
Cu-CDots nanocorals as electrocatalyst for highly efficient CO2 reduction to formate†
Sijie
Guo
,
Siqi
Zhao
,
Jin
Gao
,
Cheng
Zhu
,
Xiuqin
Wu
,
Yijun
Fu
,
Hui
Huang
,
Yang
Liu
* and
Zhenhui
Kang
*
Jiangsu Key Laboratory for Carbon-Based Functional Materials & Devices, Institute of Functional Nano & Soft Materials (FUNSOM), Soochow University, 199 Ren'ai Road, Suzhou, 215123, Jiangsu, PR China. E-mail: zhkang@suda.edu.cn
First published on 23rd November 2016
Abstract
Electrochemical reduction of CO2 is a key component of many prospective artificial technologies for renewable carbon-containing fuels, but it still suffers from the high overpotentials required to drive the process, low selectivity for diversiform products and the high cost of the catalyst. Here, we report that Cu-CDots nanocorals is a highly efficient, low-cost and stable electrocatalyst for CO2 reduction in aqueous solution. The major product of CO2 reduction on the Cu-CDots nanocorals is HCOOH with an inconceivable low overpotential of 0.13 V and a high Faraday efficiency of 79% at a moderate potential of −0.7 V vs. RHE. In the present system, CDots can increase the adsorption capacity of CO2 molecules and H+, which play important roles in CO2 reduction. The high selectivity of HCOOH for CO2 reduction may be ascribed to that CDots can greatly diminish the HCOOH desorption energy and improve the catalytic selectivity for HCOOH. Furthermore, Cu-CDots nanocorals exhibit a long-term stability during 5 h-electrolysis.
Introduction
Electroreduction of CO2 into carbon-containing fuels is an ideal artificial technology for replacing fossil feedstock and handling the excess greenhouse gas and their adverse effects on climate and environmental consequences.1–4 The critical bottlenecks for the current CO2 reduction reaction are the impractically high overpotentials required to drive the process and the low selectivity for diversiform products.5 A promising catalyst for efficient CO2 conversion would need to exhibit a high energy efficiency, a high current density as well as a high selectivity. In this field, there is an urgent need to seek a catalyst for the CO2 reduction reaction with high Faraday Efficiencies (FEs), high selectivity and low cost.Metal nanoparticles are well-known functional materials and widely investigated for their potential application in the catalysis field.6–10 Among them, silver (Ag), gold (Au) and palladium (Pd) nanoparticles (NPs) are known as promising electrocatalysts for CO2 reduction.11–14 For example, Ag and Au can selectively convert CO2 to CO, which is feedstock for the Fischer–Tropsch process to produce synthetic fuels and industrial chemicals.15 However, the high cost of precious metal catalysts is the main reason for limiting precious metal applications in industrial production. Much attention has been focused on Cu, a nonprecious metallic catalyst, which converts CO2 to hydrocarbon,16–21 but a Cu catalyst also faces a low selectivity for diversiform products and a high over potential.
Carbon dots, with easy large scale production, high chemical stability and excellent photo/electro properties, have been accepted as promising candidates for the design of photo- and electro-catalysts.22–25 Besides, the surfaces of carbon dots can be easily functionalized with different groups such as hydroxyl (–OH), carboxyl (–COOH) and amine (–NH2) groups, which could be used as functional components in catalyst design. Also, as previous work reported,26 the amine group can increase the adsorption ability of CO2, which will play an important role in CO2 reduction. Based on all the above, the combination of Cu nanomaterials with amine group functionalized carbon dots (here, the amine group functionalized carbon dots are defined as CDots for short) is desirable to fabricate promising efficient electrocatalysts for the CO2 reduction reaction.
Herein, we first synthesized Cu-CDots nanocorals which are a highly efficient, low-cost and stable electrocatalyst for CO2 reduction in aqueous solutions. The major product of CO2 reduction on the Cu-CDots nanocorals is HCOOH with inconceivable low overpotential of 0.13 V. Besides, the total FEs of CO2 reduction products (HCOOH and CH3OH) reach up to 79% at a moderate potential of −0.7 V vs. reversible hydrogen electrode (RHE, all potentials reported here are with respect to this reference). Through comparing the electrochemical characterization of Cu-CDots nanocorals with that of Cu nanocorals, we gained insight that CDots play an important role in promoting the electrocatalytic performance of Cu-CDots nanocorals. Further, Cu-CDots nanocorals exhibit a long-term stability that a constant current density of ∼4.2 mA cm−2 was obtained on the Cu-CDots nanocoral electrocatalyst during 5 h-electrolysis. The high selectivity of HCOOH for CO2 reduction may be ascribed to the surface modification with CDots, which can greatly diminish the HCOOH desorption energy and improve the catalytic selectivity for HCOOH.
Results and discussion
A transition electron microscopy (TEM) image of CDots is shown in Fig. 1a, indicating that CDots are well dispersed with diameters in the same range of 3–7 nm (size distributions shown in Fig. S1†). A high resolution TEM (HRTEM) image of CDots is shown in the inset of Fig. 1a. Here, CDots exhibit the 0.21 nm which is consistent with (100) interplanar spacing. The 0.34 nm spacing, corresponding to the graphitic (002) basal planes, is absent that may be ascribed to many defects of CDots. As shown in Fig. S2,† the Fourier transform infrared (FT-IR) spectrum of CDots displays the stretching vibrations of O–H and N–H at 3500 cm−1 and 3250 cm−1, respectively. The peak at around 1400 cm−1 and 1230 cm−1 denotes the typical C–H and C–O–C stretching vibrations, respectively, while the band at 1636 cm−1 represents the stretching vibrations of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O/CC.
O/CC.
