
Heterogeneous PEGylation of diamond nanoparticles†
Amanda S.
Barnard
Data61 CSIRO, Docklands, VIC 3008, Australia. E-mail: amanda.barnard@data61.csiro.au; Tel: +61 3 9662 7356
First published on 2nd December 2016
Abstract
Coating the surfaces of inorganic nanoparticles with polyethylene glycol (PEG) is an important step in the development of many nanoparticle-based drug delivery systems. The efficiency with which drug molecules can be loaded on to nanoparticle surfaces is contingent on the concentration, distribution and stability of the PEG coating. In this study the distribution and relative stability of PEG on diamond nanoparticles is predicted, for clean and passivated surface structures, in 3D. This is an ideal exemplar, since PEGylated diamond nanoparticles are already being trialed as carriers for doxorubicin (DOX). The results show that PEGylation is favorable near the {100} facets regardless of surface reconstructions or pre-treatment, but pre-treatment is required to increase the probability of stable and homogeneous PEGylation on other facets.
The use of nanoparticles for drug delivery is an exciting area of research that has seen a rapid increase in effort and investment. Nanoparticles that are small, non-toxic, chemically robust and biocompatible are ideal candidates, and those that are also easy to produce and process on an industrial scale are beginning to be translated into clinical and pre-clinical settings. In many cases, however, the surfaces of these particles still require modification to optimize the nanoparticle/drug interactions; facilitating both loading and unloading. For this purpose functionalization with polyethylene glycol (PEG), a process known as PEGylation, is widely used with great success.1–3 PEGylation has been shown to improve the stability and in vivo performance of gene and non-viral drug vectors4–7 and significantly improve the transport of nanoparticles.8 However, the concentration, distribution and stability of the PEG can depend sensitively on the structure of the nanoparticle surfaces.9 Unlike micelles, the surfaces of inorganic nanoparticles can be extremely heterogeneous, including a range of different crystallographic orientations, edges, corners, defects, and characteristic surface reconstructions.
Of the variety of nanoparticles used in modern drug delivery platforms, diamond nanoparticles are a perfect example that simultaneously meets many of the practical and economic requirements, while presenting many scientific challenges associated with their inherently unstable surfaces that contain a significant proportion of non-diamond carbon. Non-toxic reconstructed nanodiamonds (bucky-diamonds) have shown great promise as drug delivery vehicles;10–17 increasing selectivity18,19 and sensitivity of established treatments,20 assist in dosage moderation21 and reducing burst effusion.22 Other promising applications23 include biosensing,24in vivo imaging,25–28 tissue engineering29,30 and as ultraviolet attenuators for sunscreens.31,32 However, the heterogeneous surface speciation, which is closely related to the size and shape of each particle, impacts the efficiency, reliability and reproducibility of nanodiamond/drug interfaces33 and could lead to unacceptable batch-to-batch variation. Homogeneous covalent PEGylation of the surfaces of diamond nanoparticles is a solution to this problem,34 but to date a detailed knowledge of this interface remains elusive. Current reports average over entire samples of particles, masking the underlying structure/property relationships, and failing to provide the quantitative predictions required to identify possible improvements.
In the present study electronic structure methods are used to explore the covalent attachment of a PEG monomer to the surface of a model diamond nanoparticle with atomic level resolution. Although it is well known that the activity of the {100} and {110} surfaces are larger than the graphitized {111} facets, the results reveal that perturbation of the surface structures prior to PEGylation, or “priming”, is more important than the morphology of the nanoparticle. The probable distribution is initially heterogeneous over the surface of a reconstructed diamond nanoparticle, but the results indicate that near homogeneity can be recovered via suitable pre-processing.
