
Effect of partial PEGylation on particle uptake by macrophages†
Lucero
Sanchez
,
Yi
Yi
and
Yan
Yu
*
Department of Chemistry, Indiana University, Bloomington, Indiana 47405, USA. E-mail: yy33@indiana.edu
First published on 2nd December 2016
Abstract
Controlling the internalization of synthetic particles by immune cells remains a grand challenge for developing successful drug carrier systems. Polyethylene glycol (PEG) is frequently used as a protective coating on particles to evade immune clearance, but it also hinders the interactions of particles with their intended target cells. In this study, we investigate a spatial decoupling strategy, in which PEGs are coated on only one hemisphere of particles, so that the other hemisphere is available for functionalization of cell-targeting ligands without the hindrance effect from the PEGs. The partial coating of PEGs is realized by creating two-faced Janus particles with different surface chemistries on opposite sides. We show that a half-coating of PEGs reduces the macrophage uptake of particles as effectively as a complete coating. Owing to the surface asymmetry, Janus particles that are internalized enter macrophage cells via a combination of ligand-guided phagocytosis and macropinocytosis. By spatially segregating PEGs and ligands for targeting T cells on Janus particles, we demonstrate that the Janus particles bind T cells uni-directionally from the ligand-coated side, bypassing the hindrance from the PEGs on the other hemisphere. The results reveal a new mechanistic understanding on how a spatial coating of PEGs on particles changes the phagocytosis of particles. This study also suggests a new design principle for therapeutic particles – the spatial decoupling of PEGs and cell-targeting moieties reduces the interference between the two functions while attaining the protective effect of PEGs for macrophage evasion.
1. Introduction
Innate immune cells, including macrophages and dendritic cells, fight infection by engulfing and internalizing pathogens through a process known as phagocytosis. The phagocytosis of synthetic particles also plays a significant role in determining the fate of vaccine and drug delivery systems.1,2 On the one hand, efficient and controllable phagocytosis is desired for vaccine carriers.3 On the other hand, phagocytosis is unfortunately responsible for the failure of many drug delivery particles that target cells other than the immune cells.4,5 Those particles are removed by macrophage cells before reaching their intended destination in the body. Therefore, understanding and controlling particle phagocytosis by immune cells is critical for developing successful therapeutic particles, but it remains a significant challenge in the field.In recent years, extensive studies have revealed that, surface functionalization, in addition to size, shape and mechanical rigidity, is a key factor that influences the phagocytic fate of particles.6–8 Engineering the surface chemistry of particles to prevent their internalization by immune cells has been the focus of many studies.9–14 One widely used strategy is to coat particles with a layer of polyethylene glycol (PEG).15–17 As particles move in the blood stream, opsonins such as antibodies and serum proteins adsorb on the particles and “mark” them for phagocytic removal. A dense coating of the hydrophilic PEG shields synthetic particles from macrophage internalization by creating a hydration layer that prevents the non-specific adsorption of opsonins onto particles and reduces cell adhesion.18–22 Despite the widespread use of PEGylation in the design of therapeutic particles, this strategy has important limitations. One major problem is that the protective PEG layer hinders the binding of targeting ligands to cell surface receptors. To form a dense protective layer, long PEG polymers are often needed. But these long polymers may conceal the cell-targeting ligands that are attached to their distal ends.23–26 The PEG layer may also interfere with the release of drugs and gene transfection reagents.27 To overcome the problems associated with PEGylation, “sheddable” PEGs that are tethered onto a particle surface via cleavable linkers have been used. After particles reach the target site, the PEGs are cleaved by external stimuli, such as changes in pH, temperature, or the application of light.28–30 Enzymes, including protease enzymes from the tumor microenvironment, have also been employed to trigger the removal of the PEG layer.31 However, the development of “sheddable” PEGs requires prior knowledge of the targeted cell environment so that the proper cleavable linkers can be selected. In addition, synthesis of “sheddable” PEGs often involves nontrivial chemistries. Therefore, alternative strategies that retain the effectiveness of PEGs but reduce their interference with cell targeting functions are necessary.
Here, we report a strategy involving the spatial decoupling of PEGs and other functions on a single particle. The spatial decoupling is realized by creating two-faced particles that display different functionalities on opposite sides. These particles are known as Janus particles. In our experimental design, the Janus particles are PEGylated on one hemisphere, leaving the other hemisphere available to be functionalized with cell-targeting ligands. We show that after antibody opsonization, half-PEGylated Janus particles evade macrophage cell internalization as effectively as fully PEGylated ones. The partial PEG coating changes the mechanism under which macrophages phagocytose the particles. Owing to the asymmetry of their surfaces, the half-PEGylated particles that do enter macrophage cells do so via a combination of ligand-guided phagocytosis and macropinocytosis. Based on this observation, we designed bi-functional Janus particles that display PEGs on one hemisphere and anti-CD3 antibodies on the opposite side for targeting Jurkat T cells. We demonstrate that the bi-functional Janus particles bind to T cells uni-directionally from the anti-CD3 coated hemisphere. Although we have previously shown that phagocytosis of particles depends on the surface coverage of cell-binding ligands on particles,32,33 this study is the first to incorporate PEGs into the design of Janus particles and demonstrate the effect of a partial PEG coating on the phagocytosis of particles. Our results suggest that the spatial decoupling of PEGs from cell-targeting moieties reduces the interference between the two functions while preserving the effectiveness of PEGylation in evading macrophage clearance. This finding provides new insights into potentially using spatial segregation of surface functionalities as a new parameter to control the interactions of therapeutic particles with immune cells.
