
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exosome-like silica nanoparticles: a novel ultrasound contrast agent for stem cell imaging†
Fang
Chen
ab,
Ming
Ma
c,
Junxin
Wang
a,
Fang
Wang
d,
Shi-Xiong
Chern
c,
Eric Ruike
Zhao
a,
Anamik
Jhunjhunwala
e,
Sean
Darmadi
a,
Hangrong
Chen
c and
Jesse V.
Jokerst
*ab
aDepartment of NanoEngineering, University of California, San Diego, 9500 Gilman Drive, La Jolla, CA 92093-0448, USA. E-mail: jjokerst@ucsd.edu
bMaterials Science and Engineering Program, University of California, San Diego, 9500 Gilman Drive, La Jolla, CA 92093-0448, USA
cState Key Laboratory of High Performance Ceramics and Superfine Microstructures, Shanghai Institute of Ceramics, Chinese Academy of Sciences, Shanghai, 200050, China
dResearch Center for Bioengineering and Sensing Technology, University of Science and Technology Beijing, Beijing, 100083, China
eDepartment of Bioengineering, University of California, San Diego, 9500 Gilman Drive, La Jolla, CA 92093-0418, USA
First published on 1st December 2016
Abstract
Ultrasound is critical in many areas of medicine including obstetrics, oncology, and cardiology with emerging applications in regenerative medicine. However, one critical limitation of ultrasound is the low contrast of target tissue over background. Here, we describe a novel cup-shaped silica nanoparticle that is reminiscent of exosomes and that has significant ultrasound impedance mismatch for labelling stem cells for regenerative medicine imaging. These exosome-like silica nanoparticles (ELS) were created through emulsion templating and the silica precursors bis(triethoxysilyl)ethane (BTSE) and bis(3-trimethoxysilyl-propyl)amine (TSPA). We found that 40% TSPA resulted in the exosome like-morphology and a positive charge suitable for labelling mesenchymal stem cells. We then compared this novel structure to other silica structures used in ultrasound including Stober silica nanoparticles (SSN), MCM-41 mesoporous silica nanoparticles (MSN), and mesocellular foam silica nanoparticles (MCF) and found that the ELS offered enhanced stem cell signal due to its positive charge to facilitate cell uptake as well as inherently increased echogenicity. The in vivo detection limits were <500 cells with no detectable toxicity at the concentrations used for labelling. This novel structure may eventually find utility in applications beyond imaging requiring an exosome-like shape including drug delivery.
Introduction
Stem cells are promising in regenerative medicine as a treatment for heart diseases, diabetes, alopecia, and neurodegenerative diseases.2–7 However, the long-term efficacy of stem cells is relatively low perhaps in part due to poor cell viability after implantation and a lack of cell retention in the treatment area.9 These issues are partially due to mis-injection of stem cells or injection into highly fibrotic tissues.10,11 Fortunately, mis-injection can be prevented by real-time imaging guidance to ensure sufficient cell delivery12 to the desired locations. Ultrasound imaging13–18 in particular has shown significant potential in imaging stem cell therapy because of its high temporal resolution relative to the gold standard of magnetic resonance imaging (MRI).12,19 Ultrasound is also more affordable than alternative strategies such as MRI, positron emission tomography (PET), and computed tomography (CT). However, the use of ultrasound in stem cell imaging/tracking is limited by the low contrast of cells above the adjacent tissue.20 While Doppler imaging,21 elastography,22 and thermoacoustic techniques23 have all been validated to improve contrast, novel ultrasound contrast agents are also active area of research.24–27Silica nanoparticles are an effective in vivo ultrasound contrast agent.14,28–31 They can efficiently label stem cells because of their nano-scale size, stability,14 and biocompatibility.32–34 Moreover, silica particles are multi-functional35–37 with a tunable morphology,38,39 and these modifications in nanoparticle shape can markedly increase the echogenicity of the individual silica nanoparticles.30 Gd-tagged silica nanoparticles can also offer multimodal imaging with clear biodegradation.14,18 Other work has used rattle-type mesoporous silica nanospheres to create multiple convex/concave interfaces to increase the ultrasound contrast,30 and silica microbubbles synthesized via polystyrene templates have been used for contrast-enhanced ultrasound.40 In this study, we combine these advances and report a completely novel silica nanoparticle with a concave mesoporous structure to enhance ultrasound signal. The particles have a shape surprisingly reminiscent of exosome extracellular vesicles. These exosome-like silica (ELS) nanoparticles improve the ultrasound contrast of cells by increasing not only the echogenicity of the nanoparticles but also their affinity to stem cells.
Experimental
Chemicals
Hexadecyltrimethylammonium bromide (CTAB, ≥99%), ammonium hydroxide (NH4OH), tetraethyl orthosilicate (TEOS, ≥99%), bis(triethoxysilyl)ethane (BTSE), bis(3-trimethoxysilyl-propyl)amine (TSPA, 90%), pluronic P123, hydrochloric acid, mesitylene, and fluorescein isothiocyanate (FITC) were purchased from Sigma Aldrich Inc. Sodium hydroxide (NaOH, ACS), ammonium fluoride, and ammonium nitrate (NH4NO3, ACS) were purchased from Fisher Scientific, Acros, and Marcon, respectively. Ultrapure agarose was obtained from Life Technologies. The (3-aminopropyl)triethoxysilane (APTES) was purchased from MP Biomedicals. Ethanol was purchased from VWR. All chemicals were used as received without any further purification. The water was Millipore grade with a resistivity larger than 18.2 MΩ cm at room temperature unless specified.Synthesis of silica nanoparticles
We synthesized ELS with emulsion template method and BTSE and TSPA silica sources. First, 0.4 mmol CTAB, 0.085 ml 26% ammonia solution, 0.6 mmol decane, and 0.4 mmol dimethylhexadecylamine (DMHA) were mixed in 150 ml water. The mixture was then sonicated in a water bath at 50 °C for 2 h followed by emulsification using an ultrasonic cell disruptor (20 kHz, 600 W; Bilon92, Shanghai) at ambient temperature for 30 min. The resulting white emulsion was transferred to an oil bath at 50 °C and then stirred at 700 rpm for 30 min. Next, 2.5 mL of an ethanol solution containing 4 ml of silica sources was slowly added. The mixture was stirred for 60 min and then allowed to stand at room temperature for 12 h. After the surfactant-containing nanoparticles were spun down at 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, the solid products were washed thrice with 1 wt% NaCl in methanol for 30 min in sonication bath to remove the template. The ELS were centrifuged, washed, and then dispersed in deionized water. The effect of the ratio between BTSE and TSPA on the morphologies of ELS was also studied, and the studied ratios of BTSE:TSPA included 5:0, 4:1, and 3:2. All ELS nanoparticles used for ultrasound imaging and cellular work were done at a BTSE:TSPA ratio of 3:2.
000 rpm, the solid products were washed thrice with 1 wt% NaCl in methanol for 30 min in sonication bath to remove the template. The ELS were centrifuged, washed, and then dispersed in deionized water. The effect of the ratio between BTSE and TSPA on the morphologies of ELS was also studied, and the studied ratios of BTSE:TSPA included 5:0, 4:1, and 3:2. All ELS nanoparticles used for ultrasound imaging and cellular work were done at a BTSE:TSPA ratio of 3:2.