In this study the density functional tight-binding method with self-consistent charges (SCC-DFTB) has been used, as implemented in the DFTB+ code.35–37 SCC-DFTB is an approximate quantum chemical approach where the Kohn–Sham density functional is expanded to second order around a reference electron density, with a reference density obtained from self-consistent generalized gradient approximation (GGA) calculations of weakly confined neutral atoms. The confinement potential is optimized to anticipate the charge density and effective potential in molecules and solids. A minimal valence basis set is used to account explicitly for the two-center tight-binding matrix elements within the DFT level. The double counting terms in the Coulomb and exchange–correlation potential, as well as the intra-nuclear repulsion are replaced by a universal short-range repulsive potential. All structures have been fully relaxed with a conjugate gradient methodology until forces on each atom was minimized to be less than 10−4 a.u. (i.e. ≈5 meV Å−1). In all the calculations, the “PBC”‡ set of parameters is used to describe the contributions from diatomic interactions of carbon.38 This method has been shown to provide good and reliable results for diamond nanoparticles in the past.39–41
The present study also uses a quasi-spherical diamond nanostructure with 1502 atoms, which is equivalent to a diameter of 2.9 nm. This structure is enclosed entirely by {110}, {111}, and {100} facets (in order of prevalence) and has an effective surface-to-volume ratio of q = 1.031 × q0, where q0 is the surface to volume ratio of a sphere of equivalent mass. This structure is part of an open-access set of bucky-diamond structures provided online44 and has a total of 460 adsorption sites (surface dangling bonds). By exploiting the symmetry of the structure this has been reduced to the 26 surface sites that are geometrically and crystallographically unique, as described elsewhere.42,43 In the absence of priming (which is described later on) the site-dependent interaction energy has been calculated for the (ND)− + (PEG-OH)+ → (ND-PEG-OH) reaction, and for the (ND-H)− + (PEG-OH)+ → H + (ND-PEG-OH) reaction where priming or passivation with H was included; though other reactions could be envisaged as prescribed by given experimental conditions. The energy of H was calculated with respect to a H2O reservoir, where 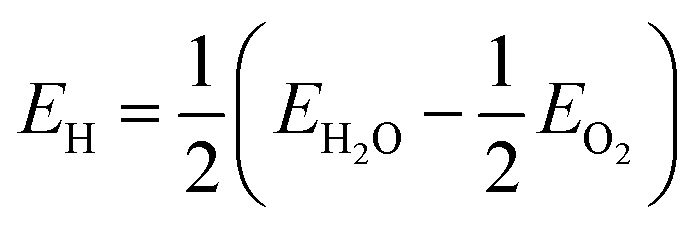 . Each result is then assigned to the symmetrically equivalent sites to provide a 3D map of the particle/PEG interaction surface. At this point it should be highlighted that the following results have been presented probabilistically, rather than absolutely, so that the relative site-dependent differences in interaction energies will be largely independent of the chosen PEGylation reaction.
. Each result is then assigned to the symmetrically equivalent sites to provide a 3D map of the particle/PEG interaction surface. At this point it should be highlighted that the following results have been presented probabilistically, rather than absolutely, so that the relative site-dependent differences in interaction energies will be largely independent of the chosen PEGylation reaction.
Simulations on the unpassivated diamond nanoparticle revealed that PEGylation is largely incompatible with the reconstructed particle, owing to the energetic penalty associated with disrupting the aromatic character of the graphitized surfaces. Although it has previously been shown that covalent bonding of foreign species on the graphitized {111} surfaces can cause buckling, and eventual recovery of the aliphatic character as the surface collapses and re-binds with the sub-surface lattice,45 this phenomenon did not accompany PEGylation on these sites and so the binding energies were highly endothermic. This is illustrated in Fig. 1 as a negative probability (or improbability, shown in blue). PEGylation is also improbable on the {110} facets due to disruption of the surface chains (that have sp2+x character), and edges and corners (that are also sp2-hybridized). The only sites that exhibit a positive probability of PEGylation are at the corners of the aliphatic {100} facets, though this is only marginally and likely to be highly susceptible to changes in the thermochemical conditions. Overall it is predicted that PEGylation of this structure will return a 7.83% success rate.
