R,R)-7, providing excellent levels of asymmetric induction (er up to 92:8), as described in Scheme 12. The enantioselectivity of the reaction appeared to be strictly dependent also on experimental parameters such as the lithiation time and the reaction temperature. Asymmetric methylation occurred smoothly at −98 °C with 1 h lithiation time, using DIANANE 33 in pentane, while lithiation and reaction with benzaldehyde furnished high enantiomeric ratios at −78 °C (lithiation time = 1 h). Eventually, in the case of acetone, asymmetric trapping required low temperature (−78 °C) and longer lithiation time (3 h).
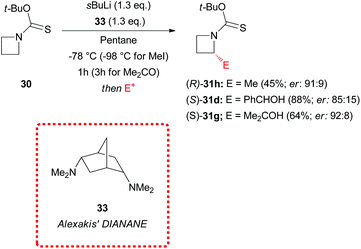 |
| | Scheme 12 Enantioselective α-electrophilic substitution of lithiated N-Botc-azetidine 30. | |
In addition, ethereal solvents proved to be unsuitable for asymmetric induction, since, as an example, the use of the same DIANANE 33 in Et2O at −78 °C (lithiation time of 1 h) gave α-methylated azetidine (R)-31h in 64% yield with poor stereoselectivity (er: 56:44). However, the authors did not provide any evidence about the configurational stability (or instability) of the lithiated intermediates and the origin of the stereoselectivity, if it was due to an asymmetric deprotonation or a dynamic resolution. According to what observed in the case of lithiated N-thiopivaloyl azetidine (Scheme 10), we can speculate that even lithiated N-Botc-azetidine could be configurationally labile and a dynamic resolution could be operative. Further study on this topic would be welcome in order to develop asymmetric synthesis of azetidines using the lithiation/trapping strategy.
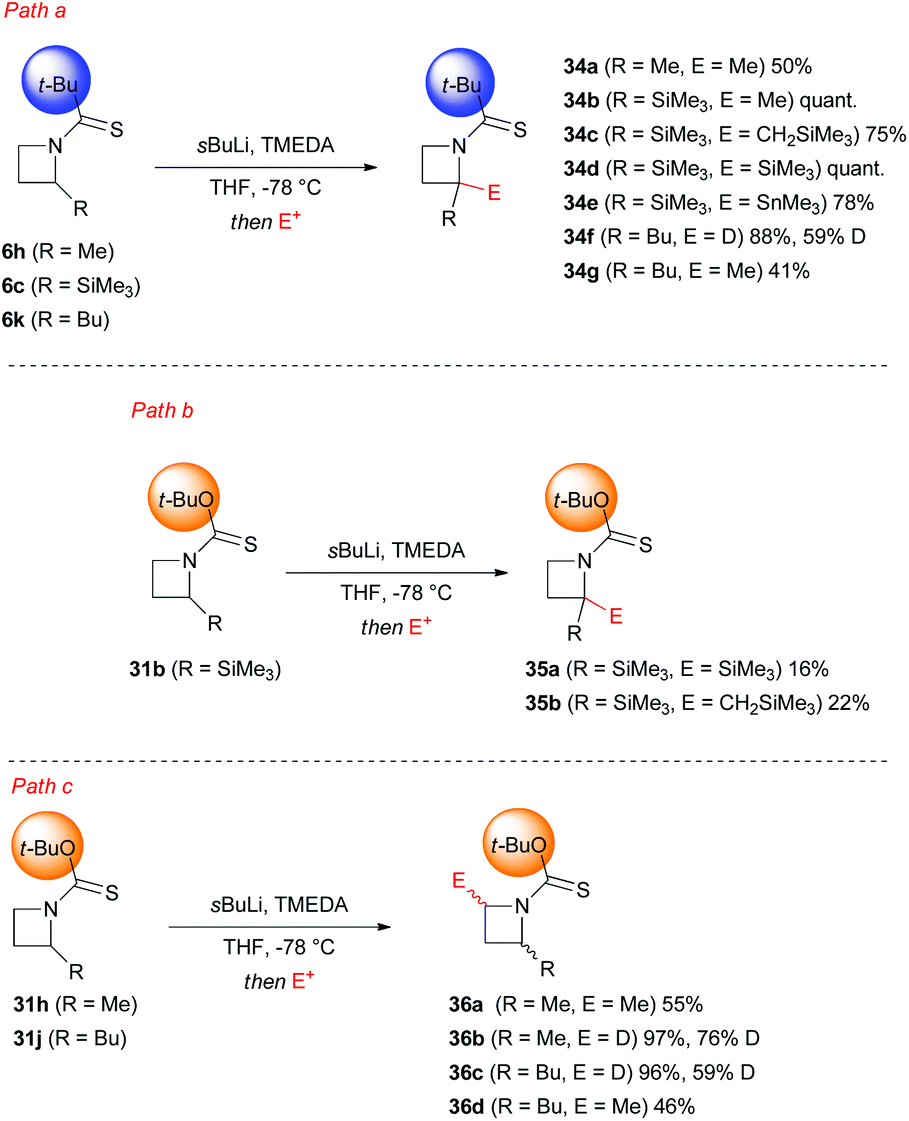
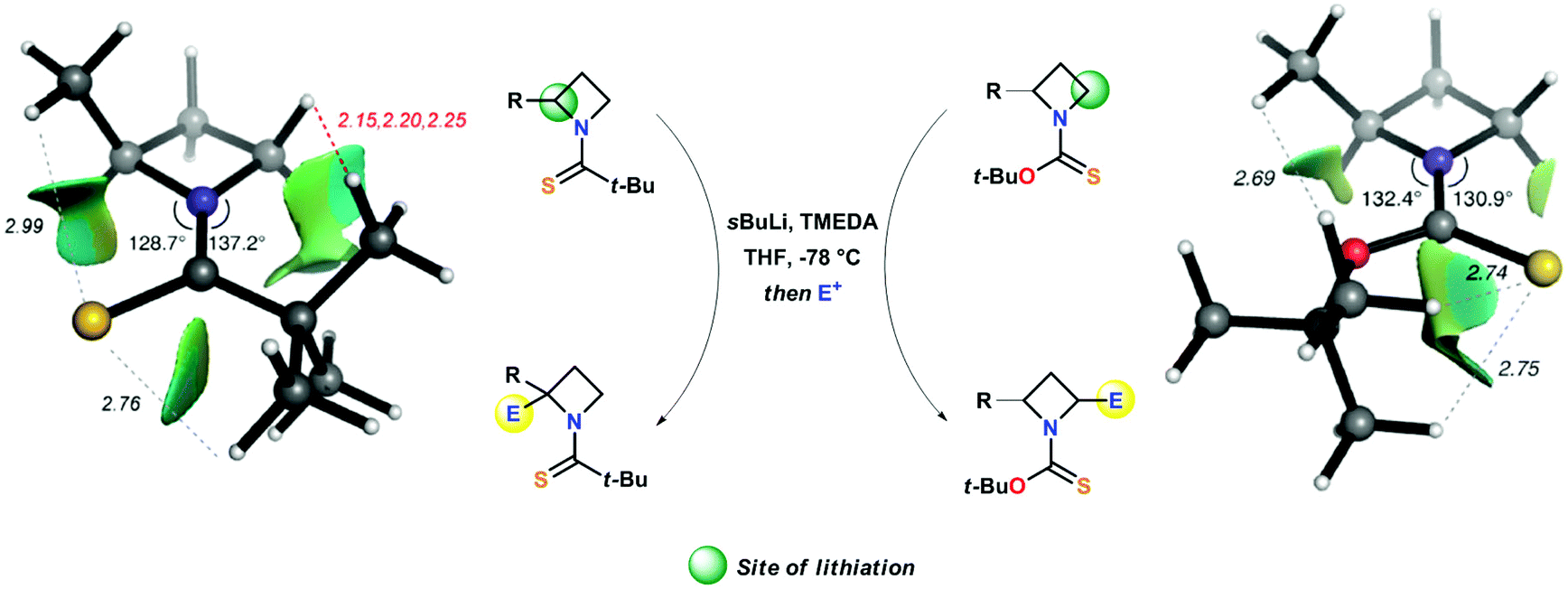
A crucial point, which requires further detailed description, is represented by the different regioselectivity in the case of a second lithiation shown respectively by N-thiopivaloyl- and N-Botc-2-substituted azetidines. Almost surprisingly, the former was found to direct lithiation to the already substituted 2-position, thus furnishing preferentially 2,2-disubstituted adduct 34a–g, even in the presence of unactivating alkyl substituent (Scheme 13, path a).31,34 On the other hand, the use of N-Botc allowed similar access to 2,2-disubstitution in considerably lower yields and only in the case of α-(trimethylsilyl)azetidine 31b, being silyl groups well known to stabilize α-anions (Scheme 13, path b). Differently, 2-unactivated N-Botc-azetidines 31h and 31j gave access, in a preferential manner, to a 2,4-disubstituted pattern, albeit with no diastereoselectivity (Scheme 13, path c).31,34 Hodgson and co-workers demonstrated that the site-selectivity (2- or 4-position, Scheme 14), observed in the lithiation/trapping sequences of N-thiopivaloyl- or N-tert-butoxythiocarbonyl-2-alkyl-substituted azetidines, likely was a result of a preferential conformation assumed by the N-substituent.34 Indeed, 1H NMR and computational studies supported the hypotheses that the regiodivergent reactivity might be ascribed to different rotamer preferences in both azetidines, since rotamer interconversion was demonstrated to be not significant under the lithiation conditions.35 In particular, DFT results suggested, in agreement with NOE studies, a preferential cis-rotamer (where the CHMe group is oriented cis to the thiocarbonyl group) for N-thiopivaloyl-2-methylazetidine (ΔGcis–trans = −6.3 kJ mol−1 in toluene and −6.1 kJ mol−1 in THF), ascribed to a relief of unfavorable steric interactions between the α-methyl and the t-Bu group, as seen for the trans conformer.
