
Synthesis engineering of iron oxide raspberry-shaped nanostructures†
O.
Gerber
ab,
B. P.
Pichon
*a,
D.
Ihiawakrim
a,
I.
Florea
ac,
S.
Moldovan
a,
O.
Ersen
a,
D.
Begin
d,
J.-M.
Grenèche
e,
S.
Lemonnier
b,
E.
Barraud
*b and
S.
Begin-Colin
*a
aInstitut de Physique et Chimie des Matériaux de Strasbourg, 23 rue du Loess, BP 43, 67037, Strasbourg Cedex 2, France. E-mail: benoit.pichon@unistra.fr; sylvie.begin@unistra.fr
bInstitut Franco-Allemand de Recherches de Saint-Louis, 5 rue du Général Cassagnou, 68300, Saint-Louis, France
cLaboratoire de Physique des Interfaces et des Couches Minces, École Polytechnique/CNRS, Route de Saclay, 91128 Palaiseau Cedex, France
dLaboratoire des Matériaux, Surfaces et Procédés pour la Catalyse, 25 rue Becquerel, 67087 Strasbourg Cedex 2, France
eInstitut des Molécules et Matériaux du Mans (IMMM), UMR CNRS 6283, Université du Maine, 72085 Le Mans Cedex 9, France
First published on 18th November 2016
Abstract
Magnetic porous nanostructures consisting of oriented aggregates of iron oxide nanocrystals display very interesting properties such as a lower oxidation state of magnetite, and enhanced saturation magnetization in comparison with individual nanoparticles of similar sizes and porosity. However, the formation mechanism of these promising nanostructures is not well understood, which hampers the fine tuning of their magnetic properties, for instance by doping them with other elements. Therefore the formation mechanism of porous raspberry shaped nanostructures (RSNs) synthesized by a one-pot polyol solvothermal method has been investigated in detail from the early stages by using a wide panel of characterization techniques, and especially by performing original in situ HR-TEM studies in temperature. A time-resolved study showed the intermediate formation of an amorphous iron alkoxide phase with a plate-like lamellar structure (PLS). Then, the fine investigation of PLS transformation upon heating up to 500 °C confirmed that the synthesis of RSNs involves two iron precursors: the starting one (hydrated iron chlorides) and the in situ formed iron alkoxide precursor which decomposes with time and heating and contributes to the growth step of nanostructures. Such an understanding of the formation mechanism of RSNs is necessary to envision efficient and rational enhancement of their magnetic properties.
Introduction
Nanomaterials with a controlled size, morphology and composition represent an important challenge in materials science because controlling these parameters is fundamental to optimize their subsequent functional properties. In particular, porous iron oxide (Fe3O4) nanostructures that combine high specific surface area, superparamagnetic behavior and high magnetization have generated tremendous interest toward the design of new magnetic nanomaterials used as supports in water treatment,1 biomedicine,2 magneto-dielectric composites3 and energy storage4 applications. These nanostructures generally consist of aggregated nanocrystals with common crystallographic orientations.5–7 The great interest of such nanostructures is that, while their mean size reaches hundreds of nanometers, they are super-paramagnetic and they display much higher saturation magnetization than the nanocrystals constituting them considered separately. Such original properties were explained by low spin canting and oxidation states of nanocrystals due to the high interface between them reducing significantly these phenomena.6 These parameters combined with strong magnetic collective properties induced by the aggregated orientation of nanocrystals ensure thus high saturation magnetization.Porous iron oxide nanostructures have been synthesized by several methods either by using templates or by controlled chemical synthesis. Hard templating leads to nanostructures with high specific surface area and easily tunable pore sizes.8 However, the use of sacrificial silica necessitates several steps, which reduces its processability. The microemulsion approach suffers from a significant lack of control on particle sizes and shapes, which also limits potential applications.9 Although the sol–gel method is easy to perform, it leads usually to disorganized aggregates and poor control of magnetic properties.10 More recently, the polyol method11 has been demonstrated to be easy to perform and extremely reliable to synthesize porous iron oxide nanostructures with well-defined sizes and porosity as well as high magnetization.12–18 Based on the reduction of Fe(III) by glycol solvents (ethylene, diethylene or polyethylene),19,20 this reaction requires an activator, such as sodium acetate, to allow precipitation of iron hydroxides, while dehydration by thermal activation results in the iron oxide phase.13,15,17 Urea can also be used as a precipitator which after decomposition accelerates the reaction kinetics.12,18 Such a polyol method was also reported to construct spherical nanostructures consisting of assemblies of iron alkoxide nanosheets, which transformed into iron oxide nanostructures after subsequent heat treatments.21–27 The amount of water and the solvothermal temperature appear to be important parameters to trigger the formation of the iron oxide phase.26
The reaction mechanism is generally simply described as a two-stage process, which consists of: (i) the nucleation of primary nanocrystals in a supersaturated solution followed by (ii) their uniform aggregation into larger secondary structures.5 It is generally accepted that it proceeds through the oriented attachment of nanocrystals in a similar direction triggered by the tendency for reducing their high surface energy, which is favored by short range interactions (van der Waals, magnetic dipolar, crystal faces… interactions) and the Brownian motion.26,28–31 This process may be followed by Ostwald ripening, which results in the growth of nanograins and formation of cavities.15,19 Finally, the nanostructure of aggregates (diameter and nanosize) can be modulated as a function of synthesis parameters such as solvents,19,32 reaction time,13,17 or stabilizing agents.15,17,33
Although the properties of such nanostructures have been investigated, their formation mechanism still remains unclear. Furthermore, as shown by the very few published papers on this topic, the homogeneous doping of such iron oxide nanostructures with elements such as Mn or Co with high contents to tune their magnetic properties remains difficult.13,34,35 A better understanding of the formation mechanism of these porous iron oxide nanostructures is thus of great interest to enhance rationally their magnetic properties.
We report herein on the synthesis mechanism of the one-pot polyol based synthesis of porous iron oxide featured by a raspberry shaped nanostructure (RSN). It has been carefully investigated in detail by carrying out a time-resolved study of samples collected at different reaction times. At first, an amorphous phase is observed, which is followed by the concomitant formation of small iron oxide aggregates of co-precipitated nanoparticles and of an intermediate iron based precursor with a plate-like lamellar structure (PLS). The thermal stability of this intermediate PLS has been investigated by performing in situ HRTEM. Building on a wide panel of characterization techniques, namely SEM, HRTEM, XPS, XRD, EELS, FTIR and Mössbauer spectroscopies, and elemental and thermogravimetric analyses, we propose that the formation mechanism of an RSN (Scheme 1) involves a PLS as an intermediate iron precursor which after decomposition with time and heating contributes to the heterogeneous growth of porous iron oxide nanostructures.