The SSN were made according to the Stober method,41 and the size was tuned by slightly changing the quantity of ethanol, water, ammonia, and/or TEOS. Specifically, aliquots of NH4OH (1, 1.8, 2.2, 2.8, and 4.4 ml) and water (4.2 or 5 ml) were added to 50 ml ethanol. The mixture was stirred for 5 minutes before adding 3.5 or 4.2 ml TEOS. This was then stirred for another 2 hours. The temperature was maintained at 30 °C throughout the procedure. Then, the SSN was centrifuged and washed with ethanol thrice followed by drying in 50 °C oven.
The MSN were prepared by CTAB-templated, base-catalyzed condensation reaction of TEOS.42 First, 40 mg CTAB were dissolved in 96 ml water and then preheated to 80 °C while stirring. Then, 0.7 ml 2 M NaOH was added to the solution with stirring for 30 minutes at 80 °C. We then added 1.4 ml TEOS to the mixture and stirred it gently for 2 hours. The product was then filtered and rinsed with water and ethanol twice and finally calcined in a furnace at 600 °C for 5 hours.
The MCF were synthesized in aqueous hydrochloric acid using P123 as a template and mesitylene as a micelle expander according to the literature43,44 with minor modifications. First, 2.43 g P123 were added into 90 mL 1.6 M HCl in an Erlenmeyer flask at room temperature, followed by adding 400.8 mg CTAB, 24.4 mg ammonium fluoride, and 1.6 ml mesitylene. The mixture was stirred at room temperature for 2 hours and then added to 5.5 ml TEOS dropwise under vigorously stirring. The mixture was stirred for another 5 minutes after the addition of TEOS. The reaction was allowed to incubate at 38 °C overnight, and then the particles were centrifuged and rinsed with ethanol and water thrice. Particles were dried and calcined at 600 °C for 5 hours.
The four silica nanoparticles were conjugated with FITC to study the interaction between silica nanoparticles and cells. An amino–silane conjugate of the dye was first made by mixing 1 mg dye and 100 μl APTES in 1 ml ethanol with overnight rotation under room temperature. Then the mixture was divided evenly into 4 tubes with 4 mg SSN, MSN, MCF, and ELS, and rotated overnight. The products were washed thrice with ethanol, dried, and stored in dark for later use. The zeta potential of the nanoparticles could also be tuned by reflect with APTES for three hours.
Characterization
Transmission electron microscopy (TEM) images of silica nanoparticles were taken with a JEOL JEM-1200 EXII operating at 120 kV or JEM-2100F operating at 210 kV. Size distributions were analyzed by TEM images as well as nanoparticle tracking analysis (NTA, LM10, Malvern) with a 532 nm laser. TEM of nanoparticles in cells was done as described previously.18,45 The particle-concentrations for 1 mg ml−1 of SSN, MSC, MCF, and ELS solutions were also determined by NTA with camera level at 14 and detect threshold at 5. The four particle solutions were diluted 100- to 10000-fold prior to analysis and studied with NTA before and after 30 minutes of sonication in bath (KENDAL ultrasonic cleaner, Model 928, Power 60 W). The N2 adsorption–desorption isotherms at 77 K were measured on a Micrometitics Tristar 3000 system. Zeta potentials of the four types of nanoparticles were measured in 50% PBS via DLS (Zetasizer, Malvern). An inductively coupled plasma optical emission spectrometer (ICP-OES, Optima 3000DV, Perkin Elmer) was used to quantify the number of silica nanoparticles endocytosed by cells after sonication in 10 N NaOH. The colloidal stability was also measured by the settling time. Here, the absorbance of a cuvette containing the nanoparticles was measured over time. As the nanoparticles settled, the absorption decreased. We monitored the sample until the absorbance dropped to 50% of the original absorbance.
Cell culture, labelling, and cytotoxicity assay
We seeded human mesenchymal stem cells (hMSCs, Lonza, PT-2501) in a T75 flask at 5000 cells per cm2 in growth media (Lonza, PT-3001). These cells were labelled with 8 ml 250 μg ml−1 silica nanoparticles in fresh cell culture media without any transfection agents. The cells were then incubated under standard conditions for 7.5 hours. We washed the cells with sterile PBS to ensure all the free silica nanoparticles were removed. Cells were then detached by adding 2 ml of TrypLE Express (Life technologies). We scanned these labelled cells with ultrasound both in vitro and in vivo. hMSCs were labelled with silica nanoparticles at different concentrations (0 to 1000 μg ml−1) and studied the cell viability by using CellTiter 96® AQueous One Solution cell proliferation assay (MTS, Promega). After incubation with silica nanoparticles for 4 hours, 20 μl of the assay reagent was pipetted into the samples in 100 μl of growth media. This was allowed to incubate under standard conditions. After 4 hours, we transferred 80 μl of the sample solutions to a new plate and read the absorbance at 490 nm. Epifluorescence microscopy used an Evos microscope (Life Technologies) and Hoechst 33342 (NucBlue® Live ReadyProbes® Reagent, Thermo Fisher Scientific) for nuclear staining.In vitro phantom preparation and ultrasound imaging
All in vitro ultrasound imaging was performed in agarose phantoms. Agarose was added to water at 1% and boiled with stirring and then held at 60 °C until use. The agarose solution was then mixed with nanoparticle solutions or cell solutions at a ratio of 1 to 1. To avoid the aggregation of nanoparticles, the nanoparticle solutions were sonicated in water bath for 30 minutes prior to mixing with agarose, and then the mixture was vortexed. The cell solutions were vortexed both before and after mixing with agarose too. We quickly and carefully pipetted 250 μl mixture into the well of 96-well plate or agarose phantom. A blank control was also made with 0.5% agarose solution for all experiments. The solidified samples were covered with fresh 1% agarose for protection. Ultrasound imaging used a VEVO 770 or 2100 (VisualSonics) with transducers having a centre frequency of 40 MHz or 25 MHz. The imaging parameters were 100% power, 71 Hz frame rate for RMV-710B, and 34 Hz frame rate for RMV-706. The distance between the imaging transducer and phantom was maintained throughout the scan. At least five fields of views (FOVs) for each sample were collected.In vivo ultrasound imaging
All animal studies were performed in accordance with the Institutional Animal Care and Use Committee at the University of California, San Diego. Human mesenchymal stem cells were incubated together with 250 μg ml−1 ELS in growth media for four hours. Then, the free particles were removed by washing with PBS thrice. The labelled cells were detached, spun down, resuspended, and then counted by hemocytometer. The pellets with 1, 0.4, 0.2, 0.05 million cells were resuspended in 150 μl of 50:100 media:matrigel and injected subcutaneously into nude mice with a 28.5 gauge catheter. Images were obtained via 3-D mode with a 40 MHz transducer at 0.0076 mm per step.