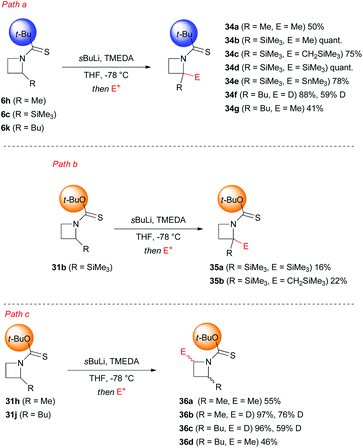 |
| | Scheme 13 Lithiation–electrophile trapping of N-thiopivaloyl- and N-(tert-butoxycarbonyl)-azetidines. | |
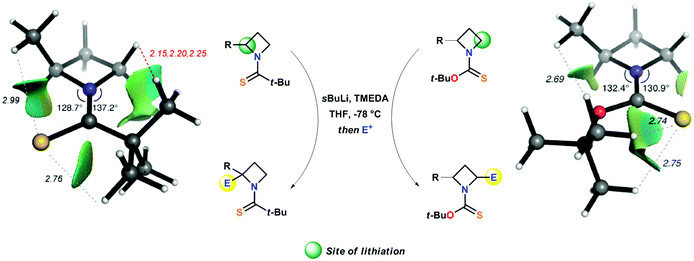 |
| | Scheme 14 Regiodivergent (C2 or C4) lithiation–electrophilic trapping sequence of N-thiopivaloyl- and N-(tert-butoxythiocarbonyl)-2-alkyl-azetidines. | |
In striking contrast, calculations identify a trans rotamer (where the CHMe group is oriented trans to the thiocarbonyl group) as the more stable in N-Boct-2-methylazetidine (ΔGcis–trans = +1.6 kJ mol−1 in toluene and +1.3 kJ mol−1 in THF). In this latter case, the presence of the oxygen atom, acting as a “spacer”, might generate this different rotamer bearing the t-Bu group effectively far from the azetidine (Scheme 14). As direct consequence, a surgical functionalization of the azetidine ring could be accomplished by choosing the suitable N-thiocarbonyl substituent (Scheme 14).
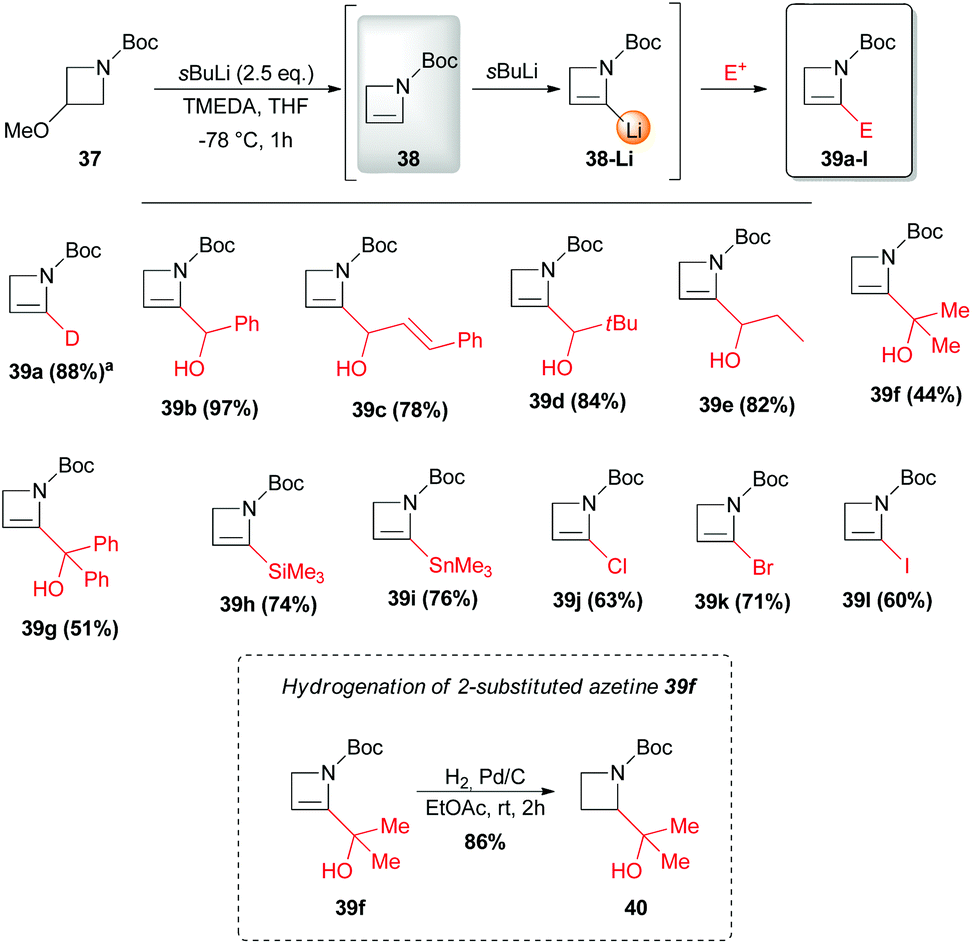
Despite the above-mentioned lack of efficiency of Boc group in promoting the lithiation–trapping of N-protected azetidines, the unsaturated and more strained analog N-Boc-2-azetine 38 was found to undergo sBuLi-mediated regioselective α-lithiation at the sp2 carbon, and effectively trapped with different electrophiles, allowing for a straightforward access to 2-substituted-2-azetines 39a–l.36 To avoid polymerization of unsubstituted N-Boc-azetine 38, which smoothly occurs at rt, the latter unsaturated system was generated in situ by a sBuLi-induced α-lithiation/elimination of LiOMe from readily available N-Boc-3-methoxyazetidine 37 (Scheme 15). Interestingly, hydrogenation of the derivate 39f provided the corresponding saturated N-Boc-2-substituted azetidine 40. Allylation and propargylation proceeded efficiently, after transmetallation of the lithiated azetine 38-Li to the corresponding organocopper, although with crotyl and prenyl bromides the desired SN2-products 41d,e were always accompanied by the competing SN2′-derived azetidines 41d′,e′ (Scheme 16). However, Neghishi coupling of lithiated azetine 38-Li, gave 2-phenylated azetine 42 in 27% yield (Scheme 16).
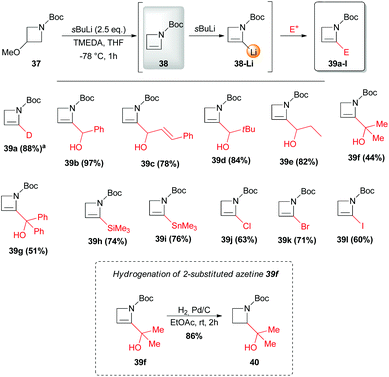 |
| | Scheme 15 Preparation, in situ-lithiation and direct electophilic trapping of N-Boc-azetine 38. Hydrogenation of azetine 39f. All presented yields are referred to the isolated product. a100% D, calculated by 1H NMR. | |
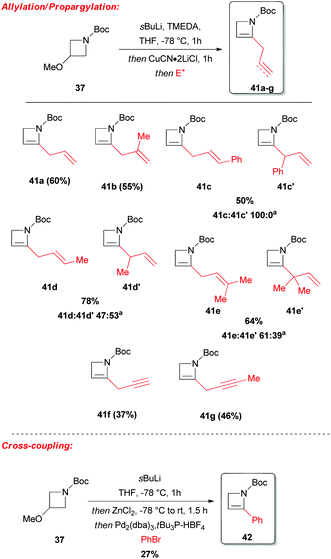 |
| | Scheme 16 Copper-mediated allylation and propargylation and Negishi cross-coupling of lithiated azetine 38-Li. aIsolated ratio. | |
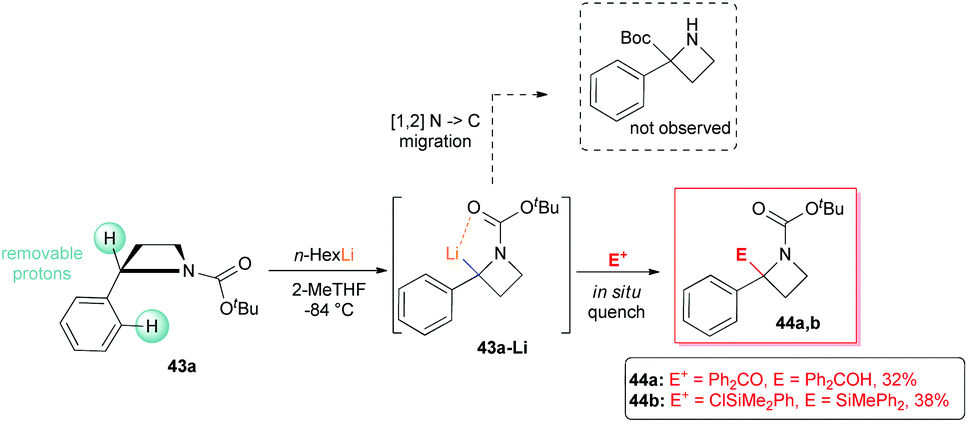
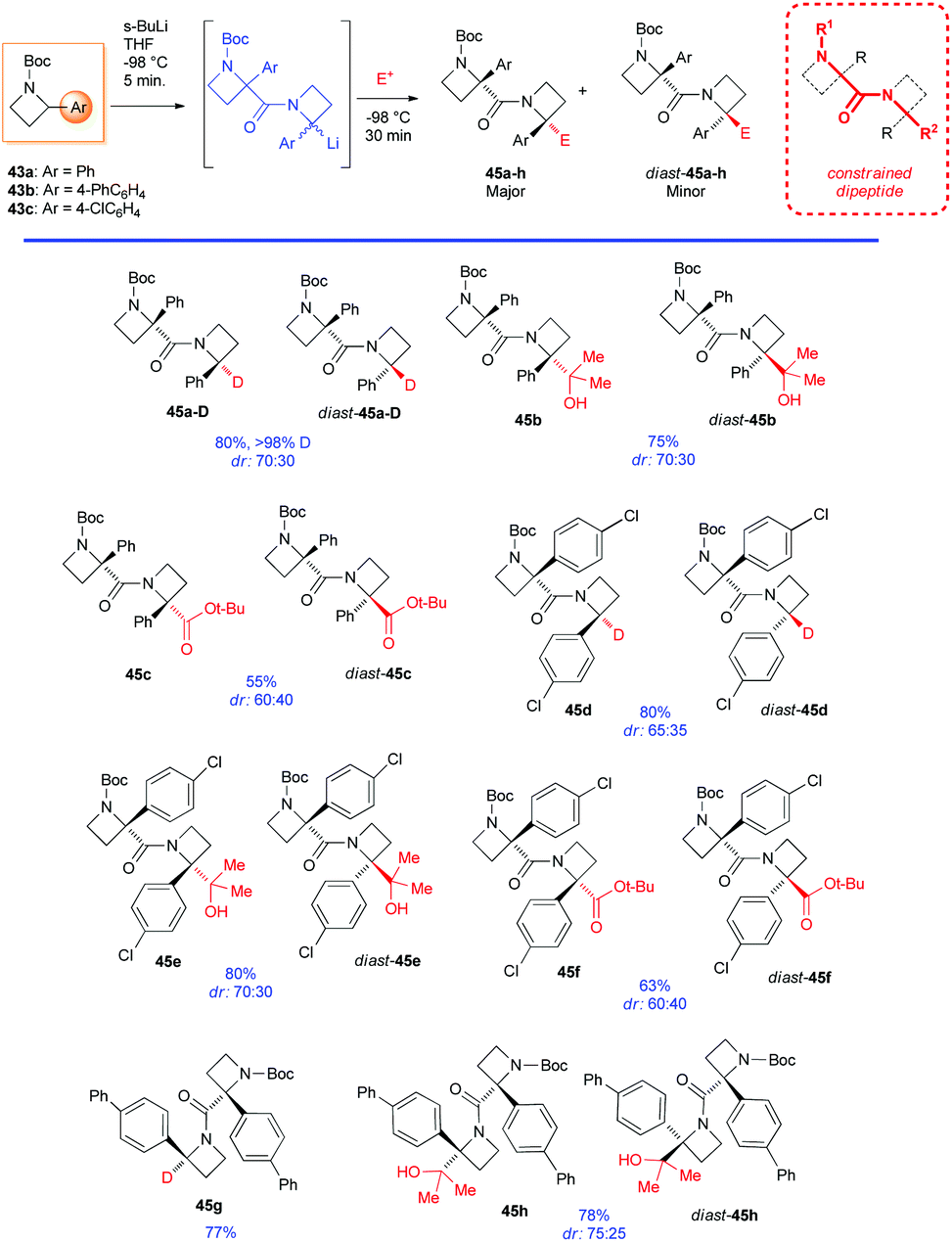
However, it is worth mentioning that it is possible to functionalize regioselectively the C2-position of N-Boc-2-phenyl azetidine 43a (although only in moderate yields), because of the increased thermodynamic acidity of its benzylic proton, as recently reported by Luisi, Degennaro and co-workers.13 By performing lithiation at low temperature (−84 °C), using n-HexLi in a polar solvent (2-MeTHF) and under in situ quench conditions, exclusively α,α-difunctionalised azetidines 44a,b were obtained (Scheme 17). Although decomposition was always observed under external quench conditions, the expected N to C [1,2]-migration product, observed in the case of lithiated N-Boc-aziridines,37 was never detected. In addition, because of the poor coordinating ability of the azetidine's nitrogen, ortho-lithiation could not occur. Intriguing, deeper investigations on N-Boc 2-arylazetidines 43a–c, displayed a peculiar reactivity scenario. Indeed, by lithiating 43a with sBuLi (3 equiv.) in THF at −98 °C for 5 minutes (0.05 M concentration), an unexpected dimerization occurred, leading to diastereomeric dimers 45a-D and diast-45a-D, after external quench with MeOD (Scheme 18).38 It should be stressed that such a dimerization, to the best of our knowledge, represents the first example of self-condensation of a stabilized α-lithiated amine, never reported before in the lithiation of higher and lower homologues such as N-Boc aziridines, pyrrolidines and piperidines. Moreover, such dimers, easily separable by flash chromatography, show peculiar structures strictly related to constrained peptidomimetics of great interest in medicinal chemistry.39 The scope of the reaction was explored with representative electrophiles (Scheme 18). By deuteration, hydroxyalkylation and carboxylation the dimers 45a–h were obtained in good yields, although with modest diastereoselectivity. Remarkably, the introduction of a tert-butoxycarbonyl unit, as in the case of 45c,f and diast-45c,f smoothly furnished constrained dipeptides, with protected N- and C-terminals (Scheme 18).