 | ||
| Fig. 1 The probability of PEGylation across the surface of a reconstructed (unpassivated) diamond nanoparticle, in the range 1 (red) to −1 (blue). Attachment is highly improbable on the graphitized {111} facets (blue). | ||
The probability of PEGylation could be improved by engineering diamond nanoparticles with a higher fraction of {100} facets and exercising precise control over the environment, but this is currently beyond the experimental capability. An alternative approach is to increase the fraction of aliphatic sp3-hybridized sites at the surface which, as mentioned above, can be achieved by disrupting the reconstructed aromatic surfaces via an initiator.45 This could be performed during a pre-processing step, or using a precursor during PEGylation, as has been demonstrated experimentally with a COOH initiator.46 This step is referred to here as “priming”, and the results for PEGylation of primed sites on the diamond nanoparticle using hydrogen as an initiator are shown in Fig. 2. These results predict that priming the surface will significantly impact the probability of PEGylation, and effectively guarantees efficient PEGylation of the corners of the {100} facets. Elsewhere, although it is insufficient to convert aromatic surface sites to sp3 sites (re-bound the to sub-surface layer below), it is sufficient convert them to sp2+x sites that have anti-bonding electrons available for reactions. The impact of this is to transform almost all sites on the {111} facets, making them similar in character to the {110} facets, and producing an almost homogeneous probability of PEGylation across the surface. Once a site has been primed the site-dependent heterogeneity is practically eliminated, and the predicted overall PEGylation success rate is increased to 97.39%.
 | ||
| Fig. 2 The probability of PEGylation across the surface of a reconstructed (unpassivated) diamond nanoparticle when sites have been primed using pre-passivation with hydrogen, in the range 1 (red) to −1 (blue). Highly probable attachment is restricted to the corners of the {100} facets (red), and almost all remaining surface sites exhibit an almost homogeneously low positive probability of attachment, with the exception of some improbable sites at the center of some graphitized {111} facets. | ||
The culmination of this endeavor would be to pre-treat the entire surface to eliminate all sp2 carbon prior to PEGylation. The results predicting the probability of PEGylation on a passivated (H-terminated) diamond nanoparticle are provided in Fig. 3, where it is shown that some of the site-dependent heterogeneity has returned. In this case attachment of PEG is supported on all sites, excluding the sites on the {111} facets that collapse and re-bind with the sub-surface lattice upon passivation (shown in black), but is now strongest surrounding the corners. This is equivalent to simultaneously priming all surface sites and so the probability reflects a competition between the stability of PEGylation at each site and the alternative stability of the passivating layer. In this case homogeneous hydrogen passivation was investigated, as this has been achieved experimentally,47 and there are a small number of sites on the {100} facets where H-termination may persist. As mentioned above, the binding energy of the hydrogen passivation layer was calculated with respect to a H2O, and the relative strength of attachment may vary if an alternative thermochemical reservoir were used. The choice of thermochemical reservoir, as well as the temperature,40 may impact the overall probability of PEGylation (with respect to the passivating layer) but is unlikely to impact the site-to-site heterogeneity. In this case the overall PEGylation success rate is predicted to be 100%.
 | ||
| Fig. 3 The probability of PEGylation across the surface of a H-terminated diamond nanoparticle that has undergone complete surface pre-passivation, in the range 1 (red) to −1 (blue). Attachment of PEG is probable across the entire surface, and is highly probable on the edges and corners of the {100} facets. Sites colored in black are inaccessible following pre-passivation, as the surface graphitization is reversed and these atoms reside in the sub-surface plane. | ||
It is clear from this study that pre-treatment of the surface with an initiator will increase the probability of covalent PEGylation. Provided a surface site has been pre-treated, replacing the initiator with PEG should be largely site-independent. The obvious exception is the corners of the {100} facets where under-coordinated sp3-hybridized sites interact strongly with PEG. For the remaining sites the issue then becomes whether the surfaces can be efficiently and homogeneously pre-treated, and this will depend upon a number of factors. The interactions and distributions of H, O, OH, N, NH, NH2, S, SH, SH2, C, CH, CH2 and COOH across the surfaces of this model nanodiamond have been reported elsewhere,42,43 all showing varying degrees of heterogeneity. However, a survey of these potential initiators show similarities in the distribution of exothermic attachment sites, and suggests that the species of initiator is not as important as the concentration.