2. Experimental
2.1 Reagents and cells
Monodisperse silica particles of 500 nm, 1.23 μm and 1.57 μm were used. Silica nanoparticles of diameter 500 nm were purchased from Cospheric LLC (Santa Barbara, CA). Monodisperse silica particles of diameters 1.23 μm and 1.57 μm (5% w/v) were purchased from Spherotech Inc. (Lake Forest, IL). (3-Aminopropyl) triethoxysilane (APTES), bovine serum albumin (BSA), biotin N-hydroxysuccinimide ester (biotin-NHS), immunoglobulin G (IgG) from rabbit serum, rhodamine b isothiocyanate (RITC)–dextran (70 kDa), RITC mixed isomers, α-cyano-4-hydroxycinnamic acid (CCA), polyethylene glycol (PEG) (MW = 1450 Da) and NaBH4 were acquired from Sigma-Aldrich (St Louis, MO). Alexa Fluor 488 succinimidyl esters, 3,3′-dioctadecyloxacarbocyanine perchlorate (DiO) and streptavidin were purchased from Invitrogen (Grand Island, NY). mPEG5k-SH and mPEG5k-biotin were purchased from Laysan Bio, Inc. (Arab, AL). 5-(N-Ethyl-N-isopropyl)-amiloride (EIPA) was purchased from Cayman Chemical (Ann Arbor, MI). IgG was biotinylated via the conjugation of biotin-NHS ester with primary amines. IgG-biotin and streptavidin used for trypan blue quenching experiments were fluorescently labeled with Alexa Fluor 488 succinimidyl esters following manufacturer instruction. RITC–APTES conjugates were synthesized by APTES (25 mM) reaction with RITC (40 mM) in 1 mL of ethanol at room temperature for 24 hours. Mus musculus macrophages (RAW 264.7) were purchased from ATCC (Manassas, VA) and cultured in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 100 units per mL penicillin, 100 μg mL−1 streptomycin, 10% fetal bovine serum (FBS) and 0.11 mg mL−1 (1 mM) sodium pyruvate. Jurkat T cells (E6.1) were a gift from Prof. Jay T. Groves (University of California, Berkeley) and originally purchased from ATCC (Manassas, VA). Jurkat T cells were cultured in RPMI 1640 complete growth media supplemented with 10% fetal bovine serum (FBS), 1 mM sodium pyruvate, 100 units per ml penicillin, and 100 μg ml−1 streptomycin. Imaging buffer (155 mM NaCl, 5 mM KCl, 2 mM CaCl2, 2 mM MgCl2·6H2O, 2 mM Na2H2PO4·H2O, 10 mM HEPES and 10 mM glucose) was adjusted to pH 7.2–7.4 and used for all cell experiments.2.2 Particle fabrication
Silica particles were treated with 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 piranha solution (H2SO4:H2O2 = 3:1 v:v) for 15 minutes at 75 °C and rinsed multiple times with milli-Q water. Silica particles were functionalized to display primary amine groups by reacting with 2% APTES in ethanol for 30 minutes at room temperature. Particles were rinsed thoroughly with ethanol and annealed in oven at 120 °C for 2 hours. After oven annealing, silanized particles were biotinylated in 1 M NaHCO3 solution (pH = 8.2) containing biotin-NHS (0.68 mg mL−1). The biotinylated silica particles were used as the no-PEG particles. To create the all-IgG particles, biotinylated silica particles were incubated with 6 μg mL−1 of streptavidin-Alexa 488 in 1× PBS buffer for 1.5 hours at room temperature and subsequently with 1.5 μg mL−1 of IgG-biotin-Alexa 488 for another 1.5 hours. To fabricate particles with uniformly distributed PEGs and anti-CD3 (referred to as “PEG-anti-CD3 uniform”), biotinylated silica particles were incubated with 6 μg mL−1 of streptavidin-Alexa 488 in 1× PBS buffer for 1.5 hours at room temperature and subsequently with a mixture of 0.23 μg mL−1 of biotin-anti-CD3 and 0.05 mg mL−1 mPEG5k-biotin for another 1.5 hours. PEG-anti-CD3 particles used for measuring T cell binding probability were fluorescently labeled with APTES–RITC prior to protein functionalization. The fluorescence labeling was carried out by incubating silica particles overnight with 5 mg mL−1 APTES–RITC in ethanol under constant rotation. In all incubation steps, 0.2 mg mL−1 of BSA was added in the solution to prevent non-specific adsorption of streptavidin on particles. Functionalized particles were rinsed multiple times with 1× PBS before use.