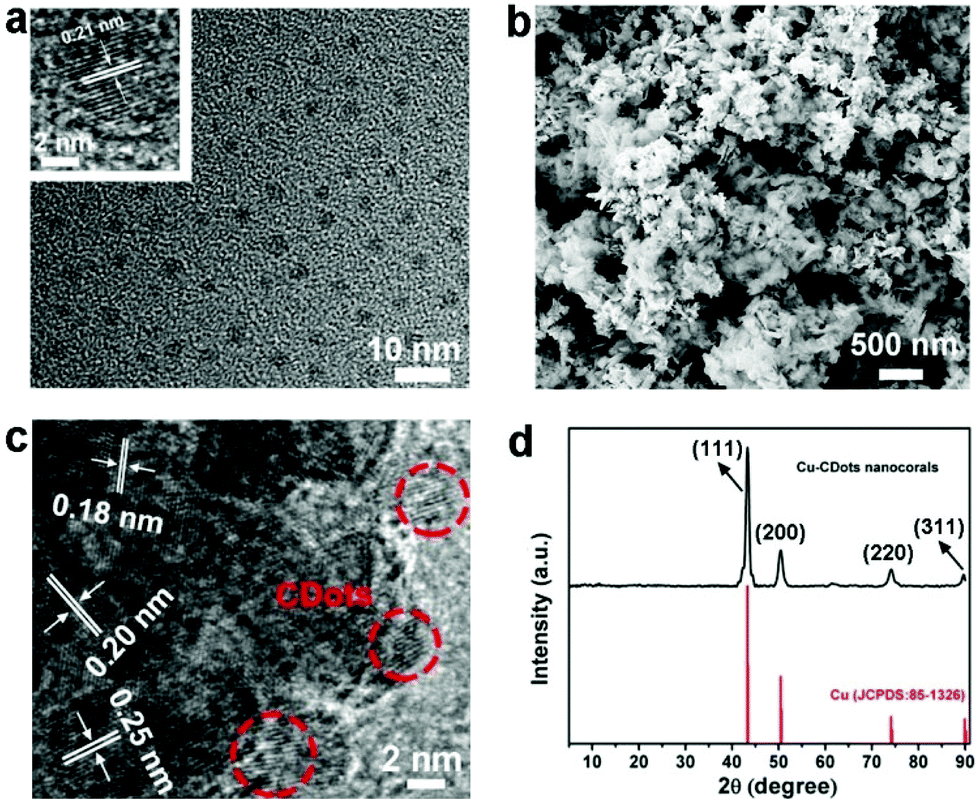 | ||
| Fig. 1 (a) The TEM image of CDots and the HRTEM image of a grain of CDots (inset); (b) the SEM image of the Cu-CDots nanocorals; (c) HRTEM image of Cu-CDots nanocorals. CDots are surrounded by red circles; (d) large-angle XRD patterns of Cu-CDots nanocorals (the black trace). | ||
Briefly, Cu-CDots nanocorals were fabricated by three steps. Firstly, Cu2O NPs (see Fig. S3†) were synthesized and then employed as templates to prepare Cu2O-CDots nanocorals (as shown in Fig. S4†). Finally, Cu2O-CDots nanocorals were reduced by applying a cathodic electrochemical potential to produce Cu-CDots nanocorals (see Fig. S5†). As shown in Fig. 1b, the morphology of Cu-CDots nanocorals appears as porous, coral-like structures. N2 adsorption–desorption isotherms (Fig. S6†) also evidence that the Brunauer–Emmett–Teller (BET) specific surface areas reach up to 91.7 m2 g−1. The HRTEM image of Cu-CDots nanocorals (see Fig. 1c) signifies that Cu nanocrystals combine with CDots to form Cu-CDots nanocorals. It also shows that the dominant d-spacings of Cu nanocorals are 0.20 and 0.18 nm, corresponding to lattice planes (111) and (200),27 respectively, and the d-spacing of CDots is 0.21 nm, consistent with lattice planes (100). The HRTEM image displays a lattice spacing of 0.25 nm on the Cu-CDots nanocorals surface, which is consistent with the lattice planes (111) of Cu2O.28 The Cu2O may be attributed to the oxidation of Cu-CDots nanocorals during the process of post-sample treatments. The FT-IR spectrum of Cu-CDots nanocorals shows that the main functional groups of CDots are reserved. To investigate the crystal structure of the Cu-CDots, XRD measurements were performed. As shown in Fig. 1d, the XRD spectrum of Cu-CDots nanocorals (black trace) only shows the characteristic peak of Cu (JCPDS card no. 85-1326). The intense and sharp peaks indicate the high crystallinity of Cu nanocrystals. The characteristic peak for CDots at 43° is not visible, probably because it has a relatively low diffraction intensity in composites and its diffractions response is covered by the Cu strong diffraction.
Cu-CDots nanocorals were further characterized by X-ray photoelectron spectroscopy (XPS) and the XPS results indicate that the main elements of Cu-CDots nanocorals are C, N, O and Cu (as shown in Fig. 2a). The low O content should mainly come from surface oxygen-containing groups (–OH and –COOH), which is further confirmed by the high-resolution XPS spectrum of C 1s (as shown in Fig. 2b). Fig. 2c shows that the N 1s peak was fitted by using three synthetic peaks positioned at BE = 397.8, 399.6 and 400.0 eV. For the N 1s peak, the first contribution is assigned to N–Cu and the second contribution is assigned to N–C,29 while the last one is related to the stable state of N–H. The high resolution XPS spectrum of Cu LMM indicates the presence of metallic Cu and trace Cu+, both of which could be distinguished from Cu LMM.30 Therefore, these results indicate the complete reduction of the Cu2O layer and the formation of Cu-CDots nanocorals. The trace Cu2O may be attributed to the oxidation of Cu-CDots nanocorals during the process of post-sample treatments.