Data analysis
Data means, standard deviations, and p values were calculated in Microsoft Excel 2016. Unless otherwise noted, all error bars represent the standard deviation. A two-tailed Student's t-test was used for significance and p values <0.05 were considered to be significant. TEM particle sizes of nanoparticles were analyzed with ImageJ 1.48v.46 For irregular particles, the Feret diameter was adopted. Ultrasound signals were also analyzed by ImageJ 1.48v. Specifically, B-mode ultrasound images were first exported from the ultrasound scanner as 8-bit images. Then, we drew a region of interest (ROI) inside the sample and managed it with ROI manager in the ImageJ. We measured the mean gray value of the same ROI in at least five different fields of views (FOVs) per sample by ImageJ 1.48v and then calculated mean and standard deviation of the mean grey values for each FOV, which ranged from 0 to 255. Detection limits were calculated at 3 standard deviations above the mean of the background signal.Results and discussion
We fabricated ELS via an emulsion soft-template method (Fig. 1A). The soft templates were an oil-in-water emulsion made of CTAB (green), DMHA (cyan), and decane (yellow) (step 1). The silica sources were then added to the emulsion and allowed to condense on the surface of the CTAB via an ammonia catalyst (step 2). After extracting the templates in a methanolic sodium chloride solution, the products were lyophilized (step 3). Electron microscopy images illustrate the shape and size similarities between the ELS nanoparticles and exosomes (Fig. 1B, C and F). When we studied the nanoparticles with TEM (Fig. 1) we found that nanoparticles have morphology remarkably reminiscent of exosomes—small extracellular vesicles (30–150 nm) secreted by cells. Like exosomes, ELS nanoparticles have a unique curvature and cup-like discoid shape. This shape offers a double scattering/reflection interface to increase echogenicity.30 | ||
| Fig. 1 Novel ELS nanoparticles around 140 nm were prepared via an emulsion template method. (A) Schematic of the ELS nanoparticle fabrication and morphology. TSPA (red) changed the overall stiffness of silica shell and render them more elastic1 to form the ELS nanoparticles. (B) TEM image of exosomes (black arrow; adapted with permission from ref. 8). (C) SEM image of typical ELS nanoparticles indicates the similarity of shape and size between ELS and exosomes. (D–G) TEM images of silica products made with (D) 0%, (E) 20%, (F) 40%, and (G) 100% TSPA (red). Hollow spheres were obtained when no TSPA was added (D); a silica gel was formed with only TSPA (G). (H) Zeta-potential and TEM size distribution of silica nanoparticles synthesized with different fractions of TSPA indicated that the 40% TSPA samples were the largest and had the most positive surface charge, and these samples were used for subsequent analysis and cell tracking. (I) Size distributions of ELS particles synthesized with 40% TSPA. NTA showed a larger mode size than TEM because NTA measures the hydrodynamic size of ELS and nanoparticles form aggregations in water. (J) N2 adsorption–desorption isotherms of ELS made with 40% TSPA indicated the existence of mesopores on the ELS nanoparticles. The BJH desorption pore size of the ELS is 5.4 nm (inset). | ||
During the fabrication, the silica source is critical to forming the exosome-like structure. Three common silica sources, TEOS, BTSE, and TSPA were investigated. We found TEOS is too stiff and brittle to produce ELS nanoparticles (data not shown). However, ELS nanoparticles were formed with the co-condensation of BTSE and TSPA. The BTSE offered a rigid framework for the particles, and TSPA allowed the nanoparticles to collapse by changing the overall stiffness—a hollow rigid sphere formed with only BTSE (Fig. 1D) and no particles formed with only TSPA (Fig. 1G). This role of TSPA is consistent with Lin et al. who found that TSPA could change the overall stiffness of silica shell and render them more elastic to form silica microbubbles.1 ELS nanoparticles prepared with different ratios of BTSE and TSPA are shown in Fig. 1E (4:1) and F (3:2) with yields of 37.2% and 35.7%, respectively.
While most silica nanoparticles are negatively charged due to the presence of hydroxyl groups, the ELS have positively charged amine groups embedded in the framework. This results in a positive nanoparticles surface charge to improve affinity with the negatively charge cell surface via a charge-dependent mechanism literature.47 The zeta-potential of final silica products (nanoparticles or gel) changed with the fraction of TSPA and was the highest at 40% TSPA (∼30 mV; Fig. 1H). The ELS made with 40% TSPA were also the largest (∼140 nm; Fig. 1H), and a preliminary study showed that larger nanoparticles have more echogenicity (Fig. S2†) Therefore, ELS nanoparticles made with 40% TSPA were characterized and selected for stem cell imaging. We tried to control the size of ELS using sonication output power, sonication time, and incubation time. However, only the ratio of TSPA had an impact on particle size and morphology (Fig. 1D–H).
Porous ELS with a mean diameter of ∼140 nm were prepared with 40% TSPA. The size of the ELS was analysed with both TEM and nanoparticle tracking analysis (NTA). Both size distributions are shown in Fig. 1I. The average TEM size is 138 nm for the ELS nanoparticles (N = 298). Additionally, a small peak near 500 nm in the NTA size distribution might indicate aggregation. The discrepancy between TEM and NTA size distributions is because that NTA measures the hydrodynamic size of the particles and nanoparticle aggregates. Unlike other biomedical applications,48,49 aggregation is actually preferred in the stem cell tracking because aggregations of nanoparticles inside the cells increase the ultrasound signal more than individual nanoparticles.18
More details about the aggregation behaviour of four types of silica nanoparticles are discussed in the ESI,† including the NTA (Fig. S1†) and settling studies (Fig. S9†) including the settling time in Fig. S9C.† We observed only slight settling (about 7%) for SSN in water even after 10 hours with only ∼20% settling for SSN in 25% FBS after 4 hours. In contrast, the MSN, MCF, and ELS settled faster in water than in 25% FBS. More specifically, they settled by 50% in water within 130.5, 15.5, and 72 minutes, and by 50% in 25% FBS within 142, 47, and 78.5 minutes, respectively. While the trend was similar in both water and 25% serum, the overall settling time of MCF and MSN was faster in water than serum suggesting that the protein corona does indeed help stabilize the nanoparticles.
The N2 adsorption–desorption isotherms of ELS indicated the existence of mesopores on the ELS nanoparticles (Fig. 1H). The BJH pore volume and desorption pore size of ELS were determined empirically to be around 1.79 cm3 g−1 and 5.4 nm (Fig. 1H inset); the BET surface area of ELS particles was 694 m2 g−1.
We then evaluated the in vitro echogenicity of the ELS nanoparticles by comparing it to that of three classical silica nanoparticles including the Stöber silica nanospheres (SSN),41 the MCM-41 mesoporous silica nanospheres (MSN),50 and the mesocellular foam silica nanoparticles (MCF)43 (Fig. 2). All nanoparticles were scanned in an agarose phantom at 40 MHz. The ELS exhibited the strongest echogenicity among four nanoparticles at identical mass concentrations (Fig. 2I). The ultrasound signal of ELS at 0.25, 0.5, and 1 mg ml−1 was 2.25-, 2.39-, and 1.76-fold of that of SSN; the signal was 1.72-, 1.85-, and 1.46-fold of the ultrasound signal of MSN; and it was 1.64-, 1.76-, and 1.62-fold of that of MCF. The theoretical LOD of ELS (3 standard deviations above background) was 0.77 μg ml−1; this value was 7.10, 3.56, and 2.61 μg ml−1 for SSN, MSN, and MCF, respectively. The higher echogenicity of ELS nanoparticles could allow a lower nanoparticle dose to produce the same ultrasound contrast and may therefore increase the biocompatibility. When the four nanoparticle types were compared at identical particle numbers, the MCF nanoparticles showed the highest signal (ELS were second highest; Fig. 2J). Importantly, however, the BJH pore volume of ELS (1.79 cm3 g−1) was higher than MCF (1.73 cm3 g−1). Thus, the echogenicity of ELS was likely stronger than MCF on a mass basis because that there were more ELS than MCF.