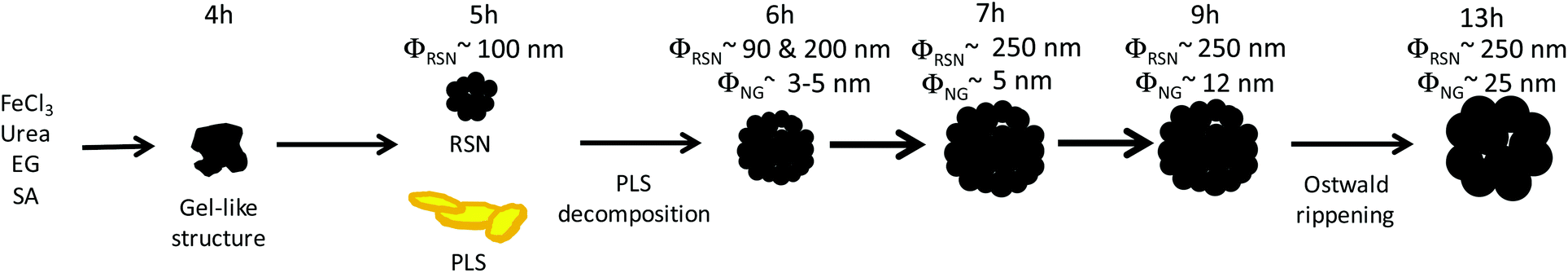 | ||
| Scheme 1 Schematic representation of the synthesis pathway. | ||
Experimental section
Materials
Hexahydrate iron(III) chloride (97%), urea (99.3%) and ethylene glycol (99%) were purchased from Alfa Aesar. Succinic acid (≥99.5%) was purchased from Sigma-Aldrich.Synthesis of raspberry-shaped nanostructures (RSNs)
In a typical synthesis of FeCl3, 6H2O (30 mmol), succinic acid (10 mmol) and urea (300 mmol) were completely dissolved in ethylene glycol (300 mL) by vigorous mechanical stirring and sonication. The solution was sealed in a Teflon lined stainless steel autoclave (600 mL capacity), slowly heated at 200 °C at 1.5 °C min−1 and kept at this temperature for 13 hours. The pressure and temperature in the autoclave were monitored during the synthesis by using a homemade acquisition device. The autoclave was cooled down to room temperature afterwards by water circulation for 1 h. The sediments were separated by centrifugation and washed 3 times with ethanol and 3 times with deionized water to eliminate organic and inorganic impurities. Finally, the precipitate was frozen and dried to remove water, giving rise to a black powder (1.5 g). The synthesis was also scaled up and performed in a larger autoclave (2 L) to produce up to 20 g of material. The time-resolved study has been conducted by collecting samples of the reaction medium, which were washed by performing the above procedure. PLSs have been collected from the reaction medium as a yellow powder after separation from the RSN under a magnetic field of 0.36 T.Characterization techniques
Samples were characterized by X-Ray diffraction (XRD) using a Bruker D8 Advance equipped with a monochromatic copper radiation source (Kα = 0.154056) and a Sol-X detector. High purity silicon powder (a = 0.543082 nm) was systematically used as an internal standard to reset the zero 2θ to calculate with accuracy the lattice parameters and the crystal domain size of the RSN. Profile matching refinements were performed through the Fullprof program36 using Le Bail's method with the modified Thompson–Cox–Hasting (TCH) pseudo-Voigt profile function.37Scanning electron microscopy (SEM) has been performed on a JEOL 6700F with a 2 nm point resolution. The inner cavity of nanostructures has been also investigated by polishing the sample previously embedded in a resin. Transmission electron microscopy (TEM) experiments have been performed on a on a JEOL 2100F electron microscope operating at 200 kV and equipped with a GIF Tridiem spectrometer allowing a spectral resolution of 0.7 eV. The in situ TEM tests have been carried out under electron diffraction mode by using a selecting area diaphragm of 200 μm and a camera length of 30 cm. A Gatan heating holder (model 652) was employed to control/carry out the progressive specimen heating from room temperature up to 500 °C, with a step of 50°. During the in situ heating experiment, the same regions were tagged and followed at each temperature.
Thermogravimetric (TG) and thermodifferential (TD) analyses were performed in the temperature range of 20 to 500 °C under a nitrogen flow with a heating rate of 5 °C min−1 by using a Texas Instruments SDT Q600. Dried powders were placed in a platinum crucible. Specific surface areas of the different samples were determined by N2 adsorption–desorption measurements at 77 K by using Micromeritics TriStar 3000 apparatus. Before the measurements, samples were outgassed at 150 °C overnight in order to desorb impurities or moisture from their surface. Elemental analysis has been performed by the analytic central service of the CNRS UMR7504 at La Vernaison by using a QqToF instrument with a precision of 5 ppm. FTIR spectroscopy was performed using a Digilab Excalibur 3000 spectrophotometer (CsI beamsplitter) in the wavenumber range of 4000–400 cm−1 on samples diluted in KBr pellets.
57Fe Mössbauer spectra were recorded at 300 and 77 K using a standard constant acceleration transmission spectrometer with a 57Co radioactive source diffused into an Rh matrix. The spectra were fitted by means of the MOSFIT program38 involving the discrete distribution of magnetic sextets and/or quadrupolar doublets based on lines with Lorentzian profiles; an α-Fe foil was used as the calibration sample. The values of the isomer shifts are quoted relative to that of α-Fe at 300 K.