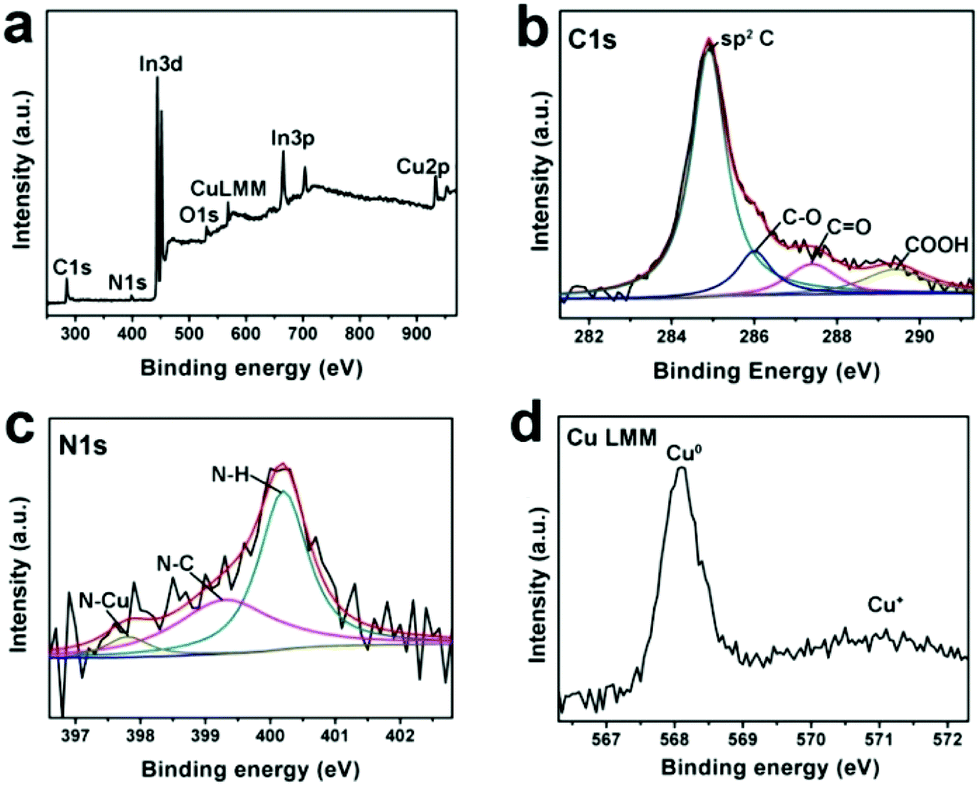 | ||
| Fig. 2 The full XPS spectrum of Cu-CDots nanocorals (a) and the high-resolution XPS spectrum of C 1s (b), N 1s (c) and Cu LMM (d) peak in Cu-CDots nanocorals. | ||
In typical experiments, the electrocatalytic activity of Cu-CDots nanocorals towards the reduction of CO2 was evaluated in a standard three-electrode configuration employing an electrolyte of high purity aqueous 0.5 M KHCO3. As shown in Fig. 3a, linear sweep voltammetry curve (LSV) of Cu-CDots nanocorals acquired in N2-saturated 0.5 M KHCO3 (pH = 8.5) shows an onset potential of −0.50 V, which can be ascribed to the hydrogen evolution reaction (HER). HER has a high overpotential (∼0.5 V). The electrolyte was purged with CO2 for at least 30 min (CO2-saturated 0.5 M KHCO3, pH = 7.2) and the electrocatalytic CO2 reduction was measured. As shown in Fig. 3a, it can be observed that the CO2 electrocatalytic reduction reaction initiates at a potential of −0.38 V, confirmed by 1H nuclear magnetic resonance (NMR, Bruker AVANCEAV III 400). That indicates a low overpotential (0.13 V) for CO2 reduced to HCOOH. In addition, the gas-phase products were measured using gas chromatography (GC) and no gas-phase products were tested except for the by-product H2.
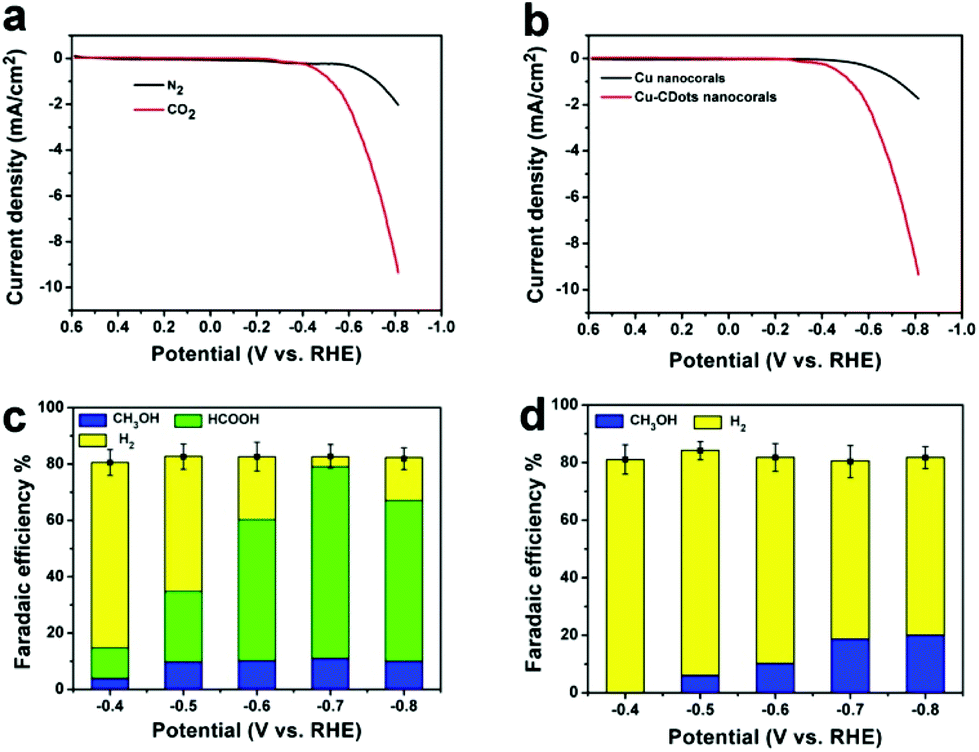 | ||
| Fig. 3 (a) LSVs for Cu-CDots nanocorals in N2- (black trace) and CO2-saturated (red trace) 0.5 M KHCO3 electrolyte, 10 mV s−1; (b) LSVs for Cu nanocorals (black trace) and Cu-CDots nanocorals (red trace) in CO2-saturated 0.5 M KHCO3 electrolyte, 10 mV s−1; (c) Faradaic efficiencies vs. potential of CH2OH, HCOOH and by-product H2 for Cu-CDots nanocorals; (d) Faradaic efficiencies vs. potential of CH3OH and by-product H2 for Cu nanocorals. | ||
To check the effect of CDots on the electrocatalytic performance of Cu-CDots nanocorals for CO2 reduction, we investigated the electrocatalytic performances of Cu nanocorals (black trace; the morphology of Cu nanocorals is shown in Fig. S7†) and Cu-CDots nanocorals (red trace) for CO2 reduction. As shown in Fig. 3b, electrocatalytic activity of Cu-CDots nanocorals is much better than that of Cu nanocorals. It can be obviously seen that the onset potential of Cu-CDots nanocorals for CO2 reduction is about −0.38 V which is much lower compared with that of Cu nanocorals. For CO2 reduction, a more positive onset potential and higher current density can be achieved by Cu-CDots nanocorals with the coral-like structures that can improve the specific surface area and active sites. The electrocatalytic performance of CDots was also tested, as shown in Fig. S9.† Pure CDots show poor electrocatalytic active for CO2 reduction. This demonstrates that CDots can significantly increase the electrocatalytic performance of Cu-CDots nanocorals.