:1 piranha solution (H2SO4:H2O2 = 3:1 v:v) for 15 minutes at 75 °C and rinsed multiple times with milli-Q water. Silica particles were functionalized to display primary amine groups by reacting with 2% APTES in ethanol for 30 minutes at room temperature. Particles were rinsed thoroughly with ethanol and annealed in oven at 120 °C for 2 hours. After oven annealing, silanized particles were biotinylated in 1 M NaHCO3 solution (pH = 8.2) containing biotin-NHS (0.68 mg mL−1). The biotinylated silica particles were used as the no-PEG particles. To create the all-IgG particles, biotinylated silica particles were incubated with 6 μg mL−1 of streptavidin-Alexa 488 in 1× PBS buffer for 1.5 hours at room temperature and subsequently with 1.5 μg mL−1 of IgG-biotin-Alexa 488 for another 1.5 hours. To fabricate particles with uniformly distributed PEGs and anti-CD3 (referred to as “PEG-anti-CD3 uniform”), biotinylated silica particles were incubated with 6 μg mL−1 of streptavidin-Alexa 488 in 1× PBS buffer for 1.5 hours at room temperature and subsequently with a mixture of 0.23 μg mL−1 of biotin-anti-CD3 and 0.05 mg mL−1 mPEG5k-biotin for another 1.5 hours. PEG-anti-CD3 particles used for measuring T cell binding probability were fluorescently labeled with APTES–RITC prior to protein functionalization. The fluorescence labeling was carried out by incubating silica particles overnight with 5 mg mL−1 APTES–RITC in ethanol under constant rotation. In all incubation steps, 0.2 mg mL−1 of BSA was added in the solution to prevent non-specific adsorption of streptavidin on particles. Functionalized particles were rinsed multiple times with 1× PBS before use.
To fabricate Janus particles, monolayers of biotinylated silica particles were first prepared on pre-cleaned glass microscope slides using the Langmuir–Blodgett transfer method as described previously.34,35 The particle monolayers were coated sequentially with thin films of chromium (5 nm) and gold (30 nm) using an Edwards thermal evaporation system at a deposition rate of 3 Å s−1. Gold-coated Janus particles were sonicated off the glass slides and immediately PEGylated in 1× PBS buffer (pH = 7.2) containing 5 mg mL−1 mPEG5k-SH for 24 hours at room temperature with constant rotation. To create IgG–PEG Janus particles, 1.5 μg mL−1 of IgG-biotin-Alexa 488 molecules were conjugated onto the biotinylated hemisphere of the Janus particles using the streptavidin–biotin conjugation procedure as described above. To create all-PEG particles, 0.09 μg mL−1 mPEG5k-biotin molecules (6:1 streptavidin-to-mPEG5k-biotin ratio) were conjugated to the biotinylated hemisphere of the Janus particles using the same procedure as described above. For fabricating IgG–BSA particles, biotinylated IgG-Alexa 488 (1.5 μg mL−1) was first conjugated onto the silica hemisphere of Janus particles via streptavidin–biotin linkage and BSA was subsequently adsorbed non-specifically onto the exposed surface of particles. To create PEG-anti-CD3 Janus particles, 0.23 μg mL−1 of biotin-anti-CD3 molecules were conjugated onto the biotinylated hemisphere of the Janus particles using the streptavidin–biotin conjugation procedure as described above.
For experiments involving the non-specific coating of IgG on particles, all-PEG, half-PEG Janus and no-PEG particles were incubated separately in 1× PBS buffer solution containing 1.5 μg mL−1 unlabeled IgG for 1.5 hours at room temperature. After incubation, particles were rinsed and re-suspended in 1× imaging buffer prior to use.