Results and discussion
Time-resolved study of raspberry shaped nanostructure formation
Iron oxide porous nanostructures have been synthesized by a modified polyol solvothermal approach, which has been detailed in ref. 6, involving iron chloride hexahydrate, urea, ethylene glycol and succinic acid as reactants. After 13 h of reaction under solvothermal conditions in a Teflon lined autoclave at 200 °C, a black powder is obtained after successive washings with water and ethanol. SEM micrographs showed particles having a spherical shape and a size distribution centered at 250 nm (Fig. 1a). They consist of aggregates of nanograins of 25 nm as shown by TEM micrographs (Fig. 1b). Nanostructures consist of the iron oxide spinel structure as shown by the XRD pattern (Fig. 1f). According to Mossbauer spectroscopy, magnetite represents 70% against maghemite (30%). Nanograins are also featured by similar crystal orientation as we have reported elsewhere.6 The saturation magnetization of the RSN is about 81 emu g−1, which is larger than that of individual 25 nm sized iron oxide nanoparticles (NPs).39 Mossbauer and magnetic characterization has been already detailed previously in ref. 6. Furthermore, SEM micrographs showed a cavity of about 90 nm in broken RSNs, which corresponds to a hollow structure (Fig. 1d). It was confirmed by studying the cross-section of these objects after their embedment in a resin followed by a polishing step (Fig. 1e).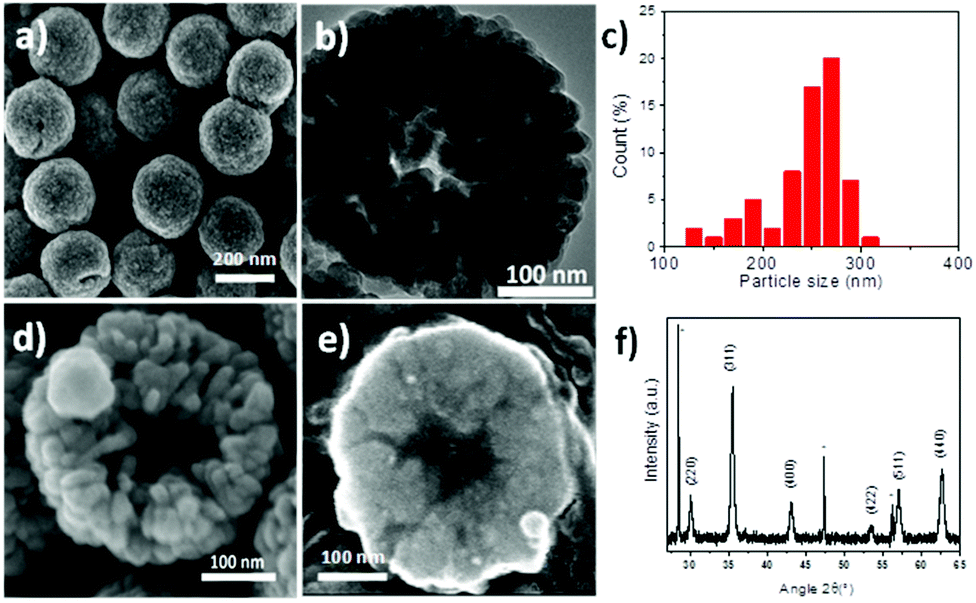 | ||
| Fig. 1 RSNs obtained after 13 h of reaction. (a) SEM micrograph. (b) TEM micrograph. (c) Size distribution. SEM micrographs of (d) the broken RSN and (e) the cross-section after embedding in a resin matrix and polishing. (f) XRD pattern, stars correspond to silicon, which is used as a reference. | ||
For a better understanding of the reaction mechanism, a time-resolved study was performed by SEM on samples collected after different reaction times (Fig. 2). After 4 hours of reaction, a gel-like structure with non-regular shape grains was observed (Fig. 2a). XRD analysis (not shown) demonstrated that this compound was amorphous. The FTIR spectrum exhibits νFe–O and νC–H bands at 500 cm−1 and 2900 cm−1, respectively (Fig. S1†), which suggests the formation of an iron–organic complex. After 5 h, the amorphous compound is no longer present and is replaced by plate-like lamellar structures (PLSs) with irregular morphologies (Fig. 2b) and some small RSNs with a mean size of about 100 nm (Fig. S2†). After 6 h, RSNs are exclusively observed and are featured by a rather large bimodal size distribution with two main sizes centered at 90 nm and 200 nm with a heterogeneous nanograin (NG) size around 3–5 nm (Fig. 2c). The mean RSN size increases up to a mean size of 250 nm after 7 h of reaction with a pretty narrow dispersion from 50 to 70 nm and then stabilizes at this mean diameter. In contrast, the nanograin size increases gradually with the reaction time to 5 nm after 9 h and up to 25 nm after 13 h.
 | ||
| Fig. 2 SEM micrographs corresponding to samples collected after (a) 4 h, (b) 5 h, (c) 6 h and (d) 9 h of reaction. Insets correspond to the size distribution measured from SEM micrographs. | ||
The porous structure has also been investigated by performing nitrogen absorption–desorption measurements (Table 1).
| Reaction time | RSN size (nm) | Nanograin size (nm) | Surface specific area (m2 g−1) |
|---|---|---|---|
| 6 h | 90 & 200 ± 10 | 5 ± 3 | 61 |
| 7 h | 250 ± 50 | 5 ± 2 | 57 |
| 9 h | 250 ± 30 | 12 ± 2 | 38 |
| 13 h | 250 ± 12 | 25 ± 3 | 27 |
Specific surface areas were estimated by the Brunauer–Emmett–Teller model and decreased gradually when the reaction time increased, which was in agreement with the increase of the nanograin size. After 9 hours of reaction, the formation of a cavity is observed. As reported by others,40,41 the increase of the nanograin size may be correlated to the formation of cavities in RSNs since, given the experimental conditions, longer reaction times favor inside–out (or inverse) Ostwald ripening. The inner part of RSNs solubilizes and recrystallizes on grains located at the surface of RSNs. The observed synthesis pathway is described in Scheme 1.
Structural study of the intermediate plate-like lamellar structures (PLSs)
The formation of PLSs is rather surprising and, to the best of our knowledge, has not been reported yet as an intermediate compound in the synthesis of magnetite based RSNs. As PLSs appear as a key step in the synthesis process, we have deeply investigated their structure with a special focus by taking advantage of HRTEM combined with other techniques.TEM micrographs evidenced a lamellar structure featured by an interspacing of 1.1 ± 0.1 nm. Alternative dark and white stripes corresponding to different electron densities are ascribed to inorganic and organic entities, respectively (Fig. 3a). The lamellar structure has also been confirmed by the XRD pattern (Fig. 3b) as reported earlier for other materials.42,43 An intense peak which is indexed to the (001) reflection is observed in the low angle region (8.31°2Θ) and corresponds to an inter-lamellar spacing of about 1.06 nm. Such a value fits well with the distance measured from TEM micrographs. This intense and very narrow peak corresponds to high periodicity of the lamellar structure on large distances. Nevertheless, only two harmonics corresponding to the (002) and (003) reflections can be observed. Additional reflection peaks can also be observed in the higher angle region and may be ascribed to the ordering of molecules within inorganic layers as previously reported for other inorganic–organic lamellar structures.42,43 None of these peaks correspond to the iron oxide spinel structure.