To further compare the electrocatalytic performance of Cu-CDots nanocorals with that of Cu nanocorals for CO2 reduction, electrocatalytic CO2 reduction reactions of the Cu-CDots nanocorals and Cu nanocorals (see Fig. S10†) were carried out in the potential range between −0.4 and −0.8 V. The FEs vs. the applied cathodic potential (−0.4, −0.5, 0.6, 0.7 and −0.80 V) of the hydrocarbon product and H2 for Cu-CDots nanocorals and Cu nanocorals are summarized in Fig. 3c and d, respectively. Quantitation of the electro-reduced products was achieved through the internal standard method and then the amounts of each group of compounds could be calculated.31 Dimethyl sulfoxide (DMSO) was added as an internal standard. As shown in Fig. S11,† the reduced products are HCOOH and CH3OH after the CO2 reduction reaction catalyzed by Cu-CDots nanocorals. Many researchers have reported the CO2 electroreduction to CH3OH at various oxidized Cu(I) electrodes,32–35 suggesting that the Cu(I) species play a critical role in electrode activity and selectivity to CH3OH. As shown in Fig. 3c, a major observation is that the FEs of the hydrocarbon products change distinctively at different applied potentials. The FEs of HCOOH continuously increase and even increase up to a maximum of 68% at the applied potential of −0.7 V. When the applied potential increases to −0.8 V, the FE of HCOOH shrinks to 57%. The FE changes of CH3OH have the same tendency as that of HCOOH and the FE of CH3OH reaches a maximum of 11% at the applied potential of −0.7 V. Besides, it is noteworthy that the total FEs of CO2 reduction products can reach 79%. Fig. 3d exhibits the obvious results that no CO2 reduction product is found at the applied potential of −0.4 V on Cu nanocorals and the total FEs of the CO2 reduction product are much lower as compared to that of Cu-CDots nanocorals. Therefore, all these results confirm that CDots can greatly improve the electrocatalytic performance of Cu nanomaterials for CO2 reduction.
To further optimize the electrocatalytic performance, different amounts of CDots (0.04 mg, 0.08 mg and 0.12 mg) were added to synthesize the Cu-CDots nanocorals and their electrocatalytic activities of CO2 reduction were investigated as shown in Fig. 4a. When the added amount of CDots is 0.08 mg, the electrocatalytic CO2 reduction reaction (red trace) occurs at an initial potential of −0.38 V. This means that the overpotential of the CO2 reduction reaction is only 0.13 V. Furthermore, the current density reaches ∼4.2 mA cm−2 at the applied potential of −0.70 V. When the added amount of CDots is more (or less) than 0.08 mg, the current density of CO2 reduction is lower. In contrast, Cu-CDots nanocorals show the best electrocatalysis activity for CO2 reduction when the amount of CDots is 0.08 mg. Based on the above results, it can be found that the incremental loading of the CDots on Cu nanocorals does not constantly improve the electrocatalytic performance of Cu-CDots nanocorals for the CO2 reduction reaction. This indicates that CDots may block some of the Cu active sites. That may be the reason for improving the catalytic selectivity for HCOOH and decreasing the catalytic activity of HER with increasing potential.
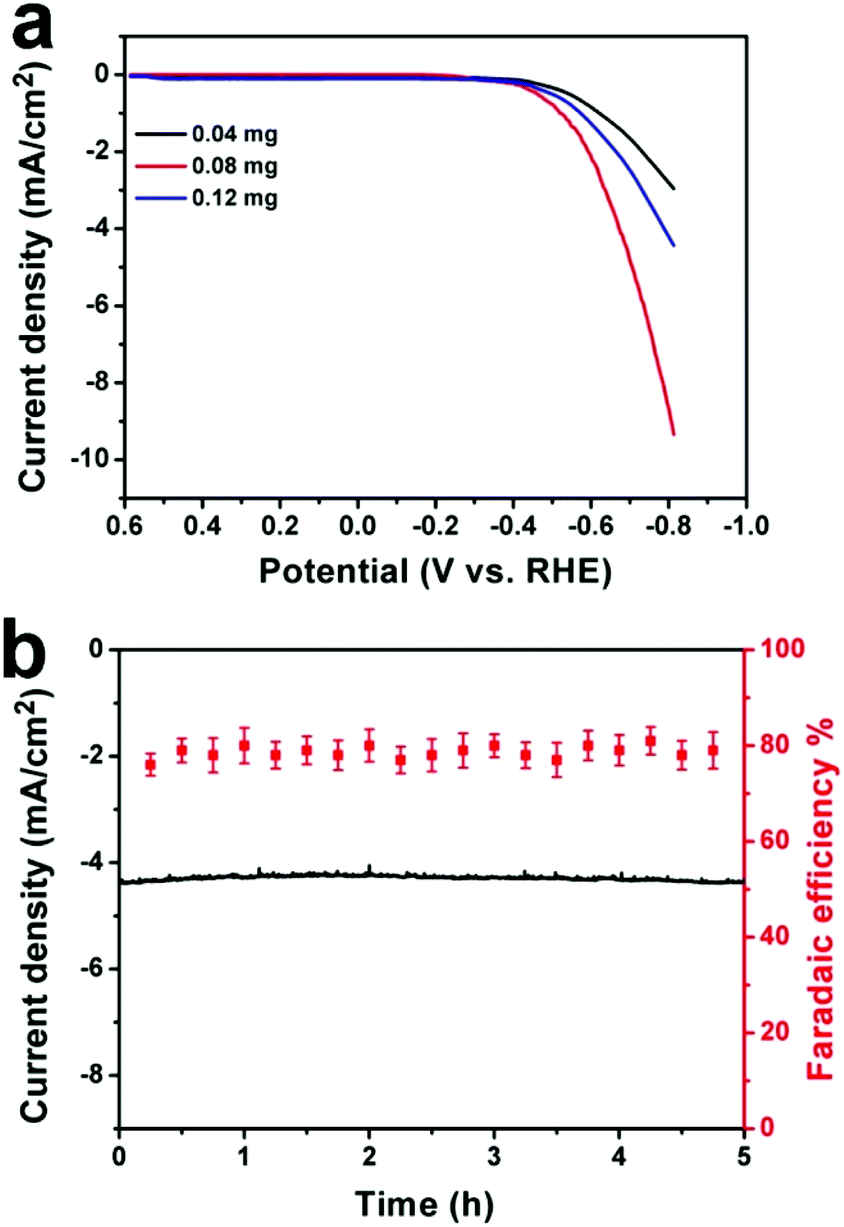 | ||
| Fig. 4 (a) LSVs for Cu-CDots nanocorals with different amounts of CDots: 0.04 (black trace), 0.08 (red trace) and 0.12 mg (blue trace), 10 mV s−1; (b) stability of Cu-CDots nanocorals for CO2 reduction operated at a potentiostatic potential of −0.70 V vs. RHE for 5 h. Geometric current density vs. time (left axis) and total FE for production (CH3OH and HCOOH) vs. time (right axis). | ||
The stability of Cu-CDots nanocorals (adding 0.08 mg CDots) was tested, as shown in Fig. 4b. A current density of ∼4.2 mA cm−2 was obtained on the Cu-CDots nanocoral electrocatalyst, which exhibits a long-term stability. Besides, the FE for the generation of hydrocarbon products (CH3OH and HCOOH) is maintained at about 79% throughout the entire process of the 5 h-electrolysis. No deactivation of Cu-CDots nanocorals for CO2 reduction can be observed over the course of 5 hours. All these results confirm that Cu-CDots nanocorals is a highly efficient and stable electrocatalyst.