Successful conjugation of PEG on the surface of diamond nanoparticles using a Br initiator has already been reported.48 Diamond nanoparticles have also been successfully PEGylated via reversible addition fragmentation chain transfer (RAFT) polymerization, where the chain transfer agent was immobilized through by reacting with hydroxyl groups on the particle surface with the carboxyl group as the initiator for surface-initiated RAFT polymerization.49 This was shown to enhance the dispersibility and the biocompatiblity of the diamond nanoparticles. This is significant because PEGylation of diamond nanoparticles has already been found to be important in interfacing the material with doxorubicin (DOX), a powerful chemotherapeutic.46,50 PEGylation of diamond nanoparticles facilitates greater control over the loading and stability of DOX for selective therapies,51 and retains the pH-sensitivity that is important for drug release.52
These results in combination with preceding studies, also suggest that facet-dependent pre-treatment may be a key to facet-dependent PEGylation; or at the very least a greater control over the concentration of PEG at the surface. It is unclear at this stage if these results hold for other types of nanoparticles, as diamond is a unique and difficult case due to the characteristic surface reconstructions. Other biologically relevant nanoparticles such as silicon and gold do not exhibit the same facet-dependent reconstructions or change in speciation, and may possibly be homogeneous to PEG attachment without an initiator to prime the surface. Complementary studies using the methodology outlined here would be needed to confirm or dispute this hypothesis.
By simulating the attachment of polyethylene glycol (PEG) on all unique binding sites on the surface of a model diamond nanoparticle, using electronic structure simulations, it has been shown that in the absence of pre-treatment the distribution of PEG on the surface will be heterogeneous, and the probability of attachment will be low; most surface sites supporting desorption. By simulating the impact of partial or complete priming of the surface with an initiator, this morphology-dependent heterogeneity can be eliminated, and the probability of attachment significantly improved. This useful finding indicates that the size- and shape-distributions present in samples of diamond nanoparticle need not hinder development, as elimination of surface heterogeneity (via surface priming and PEGylation) is akin to eliminating the impact of persistent polydispersivity. We don't need to make monodispersed samples, provides we can make them seem monodispersed to the molecules with which they interact.
At this point it should be noted that the present study utilizes a monomer of PEG (see ESI†) and in reality much longer polymer chains may be used for such small particles. This choice was pragmatic, to facilitate the large number of electronic structure relaxations required to map the surface, and considered reasonable in given that no prior evidence suggests that the variation in the interaction energy as a function of chain length would be greater than the variation due to surface attachment site. The chain length, and the concentration of PEG at the surface, does impact the configuration of longer PEG chains (whether they adopt a “brush” or “mushroom” configuration). This in turn can impact the solubility of the system, and some of the overall properties of the PEGylated particle.
Based on these reports and the results herein, there is clear evidence that greater control of PEGylation through priming with different initiators may further enhance the functionality of diamond nanoparticle-based drug delivery systems. This study is therefore the first step in our understanding of the PEGylation of diamond nanoparticles, and the impact of PEG chain length and the density of PEG at the surface are certainly worthy of further attention.
Acknowledgements
Computational resources for this project were supplied by the National Computational Infrastructure national facility under Partner Allocation Scheme, Grant q27.References
- H. Otsukaa, Y. Nagasaki and K. Kataoka, Adv. Drug Delivery Rev., 2003, 55, 403–419 CrossRef.
- J. Suh, K. L. Choy, S. K. Lai, J. S. Suk, B. C. Tang, S. Prabhu and J. Hanes, Int. J. Nanomed., 2007, 2, 735–741 CrossRef CAS PubMed.
- F. M. Veronese and A. Mero, BioDrugs, 2008, 2, 315 CrossRef.
- M. Ogris, G. Walker, T. Blessing, R. Kircheis, M. Wolschek and E. Wagner, J. Controlled Release, 2003, 91, 173–181 CrossRef CAS PubMed.
- S. Mishra, P. Webster and M. E. Davis, Eur. J. Cell Biol., 2004, 83, 97–111 CrossRef CAS PubMed.
- A. S. Zahr, M. de Villiers and M. V. Pishko, Langmuir, 2005, 21, 403–410 CrossRef CAS PubMed.
- X. Sun, R. Rossin, J. L. Turner, M. L. Becker, M. J. Joralemon, M. J. Welch and K. L. Wooley, Biomacromolecules, 2005, 6, 2541–2554 CrossRef CAS PubMed.
- S. K. Lai, E. D. O'Hanlon, S. Harrold, S. T. Man, Y.-Y. Wang, R. Cone and J. Hanes, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 1482–1487 CrossRef CAS PubMed.
- V. Cauda, C. Argyo and T. Bein, J. Mater. Chem., 2010, 20, 8693–8699 RSC.