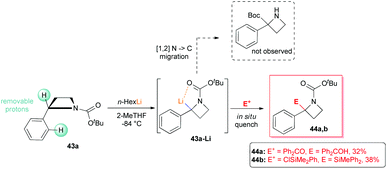 |
| | Scheme 17 Regioselective α-lithiation of N-Boc 2-phenylazetidine 43a. | |
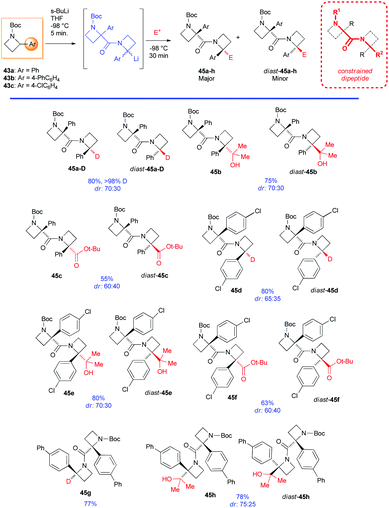 |
| | Scheme 18 Scope of the lithiation/dimerization/electrophile trapping sequence. | |
In striking contrast to 43a–c, N-Boc-2-(o-tolyl)azetidine 43d was found to behave differently when subjected to lithiation/trapping sequence with s-BuLi (2.5 equiv.) in THF at −78 °C for 10 min. Surprisingly, lithiated azetidine 43d-Li was found to be chemically stable (while lithiated azetidines 43a–c underwent full decomposition at −78 °C) and did not undergo dimerization, furnishing the corresponding 2,2-disubstituted azetidines 46a–d in high yield (up to 96%, Scheme 19). This remarkable result could be ascribed to a sort of “ortho-effect” able to hamper dimerization and, thus, to provide access to α,α-disubstituted azetidines and constrained aminoacids as in the case of 46d, a cyclic analog of phenylalanine. Such a chemical stability of 43d-Li, and its low propensity to undergo dimerization, has been ascribed to a preferential conformation retained by the o-tolyl substituent, setting syn to the α-proton of the azetidine ring, as suggested by DFT calculations and conformational analysis on 43d (Scheme 19).
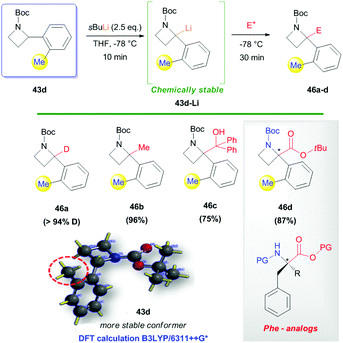 |
| | Scheme 19 Effective trapping of lithiated intermediate 43d-Li. | |
To get more insights into the mechanism of the dimerization process described above, stereochemical investigations on the lithiation/dimerization/deuteration sequence, were performed on chiral non racemic azetidine (R)-43a and diastereomerically pure dimers 45a and diast-45a. Lithiation/deuteration of 45a (dr > 95:5) or diast-45a (dr > 95:5), provided mixtures of diastereomeric dimers 45a-D and diast-45a-D having similar sense of stereoinduction, just as seen in the dimerization of 43a, although with slightly different ratios (Scheme 20, path a). These results clearly demonstrated an equilibrium between the lithiated dimers 45a-Li and diast-45a-Li. Two different reaction pathways could be envisaged for the dimerization: (a) a two steps sequence (homochiral dimerization, HD), involving the reaction of (R)-43a-Li with its neutral precursor, leading to (R,R)-diast-45a which is further deprotonated; (b) a single step dimerization of (R)-43a-Li (self-condensation, SC), directly leading to the lithiated dimer. When optically active azetidine (R)-43a (98:2 er) was lithiated under optimized conditions, quenching with MeOD furnished highly enantioenriched deuterated dimers (R,S)-45a-D (85:15 er) and (R,R)-diast-45a-D (98:2 er), in 70:30 diastereomeric ratio (Scheme 20, path b). The slight erosion observed in (R,S)-45a-D (85:15 er) might be due to the propensity of (R)-43a-Li to racemize under the reaction conditions, likely at rate similar to that of self-condensation. On the other hand, azetidine 43d, which does not dimerize, was lithiated and reacted with (R)-43a (Scheme 20, path c). No evidence for cross-condensation was found, supporting the hypothesis of a prevalent SC pathway, followed by the epimerization of the corresponding lithiated dimer, although the HD pathway cannot be completely ruled out at the moment. Remarkably, the high enantioselectivity obtained demonstrates the suitability of the process for preparing chiral azetidine-based constrained peptidomimetics.
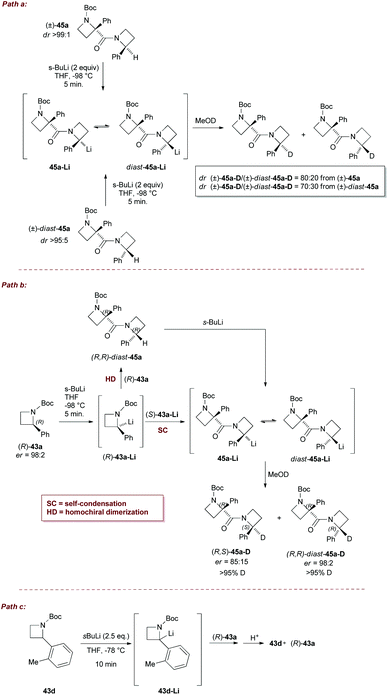 |
| | Scheme 20 Stereochemical investigations on lithiation/dimerization of N-Boc 2-phenylazetidine 43a. | |
Lithiation of N-alkyl azetidines
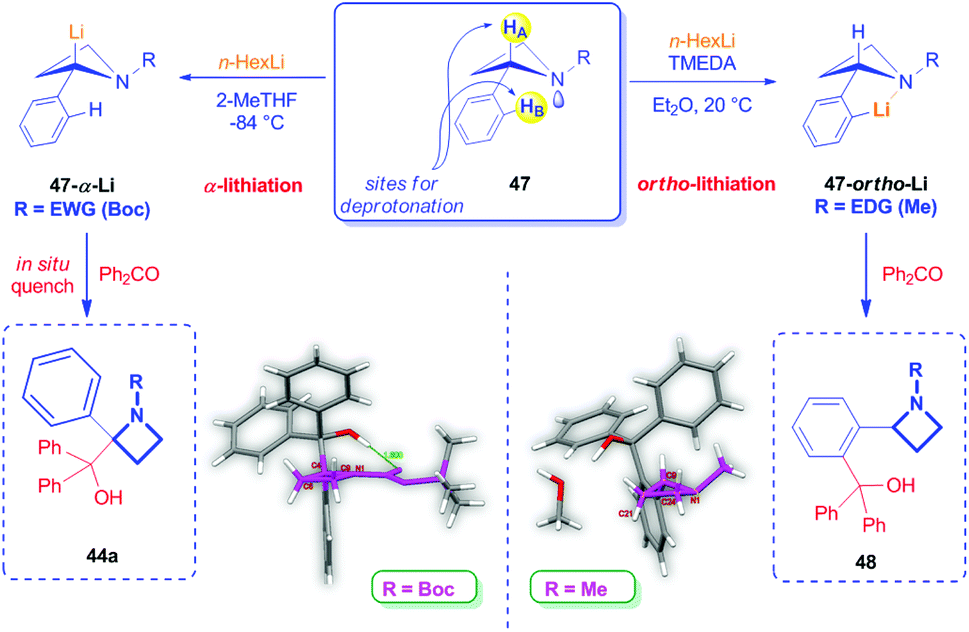
Differently from azetidines bearing an EWG group as the N-substituent, to the best of our knowledge, there are no reports dealing with the α-lithiation of unsubstituted azetidines bearing on the nitrogen atom an electron-donating group (EDG). Recently, Luisi, Degennaro and coworkers described the peculiar reactivity of N-alkyl 2-arylazetidines, which, in striking contrast to N-Boc 2-aryl azetidines, cannot be α-lithiated, but smoothly undergo ortho-lithiation upon treatment at 20 °C with n-HexLi in the presence of TMEDA (Scheme 21).13,40 In fact, in this case, the N-EDG group would increase the lone pair availability and the coordinating capability of the nitrogen. As a consequence, a drastically reduced thermodynamic acidity of the benzylic proton is expected, together with a higher capability of the nitrogen to act as an ortho-directing metallation group (DMG), just as seen in the case of 2-arylaziridines or other arenes bearing Lewis-base heteroatoms.1b
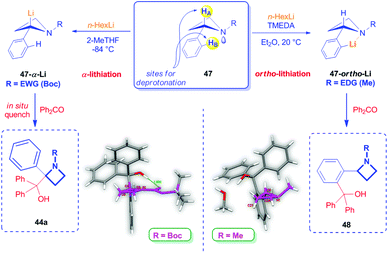 |
| | Scheme 21 Comparison of reactivity for in N-Boc- and N-alkyl-2-phenyl azetidines. | |
Structural differences between N-Boc- and N-alkyl-2-phenyl azetidines were deduced by crystallographic X-ray analysis on derivatives 44a and 48 (Scheme 21). Structure of 48 clearly revealed the nitrogen lone pair availability resulting in an almost tetrahedral nitrogen atom, likely more able to form complexes with lithiating agents, compared to the quasi-planar arrangement of the azetidine ring in 44a.