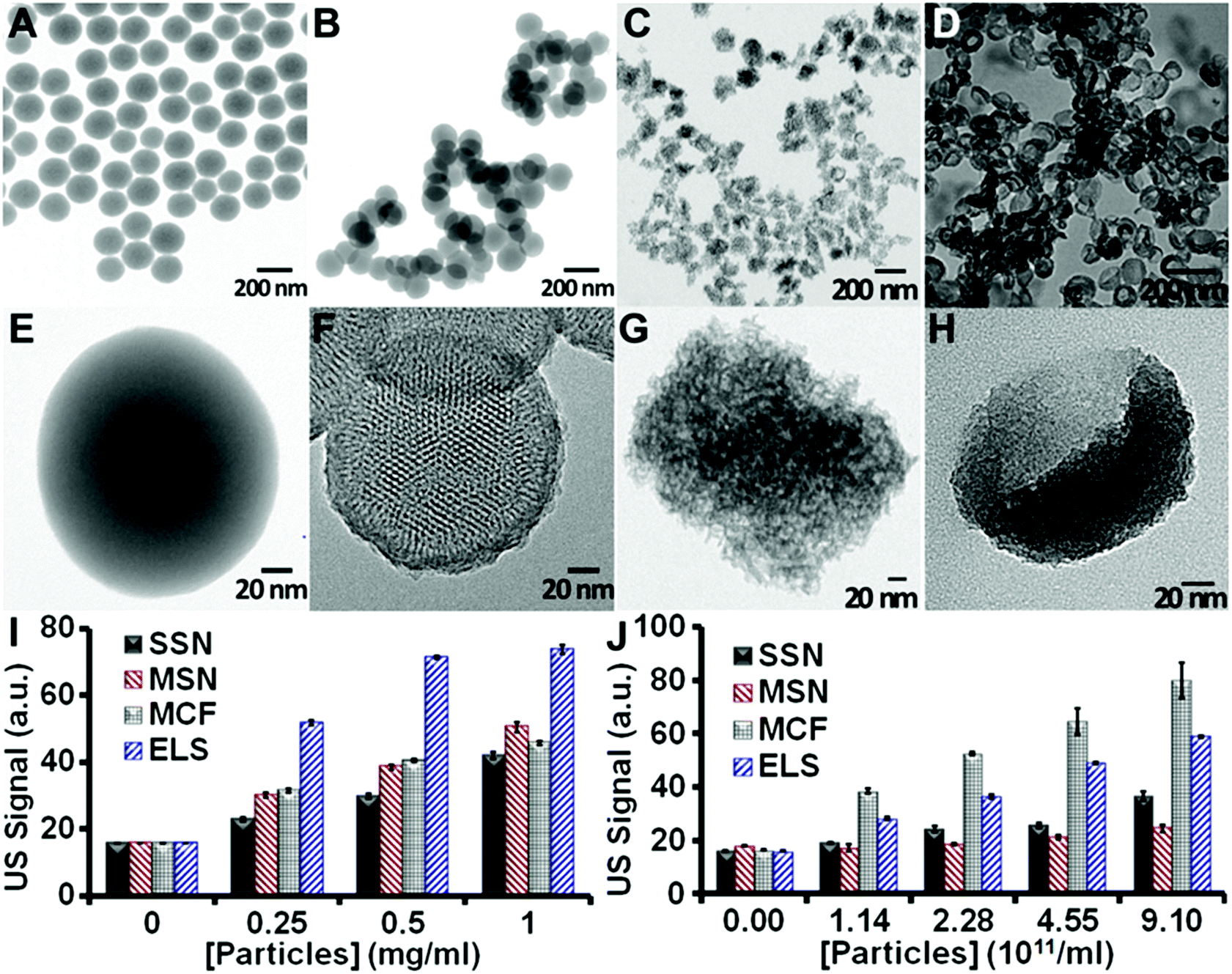 | ||
| Fig. 2 TEM images of SSN (A, E), MSN (B, F), MCF (C, G), and ELS (D, H). Both SSN and MSN are spherical, while MCF and ELS have flatter surfaces. HRTEM images (E–H) show that all particles except SSN are porous. The MSN have ordered and penetrating 2D channels, while the MCF and ELS have disordered and non-penetrating pores. (I, J) In vitro echogenicity of the four silica nanoparticles. All scans used 40 MHz with concentrations determined on (I) weight basis and (J) particle basis. Error bars represent the standard deviation of five replicate measurements. * represents p < 0.05, unpaired Student's t-test. | ||
To further understand the in vitro echogenicity of the ELS nanoparticles, we evaluated the effect of size, porosity/density, surface area, pore structure, and shape on the particle echogenicity. The morphologies of all nanoparticles are shown in Fig. 2A–H. The average TEM sizes of SSN, MSN, MCF, and ELS were 160 nm, 154 nm, 125 nm, and 138 nm respectively. The NTA size also indicated that the MCF was the smallest followed by ELS, MSN, and SSN (Fig. S1†). According to the literature51,52 as well as our studies on the effect of size on echogenicity, the ultrasound signal slightly increased as the particles became larger from 125 to 160 nm (Fig. S2†). Therefore, we conclude that the higher echogenicity of ELS and MCF compared to SSN and MSN is not due to the size. (More information of the effect of size on echogenicity is discussed in the ESI.†)
We also analyzed the echogenicity versus porosity/density, surface area, pore structure, and shape of silica nanoparticles by comparing the ultrasound signal between these four types of silica nanoparticles. As shown in Fig. 2E and F, the echogenicity of SSN was about 11% to 48% (the difference was concentration dependent) higher than that of MSN (Fig. 2J and S3†). In addition, the density of the MSN was about 50% (ESI†) lower than SSN because more of their volume is empty space. We conclude that the lower signal is because the MSNs have a lower impedance mismatch (product of density and velocity) with their surroundings and thus less echogenicity than SSN.
Fig. 2G and H show that both MCF and ELS have pores, but these are disordered and do not penetrate deep into the nanoparticle cores as in the MSN. This suggests that the ultrasound waves are unlikely to be transmitted, but are rather more likely to be backscattered. We conclude that ELS and MCF have more effective backscattering interfaces30 introduced by disordered and non-penetrating 3D pore structure and that this is responsible for their increased echogenicity. Both MCF and ELS have higher echogenicity than SSN (Fig. 2). The echogenicity of the MCF was 100%, 117%, 152%, and 118% higher than that of the SSN; the echogenicity of the ELS was 46%, 51%, 91%, and 61% higher than that of SSN. This indicated that the effective backscattering interfaces played a more important role in improving the echogenicity than impedance mismatch. More discussions on the mechanisms for improved echogenicity of ELS are provided in the ESI.†
Next, we characterized the labelling and cytotoxicity of the ELS nanoparticles with human mesenchymal stem cells (hMSCs). All silica nanoparticles were incubated separately with the hMSCs under the same conditions without any transfection treatments. We evaluated the impact of ELS on hMSC metabolism and viability by MTS, and EthD-III assays (Fig. 2D). The MTS assay indicated no decrease on cell metabolism between the cells with no ELS and those up to 1000 μg ml−1. For comparison, the other three classical silica nanoparticles were also biocompatible at a dosage up to 1 mg ml−1 (Fig. S7†) similar to the literature.14
TEM images show that ELS entered the cells and were contained in endosomes (Fig. 3A and S4†). High resolution TEM imaging clearly showed the unique curvature and discoid shape of ELS inside the hMSC (Fig. 3B); most ELS were aggregated in the cytosol. Other TEM images illustrated clusters on both the cell interior and periphery indicating endosomal uptake of ELS (Fig. S5†). Epifluorescence microscopy showed that the majority of ELS (green via fluorescein label) were bound to the hMSCs (Fig. 3C) similar to SSN, MSN, and MCF (Fig. S6†).