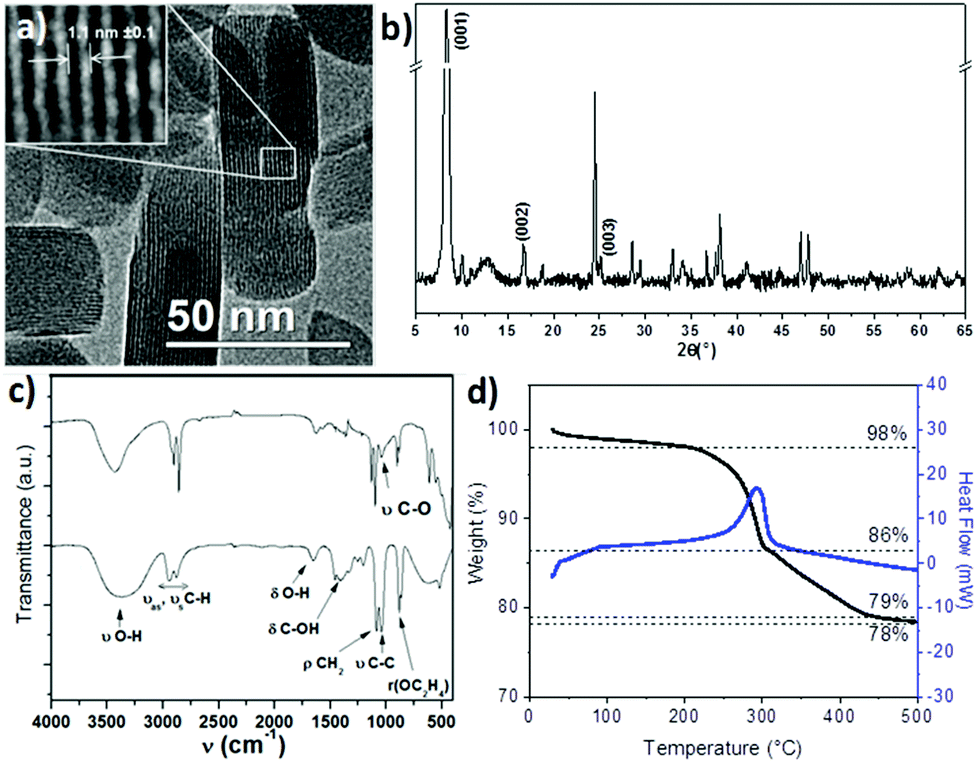 | ||
| Fig. 3 Plate-like lamellar structure (PLS) investigated by (a) TEM, (b) XRD, (c) FTIR spectroscopy of ethylene glycol (up) and PLS (down) and (d) thermogravimetry analysis. | ||
The chemical structure of the PLS was further investigated by FTIR spectroscopy (Fig. 3c). The spectrum exhibits IR bands corresponding to ethylene glycol although some differences are observed. First of all, the sharpest bands in the PLS spectrum rise from the symmetry increase within the ethylene glycol molecules. Then, most of the bands are weakened (δC-OH) or shifted (νOH, δC-OH, νC–O, ρOC2H4) which would be characteristic of the formation of the iron alkoxide complex as already reported for cobalt and manganese alkoxides.11,20,44 No band corresponding to unbound OH is observed at 3400 cm−1, which means that no iron hydroxide species are present. Moreover, bands at 800–400 cm−1 correspond to Fe–O vibration modes. Their strong intensity in comparison with νCH2 bands suggests that a large amount of iron cations interact with ethylene glycol. In addition, no bands characteristic of carboxylic acid groups interacting with iron (vasCOO at 1600 cm−1 and νsCOO at 1540 cm−1)17 are observed which suggests that succinic acid does not take part in the composition of the PLS.
The thermal behavior of the PLS has been investigated by thermogravimetry (TG) analysis under nitrogen (Fig. 3d). It revealed a 2% weight loss until 200 °C which can be attributed to the evaporation of solvents and desorption of gases. Two larger weight losses are then observed: one sharp loss of 12% around 250 °C which is correlated to an exothermic peak of the heat flow measurement and a second loss of 8%, which takes place between 300 °C and 450 °C. At 500 °C, the weight loss becomes very weak with respect to the initial mass of the PLS. The remaining weight shows that PLSs are composed of about 78% of the inorganic substructure. The XRD pattern of PLSs after heating at 500 °C showed the formation of a spinel iron oxide phase (Fig. S3†). This thermal analysis showed that the PLS decomposes at a high temperature above 200 °C.
The main observed features by XRD, IR spectroscopy and TGA are similar to those reported earlier for alkoxide structures based on cobalt and manganese ions combined with ethylene glycol in basic medium20,44 and also with those of iron alkoxides used as precursors and leading to nanoflake structures.21–23,25–27,45 These last iron alkoxide nanoflakes were then transformed into iron oxide nanostructures by a subsequent heat treatment.23,24,27 However, one may notice that their TGA curves display only one weight loss (two in our case) but the DSC curves are similar to one feature at around 250 °C. Therefore, these results suggest that the PLS would have an iron alkoxide based structure.
The chemical composition of the PLS was further investigated by elemental analysis. The following elements have been detected with the corresponding molar ratios Fe![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O:C:H/2:4.78:2.08:4.96. These results suggested the formation of an iron alkoxide lamellar structure with the formula: Fe2C2H5O5 and would be in agreement with TG measurements with an inorganic/organic ratio of about 3. Nevertheless, such a chemical composition significantly differs from that of iron alkoxides reported in other studies.22,25,27 Moreover, it is correlated to different structures (XRD pattern: peak at 8° instead of 11°) and chemical compositions (TG analysis: two weight losses instead of the only one). It has been reported earlier for the iron alkoxide formation that Fe3+ ions are reduced by EG molecules to form Fe2+ ions. These ions are then coordinated to EG molecules, which have lost protons, to form FeC2H4O2.22 However, further papers reported on the partial reduction of Fe3+ and thus on the coexistence of Fe2+ and Fe3+ in iron alkoxides with the formula Fe3(OCH2CH2O)4 or Fe(OCH2CH2O)x.22,25–27 None of these formulae agreed with the elemental analysis we have performed on our samples. Therefore, the oxidation state of iron cations and their chemical environment were further investigated by 57Fe Mössbauer spectrometry and X-ray photoemission spectroscopy (XPS) in combination with EELS measurements.