2.3 Measurement of PEG grafting density
To collect PEGs from the gold-coated Janus particles for matrix assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-TOF MS) analysis, the PEG-coated Janus particles were first rinsed thoroughly in milli-Q water to ensure complete removal of residual PEG. The particles were then resuspended in an aqueous 0.6 M NaBH4 solution for 1 hour to cleave the thiol–gold linkage.36,37 After NaBH4 reduction, the mixture was spun down and the supernatant was collected. The sedimented particles were rinsed once in milli-Q water to ensure complete collection of the cleaved PEG. A Micro Float-A-Lyzer dialysis device with a molecular weight cutoff of 0.5–1 kDa (Spectrum Laboratories, Inc., Rancho Dominguez, CA) was used to remove NaBH4 from the cleaved PEG. After 24-hour dialysis, the PEG solution was freeze-dried and analyzed by MALDI-TOF MS as described below.The mPEG5k-SH concentration was measured using MALDI-TOF MS, following a previously reported method with slight modifications.38 The design of this measurement is that PEG1.5k was used as an internal standard and mixed at a known concentration with the mPEG5k-SH to be measured. The ratio of the MALDI-TOF signal intensity of mPEG5k-SH to the signal of the internal standard PEG1.5k is proportional to the ratio of their concentrations, [mPEG5k-SH]/[PEG1.5k]. For MALDI-TOF sample preparation, mPEG5k-SH was first dissolved in methanol. For obtaining the calibration plot, a total of seven mPEG5k-SH samples (5, 6.5, 8, 10, 11.5, 13 and 15 μM) were prepared. The following reagents were subsequently added to the PEG–methanol solution: 10 mg mL−1 α-cyano-4-hydroxycinnamic acid (CCA) as the matrix, 0.16 mg mL−1 NaCl as the cationization salt and 0.06 mg mL−1 PEG1.5k as the internal standard. All concentrations listed are the final concentrations. 1 μL of each sample mixture was air-dried on the target plate for analysis. MALDI-TOF MS measurements were carried out using a Bruker Autoflex III (Bruker Daltonics, Billerica, MA) equipped with a 200 Hz frequency tripled Nd:YAG laser (355 nm). All spectra were taken with 30% laser power with 1000 shots provided per sample spot. A minimum of 5 spectra was collected from each sample spot. The signal intensity of mPEG5k-SH (IPEG5k) was obtained by summing up the intensities from 13 peaks within the range of 5270–5798 m/z, and that of the internal standard PEG1.5k was from the summed intensities from 20 peaks within the range of 1496–2155 m/z. The signal peaks of PEG5k after NaBH4 treatment shifted slightly to the smaller m/z range likely due to the loss of the end groups after the NaBH4 reduction. Therefore, the signal intensity of cleaved mPEG5k was obtained from 13 peaks within the range of 5225–5780 m/z. The signal intensity of the mPEG5k-SH and cleaved PEG5k was normalized against that of the internal standard PEG1.5k and averaged over five spectra. The calibration plot of average IPEG5k/IPEG1.5kversus [mPEG5k-SH] was fit with a linear equation: 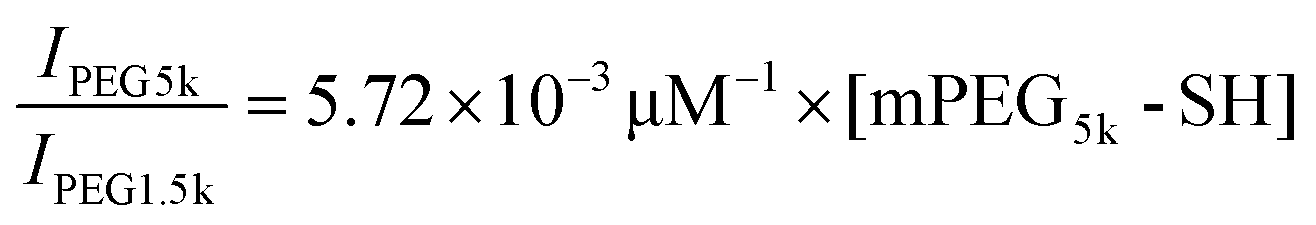 . Based on this linear equation, the concentration of cleaved mPEG5k-SH was estimated to be 10.5 μM. The total surface area of particles was calculated based on the fact that a total of 8 monolayers of particles (20 mm by 30 mm each) were used and that each Janus particle is half-coated with gold. As a result, the PEG surface density on the 500 nm Janus particles was estimated to be 0.084 ± 0.02 PEG molecules per nm2.
. Based on this linear equation, the concentration of cleaved mPEG5k-SH was estimated to be 10.5 μM. The total surface area of particles was calculated based on the fact that a total of 8 monolayers of particles (20 mm by 30 mm each) were used and that each Janus particle is half-coated with gold. As a result, the PEG surface density on the 500 nm Janus particles was estimated to be 0.084 ± 0.02 PEG molecules per nm2.