To deeply understand the mechanism of CO2 reduction reaction catalyzed by Cu-CDots nanocorals, we performed the following experiments. Generally, the process of CO2 electro-reduced to HCOOH consists of four parts:1,36 (1) CO2 molecules are captured by active sites (marked as CO2*; * represents an active site); (2) CO2* accepts one proton and one electron to form the intermediate COOH*; (3) COOH* accepts another proton and one electron to form the product HCOOH*; (4) the last part is desorption of HCOOH*.
| CO2(g) + * ↔ CO2* | (1) |
| CO2* + H+(aq) + e ↔ COOH* | (2) |
| COOH* + H+(aq) + e ↔ HCOOH* | (3) |
| HCOOH* ↔ HCOOH | (4) |
It can be found that the adsorption of CO2 and H+ play important roles in CO2 reduction. Then, the adsorption abilities of CO2 and H+ on Cu nanocorals and Cu-CDots nanocorals were investigated, as shown in Fig. 5a. Cu nanocorals and Cu-CDots nanocorals show CO2 adsorption capacities of 0.05 and 0.22 mmol g−1 at 25 °C, respectively, indicating that the Cu-CDots nanocorals possess a more excellent CO2 adsorption capability than the Cu nanocorals. The dependence of adsorption time on the removal of H+ by Cu nanocorals and Cu-CDots nanocorals is shown in Fig. 5b. The amount of adsorbed H+ (based on the quantity of HCl; for details see the Experiment section) is about 10.03 mg g−1, while the amount of adsorbed H+ is only 4.46 mg g−1 for Cu nanocorals. This indicates that CDots significantly increase the adsorbed capacity of CO2 and H+.
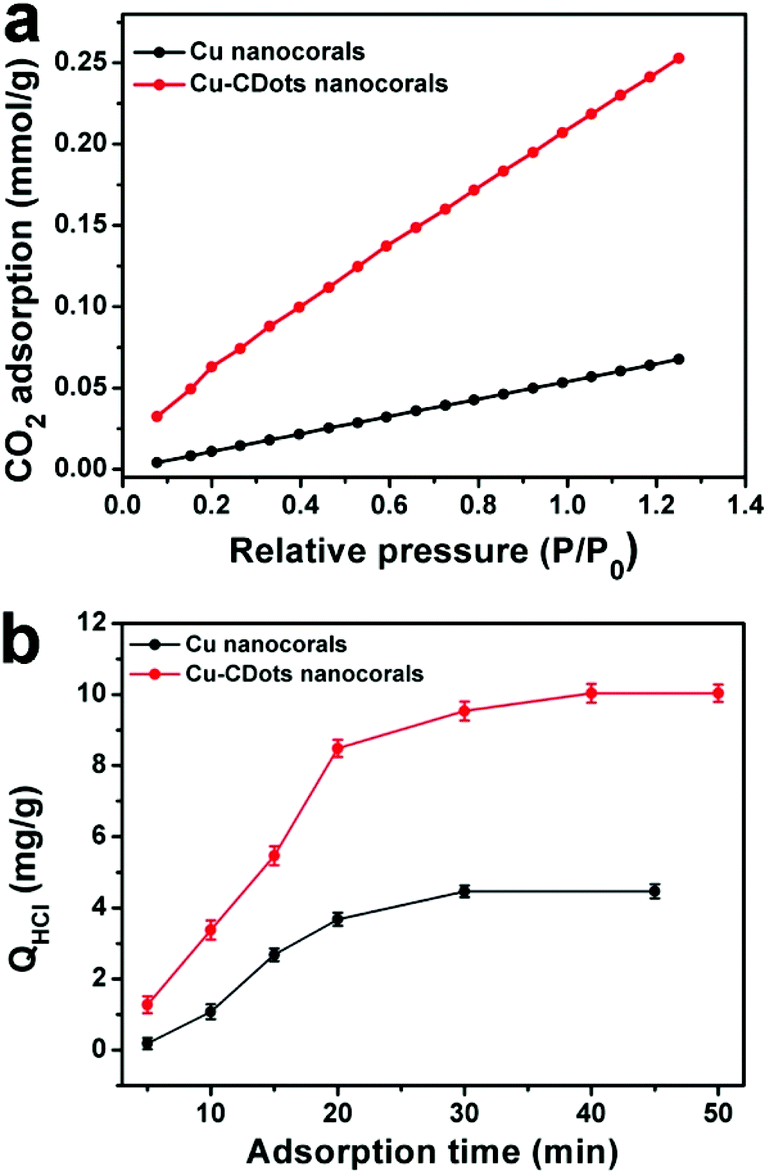 | ||
| Fig. 5 (a) CO2 adsorption isotherm of Cu nanocorals (black trace) and Cu-CDots nanocorals (red trace); (b) dependence of adsorption time on the adsorption of H+ by Cu nanocorals (black trace) and Cu-CDots nanocorals (red trace). | ||
Theoretically, the formation of HCOOH should always be more favorable in comparison with the formation of other CO2 reduction products in the process of the electrocatalytic CO2 reduction reaction using Cu nanomaterials as the catalyst.37 However, many works have reported that by using Cu as the catalyst, the main product is not HCOOH,38–41 which is attributed to the high desorption energy of HCOOH on Cu-based catalysts. Here, the surface of Cu nanocorals modified with CDots can greatly diminish the HCOOH desorption energy on the Cu electrode and improve the catalytic selectivity for HCOOH.37,42 On the basis of the above results, we further proposed a mechanism for the CO2 reduction reaction on Cu-CDots nanocorals as shown in Fig. 6. CO2 and H+ can be easily adsorbed on the surface of Cu-CDots nanocorals, which can promote CO2 reduction to HCOOH* on active sites.43,44 The Cu nanocorals modified with CDots can alter the coordination environment of surface copper atoms. Thus, the HCOOH desorption energy on Cu-CDots nanocorals can be diminished. HCOOH can escape from the surface of the Cu-CDots nanocorals, which prevents HCOOH from being further reduced. Therefore, the main product of CO2 reduction is HCOOH.