- D. Ho, ACS Nano, 2009, 7, 3825–3829 CrossRef PubMed.
- H. Huang, E. Pierstorff, E. Ōsawa and D. Ho, Nano Lett., 2007, 7, 3305–3314 CrossRef CAS PubMed.
- H. Huang, E. Pierstorff and D. Ho, ACS Nano, 2008, 2, 203–212 CrossRef CAS PubMed.
- E. Chow, E. Pierstorff, G. Cheng and D. Ho, ACS Nano, 2008, 2, 33–40 CrossRef CAS PubMed.
- R. Lam and D. Ho, Expert Opin. Drug Delivery, 2009, 6, 883–895 CrossRef CAS PubMed.
- M. Chen, X. Zhang, H. Man, R. Lam, E. Chow and D. Ho, J. Phys. Chem. Lett., 2010, 1, 3167–3171 CrossRef CAS.
- X.-Q. Zhang, M. Chen, R. Lam, X. Xu, E. Ōsawa and D. Ho, ACS Nano, 2009, 3, 2609–2616 CrossRef CAS PubMed.
- M. Chen, E. D. Pierstorff, R. Lam, S. Y. Li, H. Huang, E. Ōsawa and D. Ho, ACS Nano, 2009, 3, 2016–2022 CrossRef CAS PubMed.
- A. Smith, E. Robinson, X. Zhang, E. Chow, E. Osawa and D. Ho, Nanoscale, 2011, 3, 2844–2848 RSC.
- X. Zhang, R. Lam, X. Xu, E. K. Chow, H. Kim and D. Ho, Adv. Mater., 2011, 23, 4770–4775 CrossRef CAS PubMed.
- R. Shimkunas, E. Robinson, X. Zhang, R. Lam, X. Xu, E. Osawa and D. Ho, Biomater., 2009, 30, 5720–5728 CrossRef CAS PubMed.
- M. Chen, E. Robinson, H. Huang, E. Pierstorff and D. Ho, Ann. Biomed. Eng. Soc., 2009, 37, 2003–2017 CrossRef PubMed.
- R. Lam, M. Chen, E. Pierstorff, H. Huang, E. Ōsawa and D. Ho, ACS Nano, 2008, 2, 2095–2102 CrossRef CAS PubMed.
- A. Krueger, Chem. – Eur. J., 2008, 14, 1382–1390 CrossRef CAS PubMed.
- W. Zhang, K. Patel, A. Schexnider, S. Banu and A. D. Radadia, ACS Nano, 2014, 2, 1419–1428 CrossRef PubMed.
- L. P. McGuinness, Y. Yan, A. Stacey, D. A. Simpson, L. T. Hall, D. Maclaurin, S. Prawer, P. Mulvaney, J. Wrachtrup, F. Caruso, R. E. Scholten and L. C. L. Hollenberg, Nat. Nanotechnol., 2011, 6, 358–363 CrossRef CAS PubMed.
- N. Mohan, C.-S. Chen, H.-H. Hsieh, Y.-C. Wu and H.-C. Chang, Nano Lett., 2010, 10, 3692–3699 CrossRef CAS PubMed.
- T. J. Wu, Y.-K. Tzeng, W.-W. Chang, C.-A. Cheng, Y. Kuo, C.-H. Chien, H.-C. Chang and J. Yu, Nat. Nanotechnol., 2013, 8, 682–689 CrossRef CAS PubMed.
- Y.-R. Chang, H.-Y. Lee, K. Chen, C.-C. Chang, D.-S. Tsai, C.-C. Fu, T.-S. Lim, Y.-K. Tzeng, C.-Y. Fang, C.-C. Han, H.-C. Chang and W. S. Fann, Nat. Nanotechnol., 2008, 3, 284–288 CrossRef CAS PubMed.
- Q. Zhang, V. N. Mochalin, I. Neitzel, I. Y. Knoke, J. Han, C. A. Klug, J. G. Zhou, P. I. Lelkes and Y. Gogotsi, Biomaterials, 2011, 32, 87–94 CrossRef CAS PubMed.
- F. Ostadhossein, N. Mahmoudi, G. Morales-Cid, E. Tamjid, F. J. Navas-Martos, B. Soriano-Cuadrado, J. M. López Paniza and A. Simch, Materials, 2015, 8, 6401–6418 CrossRef.