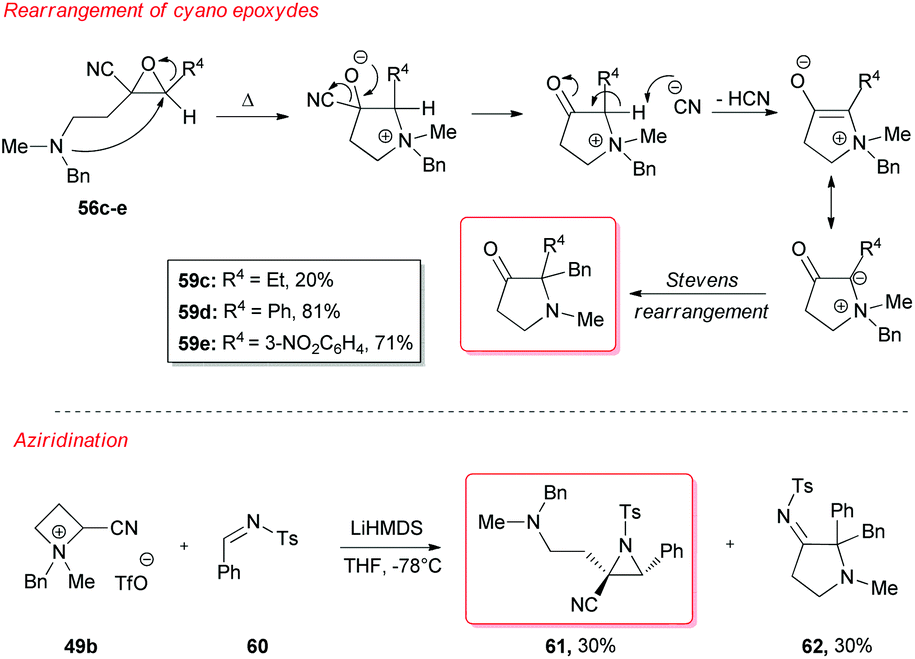
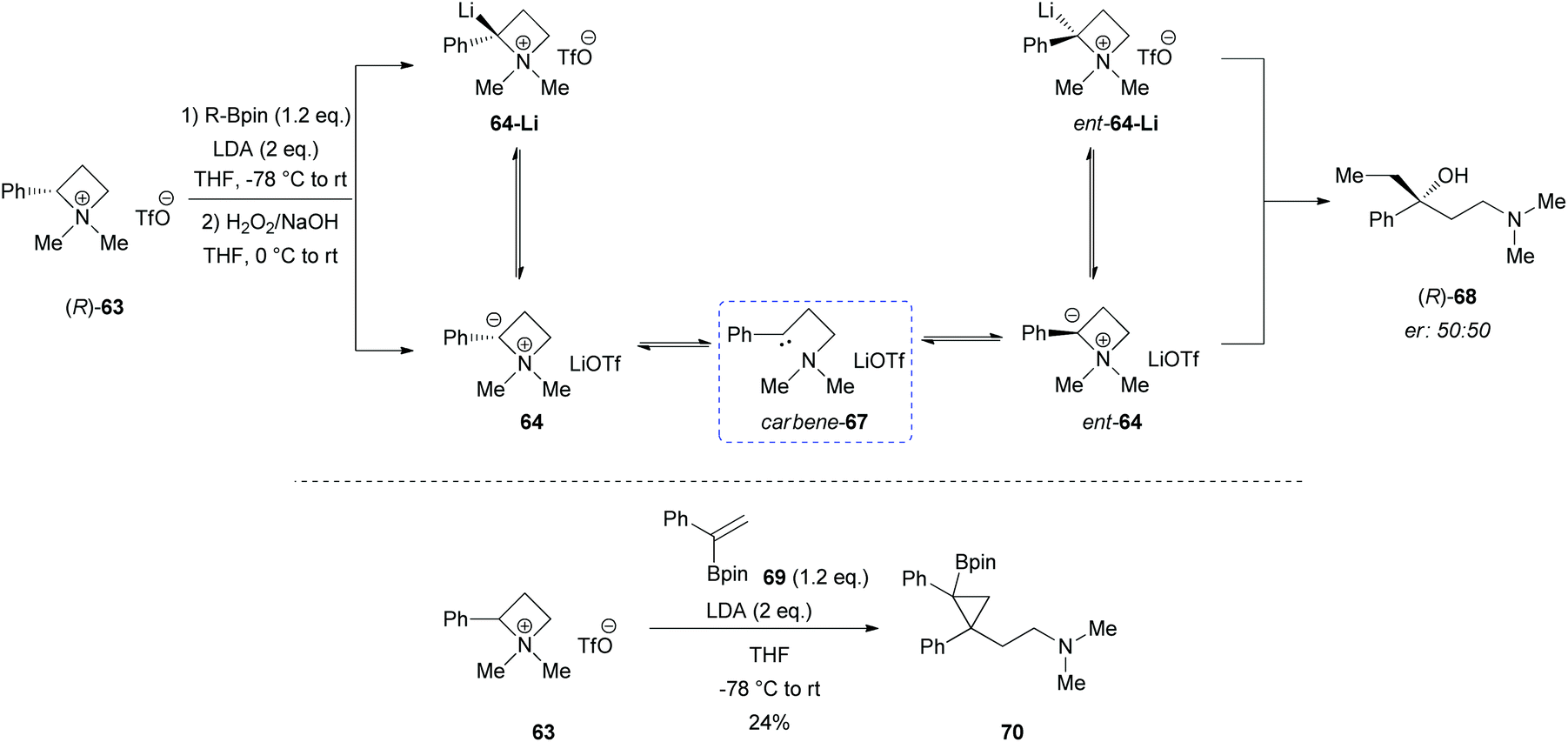
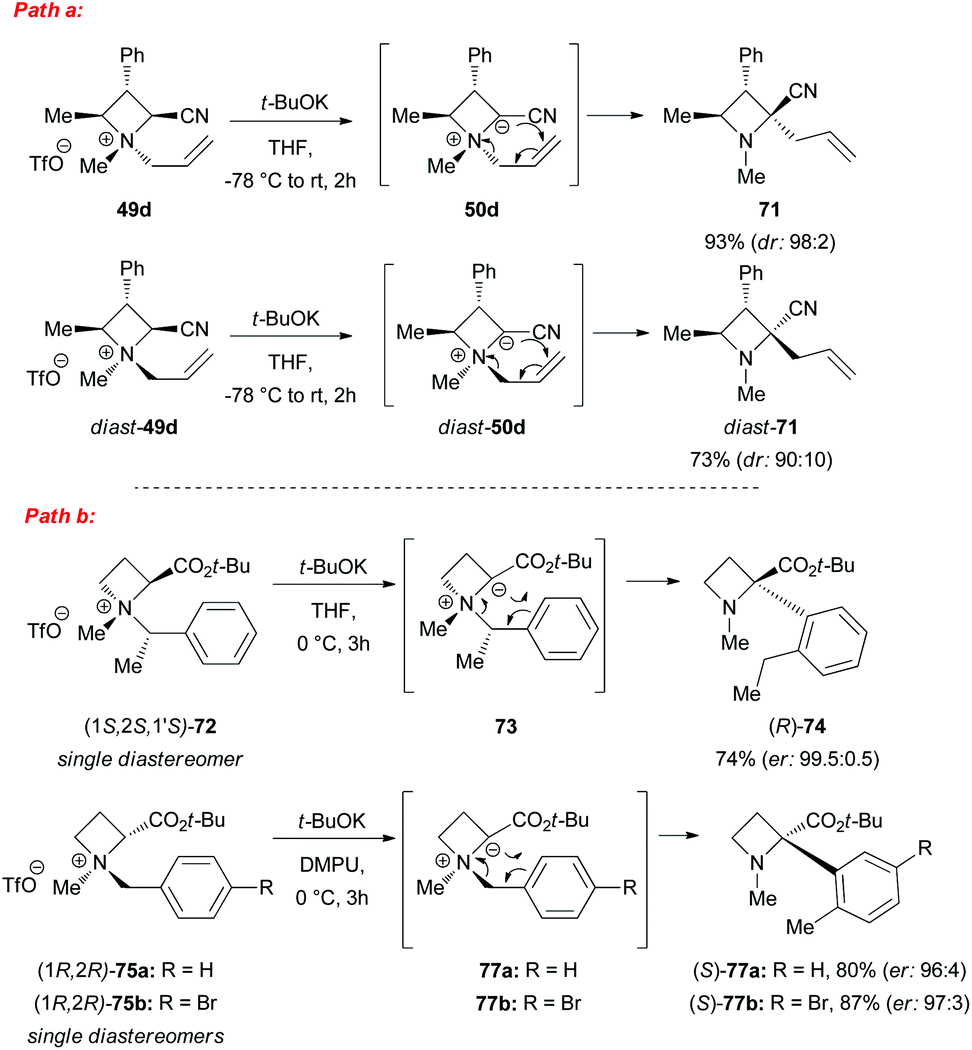
However, a convenient alternative to perform α-deprotonation of N-alkyl azetidines lies in their conversion into quaternary azetidinium ions, by alkylation (Scheme 22). Couty and David displayed how 2-cyano-azetidinium ions 49a–c could be efficiently α-deprotonated at −78 °C using a strong non-nucleophilic base (to prevent any competitive ring opening) such as lithium hexamethyldisilazide (LiHMDS).41 The corresponding azetidinium ylide 50a was found to have a remarkable ability to promote cyclopropanation when trapped in situ with Michael acceptors, giving rise to substituted cyclopropanes in good yields and with a high level of stereocontrol (Scheme 22, path a).41a Configurational instability of the anionic centre of the ammonium ylide 50a was addressed as the reason for the invariable absolute R-configuration of the carbon bearing the cyano group. Indeed, it was suggested that diasteromeric ylides 50a and diast-50a undergo rapid epimerization in which only less hindered 50a likely attacks alkenes to provide the zwitterionic ammonium enolates 52, which finally evolves to cyclopropanes 53, via intramolecular SN2 reaction. In a similar fashion, ylide 50b was effectively employed to perform facile epoxydation of various aldehydes and ketones with excellent diastereoselectivity (Scheme 22, path b).41b In both pathways the chemical instability of ammonium ylides, even at low temperatures, requires the electrophile to be present during their formation. It is important to underline also that such reactions proved to be ineffective on the larger homologue 2-cyano pyrrolidinium ion, highlighting the crucial role played by the ring strain present in the azetidinium ylides to achieve reaction efficiency, likely ascribable to an enhanced leaving group capability of the ammonium group associated to the final ring strain relief. Additionally, azetidinium ylides, derived from deprotonation of 2-cyano-azetidinium salts 49a,c as precursors, could efficiently be converted into differently substituted α,β-unsatured nitriles 58a–h, through a one-pot two-steps sequence involving trapping, with different alkyl halides, of in situ generated azetidinium ylides, followed by a final regioselective elimination (Scheme 22, path c).41c Remarkably, the latter reaction was reported to be strictly sensitive to both steric hindrance around the nitrogen atom (as displayed by low yields of N-cyclohexyl derivatives 58g,h) and nature of the starting halide (propargyl bromide gave product 58f in only 14% yields, while aliphatic halides were ineffective at all). Remarkably, epoxides 56c–e, obtained from aldehydes, cleanly evolved into the corresponding 2-disubstituted pyrrolidin-3-one adducts 59c–e upon warming at 50 °C. The proposed mechanism involves elimination of hydrogen cyanide with the formation of a different ammonium ylide, which undergoes a classical Stevens rearrangement. This interesting rearrangement was observed also in the case of aziridination of tosylimine 60 (Scheme 23).41b Further insights in the chemistry of azetidinium ylides came recently from Aggarwal and coworkers.42 They described the reaction of in situ generated 2-phenyl azetidinium ylide 64 with primary and secondary boronic esters (including alkyl-, allyl- and aryl-ones) to give acyclic γ-dimethylamino tertiary boronic esters 66a–i. Specifically, it was suggested that the transformation proceeded through the trapping of the lithiated