 | ||
| Fig. 3 hMSCs labeling and cytotoxicity of ELS. (A) TEM images of ELS-labeled hMSCs indicated aggregation of ELS inside the cells. ELS located both inside and on the cells. Arrows indicate the ELS nanoparticles, and Nuc indicates nucleus. (B) Higher magnification TEM image indicated that the ELS retained the unique curvature after entering the hMSCs. (C) Epifluorescence microscopy with hMSCs nucleus in blue and ELS nanoparticles fluorescently tagged in green. The majority of ELS were specifically bound to the hMSCs. (D) Cytotoxicity of ELS to hMSCs was determined by MTT and EthD-III assays. MTT assays indicate no decrease on ELS-labeled cell metabolism. “POS” indicated positive control of hMSCs with cytotoxic agent. (Data presented as mean with standard deviations; * represents p < 0.05, unpaired Student's t-test.) | ||
Next, we quantified the numbers of silica nanoparticles per vesicle inside hMSCs observed by TEM. The average ELS, SSN, MSN, and MCF numbers per vesicle were 285, 6, 4, and 30. For a more global analysis, we measured the Si content per cell via ICP-OES after the labelled cells were dissolved in concentrated base. We measured 1.11 ng Si per cell which equals to 4.14 million ELS particles per ELS-labelled hMSC. The Si content per cell for ELS-labelled hMSCs was 2.8-, 22.2-, and 1.5-times higher than that in SSN-, MSN-, and MCF-labelled hMSCs, respectively (Table S1†). Also, the particle number per cell in ELS-labelled hMSCs was 6.3-, 21.8-, and 2.02-times larger than that in SSN-, MSN-, and MCF-labelled hMSCs, which were 0.66, 0.19, and 2.05 million nanoparticles per cell, respectively (the calculations for nanoparticles per cell were based on the ICP-OES and NTA data; see ESI†). We rationalize that these differences were due to the differences in surface charge. Zeta-potentials of ELS, SSN, MSN, and MCF were +30.0 mV, −38.7 mV, −32.0 mV, and −23.1 mV, respectively. The unique positive charge on a silica particle seen here via the novel TSPA chemistry is what facilitates this increased cell labelling.
Importantly, the echogenicity of ELS-labelled hMSCs was increased versus unlabelled cells and cells labelled with other silica nanoparticles. After the hMSCs were incubated with nanoparticles for four hours at 250 μg ml−1, an agarose phantom with the same cell number was scanned with ultrasound at both 25 and 40 MHz. B-mode ultrasound image of ELS-labelled hMSCs was much brighter than unlabelled hMSCs with the same cell number with both frequencies (Fig. 4A). We then analysed the mean grey value of these ultrasound images using five different FOVs for each sample with ImageJ software.46 The ELS increased the echogenicity of hMSCs by 3.63-fold. While all four silica nanoparticles increased the echogenicity of hMSCs (Fig. 4B), the ELS increased it the most. Moreover, the echogenicity improvement was related to the nanoparticles endocytosed/bound by cells (Fig. 4C).
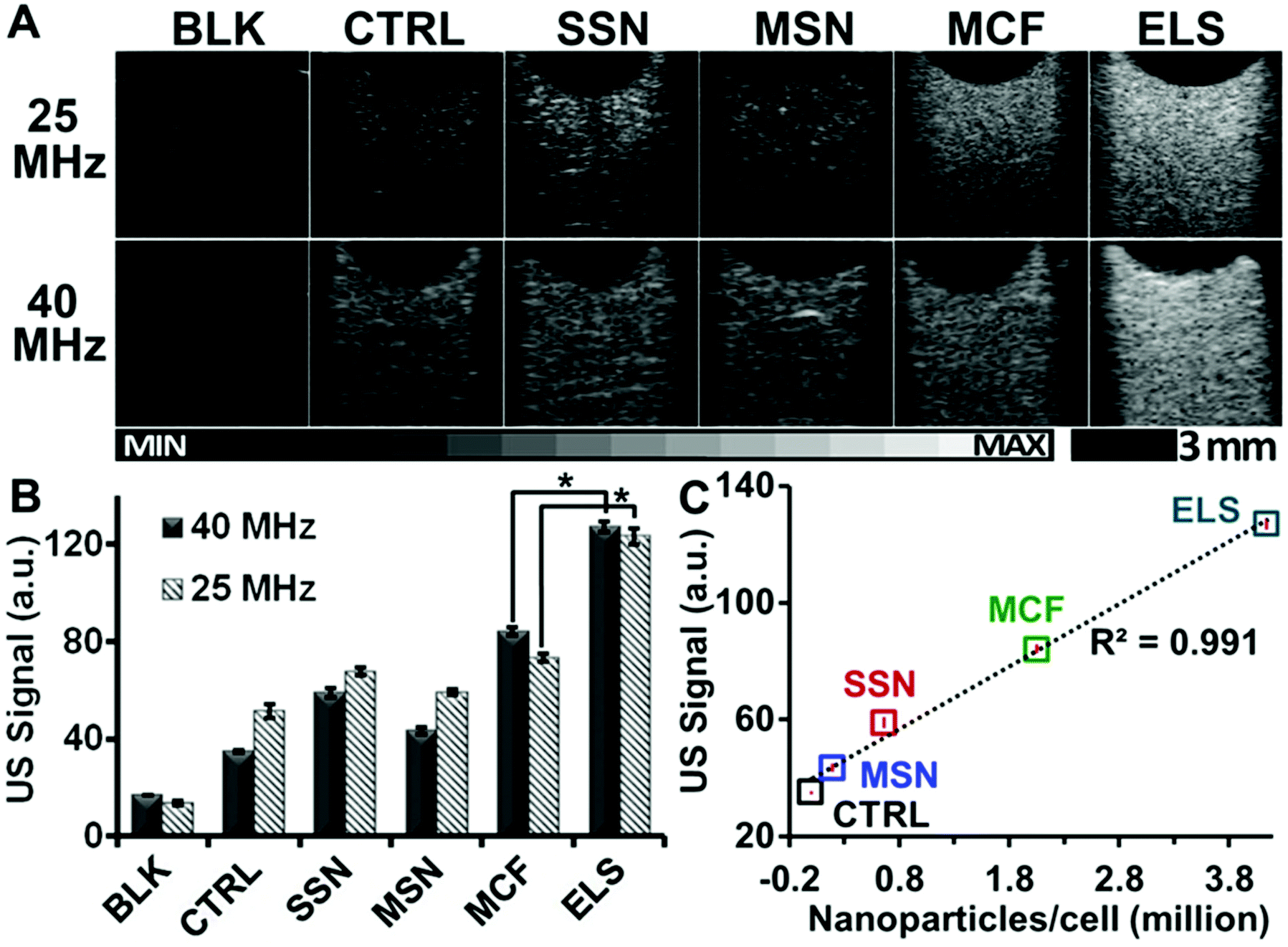 | ||
| Fig. 4 ELS significantly increased the echogenicity of hMSCs. Cells were incubated with different types of silica nanoparticles under identical conditions. The labeled cells with the same number were then scanned with ultrasound. Both ultrasound images (A) and ultrasound intensity analysis (B) show that ELS increase the echogenicity of hMSCs. The signal of ELS labeled cells was 3.63-fold of unlabeled cells. Other silica nanoparticles including SSN, MSN, and MCF also increased the echogenicity of hMSCs but not as strong as the ELS nanoparticles. The differences in ultrasound signals between ELS- and MCF-labeled cells were significant. (C) The ultrasound signals (40 MHz) of cells were positively related to the nanoparticles endocytosed by or bound to the hMSCs. The average nanoparticles per cell was determined by ICP-OES after the cells were incubated in different nanoparticles with the incubation conditions. | ||
The ELS were positively charged while the other nanoparticles were negative. Thus, we next modified the SSN, MSN, and MCF with the APTES to make them positive and use these modified nanoparticles for a control experiment (Table S2†). We incubated these modified nanoparticles with hMSCs and compared the hMSC ultrasound signal to the cells labelled with unmodified nanoparticles. APTES-modified MCF increased the ultrasound signal of hMSCs more than unmodified MCF, but MSN and SSN showed no difference. This result may be because of the difference in zeta potential after modification. We checked the zeta potential of the modified and unmodified SSN, MSN, and MCF. Previous work47 has shown that the uptake of silica nanoparticles by hMSCs can be regulated by surface charge but that the increase was only significant at high (+19 mV) surface charge. This reflects our data—the biggest difference between modified and unmodified NPs was seen with MCF, and this nanoparticle had the biggest change in zeta potential (Table S2†). Regardless, even with these controls for surface charge, the ELS still had the highest ultrasound signal (Fig. S8†).