:O:C:H/2:4.78:2.08:4.96. These results suggested the formation of an iron alkoxide lamellar structure with the formula: Fe2C2H5O5 and would be in agreement with TG measurements with an inorganic/organic ratio of about 3. Nevertheless, such a chemical composition significantly differs from that of iron alkoxides reported in other studies.22,25,27 Moreover, it is correlated to different structures (XRD pattern: peak at 8° instead of 11°) and chemical compositions (TG analysis: two weight losses instead of the only one). It has been reported earlier for the iron alkoxide formation that Fe3+ ions are reduced by EG molecules to form Fe2+ ions. These ions are then coordinated to EG molecules, which have lost protons, to form FeC2H4O2.22 However, further papers reported on the partial reduction of Fe3+ and thus on the coexistence of Fe2+ and Fe3+ in iron alkoxides with the formula Fe3(OCH2CH2O)4 or Fe(OCH2CH2O)x.22,25–27 None of these formulae agreed with the elemental analysis we have performed on our samples. Therefore, the oxidation state of iron cations and their chemical environment were further investigated by 57Fe Mössbauer spectrometry and X-ray photoemission spectroscopy (XPS) in combination with EELS measurements.
Mössbauer spectra recorded at 300 K and 77 K display a hyperfine structure with two weakly broadened lines, which can be rather well described by a single quadrupolar component, which is unambiguously attributed to a ferric species (Fig. S4†). Nevertheless, to obtain a good fit of the Mössbauer spectra, it was necessary to introduce two quadrupolar doublets ascribed to two different iron cations. Both sites are featured by very close values of the isomer shift (0.38 and 0.50 ± 0.01 mm s−1 at 300 K and 77 K, respectively) but significantly different values of quadrupolar splitting of 0.39 and 0.59 ± 0.02 mm s−1. In addition, similar widths for both contributions indicate that there are similar amounts of both iron species, assuming the same recoilless Lamb–Mössbauer factor. Such a description is fairly consistent with the presence of two iron(III) cations with slightly different chemical environments. Such an oxidation state of iron ions disagrees with the above reported studies on iron alkoxides.22,25,27 Because PLSs have been studied by Mössbauer spectroscopy after being exposed to air for several weeks, XPS measurements combined with EELS have been performed just after the PLS synthesis. The spectrum showed a broad peak centered at 707.1 eV corresponding to the Fe 2p signal (Fig. S5†). Deconvolution of this peak reveals two contributions centered at 707.8 eV and 709.5 eV, which can be ascribed to Fe(II) and Fe(III), respectively.46 The presence of Fe(II) is in agreement with the partial reduction of Fe(III) by EG.25–27 Furthermore, this peak becomes narrow and shifts to higher eV values corresponding to Fe(III) after heating the sample at 500 °C. These results confirm that PLSs formed during the RSN synthesis consist of a mixture of Fe(II) and Fe(III) and that they are highly sensitive to oxidation when exposed to air.
Therefore from all the above results, PLSs may be assumed to consist of an iron alkoxide complex with the following chemical formula: Fe2O3(OC2H4OH) with Fe(III) and Fe(II) cations bound to O and OC2H4O alkoxides. The EG molecules would not be fully deprotonated as reported in earlier published results but partially deprotonated.
Thermal stability study of the intermediate plate-like lamellar structures (PLSs)
In a second approach, a real time study of PLSs has been performed by TEM under electron irradiation that induces a thermal gradient within the sample (Fig. 4 and S6†). The global size of the PLS (about 25 to 100 nm) remains roughly the same, and one can easily identify the formation of a granular structure after 20 minutes of exposition (Fig. 4a and b). The contrast change during and after irradiation suggests a certain variation of the atomic mass at the nanometer scale. Micrographs recorded at higher magnification show that in the final stage, the PLSs consist of nanograins with a mean size of about 2–3 nm (inset Fig. 4b), embedded most likely in an amorphous structure. Such an evolution is similar to that reported with alkoxide nanoflakes after heat treatment above 500 °C.23 High resolution TEM micrographs show that each nanograin is a single crystal (Fig. 4c and S6†). Lattice fringes correspond to (hkl) orientations, which are correlated to the spinel structure of iron oxide (magnetite JCPDS file 19-629 or maghemite JCPDS file 39-1346). These nanograins are randomly oriented and exhibit different crystal orientations as confirmed by electron diffraction patterns, which display rings indexed to the spinel structure (Fig. 4d). Electron diffraction patterns have been captured upon exposure time to the electron beam over the course of 20 minutes. Indeed the evolution of the crystal structure of the PLS could be observed from the very early stages on the same area. Electron diffraction patterns taken progressively during irradiation showed that rings could be already observed after only 8 minutes (Fig. S7†). These results demonstrated that local heating by the electron beam induced an evolution of the PLS into 2D aggregated nanocrystals, which confirmed that the PLS decomposed with temperature and can be considered as a precursor of iron oxide nanoparticles. | ||
| Fig. 4 TEM micrographs of PLS (a) before and (b) after heating at 500 °C, inset is higher magnification of (b). (c) High resolution TEM micrograph and (d) the electron diffraction pattern of PLS after heating at 500 °C. | ||
To better understand the decomposition mechanisms of PLSs and how they are involved in the synthesis of the RSNs (Scheme 1), we have conducted an in situ TEM temperature study of the evolution of a PLS crystal structure with a precise control of temperature from 20 °C to 500 °C with a heating rate of 5 °C min−1 (Fig. S9†). Electron diffraction patterns (Fig. 5) and the corresponding TEM micrographs (Fig. S9†) have been recorded at 50 °C after 5 minutes of stabilization. As the electron beam generates a local increase in temperature that accelerates the transformation of the PLS, electron diffraction patterns have been recorded with the shortest exposition time as possible, few seconds in contrast to several minutes for the pure electron irradiation test mentioned above. Although PLSs are featured by a long range lamellar structure, the electron diffraction pattern recorded at 20 °C only displays diffuse and broad rings which are commonly ascribed to the carbon membrane. This observation agrees with the low density of PLSs and the lamellar structure whose (00l) reflection is oriented along their thickness. The intensity profiles of rings integrated radially show a significant variation in the crystal structure of the PLS with the increase of temperature (see the ESI†). Between 100 and 300 °C, the diffuse rings observed correspond to the (220) and (440) reflections of the spinel phase of iron oxide. A new ring corresponding to the (311) reflection is clearly observed after heating at 350 °C. Starting from 400 °C the (311) peak sharpens, whereas a new peak corresponding to the (422) orientation rises. Moreover, individual additional spots corresponding to crystals with well-defined orientations are observed above 450 °C and increase considerably in number at 500 °C.