2.4 Live cell imaging
RAW 264.7 mus musculus macrophages were seeded on glass coverslips at a concentration of 0.1 million cells per mL in DMEM and allowed to grow overnight. Cells were serum starved for 3 hours prior to imaging and maintained at 37.0 °C in 1× imaging buffer during imaging. Jurkat T cell membrane was labeled with DiO via incubation with 5 μM DiO at 37 °C for 2 minutes. Live cell images were acquired at a frame interval time of 2 seconds on a Nikon Eclipse-Ti epi-fluorescence microscope equipped with an Andor iXon3 EMCCD camera and a Nikon Plan Apo 100×/1.49 N.A. objective. Laser scanning confocal fluorescence imaging was done using a Nikon A1R-A1 confocal microscope system equipped with a Nikon 100× oil-immersed objective and a Hamamatsu C11440 camera.2.5 Measurement of particle internalization efficiency
The trypan blue quenching method was used to differentiate internalized particles from non-internalized ones.39,40 Particles were conjugated with streptavidin-Alexa 488 in the trypan blue quenching experiments. Particles were gently mixed with cells in 1× imaging buffer and incubated for 30 minutes at 37.0 °C under static conditions (no constant shaking). The 30-minute incubation time was chosen to provide sufficient time for phagocytosis, based on our previous studies showing that most microparticles coated with IgG are internalized by macrophages within 10 minutes.41 To minimize cell endocytosis of trypan blue dyes, 0.4% trypan blue solution was incubated with cells on ice for 20 minutes to thoroughly quench the fluorescence from non-internalized particles. Samples were imaged at room temperature immediately after the trypan blue quenching step. Bright-field and fluorescence images were acquired at different z-positions to clearly differentiate particles that were internalized from ones bound to the cell surface. Particles that remained fluorescent were considered internalized, whereas particles that were in contact with cells in bright-field images but exhibited no fluorescence were considered membrane-bound. We defined internalization probability as: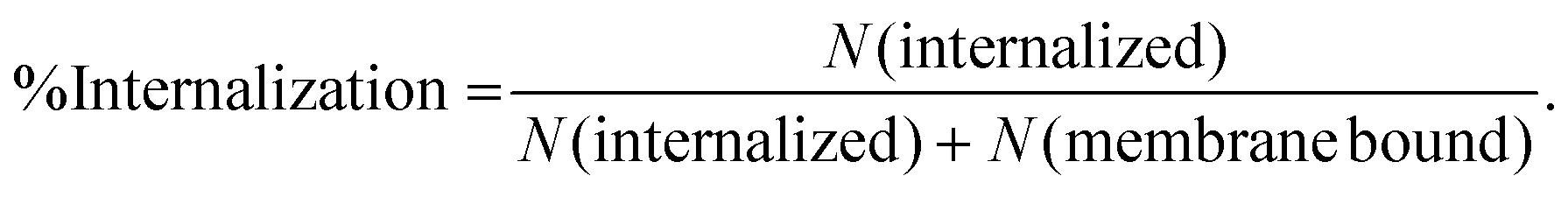
2.6 SEM imaging
Macrophage cells were seeded on glass coverslips at a concentration of 0.04 million cells per mL. After serum starvation, cells were incubated with particles for 30 minutes at 37.0 °C (5% CO2). After particle incubation, cells were fixed for 30 minutes on ice using ice-cold 0.1 M PIPES buffer (pH = 7.0) containing 2.5 v/v% glutaraldehyde. Fixed cells were rinsed with 1× imaging buffer to completely remove the chemical fixatives. After fixation, samples were submerged sequentially into ice-cold solutions containing 30% (v/v), 50%, 75%, 90%, 95% and 100% ethanol for 5 min each. Dehydration in 100% ethanol was repeated twice at room temperature. After dehydration in ethanol, fixed cell samples were subsequently dried using the critical point drying method. Fixed cell samples were imaged using low vacuum SEM (FEI Quanta 600F) with 2 kV voltage, 60 Pa pressure, 2–3 point size and 6–8 mm working distance.2.7 TEM imaging
Particles were incubated with cells for 15 minutes at 37.0 °C (5% CO2) before fixation. This short incubation time was necessary to capture the early stage of phagocytosis. Cells were gently rinsed with 1× PBS buffer three times and trypsinized to detach from the coverslips. Cells were fixed in three steps. First, cells were incubated with an ice-cold fixative mixture containing 1% OsO4 and 2.5% glutaraldehyde in 1× PBS for 2 minutes at room temperature. Second, the fixative mixture was replaced with fresh solution of the same composition and incubated with cells for 1 hour on ice. Last, the fixative mixture was replaced with 2% aqueous uranyl acetate and incubated with cells for 30 minutes on ice. After the uranyl acetate treatment, cells were rinsed with milli-Q water twice for 10 minutes each. Cell pellets were dehydrated sequentially in a series of ice-cold aqueous solutions containing 30% (v/v), 50%, 75%, 90%, 95% and 100% ethanol for 5 minutes each. Dehydration in 100 v/v% ethanol was repeated three times at room temperature. The dehydrated cell pellet was immersed sequentially in resin infiltration solutions that contain ethanol and Spurr's resin at various concentrations (2:1 (v:v), 1:1 and 1:2) for 30 minutes each at room temperature. Cells were then mixed with 100% Spurr's resin overnight. The cell pellet in Spurr's resin was cured at 60 °C for 18 hours prior to microtome sectioning. Sections were stained with 8% uranyl acetate and lead citrate prior to TEM imaging.