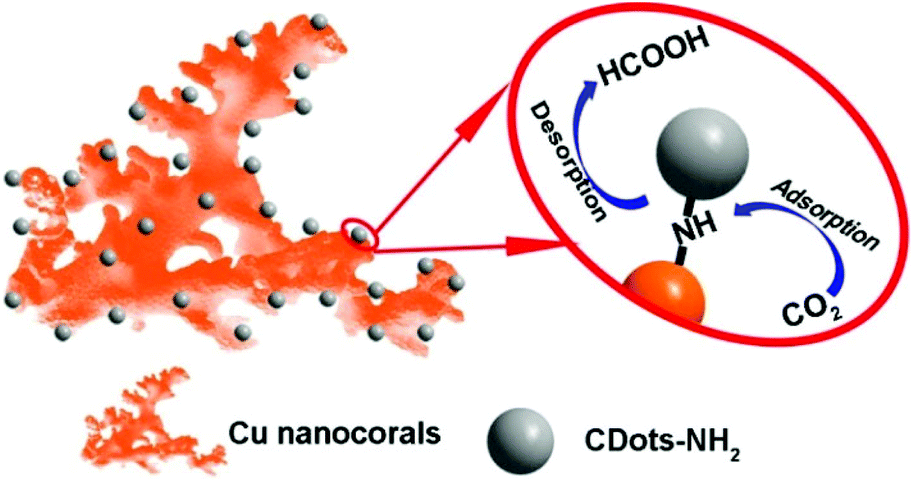 | ||
| Fig. 6 The proposed reaction mechanism of electrocatalytic CO2 reduction by Cu-CDots nanocorals. | ||
Conclusions
We have first successfully synthesized a highly efficient, low-cost and stable Cu-CDots nanocorals electrocatalyst for CO2 reduction in aqueous solutions. The major product of CO2 reduction on the Cu-CDots nanocorals is HCOOH with an inconceivable low overpotential of 0.13 V and FEs of 79% at a moderate potential of −0.7 V. Cu-CDots nanocorals exhibit a long-term stability during 5 h-electrolysis. We propose insights into the role of CDots on the increased activity of the Cu-CDots nanocorals electrocatalyst for CO2 reduction. This may be ascribed to the fact that CDots have an enhanced adsorption capacity for the CO2 molecule and the HCOOH desorption energy on Cu-CDots nanocorals can be diminished because of the modification to the CDots. Thus, the appropriate morphology and surface modification can greatly improve selectivity and decrease the overpotential for CO2 electroreduction, indicating an acceptable strategy for developing efficient CO2 electroreduction catalysts. Future work is to contribute to selectively reducing CO2 to a higher energy density product, CH3OH.Instruments
TEM and HRTEM images were obtained using a FEI/Philips Tecnai G2 F20 TWIN transmission electron microscope. SEM images were characterized by a Carl Zeiss Supra 55 scanning electron microscope. The crystal structure of the resultant products was characterized by X-ray diffraction using an X'Pert-ProMPD (Holland) D/max-γAX-ray diffractometer with Cu Kα radiation (λ = 0.154178 nm). FT-IR spectra were recorded on a FTIR spectrometer (Spectrum One, Perkin Elmer) using a standard KBr pellet technique. BET surface area was determined by plotting the adsorption isotherm of N2 at liquid N2 temperature (77 K) obtained using a Micromeritics ASAP 2050 instrument. XPS spectra were obtained by using a KRATOS Axis ultra-DLD X-ray photoelectron spectrometer with a monochromatized Mg Kα X-ray source (hν = 1283.3 eV). CO2 adsorption was determined by plotting the adsorption isotherm of CO2 at 25 °C obtained using a Micromeritics ASAP 2050 instrument. The electro-catalysis actions were tested by a Model CHI 660C workstation (CH Instruments, Chenhua, Shanghai, China).Fabrication of the catalysts
Electrochemical measurements
Linear sweep voltammetry experiments were performed using a standard three-electrode configuration. A platinum wire was used as an auxiliary electrode and a saturated calomel electrode (SCE) was used as a reference electrode. The working electrode was either a catalyst modified glassy carbon disk electrode (GCE, 3.0 mm diameter CH Instruments), or a catalyst modified carbon fiber paper electrode (0.7 cm × 0.7 cm). For product analysis, the bulk electrolysis was performed in an airtight electrochemical H-type cell with a catalyst modified carbon fiber paper electrode (0.7 cm × 0.7 cm) as the work electrode. In addition, an electrochemical test was used with the catalyst modified glassy carbon disk electrode as the work electrode. Initially, polarization curves for the modified electrode were carried out under an inert N2 (gas) atmosphere. After this, the solution was purged with CO2 (99.999%) for at least 30 min (CO2-saturated high purity aqueous 0.5 M KHCO3) and the electrocatalytic CO2 reduction was measured. The constant-potential electrolysis experiments were performed in an airtight electrochemical H-type cell with three electrodes and two compartments (volume of each part is 115 mL) separated by a Nafion®212 anion exchange membrane with 75 mL 0.5 M KHCO3 electrolyte in each chamber.Product analysis