carbon of the ylide with the boronic esters to form zwitterionic boronates 65, which in turn could undergo ring-opening 1,2-migration, likely favored by ring stain release (Scheme 24). Deprotonation of azetidinium ion 63 was performed with LDA at −78 °C in THF, and trapping with alkyl boronates furnished the desired products in moderate to good yields. The targeted boronic esters are particularly attractive owing to the their great synthetic versatility. Indeed, the C–B bond can be easily functionalized and transformed under mild conditions into a variety of different moieties such as C–OH, C–vinyl, C–H and C–BF3.43 These authors found the reaction to be not stereospecific (Scheme 25), due to the configurational instability of the initially formed lithium-stabilized ylide 64-Li (or of ylide 64, generated directly by deprotonation). They postulated that the rapid inversion might be ascribed to the transient formation of the open-chain carbene 67, being this hypothesis supported by the formation of cyclopropane 70 when vinyl boronic ester 69 was employed (Scheme 25). Indeed, in the latter case, the targeted boronate did not form or undergo 1,2 migration, probably because of higher steric hindrance, and alternatively carbene 67 could perform cyclopropanation on the starting alkene. However, the rich reactivity displayed by azetidinium ylides is not limited to the above described intermolecular reaction with different kind of electrophiles. Alternatively they can react intramolecularly through a [2,3] sigmatropic rearrangement.44 The latter pathway easily led to optically active azetidines with an α-quaternary centre, through high level of N-to-C chirality transfer, starting both from 2-cyano azetidinium ylides 49d and diast-49d (Scheme 26, path a)44a and azetidine-2-carboxylic acid ester derived ammonium salts 72 and 75a,b (Scheme 26, path b).44b Remarkably, only the targeted products derived from Sommelet–Hauser (S–H) rearrangement were observed, with no traces of the competitive [1,2] Stevens rearrangement products.45
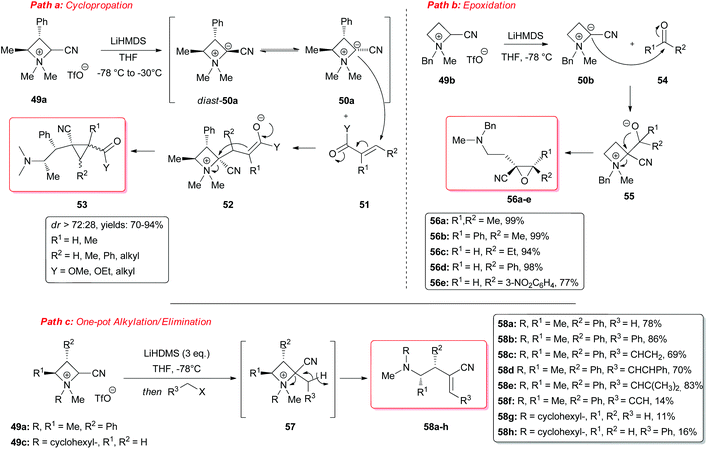 |
| | Scheme 22 Strained azetidinium ylides as reagents for cyclopropanation, epoxydation and synthesis of different acrylonitriles. | |
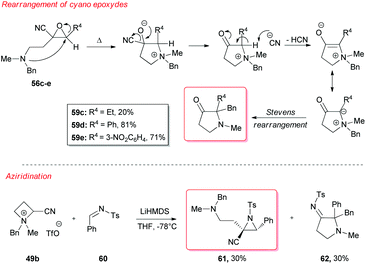 |
| | Scheme 23 Intrinsic reactivity of cyano epoxydes and aziridines. | |
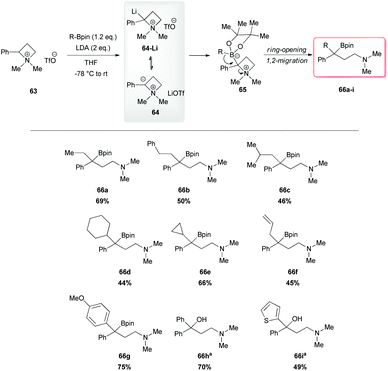 |
| | Scheme 24 Lithiation–borylation of ammonium ion 63. aIsolation of the boronic ester was not possible owing to portodeboronation; in situ oxidation with H2O2/NaOH allowed the isolation of the corresponding tertiary alcohols. | |
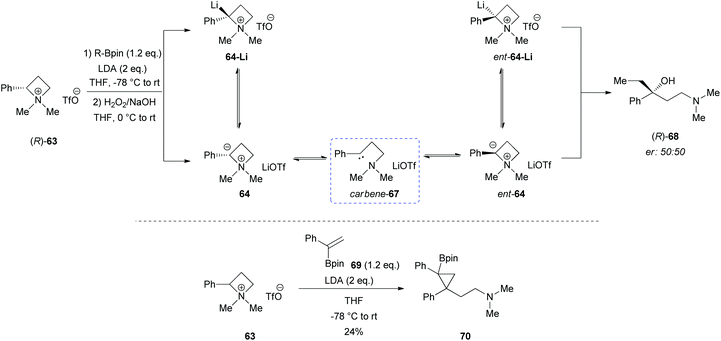 |
| | Scheme 25 Proposed mechanism of racemization of ylide 64. | |
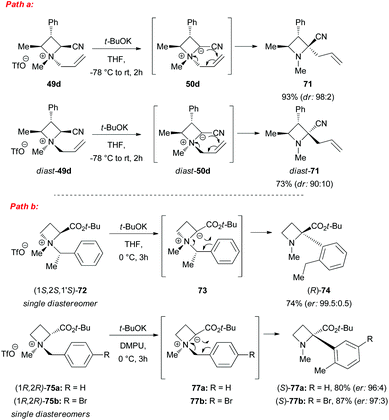 |
| | Scheme 26 Asymmetric base-induced [2,3] sigmatropic rearrangement of α-stabilized azetidinium ylides. DMPU = N,N′-dimethylpropyleneurea. | |
C3-Metallation of azetidines
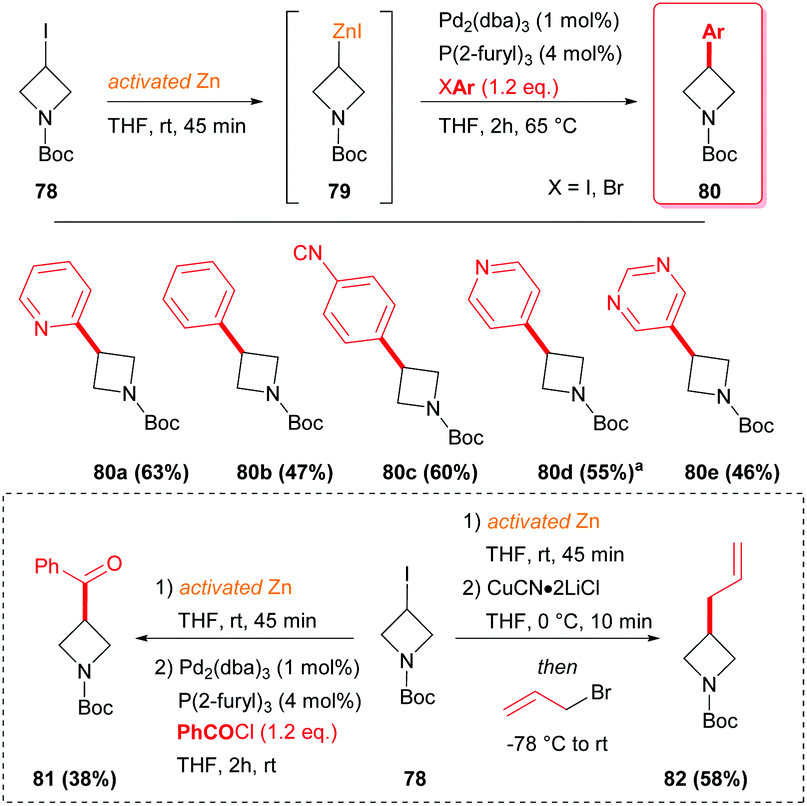
The most typical strategy to synthesize 3-substituted azetidines, by the corresponding organometallic intermediates, is the cross-coupling through the use of palladium catalysts and phosphine ligands, starting from the corresponding 3-iodo-azetidine.46 In such a protocol, an azetidine–zinc complex has to be generated before the subsequent reaction with a suitable aryl halide or with benzyl chloride (Scheme 27). The organozinc species 79 could readily transmetallate with CuCN·2LiCl in THF and by reacting with allyl bromide, furnished azetidine 82 in moderate yield.