- O. Shenderova, V. Grichko, S. Hens and J. Walch, Diamond Relat. Mater., 2007, 16, 2003–2008 CrossRef CAS.
- M.-S. Wu, D.-S. Sun, Y.-C. Lin, C.-L. Cheng, S. C. Hung, P. K. Chen, J. H. Yang and H. H. Chang, J. Nanobiotechnol., 2015, 13, 1–12 CrossRef CAS PubMed.
- A. Adnan, R. Lam, C. Hanning, J. Lee, D. J. Schaffer, A. S. Barnard, G. C. Schatz, D. Ho and W. K. Liu, Mol. Pharmaceutics, 2011, 368–374 CrossRef CAS PubMed.
- L. Zhao, Y.-H. Xu, T. Akasaka, S. Abe, N. Komatsu, F. Watari and X. Chen, Biomaterials, 2014, 35, 5393–5406 CrossRef CAS PubMed.
- D. Porezag, Th. Frauenheim, Th. Köhler, G. Seifert and R. Kaschner, Phys. Rev. B: Condens. Matter, 1995, 51, 12947–12957 CrossRef CAS.
- Th. Frauenheim, G. Seifert, M. Elstner, Th. Niehaus, C. Köhler, M. Amkreutz, M. Sternberg, Z. Hajnal, A. Di Carlo and S. Suhai, J. Phys.: Condens. Matter, 2002, 14, 3015 CrossRef CAS.
- B. Aradi, B. Hourahine and Th. Frauenheim, J. Phys. Chem. A, 2007, 111, 5678–5684 CrossRef CAS PubMed.
- C. Kohler and T. Frauenheim, Surf. Sci., 2006, 600, 453–460 CrossRef.
- L. Lai and A. S. Barnard, Nanoscale, 2011, 3, 2566–2575 RSC.
- L. Lai and A. S. Barnard, J. Mater. Chem., 2012, 22, 16774–16780 RSC.
- L. Lai and A. S. Barnard, Phys. Chem. Chem. Phys., 2013, 15, 9156–9162 RSC.
- L. Lai and A. S. Barnard, Nanoscale, 2014, 6, 14185–14189 RSC.
- L. Lai and A. S. Barnard, Nanoscale, 2016, 8, 7899–7905 RSC.
- A. Barnard, Nanodiamond Data Set. v1. CSIRO. Data Collection, 2016, DOI:10.4225/08/571F076D050B1.
- A. S. Barnard, Phys. Chem. Chem. Phys., 2012, 14, 10080–10093 RSC.
- D. Wang, Y. Tong, Y. Li, Z. Tian, R. Cao and B. Yang, Diamond Relat. Mater., 2013, 36, 26–34 CrossRef CAS.
- O. A. Williams, J. Hees, C. Dieke, W. Jäger, L. Kirste and C. E. Nebel, ACS Nano, 2010, 4, 4824–4830 CrossRef CAS PubMed.
- X. Zhang, C. Fu, L. Feng, Y. Ji, L. Tao, Q. Huang, S. Li and Y. Wei, Polymer, 2012, 53, 3178–3184 CrossRef CAS.
- Y. Shi, M. Liu, K. Wang, H. Huang, Q. Wan, L. Tao, L. Fu, X. Zhang and Y. Wei, Appl. Surf. Sci., 2015, 357, 2147–2153 CrossRef CAS.
- D. Passeri, F. Rinaldi, C. Ingallina, M. Carafa, M. Rossi, M. L. Terranova and C. Marianecci, J. Nanosci. Nanotechnol., 2015, 15, 972–988 CrossRef CAS PubMed.
- L. Li, L. Tian, W. Zhao, Y. Li and B. Yang, Integr. Biol., 2016, 8, 956–967 RSC.
- L. Li, L. Tian, W. Zhao, F. Cheng, Y. Li and B. Yang, RSC Adv., 2016, 6, 36407–36417 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Visualization of polyethylene glycol attachment on unique nanodiamond surface sites. See DOI: 10.1039/C6NR08315C |
| ‡ “PBC” is the name of the set of parameters, not an indication that periodic boundary conditions were used in this study. |
| This journal is © The Royal Society of Chemistry 2017 |