The ELS also increased the in vivo echogenicity and ultrasound sensitivity of hMSCs. The ELS-labelled hMSCs were subcutaneously injected with a matrigel carrier into nude mice. PBS and unlabelled cells were also injected as controls. In vivo ultrasound images demonstrated significant increase of echogenicity of transplanted ELS-labelled stem cells compared to unlabelled cells (Fig. 5A–D). ELS increased the in vivo echogenicity of hMSCs 3.3-fold times with 200000 cells (Fig. 4E). Therefore, the ELS nanoparticles increased the sensitivity of stem cells via ultrasound. The theoretical limit of detection (LOD) of ELS labelled hMSCs was 475 cells—nearly 50-fold higher than the LOD of unlabelled cells.
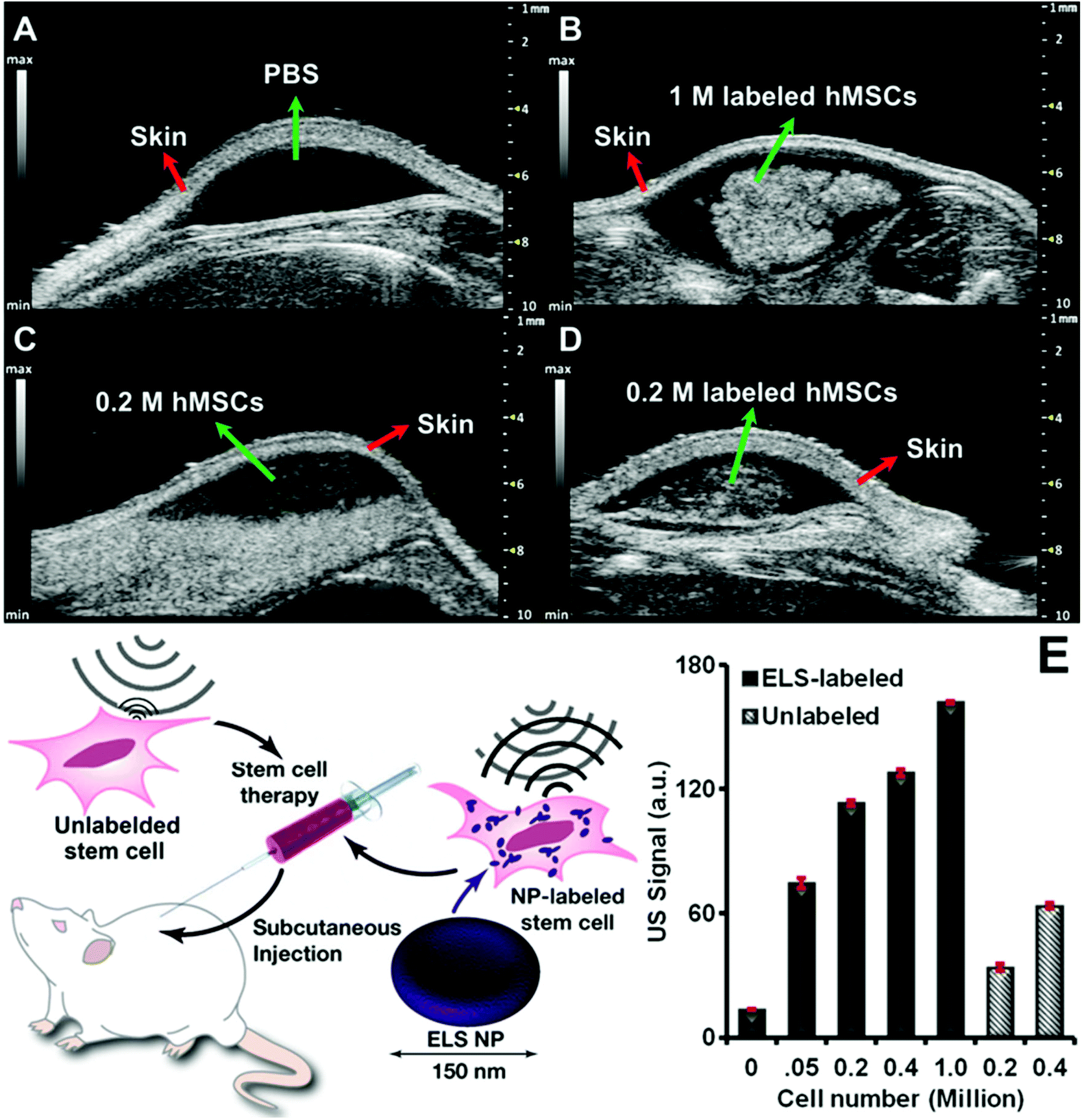 | ||
| Fig. 5
In vivo ultrasound images and quantification of cells echogenicity. In vivo ultrasound images of (A) PBS, (B) 1 million ELS-labeled hMSCs, (C) 0.2 million unlabeled hMSCs, and (D) 0.2 million ELS-labeled hMSCs. (E) Quantification of In vivo echogenicity of hMSCs shows ultrasound signal of both unlabeled and ELS-labeled cells were cell number dependent. Bars stands for standard deviation. The LOD of ELS labeled and unlabeled hMSCs was 475 and 21364 cells respectively. | ||
Conclusions
In summary, ELS silica nanoparticles increased both the in vitro and in vivo echogenicity of stem cells via strengthened particle echogenicity as well as improved cell labelling efficiency. The incorporation of TSPA—an elastic silica source containing an amine group—to the ELS nanoparticles not only provide the exosome-like shape but also increases the zeta-potential, which increases the cell uptake. The unique curvature and discoid shape provides more effective ultrasound backscattering interfaces to enhance the ultrasound contrast of individual ELS nanoparticle. This novel ELS nanoparticle increases the cell contrast and enables real-time cell tracking/imaging by affordable ultrasound. Noticeably, for stem cell treatment, injected cells tend to die because of hostile environment.53,54 As mesoporous silica nanoparticles have been demonstrated to load and control release various drugs,42,55,56 the ELS nanoparticles could also be loaded with prosurvival reagents57–59 to increase the cell viability. In addition, these ELS particles are easily chemically modified. While we labelled them with fluorescein here, they could also be coated with a radionuclide or a gadolinium ion for multimodal imaging.Acknowledgements
F. Chen would like to thank T. Hu on TEM imaging, J. Naughton, L. Lisowski, and C. Deighan for help on NTA, C. Barback and J. Corbeil on ultrasound imaging, P. Castillo and C. MacIsaac on ICP-OES. Jesse V. Jokerst acknowledges funding from the NIH R00 HL117048 and DP2 HL137187 and infrastructure from S10 OD021821. The authors also thank the American Cancer Society Institutional Research [grant number 14-250-42] provided through the Moores Cancer Center, University of California, San Diego. M. Ma acknowledges the support of the National Natural Science Foundation of China [grant number 51402329].Notes and references
- P. L. Lin, R. J. Eckersley and E. A. Hall, Adv. Mater., 2009, 21, 3949–3952 CrossRef CAS.