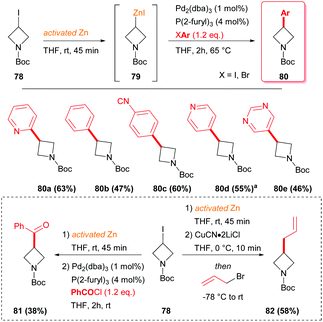 |
| | Scheme 27 Pd(0) mediated cross coupling and transmetallation with CuCN·2LiCl of azetidine-derived organozinc species 79. aReaction carried out at room temperature. | |
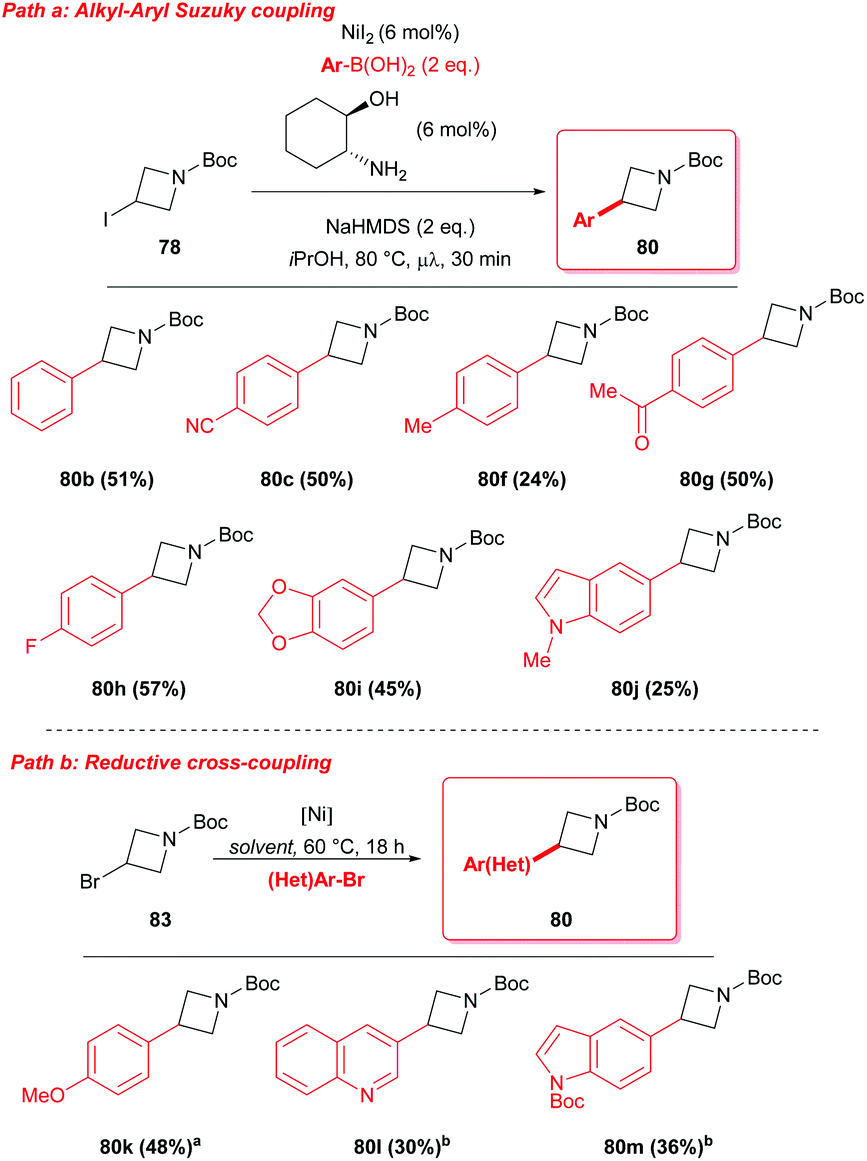
Alternatively, nickel catalyzed procedures in the presence of a ligand have been also reported, allowing the synthesis of 3-functionalised azetidines although with limited success (Scheme 28).47 In 2008, Duncton reported an interesting microwaved Suzuki coupling to functionalize 3-iodo-azetidine 78 at the C-3 position.47a The reaction with different aryl-boronic acids proceeded in the presence of catalytic amounts of nickel(II) iodide and trans-2-aminocyclohexanol as ligand (Scheme 28, path a).
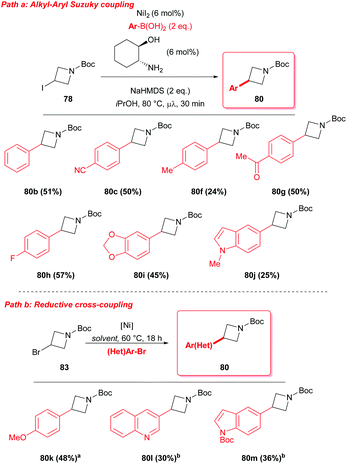 |
| | Scheme 28 Ni based strategies for the synthesis of 3-aryl azetidines. aNiCl2·glyme (10 mol%), phenanthroline (20 mol%), 4-ethylpirydine (50 mol%), NaBF4 (50 mol%), Mn (2 eq.), MeOH (0.2 M). bNiI2 (10 mol%), di-tBu-bipyridine (10 mol%), 4-ethylpirydine (1 eq.), MgCl2 (1 eq.), Mn (2 eq.), DMA (0.2 M). | |
More recently, Molander showed the possibility to perform a reductive cross coupling, using air-stable Ni(II) sources in the presence of a diamine ligand, an inorganic salt as additive, and a reducing metal, to obtain the incorporation of (hetero)aryl halides at C-3 of the azetidine ring (Scheme 28, path b).47b
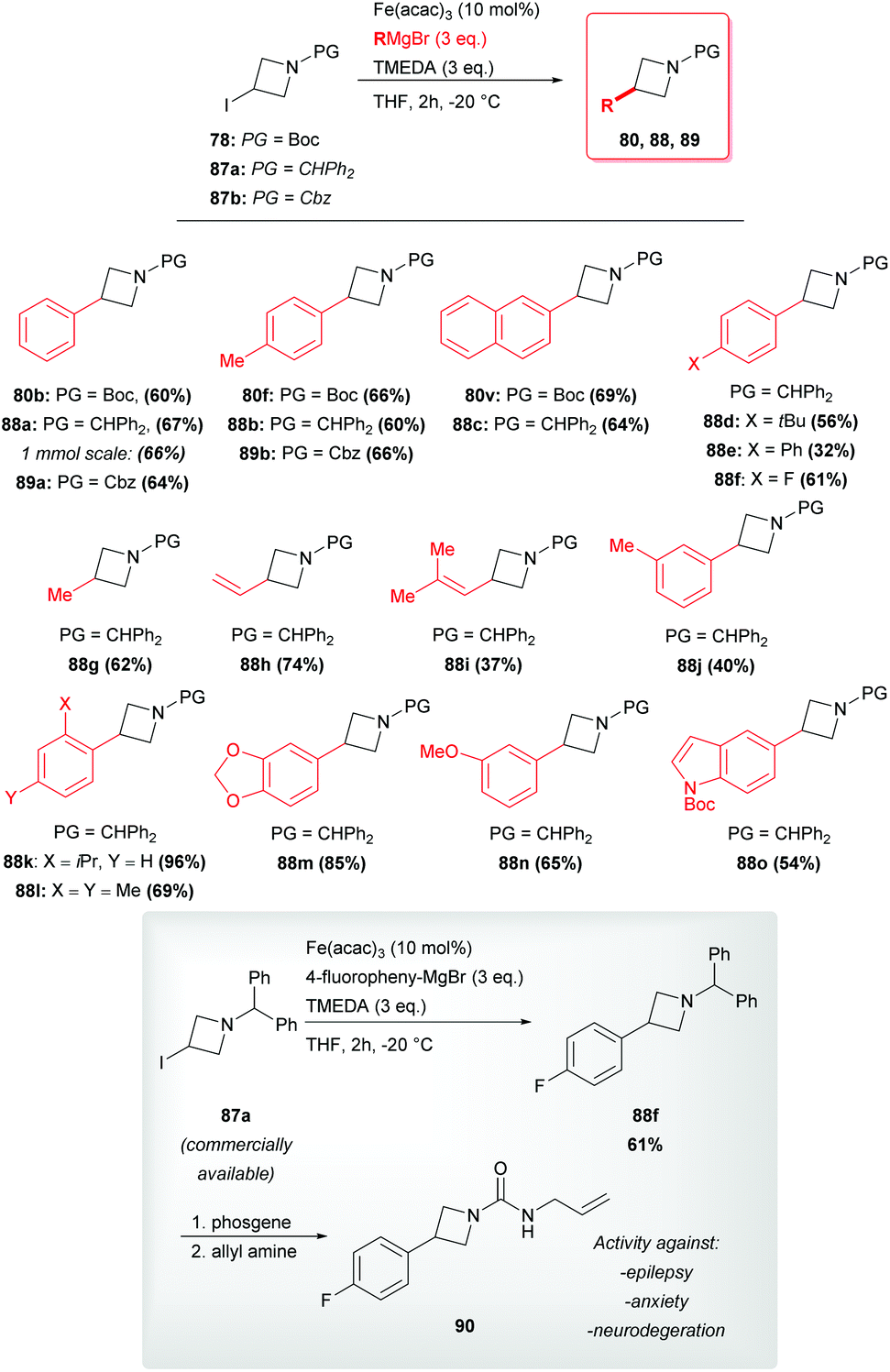
Recently, highly appealing methods for the coupling of azetidine 78 have featured in reports dealing with the employment of cheap iron or cobalt catalysts and readily available organomagnesium reagents in the absence of any additional phosphine ligands.48,49 Cossy and coworkers disclosed that both CoCl2 and FeCl2 catalysts allow the smooth reaction between azetidine 78 and a large variety of (hetero)aryl Grignard reagents, generally exhibiting similar performances (Scheme 29).48 Products were obtained in high yields and the protocol seemed to tolerate a large variety of functional groups, but required the use of ligand 84 ((R,R)-tetramethylcyclohexan-1,2-diamine). Interestingly, when 2,3-disubstituted iodo-azetidine 85 (dr: 75:25 cis/trans) was treated with PhMgBr in the presence of both catalytic systems the desired product 86 was obtained with a good trans diastereoselectivity (Scheme 29). The authors postulated that the inversion of stereochemistry might be ascribed to the formation of a radical intermediate at C-3.