The bulk electrolysis was further performed in an airtight electrochemical H-type cell with 75 mL 0.5 M KHCO3 electrolyte in each chamber. For the detection of the gas products, hydrocarbons (CO, CH4, C2H4, and C2H6) were tested by a flame ionization detector (FID) with helium as the carrier gas. A thermal conductivity detector (TCD) was used to detect hydrogen with nitrogen as the carrier gas. The liquid products were collected from the cathode chambers after electrolysis and quantified by NMR (Bruker AVANCEAV III 400) spectroscopy, in which 0.5 ml electrolyte was mixed with 0.1 mL D2O (deuterated water) and 0.1 μL dimethyl sulfoxide (DMSO, Sigma, 99.99%) was added as an internal standard. The calculation of Faradaic efficiency:For HCOOH,
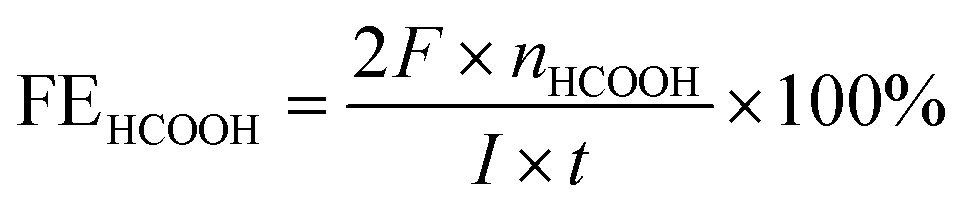
For CH3OH,
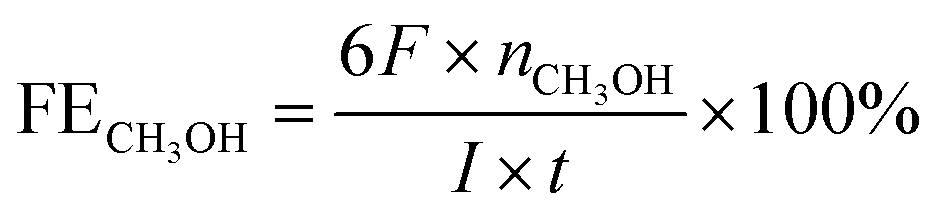
H+ adsorption measurements
To study the proton (H+) adsorption capacity of Cu-CDots nanocorals and Cu nanocorals, the concentration of 5 mM HCl solution was selected and the adsorption experiments were conducted with a dialysis method. 1 g of catalyst (Cu-CDots nanocorals or Cu nanocorals) was added to a 50 mL HCl solution (5 mM). Then, the Cu-CDots nanocoral or Cu nanocoral solution was dialyzed using a semi-permeable membrane (MWCO 1000) in a 600 mL beaker, and the dialysate was 5 mM HCl (500 mL). After stirring on a shaker for predetermined time intervals, 2 mL of dialysate was taken out and the concentration of HCl solution was determined by titrating with 5 mM NaOH solution. The concentration of HCl solution vs. time can be obtained.CO2 adsorption measurements
CO2 adsorption was determined by plotting the adsorption isotherm of CO2 at 25 °C obtained using a Micromeritics ASAP 2050 instrument. 0.1 g of catalyst was added into the test chamber. After two cycles of gas desorption, the adsorption of CO2 was performed at 25 °C. The adsorbed CO2 molecules on the catalyst increase with the incremental pressure of CO2, the adsorbed CO2 on the catalyst at 25 °C. Therefore, the adsorption isotherm of CO2 at 25 °C can be obtained.Acknowledgements
This work is supported by the Collaborative Innovation Center of Suzhou Nano Science and Technology, the National Natural Science Foundation of China (51422207, 51132006, 51572179, 21471106, 21501126), the Specialized Research Fund for the Doctoral Program of Higher Education (20123201110018), a Suzhou Planning Project of Science and Technology (ZXG2012028), and a project funded by the Priority Academic Program Development of the Jiangsu Higher Education Institutions (PAPD).References
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- J. Albo, M. Alvarez-Guerra, P. Castañoa and A. Irabien, Green Chem., 2015, 17, 2304–2324 RSC.
- S. Ma and P. J. A. Kenis, Curr. Opin. Chem. Eng., 2013, 2, 191–199 CrossRef.
- C. Costentin, M. Robert and J. M. Savéant, Chem. Soc. Rev., 2013, 42, 2423–2436 RSC.
- A. Goeppert, M. Czaun, J. P. Jones, G. K. S. Prakash and G. A. Olah, Chem. Soc. Rev., 2014, 43, 7995–8048 RSC.
- D. Gao, H. Zhou, J. Wang, S. Miao, F. Yang, G. Wang, J. Wang and X. Bao, J. Am. Chem. Soc., 2015, 137, 4288–4291 CrossRef CAS PubMed.
- X. Jin, M. Li, J. Wang, C. Marambio-Jones, F. Peng, X. Huang, R. Damoiseaux and E. M. Hoek, Environ. Sci. Technol., 2010, 44, 7321–7328 CrossRef CAS PubMed.
- R. Lü, K. Shi, W. Zhou, L. Wang, C. Tian, K. Pan, S. Li and H. Fu, J. Mater. Chem., 2012, 22, 24814–24820 RSC.
- H. I. Park, S. Lee, J. M. Lee, S. A. Nam, T. Jeon, S. W. Han and S. O. Kim, ACS Nano, 2014, 8, 10305–10312 CrossRef CAS PubMed.
- G. L. Xu, Q. Wang, J. C. Fang, Y. F. Xu, J. T. Li, L. Huang and S. G. Sun, J. Mater. Chem. A, 2014, 2, 19941–19962 CAS.
- Z. Wang, G. Yang, Z. Zhang, M. Jin and Y. Yin, ACS Nano, 2016, 10, 4559–4564 CrossRef CAS PubMed.
- Q. Lu, J. Rosen, Y. Zhou, G. S. Hutchings, Y. C. Kimmel, J. G. Chen and F. Jiao, Nat. Commun., 2014, 5, 1–6 Search PubMed.
- W. Zhu, R. Michalsky, O. Metin, H. Lv, S. Guo, C. J. Wright, X. Sun, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2013, 135, 16833–16836 CrossRef CAS PubMed.
- D. Gao, H. Zhou, J. Wang, S. Miao, F. Yang, G. Wang, J. Wang and X. Bao, J. Am. Chem. Soc., 2015, 137, 4288–4291 CrossRef CAS PubMed.