2.8 EIPA treatment
Cells were incubated with 10 μM EIPA in imaging buffer for 30 min at 37.0 °C (5% CO2) before the incubation with particles for another 30 min. Subsequent live cell and fixed cell TEM imaging were performed as described above.2.9 Dextran colocalization measurement
Cells were incubated with particles and 1 mg mL−1 RITC–dextran in imaging buffer for 30 minutes at 37.0 °C (5% CO2). The incubation time was the same as reported in previous studies that used dextran of the same molecular weight (70 kDa) for macropinocytosis studies.42–45 Prior to chemical fixation, cells were rinsed with ice-cold 1× PBS buffer to remove particles in the solution and then ice-cold low-pH buffer (0.1 M CH3COONa and 0.05 M NaCl, pH = 5.5) to remove residual dextran.46 Cells were imaged immediately after the removal of residual dextran.2.10 Measurement of particle binding to Jurkat T cells
Particles coated with anti-CD3 were incubated with T cells in 1× imaging buffer for 30 minutes at 37.0 °C. The particle–cell solution was gently mixed every 5 minutes. To measure the fraction of particles bound to T cells, particles that were fluorescently labeled with RITC were used. Membrane bound particles were distinguished from internalized ones by using a secondary antibody labeling method.47 For the secondary antibody labeling, T cells after incubation with particles were rinsed in 1× imaging buffer containing 1% BSA and kept on ice for 20 minutes. Next, chicken anti-mouse IgG-Alexa 647 was added to the T cell samples at a final concentration of 0.1 mg mL−1 and incubated on ice for 30 minutes. Finally, the T cell samples were rinsed three times with 1×PBS and kept on ice until imaging. For imaging, bright-field and epi-fluorescence images were acquired at different z-positions. Because secondary antibodies only label anti-CD3 molecules outside of cells, particles that were labeled by the secondary antibody (Alexa 647) but not attached to cells in bright-field images were considered unbound (Nunbound). Particles that were labeled with the secondary antibody and also overlapped with cells were counted as membrane-bound ones (Nmembrane-bound). Particles without antibody labeling but appearing in contact with cells were considered internalized (Ninternalized). The fraction of membrane-bound particles was calculated as: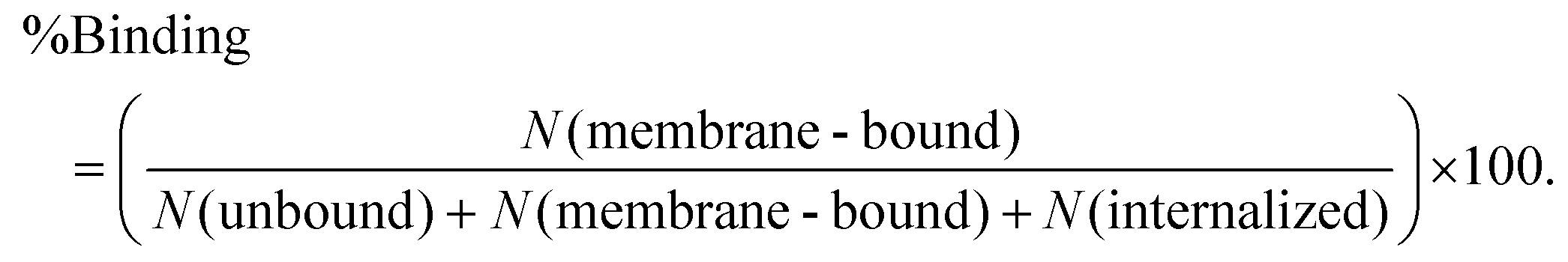
3. Results and discussion
3.1 Effect of half-PEGylation on the internalization probability of particles by macrophages
We first sought to investigate the effect of a partial coating of PEGs on the particle phagocytosis by comparing it with the effects of a complete PEG coating. Janus particles with asymmetric surface chemistries were made by depositing a thin layer of gold onto one hemisphere of silica particles. To make Janus particles that were half-coated with PEGs, PEGs (Mw = 5 kg mol−1) were conjugated on the gold caps via thiol–gold chemistry. The surface density of mPEG5k-SH on the gold-coated Janus nanoparticles was 0.08 ± 0.02 PEG molecules per nm2, measured from MALDI-TOF MS measurements (Fig. S1, ESI†). The mPEG5k molecules at this grafting density are expected to exhibit the “brush” conformation, based on values reported previously on PEGs of the same molecular weight.48 This “brush” confirmation is known to reduce non-specific protein adsorption more significantly than other chain conformation.49,50 To prepare the fully PEGylated particles (all-PEG) without altering the material composition of the Janus particles, we used streptavidin–biotin linkers to conjugate a second mPEG5k coating onto the silica hemisphere of particles whose gold side had already been PEGylated (Fig. 1a). To mimic the adsorption of opsonins on particles, polyclonal immunoglobulin G (IgG) antibodies were allowed to non-specifically adsorb onto the particle surfaces. We chose IgG because it is one of the four major