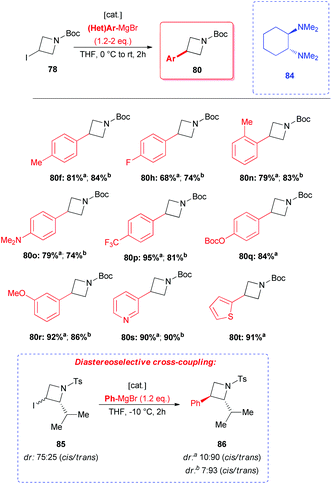 |
| | Scheme 29 Iron- and cobalt-mediated arylation of azetidines. a[cat.] = CoCl2 (5 mol%)/84 (6 mol%). b[cat.] = FeCl2 (10 mol%)/84 (10 mol%). Ts = Tosyl. | |
Moreover, Rueping reported a very mild procedure involving Fe(acac)3 as the catalyst. In addition, this strategy proved to be efficient and chemoselective and to tolerate a variety of (hetero)aryl, vinyl and alkyl Grignard reagents, as well as different protecting groups on the nitrogen atom (Scheme 30).49 Remarkably, these authors reported the possibility to scale up the reaction with no decrease of the yield. The synthetic potential of this route could be pointed out in the light of its successful application to the short formal synthesis of the challenging molecule 90 possessing pharmacological activity against central nervous system disorders (Scheme 30).11a The patented reported route for the preparation of this molecule (and structurally related compounds) requires three steps, two days of work and two chromatographic separations to obtain intermediate 88f in an overall combined yield of 42%.11a Remarkably, by using the developed iron-catalyzed cross-coupling directly on the commercial iodide 87a, Rueping and coworkers yield compound 88f in 61%. The latter can be easily converted into the desired molecule 90 through a reported protecting group exchange step.11a
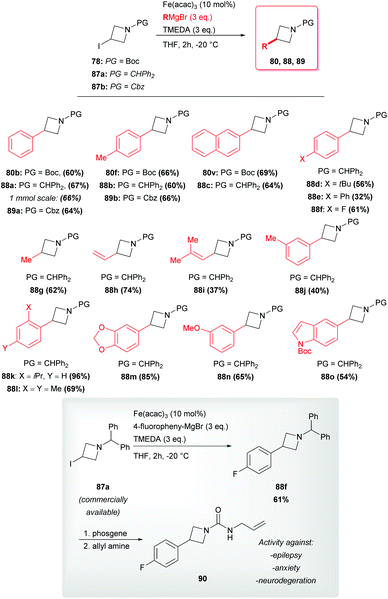 |
| | Scheme 30 Rueping's azetidine cross-coupling with Grignard reagents and synthetic application of pharmaceutical interest. | |
Conclusions and outlooks
Despite some limitations, the most significant conceptual advances over the past decade in the area of synthesis of differently functionalized azetidine rings, are represented by metallation reactions that led to the direct substitution at C2 and/or C3 of this heterocycle. These strategies are emerging to have an even growing impact in modern medicinal chemistry. The reactivity of the corresponding metallated systems were demonstrated to be extremely dependent on ring strain and on substitutions at both nitrogen and carbon atoms, that, by deeply influencing conformational preferences and coordinative phenomena, allow surgical site-selective functionalization.50 Undoubtedly, the most difficult challenge that chemists have to overcome in this scenario, remains the addressing of optimal enantioselective lithiation/trapping sequence, mainly because of the configurational instability of the lithiated azetidine intermediates, which does not allow facile predictability of the stereochemical outcome. The ideal metallation reaction would be easily predictable, in terms of regio- and stereoselectivity, general and functional-group tolerant. Future research in the field of metallated four-membered azetidines should focus on the use of chiral diamines working in dynamic resolutions and on getting insights of their structure-dependent reactivity by spectroscopic methods, computational studies and X-ray analysis. This will lead to a better and extensive application in synthetic organic chemistry, pharmaceuticals, agrochemicals and advanced materials of such heterocyclic intermediates.
Acknowledgements
We thank University of Bari and the Consortium CINMPIS for support.
Notes and references
- For selected reviews on the synthesis of saturated azacycles derivatives see:
(a) C.-V. T. Vo and J. W. Bode, J. Org. Chem., 2014, 79, 2809–2815 CrossRef CAS PubMed;
(b) V. Capriati, S. Florio and R. Luisi, Eur. J. Org. Chem., 2014, 5397–5417 CrossRef CAS;
(c)
L. Degennaro, B. Musio and R. Luisi, in Lithium Compounds in Organic Synthesis – From Fundamentals to Applications, ed. R. Luisi and V. Capriati, Wiley-VCH, Weinheim, 1st edn, 2014, ch. 7, pp. 191–223 Search PubMed;
(d) E. A. Mitchell, A. Peschiulli, N. Lefevre, L. Meerpoel and B. U. W. Maes, Chem. – Eur. J., 2012, 18, 10092–10142 CrossRef CAS PubMed.
-
(a) A. Nadin, C. Hattotuwagama and I. Churcher, Angew. Chem., Int. Ed., 2012, 51, 1114–1122 (
Angew. Chem.
, 2012
, 124
, 1140
) CrossRef CAS PubMed;
(b) S. J. Teague, A. M. Davis, P. D. Leeson and T. Oprea, Angew. Chem., Int. Ed., 1999, 38, 3743–3748 (
Angew. Chem.
, 1999
, 111
, 3962
) CrossRef CAS;
(c) F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed;
(d) F. Lovering, Med. Chem. Commun., 2013, 4, 515–519 RSC;
(e) M. Ishikawa and Y. Hashimoto, J. Med. Chem., 2011, 54, 1539–1554 CrossRef CAS PubMed;
(f) B. Over, S. Wetzel, C. Grütter, Y. Nakai, S. Renner, D. Rauh and H. Waldmann, Nat. Chem., 2013, 5, 21–28 CrossRef CAS PubMed.
-
(a) T. J. Ritchie, S. J. F. Macdonald, R. J. Young and S. D. Pickett, Drug Discovery Today, 2011, 16, 164–171 CrossRef CAS PubMed;
(b) T. J. Ritchie, S. J. F. Macdonald, S. Peace, S. D. Pickett and C. N. Luscombe, Med. Chem. Commun., 2013, 4, 673–680 RSC;
(c) M. M. Hann, Med. Chem. Commun., 2011, 2, 349–355 RSC;
(d) A. D. Morley, A. Pugliese, K. Birchall, J. Bower, P. Brennan, N. Brown, T. Chapman, M. Drysdale, I. H. Gilbert, S. Hoelder, A. Jordan, S. V. Ley, A. Merritt, D. Miller, M. E. Swarbrick and P. G. Wyatt, Drug Discovery Today, 2013, 18, 1221–1227 CrossRef PubMed.
-
(a) N. H. Cromwell and B. Phillips, Chem. Rev., 1979, 79, 331 CrossRef CAS;
(b) A. Brandi, S. Cicchi and F. M. Cordero, Chem. Rev., 2008, 108, 3988 CrossRef CAS PubMed;
(c) F. Couty and G. Evano, Synlett, 2009, 3053 CrossRef CAS.
-
(a) E. M. Carreira and T. C. Fessard, Chem. Rev., 2014, 114, 8257 CrossRef CAS PubMed;
(b) B. H. Rotstein, S. Zaretsky, V. Rai and A. K. Yudin, Chem. Rev., 2014, 114, 8323 CrossRef CAS PubMed;
(c) M. Han, C. Song, N. Jeong and H.-G. Hahn, ACS Med. Chem. Lett., 2014, 5, 999 CrossRef CAS PubMed;
(d) L. Carroccia, L. Degennaro, G. Romanazzi, C. Cuocci, L. Pisano and R. Luisi, Org. Biomol. Chem., 2014, 12, 2180–2184 RSC;
(e)
J. Royer, Asymmetric Synthesis of Nitrogen Heterocycles, Wiley-VCH, Weinheim, 2009 Search PubMed;
(f) A. H. Lipkus, Q. Yuan, K. A. Lucas, S. A. Funk, W. F. Bartelt, R. J. Schenck and A. J. Trippe, J. Org. Chem., 2008, 73, 4443 CrossRef CAS PubMed.
-
(a) F. Matsuura, Y. Hamada and T. Shioiri, Tetrahedron, 1994, 50, 265–274 CrossRef CAS;
(b) S. Raghavan and V. Krishnaiah, J. Org. Chem., 2010, 75, 748–861 CrossRef CAS PubMed.
-
(a) Y. Han, M. Han, D. Shin, C. Song and H.-G. Hahn, J. Med. Chem., 2012, 55, 8188–8192 CrossRef CAS PubMed;
(b) H. Bräuner-Osborne, L. Bunch, N. Chopin, F. Couty, G. Evano, A. A. Jensen, M. Kusk, B. Nielsen and N. Rabasso, Org. Biomol. Chem., 2005, 3, 3926 RSC;
(c) D. Honcharenko, C. Zhou and J. Chattopadhyaya, J. Org. Chem., 2008, 73, 2829 CrossRef CAS PubMed;
(d) T. Hart, A. T. Macias, K. Benwell, T. Brooks, J. D'Alessandro, P. Dokurno, G. Francis, B. Gibbons, T. Haymes, G. Kennett, S. Lightowler, H. Mansell, N. Matassova, A. Misra, A. Padfield, R. Parsons, R. Pratt, A. Robertson, S. Walls, M. Wong and S. Roughley, Bioorg. Med. Chem. Lett., 2009, 19, 4241–4244 CrossRef CAS PubMed;
(e)
K. Gerlach, H. Priepke, W. Weinen, A. Schuler-Metz and H. Nar, WIPO Pat, WO2008/135525A2, 2008 Search PubMed
Chem. Abstr.
2008
149
556457
Search PubMed.
-
(a)
G. S. Singh, M. D'hooghe and N. De Kimpe, in Comprehensive Heterocyclic Chemistry III, ed. C. V. Stevens, Elsevier, Oxford, 3rd edn, 2008, vol. 2, pp. 1–110 Search PubMed;
(b)
F. Couty, in Science of Synthesis, ed. E. Schaumann and D. Enders, Thieme, Stuttgart, 2009, vol. 40a, pp. 773–816 Search PubMed;
(c)
G. Rousseau and S. Robin, in Modern Heterocyclic Chemistry, ed. J. Alzerez-Builla, J. J. Vaquero and J. Barluenga, Wiley-VCH, Weinheim, 1st edn, 2011, vol. 1, pp. 163–268 Search PubMed.
- Y. Yagil, M. Miyamoto, L. Frasier, K. Oizumi and H. Koike, Am. J. Hypertens., 1994, 7, 637 CAS.
-
(a) B. I. Ericksson, S. Carlsson, M. Halvarsson, B. Risberg and C. Mattsson, Thromb. Haemostasis, 1997, 78, 1404 Search PubMed;
(b)
AstraZeneca, PCT Int. Appl, WO00/41716, 2000 Search PubMed.