- A. de Klerk, Green Chem., 2008, 10, 1249–1279 RSC.
- H. Mistry, F. Behafarid, R. Reske, A. S. Varela, P. Strasser and B. R. Cuenya, ACS Catal., 2016, 6, 1075–1080 CrossRef CAS.
- I. Shown, H. C. Hsu, Y. C. Chang, C. H. Lin, P. K. Roy, A. Ganguly, C. H. Wang, J. K. Chang, C. I. Wu, L. C. Chen and K. H. Chen, Nano Lett., 2014, 14, 6097–6103 CrossRef CAS PubMed.
- R. Reske, H. Mistry, F. Behafarid, B. R. Cuenya and P. Strasser, J. Am. Chem. Soc., 2014, 136, 6978–6986 CrossRef CAS PubMed.
- J. Cheng, M. Zhang, J. Liu, J. Zhou and K. Cen, J. Mater. Chem. A, 2015, 3, 12947–12957 CAS.
- D. Raciti, K. J. Liviand and C. Wang, Nano Lett., 2015, 15, 6829–6835 CrossRef CAS PubMed.
- M. Ma, K. Djanashvili and W. A. Smith, Phys. Chem. Chem. Phys., 2015, 17, 20861–20867 RSC.
- R. Liu, H. Huang, H. Li, Y. Liu, J. Zhong, Y. Li, S. Zhang and Z. Kang, ACS Catal., 2014, 4, 328–336 CrossRef CAS.
- D. Tang, J. Liu, X. Wu, R. Liu, X. Han, Y. Han, H. Huang, Y. Liu and Z. Kang, ACS Appl. Mater. Interfaces, 2014, 6, 7918–7925 CAS.
- L. Zhou, J. Liu, X. Zhang, R. Liu, H. Huang, Y. Liu and Z. Kang, Nanoscale, 2014, 6, 5831–5837 RSC.
- Y. Yang, J. Liu, S. Guo, Y. Liu and Z. Kang, J. Mater. Chem. A, 2015, 3, 18598–18604 CAS.
- (a) K. Sim, N. Lee, J. Kim, E. B. Cho, C. Gunathilake and M. Jaroniec, ACS Appl. Mater. Interfaces, 2015, 7, 6792–6802 CrossRef CAS PubMed; (b) C. J. Yoo, L. C. Lee and C. W. Jones, Langmuir, 2015, 31, 13350–13360 CrossRef CAS PubMed; (c) S. Chaemchuen, N. A. Kabir, K. Zhou and F. Verpoort, Chem. Soc. Rev., 2013, 42, 9304–9332 RSC.
- A. D. Brumbaugh, K. A. Cohen and S. K. St. Angelo, ACS Sustainable Chem. Eng., 2014, 2, 1933–1939 CrossRef CAS.
- W. Yuan, J. Yuan, J. Xie and C. M. Li, ACS Appl. Mater. Interfaces, 2016, 8, 6082–6092 Search PubMed.
- S. Seifert, S. Höhne, F. Simon, C. Hanzelmann, R. Winkler, T. Schmidt, R. Frenzel, P. Uhlmann and S. Spange, Langmuir, 2012, 28, 14935–14943 CrossRef CAS PubMed.
- H. Tian, X. L. Zhang, J. Scott, C. Ng and R. Amal, J. Mater. Chem. A, 2014, 2, 6432–6438 CAS.
- S. Gao, Y. Lin, X. Jiao, Y. Sun1, Q. Luo, W. Zhang1, D. Li, J. Yang and Y. Xie, Nature, 2016, 529, 68–71 CrossRef CAS PubMed.
- S. Ohya, S. Kaneco, H. Katsumata, T. Suzuki and K. Ohta, Catal. Today, 2009, 148, 329–334 CrossRef CAS.
- J. F. Brito, A. A. Silva, A. J. Cavalheiro and M. V. B. Zanoni, Int. J. Electrochem. Sci., 2014, 9, 5961–5973 Search PubMed.
- J. Albo, A. Sáez, J. Solla-Gullón, V. Montiel and A. Irabien, Appl. Catal., B, 2015, 176–177, 709–717 CrossRef CAS.
- E. L. Uzunova, N. Serianib and H. Mikosch, Phys. Chem. Chem. Phys., 2015, 17, 11088–11094 RSC.
- S. Lee, H. K. Ju, R. Machunda, S. Uhm, Ja. K. Lee, H. J. Lee and J. Lee, J. Mater. Chem. A, 2015, 3, 3029–3034 CAS.
- T. N. Huan, E. S. Andreiadis, J. Heidkamp, P. Simon, E. Derat, S. Cobo, G. Royal, A. Bergmann, P. Strasser, H. Dau, V. Artero and M. Fontecave, J. Mater. Chem. A, 2015, 3, 3901–3907 CAS.
- L. C. Grabow and M. Mavrikakis, ACS Catal., 2011, 1, 365–384 CrossRef CAS.
- A. Karelovic and P. Ruiza, Catal. Sci. Technol., 2015, 5, 869–881 CAS.
- S. Velu, K. Suzuki, M. Okazaki, M. P. Kapoor, T. Osaki and F. Ohashi, J. Catal., 2000, 194, 373 CrossRef CAS.
- M. Le, M. Ren, Z. Zhang, P. T. Sprunger, R. L. Kurtz and J. C. Flake, J. Electrochem. Soc., 2011, 158, 45–49 CrossRef.
- P. Xiao, A. Kuc, T. Frauenheim and T. Heine, J. Mater. Chem. A, 2014, 2, 4885–4488 Search PubMed.
- H. Wang, Y. Chen, X. Hou, C. Ma and T. Tan, Green Chem., 2016, 18, 3250–3256 RSC.
- S. Zhang, P. Kang, S. Ubnoske, M. K. Brennaman, N. Song, R. L. House, J. T. Glass and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 7845–7848 CrossRef CAS PubMed.
- H. Li, R. Liu, Y. Liu, H. Huang, H. Yu, H. Ming, S. Lian, S. T. Lee and Z. Kang, J. Mater. Chem., 2012, 22, 17470–17475 RSC.
- H. Ming, Z. Ma, Y. Liu, K. Pan, Ha. Yu, F. Wang and Z. Kang, Dalton Trans., 2012, 41, 9526–9531 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary figures. See DOI: 10.1039/c6nr08104e |
| This journal is © The Royal Society of Chemistry 2017 |