- S. Suarez, A. Almutairi and K. L. Christman, Biomater. Sci., 2015, 3, 564–580 RSC.
- W. J. M. Mulder and Z. A. Fayad, Arterioscler., Thromb., Vasc. Biol., 2008, 28, 801–802 CrossRef CAS PubMed.
- J. H. Tai, P. Foster, A. Rosales, B. Feng, C. Hasilo, V. Martinez, S. Ramadan, J. Snir, C. W. J. Melling, S. Dhanvantari, B. Rutt and D. J. G. White, Diabetes, 2006, 55, 2931–2938 CrossRef CAS PubMed.
- Z. Ma, T. M. Lim and L.-Y. Lim, Int. J. Pharm., 2005, 293, 271–280 CrossRef CAS PubMed.
- R. J. Morris, Y. Liu, L. Marles, Z. Yang, C. Trempus, S. Li, J. S. Lin, J. A. Sawicki and G. Cotsarelis, Nat. Biotechnol., 2004, 22, 411–417 CrossRef CAS PubMed.
- W.-B. Shen, C. Plachez, O. Tsymbalyuk, N. Tsymbalyuk, S. Xu, A. Smith, S. Michel, D. Yarnell, R. Mullins, R. Gulapalli, A. Puche, J. M. Simard, P. S. Fishman and P. Yarowsky, Cell Transplant., 2016, 25, 1085–1099 Search PubMed.
- C. Théry, S. Amigorena, G. Raposo and A. Clayton, in Current Protocols in Cell Biology, John Wiley & Sons, Inc., 2001, DOI:10.1002/0471143030.cb0322s30.
- S. K. Sanganalmath and R. Bolli, Circ. Res., 2013, 113, 810–834 CrossRef CAS PubMed.
- M. Rodriguez-Porcel, O. Gheysens, I. Y. Chen, J. C. Wu and S. S. Gambhir, Mol. Ther., 2005, 12, 1142–1147 CrossRef CAS PubMed.
- G. Vunjak-Novakovic, K. O. Lui, N. Tandon and K. R. Chien, Annu. Rev. Biomed. Eng., 2011, 13, 245–267 CrossRef CAS PubMed.
- D. L. Kraitchman, A. W. Heldman, E. Atalar, L. C. Amado, B. J. Martin, M. F. Pittenger, J. M. Hare and J. W. M. Bulte, Circulation, 2003, 107, 2290–2293 CrossRef PubMed.
- M. Rodriguez-Porcel, O. Gheysens, I. Y. Chen, J. C. Wu and S. S. Gambhir, Mol. Ther., 2005, 12, 1142–1147 CrossRef CAS PubMed.
- P. J. Kempen, S. Greasley, K. A. Parker, J. L. Campbell, H.-Y. Chang, J. R. Jones, R. Sinclair, S. S. Gambhir and J. V. Jokerst, Theranostics, 2015, 5, 631 CrossRef CAS PubMed.
- C. Bara, A. Ghodsizad, M. Niehaus, M. Makoui, C. Piechaczek, U. Martin, G. Warnecke, M. Karck, E. Gams, H. M. Klein, A. Haverich and A. Ruhparwar, J. Am. Soc. Echocardiogr., 2006, 19, 563–568 CrossRef PubMed.
- A. V. Naumova, M. Modo, A. Moore, C. E. Murry and J. A. Frank, Nat. Biotechnol., 2014, 32, 804–818 CrossRef CAS PubMed.
- W. Cui, S. Tavri, M. J. Benchimol, M. Itani, E. S. Olson, H. Zhang, M. Decyk, R. G. Ramirez, C. V. Barback, Y. Kono and R. F. Mattrey, Biomaterials, 2013, 34, 4926–4935 CrossRef CAS PubMed.
- J. V. Jokerst, C. Khademi and S. S. Gambhir, Sci. Transl. Med., 2013, 5, 177ra135 Search PubMed.
- R. T. Castaneda, A. Khurana, R. Khan and H. E. Daldrup-Link, J. Visualized Exp., 2011, 57, e3482–e3482 Search PubMed.
- J. I. Park, D. Jagadeesan, R. Williams, W. Oakden, S. Chung, G. J. Stanisz and E. Kumacheva, ACS Nano, 2010, 4, 6579–6586 CrossRef CAS PubMed.
- S. Ommen, R. Nishimura, C. Appleton, F. Miller, J. Oh, M. Redfield and A. Tajik, Circulation, 2000, 102, 1788–1794 CrossRef CAS PubMed.
- L. Sandrin, B. Fourquet, J.-M. Hasquenoph, S. Yon, C. Fournier, F. Mal, C. Christidis, M. Ziol, B. Poulet and F. Kazemi, Ultrasound Med. Biol., 2003, 29, 1705–1713 CrossRef PubMed.
- G. Ku, B. D. Fornage, X. Jin, M. Xu, K. K. Hunt and L. V. Wang, Technol. Cancer Res. Treat., 2005, 4, 559–565 CrossRef PubMed.
- A. L. Klibanov, Invest. Radiol., 2006, 41, 354–362 CrossRef PubMed.
- R. F. Mattrey, G. Strich, R. E. Shelton, B. B. Gosink, G. R. Leopold, T. Lee and J. Forsythe, Radiology, 1987, 163, 339–343 CrossRef CAS PubMed.
- K. E. Wilson, T. Y. Wang and J. K. Willmann, J. Nucl. Med., 2013, 54, 1851–1854 CrossRef PubMed.
- F. Foroutan, J. V. Jokerst, S. S. Gambhir, O. Vermesh, H.-W. Kim and J. C. Knowles, ACS Nano, 2015, 9, 1868–1877 CrossRef CAS PubMed.