-
(a)
D. R. Adams, J. Bentley, C. D. Bodkin, I. A. Cliffe, J. E. P. Davidson, H. L. Mansell, N. J. Monck, R. G. Shepherd and J. M. Shepherd, US Pat, 6831078, 2004 Search PubMed;
(b)
S. R. Brunette, A. Abeywardane, M. J. Burke, S. R. Kapadia, T. M. Kirrane Jr., M. R. Netherton, H. Razavi, S. Rodriguez, A. Saha, R. Sibley, K. Smith Keenan, L. Lana, H. Takahashi, M. R. Turner, J.-P. Wu, E. R. R. Young, Q. Zhang, Q. Zhang and R. M. Zindell, WOWO2013/134226A1, 2013 Search PubMed;
(c) D. W. Kung, S. B. Coffey, R. M. Jones, S. Cabral, W. Jiao, M. Fichtner, P. A. Carpino, C. R. Rose, R. F. Hank, M. G. Lopaze, R. Swartz, H. T. Chen, Z. Hendsch, B. Posner, C. F. Wielis, B. Manning, J. Dubins, I. A. Stock, S. Varma, M. Campbell, D. DeBartola, R. Kosa-Maines, S. J. Steyn and K. F. McClure, Bioorg. Med. Chem. Lett., 2012, 22, 4281 CrossRef CAS PubMed;
(d) J. M. Keith, W. M. Jones, J. M. Pierce, M. Seierstad, J. A. Palmer, M. Webb, M. J. Karbarz, B. P. Scott, S. J. Wilson, L. Luo, M. L. Wennerholm, L. Chang, S. M. Brown, M. Rizzolio, R. Rynberg, S. R. Chaplan and J. G. Breitenbucher, Bioorg. Med. Chem. Lett., 2014, 24, 737 CrossRef CAS PubMed.
- For an example of anodic acetoxylation of N-tosylazetidine, see: T. Shono, Y. Matsumura, K. Uchida and F. Nakatani, Bull. Chem. Soc. Jpn., 1988, 61, 3029 CrossRef CAS.
- L. Degennaro, M. Zenzola, P. Trinchera, L. Carroccia, A. Giovine, G. Romanazzi, A. Falcicchio and R. Luisi, Chem. Commun., 2014, 50, 1698–1700 RSC.
-
(a) A. Münch, B. Wendt and M. Christmann, Synlett, 2004, 2751–2755 Search PubMed;
(b) R. M. de Figueiredo, R. Fröhlich and M. Christmann, J. Org. Chem., 2006, 71, 4147–4154 CrossRef CAS PubMed.
-
(a) D. Enders, J. Gries and Z.-S. Kim, Eur. J. Org. Chem., 2004, 4471 CrossRef CAS;
(b) M. Tiecco, L. Testaferri, A. Temperini, R. Terlizzi, L. Bagnoli, F. Marini and C. Santi, Org. Biomol. Chem., 2007, 5, 3510 RSC;
(c) M. Medjahdi, J. C. Gonzáles-Gómez, F. Foubelo and M. Yus, J. Org. Chem., 2009, 74, 7859 CrossRef CAS PubMed.
- N. Kern, A.-S. Felten, J.-M. Weibel, P. Pale and A. Blanc, Org. Lett., 2014, 16, 6104 CrossRef CAS PubMed.
- K. L. Jensen, D. U. Nielsen and T. F. Jamison, Chem. – Eur. J., 2015, 21, 7379–7383 CrossRef CAS PubMed.
-
(a) D. Seebach, D. Enders and B. Renger, Chem. Ber., 1977, 110, 1852–1865 CrossRef CAS;
(b) W. Wykypiel, J. J. Lohmann and D. Seebach, Helv. Chim. Acta, 1981, 64, 1337–1346 CrossRef CAS;
(c) D. Seebach, T. Vettiger, H.-M. Mueller, D. A. Plattner and W. Petter, Liebigs Ann. Chem., 1990, 687 CrossRef CAS.
-
D. M. Hodgson, Organolithiums in Enantioselective Synthesis: Topics in Organometallic Chemistry, Springer, Berlin, 2003 Search PubMed.
-
(a) D. Seebach and W. Lubosch, Angew. Chem., 1976, 88, 339–340 (
Angew. Chem., Int. Ed. Engl.
, 1976
, 15
, 313–314
) CrossRef CAS;
(b) W. Lubosch and D. Seebach, Helv. Chim. Acta, 1980, 63, 102–116 CrossRef CAS.
- D. M. Hodgson and J. Kloesges, Angew. Chem., Int. Ed., 2010, 49, 2900–2903 CrossRef CAS PubMed.
-
R. E. Gawley, S. O'Connor and R. Klein, in Science of Synthesis, ed. V. Snieckus and M. Majewski, Thieme, Stuttgart, 2006, vol. 8a, pp. 677–757 Search PubMed.
- For a comprehensive study on the stereochemistry of this lithiation/trapping sequence see ref. 29.
- D. M. Hodgson, S. P. Hughes, A. L. Thompson and T. D. Heightman, Org. Lett., 2008, 10, 3453–3456 CrossRef CAS PubMed.
- J.-C. Kizirian, J.-C. Caille and A. Alexakis, Tetrahedron Lett., 2003, 44, 8893–8895 CrossRef CAS.
-
(a) P. Beak, S. T. Kerrick, S. Wu and J. Chu, J. Am. Chem. Soc., 1994, 116, 3231 CrossRef CAS;
(b) W. F. Bailey, P. Beak, S. T. Kerrick, S. Ma and K. B. Wiberg, J. Am. Chem. Soc., 2002, 124, 1889 CrossRef CAS PubMed;
(c) D. Stead, P. O'Brien and A. Sanderson, Org. Lett., 2008, 10, 1409 CrossRef CAS PubMed;
(d) I. Coldham, P. O'Brien, J. J. Patel, S. Raimbault, A. J. Sanderson, D. Stead and D. T. E. Whittaker, Tetrahedron: Asymmetry, 2007, 18, 2113 CrossRef CAS.
- D. M. Hodgson, C. I. Pearson and A. L. Thompson, J. Org. Chem., 2013, 78, 1098–1106 CrossRef CAS PubMed.
- A. I. Meyers and D. A. Dickman, J. Am. Chem. Soc., 1987, 109, 1263–1265 CrossRef CAS.
- P. J. Rayner, J. C. Smith, C. Denneval, P. O'Brien, P. A. Clarke and R. A. J. Horan, Chem. Commun., 2016, 52, 1354–1357 RSC.
- G. Gelardi, G. Barker, P. O'Brien and D. C. Blackemore, Org. Lett., 2013, 15, 5424 CrossRef CAS PubMed.
- D. M. Hodgson, C. L. Mortimer and J. M. McKenna, Org. Lett., 2015, 17, 330–333 CrossRef CAS PubMed.
- G. C. Barrett and C. M. O. A. Martins, J. Chem. Soc., Chem. Commun., 1972, 638–639 RSC.
- J. Praz, L. Guénée, S. Aziz, A. Berkessel and A. Alexakis, Adv. Synth. Catal., 2012, 354, 1780–1790 CrossRef CAS.
- K. E. Jackson, C. L. Mortimer, B. Odell, J. M. McKenna, T. D. W. Claridge, R. S. Paton and D. M. Hodgson, J. Org. Chem., 2015, 80, 9838–9846 CrossRef CAS PubMed.
- The NMR-determined rotational barriers for N-thiopivaloyl-2-methylazetidine and N-Boct-2-methylazetidine at 25 °C (70.7 and 75.0 kJ mol−1, respectively) indicate rotation half-lives (for the major conformer) of 0.3 and 1.6 s. However, these extend to 2.1 and 116 days at −78 °C, strongly suggesting that rotamer interconversion is not significant under lithiation condition.
- D. M. Hodgson, C. I. Pearson and M. Kazmi, Org. Lett., 2014, 16, 856–859 CrossRef CAS PubMed.
-
(a) D. M. Hodgson, P. G. Humphreys, Z. Xu and J. G. Ward, Angew. Chem., Int. Ed., 2007, 46, 2245 CrossRef CAS PubMed;
(b) B. Musio, G. J. Clarkson, M. Shipman, S. Florio and R. Luisi, Org. Lett., 2009, 11, 325 CrossRef CAS PubMed;
(c) R. Luisi and S. Florio, Chem. Rev., 2010, 110, 5128 CrossRef PubMed.
- G. Parisi, E. Capitanelli, A. Pierro, G. Romanazzi, G. J. Clarkson, L. Degennaro and R. Luisi, Chem. Commun., 2015, 51, 15588–15591 RSC.
- Strictly related oxetane-containing tripeptides have recently been reported: N. H. Powell, G. J. Clarkson, R. Notman, P. Raubo, N. G. Martin and M. Shipman, Chem. Commun., 2014, 50, 8797 RSC.
- M. Zenzola, L. Degennaro, P. Trinchera, L. Carroccia, A. Giovine, G. Romanazzi, P. Mastrorilli, R. Rizzi, L. Pisano and R. Luisi, Chem. – Eur. J., 2014, 20, 12190–12200 CrossRef CAS PubMed.
-
(a) F. Couty, O. David, B. Larmanjat and J. Marrot, J. Org. Chem., 2007, 72, 1058–1061 CrossRef CAS PubMed;
(b) A. Alex, B. Larmanjat, J. Marrot, F. Couty and O. David, Chem. Commun., 2007, 2500–2502 RSC;
(c) C. Lo, O. David and F. Couty, Tetrahedron Lett., 2014, 55, 535–537 CrossRef CAS.
- G. Casoni, E. L. Myers and V. K. Aggarwal, Synthesis, 2016, 48, 3241–3253 CrossRef CAS.
-
(a)
D. G. Hall, Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials, Wiley-VCH, Weinheim, 2nd edn, 2011, vol. 1 and 2 Search PubMed;
(b)
E. Fernandez and A. Whiting, Synthesis and Application of Organoboron Compounds, Springer International Publishing, Cham, 2015 Search PubMed;
(c)
M. Davison, A. K. Hughes, T. B. Marder and K. Wade, Contemporary Boron Chemistry, RSC, Cambridge, UK, 2000 Search PubMed.
-
(a) B. Drouillat, F. Couty and J. Marrot, Synlett, 2009, 767–770 CAS;
(b) E. Tayama, K. Watanabe and Y. Matano, Eur. J. Org. Chem., 2016, 3631–3641 CrossRef CAS.
- For recent examples of base-induced Stevens rearrangement on azetidinium ions see: B. Drouillat, E. d'Aboville, F. Bourdreux and F. Couty, Eur. J. Org. Chem., 2014, 1103–1109 CrossRef CAS.
- S. Billotte, Synlett, 1998, 379 CrossRef CAS.
-
(a) M. A. J. Duncton, M. A. Estiarte, D. Tan, C. Kaub, D. J. R. O'Mahony, R. J. Johnson, M. Cox, W. T. Edwards, M. Wan, J. Kincaid and M. G. Kelly, Org. Lett., 2008, 10, 3259 CrossRef CAS PubMed;
(b) G. A. Molander, K. M. Traister and B. T. O'Neill, J. Org. Chem., 2014, 79, 5771 CrossRef CAS PubMed.
- B. Barré, L. Gonnard, R. Campagne, S. Reymond, J. Marin, P. Ciapetti, M. Brellier, A. Guérinot and J. Cossy, Org. Lett., 2014, 16, 6160 CrossRef PubMed.
- D. Parmar, L. Henkel, J. Dib and M. Rueping, Chem. Commun., 2015, 51, 2111–2113 RSC.
- G. Parisi, M. Zenzola, E. Capitanelli, C. Carlucci, G. Romanazzi, L. Pisano, L. Degennaro and R. Luisi, Pure Appl. Chem., 2016, 88, 631–648 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here.