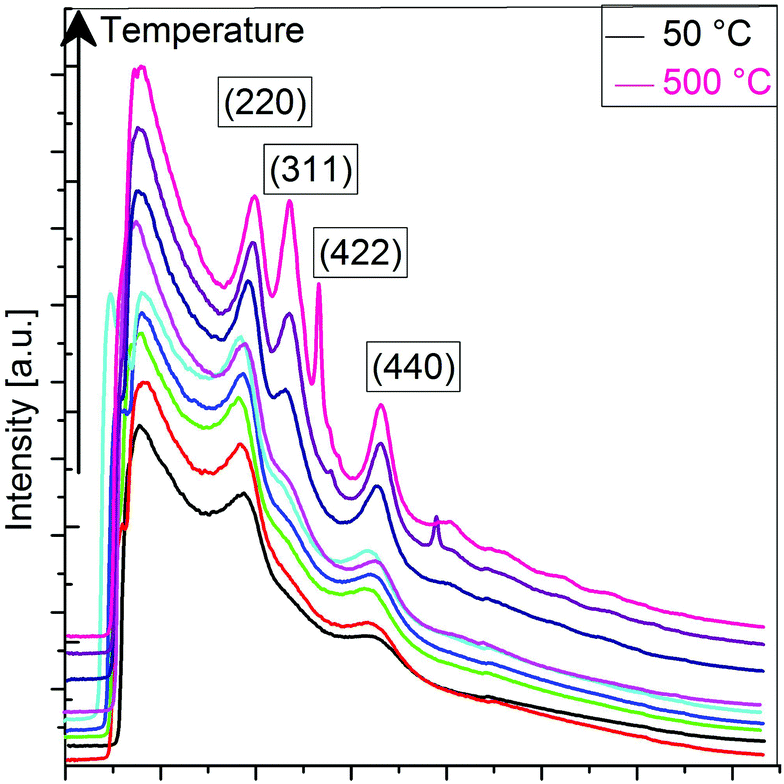 | ||
| Fig. 5 Diffraction rings’ intensity as evaluated from the SAED patterns acquired during the in situ thermal treatment from 50 °C to 500 °C. | ||
These results combined with TG and TD measurements confirmed unambiguously that PLSs decompose in iron oxide nanocrystals at a high temperature above 200 °C.
Furthermore, prolonged heating at 500 °C for 20 minutes shows some variations in intensity within the already existing rings (Fig. S9g†). Obviously, a longer exposure time to the electron beam favors the selective orientation of nanocrystals, which may be favored by the bidimensionality of plates.
Such a change in the structure is irreversible and no subsequent rearrangement in other polymorph crystals such as hematite is observed. High resolution TEM micrographs show that these clusters are single crystals and their size is about 3 nm. These results are confirmed by XRD patterns performed on PLSs after heat treatment at 500 °C, which display peaks indexed to the spinel structure of iron oxide (Fig. S3†). Rietveld refinement resulted in calculation of the crystal domain size of 3 nm. The value of the cell parameter (8.358 Å) is very close to maghemite and is correlated to the oxidation of Fe2+ upon exposure to air although the material has been exposed to possible electron beam induced reduction. These results are confirmed by EELS measurements performed on the same region prior to and after the thermal treatment (Fig. S5†). By evaluating the fine structure on the Fe L-edge, one can identify the presence of both Fe2+–Fe3+ within the native specimen at room temperature. The thermal treatment induces an energy loss shift within the Fe peak towards higher values, which indicates the partial transition from Fe2+ to Fe3+ under thermal constraints.
Discussion: reaction mechanism
The formation of RSNs is rather complex since the reaction proceeds through a multistep process (Scheme 1). The observation of the RSN synthesis at different reaction times showed that an amorphous phase is formed after 4 hours. Then, PLSs are observed after 5 h together with small RSNs. When the reaction time increases, the PLSs disappear while the RSN size increases until 7 h of reaction is reached. Interestingly, in the meantime, the nanograin size does not evolve that much but increases significantly later, between 7 and 13 h of reaction, which is correlated to the formation of a core cavity.In order to enhance our understanding of the synthesis mechanism, temperature and pressure have been both recorded during the reaction (Fig. 6). One may notice that the pressure is stable during the first two hours. Indeed, the pressure increases when a slower increase in temperature is observed at 130 °C. Therefore, it is correlated to an endothermic reaction corresponding to the decomposition of urea in ammonia leading to the formation of OH− + NH4+.19,47 These conditions are well known to favor the co-precipitation of iron hydroxides and further formation of iron oxide.
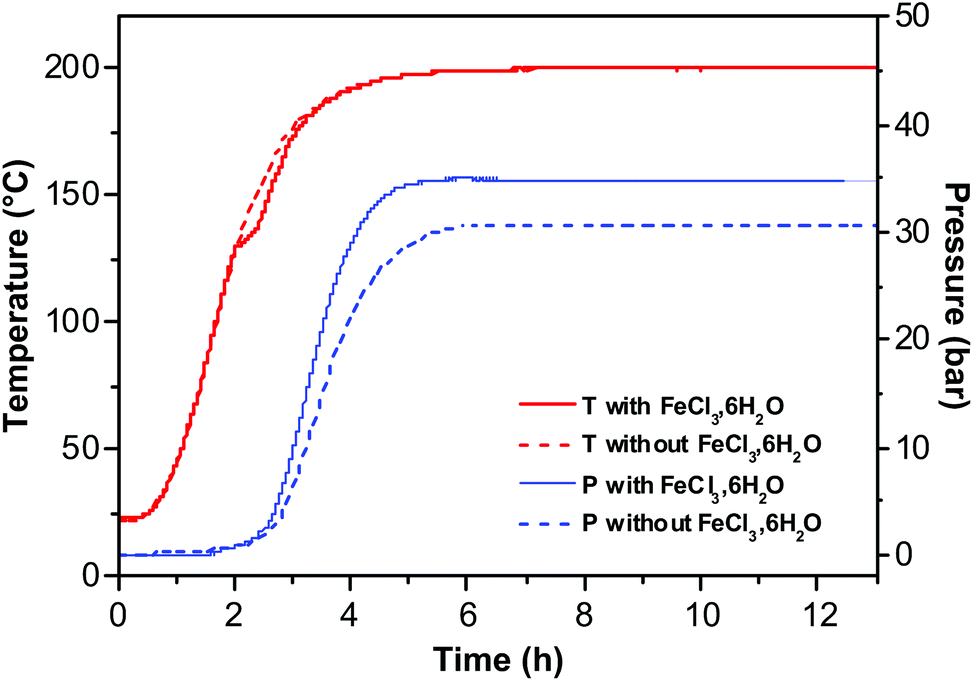 | ||
| Fig. 6 Temperature and pressure measurements as a function of reaction time with (solid line) and without (dotted line) an iron chloride precursor. | ||
Besides the formation of hydroxides and then oxides, ammonia also favors the deprotonation of ethylene glycol, which coordinates with Fe species and forms iron alkoxides.27,44 These results are correlated to the observation of PLSs and small RSNs after 5 h of reaction. Indeed, the non-observation of such an endothermic peak when no iron chloride is added in the reaction medium confirms the formation of iron hydroxide and iron alkoxides. The increase of pressure requires the same incubation time despite the presence of iron chloride. However, iron chlorides fasten the increase of pressure and higher maximum, which means that this reactant (introducing water) favors the decomposition of urea in ammoniac. The amorphous phase observed after 4 h of reaction may result from competitive interactions of ammonia with water and EG leading thus to amorphous intermediate iron alkoxide and iron hydroxide phases which are formed under kinetic control. Then, the observation of the well-structured PLS agrees with some organization process of the amorphous phase under thermodynamic control.