- H. Hu, H. Zhou, J. Du, Z. Wang, L. An, H. Yang, F. Li, H. Wu and S. Yang, J. Mater. Chem., 2011, 21, 6576–6583 RSC.
- A. Milgroom, M. Intrator, K. Madhavan, L. Mazzaro, R. Shandas, B. Liu and D. Park, Colloids Surf., B, 2014, 116, 652–657 CrossRef CAS PubMed.
- K. Zhang, H. Chen, X. Guo, D. Zhang, Y. Zheng, H. Zheng and J. Shi, Sci. Rep., 2015, 5, 8766 CrossRef CAS PubMed.
- M. Ma, H. Chen and J. Shi, Sci. Bull., 2015, 60, 1170–1183 CrossRef CAS.
- M. Benezra, O. Penate-Medina, P. B. Zanzonico, D. Schaer, H. Ow, A. Burns, E. DeStanchina, V. Longo, E. Herz, S. Iyer, J. Wolchok, S. M. Larson, U. Wiesner and M. S. Bradbury, J. Clin. Invest., 2011, 121, 2768–2780 CAS.
- H. Y. Zhang, D. R. Dunphy, X. M. Jiang, H. Meng, B. B. Sun, D. Tarn, M. Xue, X. Wang, S. J. Lin, Z. X. Ji, R. B. Li, F. L. Garcia, J. Yang, M. L. Kirk, T. Xia, J. I. Zink, A. Nel and C. J. Brinker, J. Am. Chem. Soc., 2012, 134, 15790–15804 CrossRef CAS PubMed.
- A. Liberman, N. Mendez, W. C. Trogler and A. C. Kummel, Surf. Sci. Rep., 2014, 69, 132–158 CrossRef CAS PubMed.
- M. Colilla, B. Gonzalez and M. Vallet-Regi, Biomater. Sci., 2013, 1, 114–134 RSC.
- M. D. Yilmaz, M. Xue, M. W. Ambrogio, O. Buyukcakir, Y. Wu, M. Frasconi, X. Chen, M. S. Nassar, J. F. Stoddart and J. I. Zink, Nanoscale, 2014, 7, 1067–1072 RSC.
- J. G. Croissant, X. Cattoen, M. Wong Chi Man, J.-O. Durand and N. M. Khashab, Nanoscale, 2015, 7, 20318–20334 RSC.
- K. Zhang, L.-L. Xu, J.-G. Jiang, N. Calin, K.-F. Lam, S.-J. Zhang, H.-H. Wu, G.-D. Wu, B. Albela, L. Bonneviot and P. Wu, J. Am. Chem. Soc., 2013, 135, 2427–2430 CrossRef CAS PubMed.
- M. Kruk, Acc. Chem. Res., 2012, 45, 1678–1687 CrossRef CAS PubMed.
- A. Liberman, H. P. Martinez, C. N. Ta, C. V. Barback, R. F. Mattrey, Y. Kono, S. L. Blair, W. C. Trogler, A. C. Kummel and Z. Wu, Biomaterials, 2012, 33, 5124–5129 CrossRef CAS PubMed.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- F. Chen and Y. Zhu, Microporous Mesoporous Mater., 2012, 150, 83–89 CrossRef CAS.
- P. Schmidt-Winkel, W. W. Lukens, D. Zhao, P. Yang, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1999, 121, 254–255 CrossRef CAS.
- Y. Zhang, T. Jiang, Q. Zhang and S. Wang, Eur. J. Pharm. Biopharm., 2010, 76, 17–23 CrossRef CAS PubMed.
- P. J. Kempen, M. F. Kircher, A. de la Zerda, C. L. Zavaleta, J. V. Jokerst, I. K. Mellinghoff, S. S. Gambhir and R. Sinclair, Micron, 2015, 68, 70–76 CrossRef CAS PubMed.
- M. D. Abràmoff, P. J. Magalhães and S. J. Ram, Biophotonics Int., 2004, 11, 36–42 Search PubMed.
- T.-H. Chung, S.-H. Wu, M. Yao, C.-W. Lu, Y.-S. Lin, Y. Hung, C.-Y. Mou, Y.-C. Chen and D.-M. Huang, Biomaterials, 2007, 28, 2959–2966 CrossRef CAS PubMed.
- Y.-S. Lin, N. Abadeer, K. R. Hurley and C. L. Haynes, J. Am. Chem. Soc., 2011, 133, 20444–20457 CrossRef CAS PubMed.
- P. N. Durfee, Y.-S. Lin, D. R. Dunphy, A. J. Muñiz, K. S. Butler, K. R. Humphrey, A. J. Lokke, J. O. Agola, S. S. Chou, I. M. Chen, W. Wharton, J. L. Townson, C. L. Willman and C. J. Brinker, ACS Nano, 2016, 10, 8325–8345 CrossRef CAS PubMed.
- M. E. L. C. T. Kresge, W. J. Roth, J. C. Vartuli and J. S. Beck, Nat. Mater., 1992, 359, 710–712 CrossRef.
- S. Casciaro, F. Conversano, A. Ragusa, M. A. Malvindi, R. Franchini, A. Greco, T. Pellegrino and G. Gigli, Invest. Radiol., 2010, 45, 715–724 CrossRef PubMed.
- F. Chiriacò, F. Conversano, G. Soloperto, E. Casciaro, A. Ragusa, E. Sbenaglia, L. Dipaola and S. Casciaro, J. Nanopart. Res., 2013, 15, 1–13 CrossRef.
- C. W. Don and C. E. Murry, J. Cell. Mol. Med., 2013, 17, 1355–1362 CrossRef CAS PubMed.
- I. Kutschka, T. Kofidis, I. Y. Chen, G. von Degenfeld, M. Zwierzchoniewska, G. Hoyt, T. Arai, D. R. Lebl, S. L. Hendry, A. Y. Sheikh, D. T. Cooke, A. Connolly, H. M. Blau, S. S. Gambhir and R. C. Robbins, Circulation, 2006, 114, I-174–I-180 Search PubMed.
- S. Niedermayer, V. Weiss, A. Herrmann, A. Schmidt, S. Datz, K. Müller, E. Wagner, T. Bein and C. Bräuchle, Nanoscale, 2015, 7, 7953–7964 RSC.
- L. Palanikumar, E. S. Choi, J. Y. Cheon, S. H. Joo and J. H. Ryu, Adv. Funct. Mater., 2015, 25, 957–965 CrossRef CAS.
- M. A. Laflamme, K. Y. Chen, A. V. Naumova, V. Muskheli, J. A. Fugate, S. K. Dupras, H. Reinecke, C. Xu, M. Hassanipour and S. Police, Nat. Biotechnol., 2007, 25, 1015–1024 CrossRef CAS PubMed.
- M. E. Davis, P. C. Hsieh, T. Takahashi, Q. Song, S. Zhang, R. D. Kamm, A. J. Grodzinsky, P. Anversa and R. T. Lee, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8155–8160 CrossRef CAS PubMed.
- H. K. Haider, S. Jiang, N. M. Idris and M. Ashraf, Circ. Res., 2008, 103, 1300–1308 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterizations of silica nanoparticles, discussions on the effect of particles morphologies on the echogenicity, and calculations for the density of spherical silica nanoparticles as well as number of particles per cell. See DOI: 10.1039/c6nr08177k |
| This journal is © The Royal Society of Chemistry 2017 |