corona-forming proteins that trigger phagocytosis.51,52 As shown in fluorescence images (Fig. 1b), IgG was adsorbed predominantly on the entire surface of no-PEG particles and the exposed silica surface of half-PEG particles, but it exhibited negligible accumulation on the all-PEG particles or the PEGylated side of the half-PEG particles. This confirms that the PEGylation effectively reduces non-specific IgG adsorption. The negligible adsorption of IgG on the all-PEG particles also indicates a complete coating of PEGs even though the PEGs on the two hemispheres are conjugated via two different types of linkers.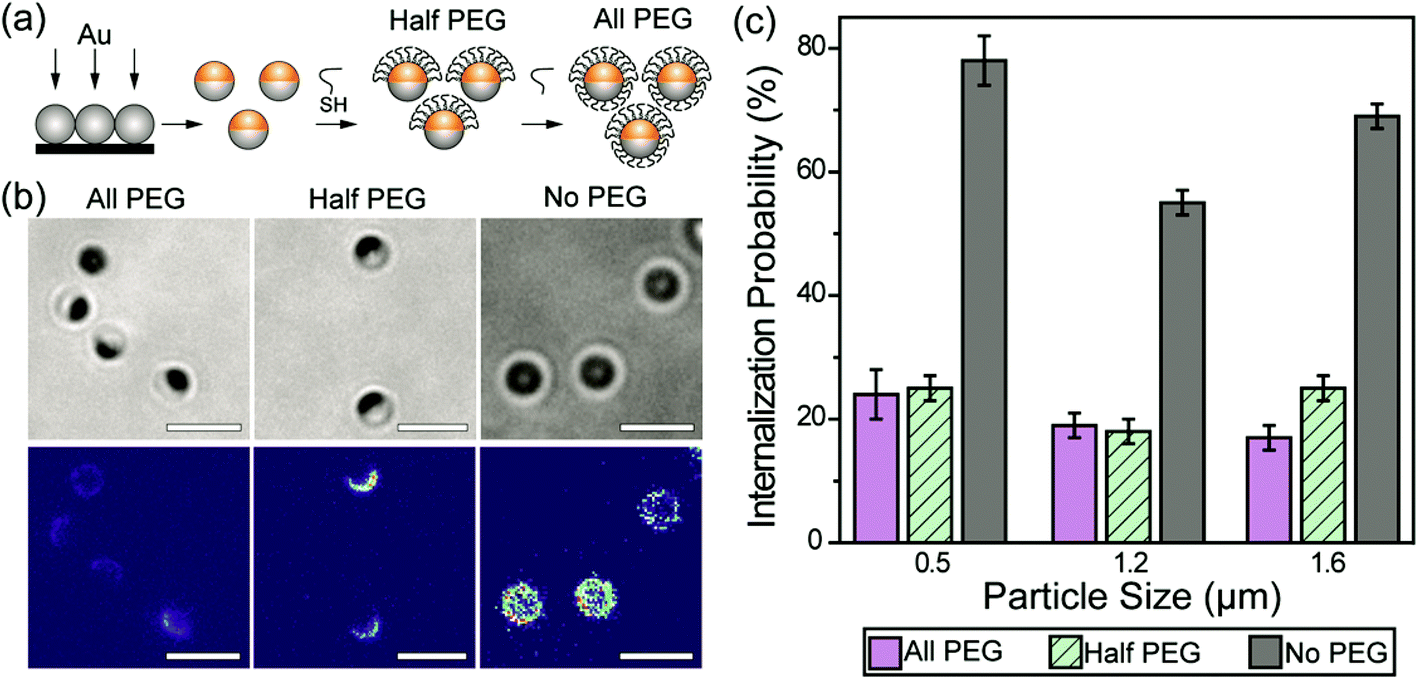 | ||
| Fig. 1 (a) Schematic illustration of the Janus particle fabrication procedure. (b) Bright-field and fluorescence images showing three types of Janus particles as indicated, and the non-specifically adsorbed IgG-Alexa488 (fluorescent). The fluorescence intensity of IgG is shown on the same color scale in all three images to highlight the difference in intensity. All particles are 1.6 μm in diameter. (c) Internalization probability of IgG-opsonized particles by RAW 264.7 macrophage cells. The 500 nm and 1.2 μm all-PEG and half-PEG Janus particles were statistically similar at the p < 0.05 level. The internalization of 1.6 μm all-PEG and half-PEG Janus particles were statistically different at the p < 0.05 level. Results for each type of particle were from 25–48 images in 2–6 independent samples. Scale bars: 3 μm. | ||
We next measured and compared the internalization probability of particles with different surface functionalization. The internalization probability was defined as the ratio of the number of internalized particles to the number of particles making contact with the cell membrane, which includes both internalized and membrane-bound particles. Based on this definition, the internalization probability does not change with the total number of particles bound to cell surfaces or added to cell samples (Fig. S2, ESI†). We found that the internalization probability of half-PEG particles by macrophages was as low as that of the all-PEG particles (Fig. 1c). The 500 nm and 1.2 μm all-PEG and half-PEG Janus particles were internalized at statistically similar probability (student T-test: p < 0.05). The internalization probability of 1.6 μm half-PEG Janus particles is low, but slightly higher than that of all-PEG particles (statistically different at the p < 0.05 level). In comparison, particles without any PEGylation exhibited a significantly higher internalization probability. These results show that a half-coating of PEGs is as effective in reducing macrophage clearance of IgG-opsonized particles. We have shown previously that the internalization probability of particles decreases with reduced surface coverage of IgG; particles with a partial coating of IgG are less likely to be internalized than fully coated particles.33 In order to differentiate the effect of the half-PEGylation from that of the half-coating of IgG, we prepared particles that were coated with IgG on one hemisphere and simply passivated with BSA on the other side. We refer to those particles as IgG–BSA particles. The half-PEG particles exhibited lower internalization probability than that of the IgG–BSA particles (Fig. S3, ESI†), indicating that the half coating of PEG plays an important role in reducing the macrophage uptake of the Janus particles. It is important to point out that we used optical imaging instead of flow cytometry to measure the internalization probability of particles, because the metal cap on Janus particles blocks fluorescence from the silica hemisphere differently depending on the particle orientation, which interferes with the cell sorting in flow cytometry. A large number of particles (typically a few hundreds to thousands) were analyzed for each sample to ensure the rigor of the image analysis.