Finally, after 5 h, the transformation of the PLS and further growth of the RSN occur at a constant temperature and pressure. Therefore, urea is fully decomposed and all iron cations are coprecipitated or involved in the PLS which means that RSNs grow from the decomposition of the PLS as shown by some PLSs which are incorporated in RSNs (Fig. S2†). Indeed, the PLS acted as an intermediate iron precursor (or iron reservoir), which decomposes at higher temperature and is expected to induce heterogeneous nucleation of nanocrystals on previously formed RSNs by coprecipitation. Pressure and temperature conditions emphasized the oriented aggregation of nanograins and led to well-shaped RSNs with a narrow size distribution after 7 h of reaction. Such a mechanism may explain the increase of the mean RSN size whereas nanograins’ size remains quite constant between 5 and 7 h of reaction. After 7 h, there are no more PLSs available in the media and RSNs reach their maximum size. As the reaction time increases under these high pressure and temperature conditions, digestive inside–out Ostwald ripening proceeds to solubilization–recrystallization of grains.40 Nanograins located at the centre of RSNs undergo solubilization while the ones located close to the surface grow with the reaction time.
Conclusion
The preparation of raspberry-shaped nanostructures (RSNs) by a modified polyol method has been reported: the nanograin size of the RSN can be easily controlled by adjusting the reaction time and long reaction times were shown to favor inside–out Ostwald ripening leading to the core cavity. The detailed time-resolved study of the RSN formation revealed that the reaction mechanism involved an intermediate plate-like lamellar structure (PLS) which is different from those reported in other studies and acts as a second iron precursor/reservoir. To the best of our knowledge, such PLSs have never been observed in the course of only one-step RSN synthesis. The in situ HRTEM study of PLSs has shown that they decompose above 200 °C and that upon heating up to 500 °C, the alkoxide lamellar structure turns into iron oxide-clusters with selective crystal orientation. To summarize, the formation mechanism consists of, at first, the formation of an amorphous gel, then of small aggregates of coprecipitated nanograins triggered by the decomposition of urea, and finally the oriented attachment and coalescence of single crystal nanograins in RSNs upon the thermal decomposition of the intermediate PLS. The main difficulties in doping iron oxides in such RSNs are therefore correlated to the capacity of the doping element to form such intermediate phases. These results on the formation mechanism of RSNs will afford to better design RSNs, which are very promising for a wide range of applications such as waste recovering or catalysis which require magnetic materials with a large surface area. A first scaling test was shown very promising (Fig. S10†) and demonstrated that the synthesis process could be easily scaled up to produce up to 20 g of RSNs which is very interesting for future industrial applications.Acknowledgements
The authors acknowledge the funding from the Direction Générale de l'Armement (DGA) and French-German Research Institute of Saint-Louis (ISL). This material is also based on research funding supported by the Asian Office of Aerospace Research and Development, Grant # FA2386-15-1-4112.Notes and references
- M. Takafuji, S. Ide, H. Ihara and Z. Xu, Chem. Mater., 2004, 16, 1977 CrossRef CAS.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. Vander Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064 CrossRef CAS PubMed.
- M. Vural, O. Gerber, B. P. Pichon, S. Lemonnier, E. Barraud, L. C. Kempel, S. Begin-Colin and P. Kofinas, J. Mater. Chem. C, 2016, 4, 2345 RSC.
- Q. Q. Xiong, J. P. Tu, Y. Lu, J. Chen, Y. X. Yu, Y. Q. Qiao, X. L. Wang and C. D. Gu, J. Phys. Chem. C, 2012, 116, 6495 CAS.
- J. Ge, Y. Hu, M. Biasini, W. P. Beyermann and Y. Yin, Angew. Chem., Int. Ed., 2007, 46, 4342 CrossRef CAS PubMed.
- O. Gerber, B. P. Pichon, C. Ulhaq, J.-M. Grenèche, C. Lefevre, I. Florea, O. Ersen, D. Begin, S. Lemonnier, E. Barraud and S. Begin-Colin, J. Phys. Chem. C, 2015, 119, 24665 CAS.
- A. Kostopoulou, K. Brintakis, M. Vasilakaki, K. N. Trohidou, A. P. Douvalis, A. Lascialfari, L. Manna and A. Lappas, Nanoscale, 2014, 6, 3764 RSC.
- J. Ming, Y. Wu, L. Wang, Y. Yu and F. Zhao, J. Mater. Chem., 2011, 21, 17776 RSC.
- G. Ghosh, A. Vilchez, J. Esquena, C. Solans and C. Rodriguez-Abreu, Mater. Chem. Phys., 2011, 130, 786 CrossRef CAS.
- A. E. Gash, T. M. Tillotson, J. H. Satcher, J. F. Poco, L. W. Hrubesh and R. L. Simpson, Chem. Mater., 2001, 13, 999 CrossRef CAS.
- F. Fiévet, J.-P. Lagier and M. Figlarz, MRS Bull., 1989, 14, 29 CrossRef.
- W. Cheng, K. Tang, Y. Qi, J. Sheng and Z. Liu, J. Mater. Chem., 2010, 20, 1799 RSC.
- H. Deng, X. Li, Q. Peng, X. Wang, J. Chen and Y. Li, Angew. Chem., Int. Ed., 2005, 117, 2842 CrossRef.
- P. Hu, L. Yu, A. Zuo, C. Guo and F. Yuan, J. Phys. Chem. C, 2008, 113, 900 Search PubMed.
- M. Lin, H. Huang, Z. Liu, Y. Liu, J. Ge and Y. Fang, Langmuir, 2013, 29, 15433 CrossRef CAS PubMed.
- D. T. Nguyen and K.-S. Kim, J. Nanosci. Nanotechnol., 2013, 13, 5773 CrossRef CAS PubMed.
- S. Xuan, Y.-X. J. Wang, J. C. Yu and K. Cham-Fai Leung, Chem. Mater., 2009, 21, 5079 CrossRef CAS.
- Y. Zhu, W. Zhao, H. Chen and J. Shi, J. Phys. Chem. C, 2007, 111, 5281 CAS.
- C. Cheng, F. Xu and H. Gu, New J. Chem., 2011, 35, 1072 RSC.
- D. Larcher and R. Patrice, J. Solid State Chem., 2000, 154, 405 CrossRef CAS.
- S.-W. Cao and Y.-J. Zhu, J. Phys. Chem. C, 2008, 112, 12149 CAS.
- S.-W. Cao, Y.-J. Zhu, M.-Y. Ma, L. Li and L. Zhang, J. Phys. Chem. C, 2008, 112, 1851 CAS.
- S. Jin, H. Deng, D. Long, X. Liu, L. Zhan, X. Liang, W. Qiao and L. Ling, J. Power Sources, 2008, 196, 3887 CrossRef.
- M. Khosravi and S. Azizian, Adv. Powder Technol., 2014, 25, 1578 CrossRef CAS.
- X. Li, B. Zhang, C. Ju, X. Han, Y. Du and P. Xu, J. Phys. Chem. C, 2011, 115, 12350 CAS.
- D. Zhang, C. Lu, Y. Ni, Z. Xu and W. Zhang, CrystEngComm, 2013, 15, 4755 RSC.
- J.-J. Zhang, Y.-L. Chen, Y.-F. Sun, T. Huang and A.-S. Yu, RSC Adv., 2013, 3, 20639 RSC.
- A. P. Alivisatos, Science, 2000, 289, 736 CrossRef CAS PubMed.
- J. F. Banfield, S. A. Welch, H. Zhang, T. T. Ebert and R. L. Penn, Science, 2000, 289, 751–754 CrossRef CAS PubMed.
- S.-W. Cao, Y.-J. Zhu and J. Chang, New J. Chem., 2008, 32, 1526 RSC.
- T. He, D. Chen and X. Jiao, Chem. Mater., 2004, 16, 737 CrossRef CAS.
- H. Yuan, Y. Wang, S.-M. Zhou and S. Lou, Chem. Eng. J., 2011, 175, 555 CrossRef CAS.
- C. Cheng, Y. Wen, X. Xu and H. Gu, J. Mater. Chem., 2009, 19, 8782 RSC.
- W. Baaziz, B. P. Pichon, Y. Liu, J.-M. Greneche, C. Ulhaq-Bouillet, E. Terrier, N. Bergeard, V. Halte, C. Boeglin, F. Choueikani, M. Toumi, T. Mhiri and S. Begin-Colin, Chem. Mater., 2014, 26, 5063 CrossRef CAS.
- A. Sathya, P. Guardia, R. Brescia, N. Silvestri, G. Pugliese, S. Nitti, L. Manna and T. Pellegrino, Chem. Mater., 2016, 28, 1769 CrossRef CAS.
- J. Rodriguez-Carvajal, Physica B, 1993, 192, 55 CrossRef CAS.
- P. Thompson, D. E. Cox and J. B. Hastings, J. Appl. Crystallogr., 1987, 20, 79 CrossRef CAS.
- J. Teillet and F. Varret, Homemade MOSFIT program, Université du Maine Search PubMed.
- W. Baaziz, B. P. Pichon, S. Fleutot, Y. Liu, C. Lefevre, J.-M. Greneche, M. Toumi, T. Mhiri and S. Begin-Colin, J. Phys. Chem. C, 2014, 118, 3795 CAS.
- N. Guan, Y. Wang, D. Sun and J. Xu, Nanotechnology, 2009, 20, 105603 CrossRef PubMed.
- L.-P. Zhu, H.-M. Xiao, W.-D. Zhang, G. Yang and S.-Y. Fu, Cryst. Growth Des., 2008, 8, 957 CAS.
- J. J. E. Moreau, B. P. Pichon, M. Wong Chi Man, C. Bied, H. Pritzkow, J.-L. Bantignies, P. Dieudonné and J.-L. Sauvajol, Angew. Chem., Int. Ed., 2004, 43, 203 CrossRef CAS PubMed.
- B. P. Pichon, C. Leuvrey, D. Ihiawakrim, D. Tichit and C. Gérardin, J. Phys. Chem. C, 2011, 115, 23695 CAS.
- N. Chakroune, G. Viau, S. Ammar, N. Jouini, P. Gredin, M. J. Vaulay and F. Fievet, New J. Chem., 2005, 29, 355 RSC.
- L. S. Zhong, J. S. Hu, H. P. Liang, A. M. Cao, W. G. Song and L. J. Wan, Adv. Mater., 2006, 18, 2426 CrossRef CAS.
- R. Sinmyo, K. Hirose, H. St. C. O'Neill and E. Okunishi, Geophys. Res. Lett., 2006, 33, L12S13 CrossRef.
- T. Fan, D. Pan and H. Zhang, Ind. Eng. Chem. Res., 2011, 50, 9009 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: FTIR spectra of a gel-like structure and succinic acid; SEM image of RSN5 showing a PLS incorporated in an RSN; XRD pattern of the PLS after heating at 500 °C under nitrogen. Peaks are indexed to the spinelle phase of iron oxide; Mössbauer spectra of PLSs at (a) 300 K and (b) 77 K; EELS spectra of PLS (a) before and (b) after heat treatment at 500 °C; TEM micrographs of PLS after heat treatment at 500 °C. (a) and (b) At different magnification. (c) High resolution image showing lattice fringes of clusters; electron diffraction patterns of PLS after being exposed to an electron beam for (a) 0 min, (b) 0.5 min, (c) 1 min, (d) 2 min, (e) 4 min, (f) 6 min, (g) 10 min and (h) 20 min; heating rate of PLS from 20 °C to 500 °C; electron diffraction patterns of PLS and the corresponding area in TEM micrographs upon heating from 20 °C to 500 °C, SEM image after the first scaling up process test. See DOI: 10.1039/c6nr07567c |
| This journal is © The Royal Society of Chemistry 2017 |