3.2 Effect of half-PEGylation on particle–macrophage cell binding
We next investigated the effect of the asymmetric surface functionalization on the particle–macrophage interaction. To avoid the potential effect of IgG desorption, IgG-biotin was conjugated on the biotinylated-silica hemisphere via streptavidin linkers. In live cell imaging, we observed that the dynamics of binding between Janus particles and macrophage cells depend on the initial particle–cell orientation. Particles that faced the cells with their IgG-coated side were directly pulled to the cell by thin filopodia-like membrane protrusions (Video 1, ESI†). Particles that were initially in other orientations were rapidly rotated while they were being pulled and finally bound to the cell from the IgG coated side (Fig. 2a; Video 2, ESI†). This uni-directionality in the particle–cell binding is likely caused by the asymmetric presentation of ligands. Interestingly, we also observed that while the majority of the internalized IgG–PEG Janus particles entered cells individually, particles were occasionally gathered into clusters by macrophages during their cell entry. A representative process is shown in Fig. 2b. As multiple particles in close proximity were being pulled toward the cell body, they were clustered and rotated in such a way that their PEGylated hemispheres were oriented toward the inside of the cluster while the IgG-coated hemispheres were oriented toward the outside. This orientation of particles within a cluster effectively shielded their PEGylated hemispheres from the cell. This led to higher internalization probabilities for the particle clusters than that for single IgG–PEG particles (Fig. 2c). The particles were clustered only occasionally when their local concentration was high, so it has negligible effect on the overall internalization probability of the IgG–PEG particles. Nevertheless, it represents an interesting process through which cells were able to bypass the PEG coating that hinders cell–particle adhesion. | ||
| Fig. 2 Dynamics of cell-binding and internalization of single IgG–PEG Janus particles as well as clusters of IgG–PEG Janus particles. Bright-field images showing the rotation of a 1.6 μm IgG–PEG Janus particle during cell binding (a) and the clustering of 1.2 μm IgG–PEG Janus particles (b) during the binding and internalization process. The PEGylated hemisphere of Janus particles appears dark in all images due to the gold coating. (c) A graph showing the internalization efficiency of single and clustered Janus particles in comparison to all-IgG control particles. Internalization probability was determined from live cell images of 10–18 videos from 2–6 independent samples. Scale bars: 3 μm. | ||
3.3 Effect of half-PEGylation on the phagocytosis mechanism
A small fraction of the half-PEG particles still managed to enter macrophages, as indicated by our results of internalization probability. We then investigated how the half-PEGylation affects the mechanism of phagocytosis of particles. Macrophages are known to internalize particles using two different mechanisms. One is zipper-like phagocytosis, in which ligand–receptor binding drives membrane engulfment around particles.53 This mechanism is expected to result in tight phagosomes that follow the shape of the ingested particles. IgG-triggered phagocytosis has been reported to follow the zipper mechanism.54 The second mechanism is macropinocytosis, in which induced large cell membrane ruffles engulf particles in a non-specific “cell drinking” process.55 Different from the tight membrane around particles as expected in the zipper mechanism, membrane ruffles in macropinocytosis do not follow the shape of the engulfed particle. The resulted phagosomes are also more spacious than the size of the particles inside.56 These characteristics are typically used to identify the phagocytosis mechanism for particles. To investigate the morphology of membrane protrusion around the IgG–PEG Janus particles, we resolved the fine membrane structures using scanning and transmission electron microscopies separately. The membrane morphology on the two hemispheres of the IgG–PEG Janus particles was found to be dramatically different (Fig. 3 and S4, ESI†). Protruded membranes tightly enveloped the IgG-coated side, forming “phagocytic cups” that are characteristic of the zipper-like phagocytosis.56–58 In contrast, the cell membrane had little to no contact with the PEG-coated side of particles, but rather exhibited large membrane ruffles (Fig. 3). The membrane ruffles indicate macropinocytosis. This result suggests the IgG–PEG Janus particles likely employ a combination of zipper-like phagocytosis and macropinocytosis to enter macrophages due to the different surface properties on the two hemispheres.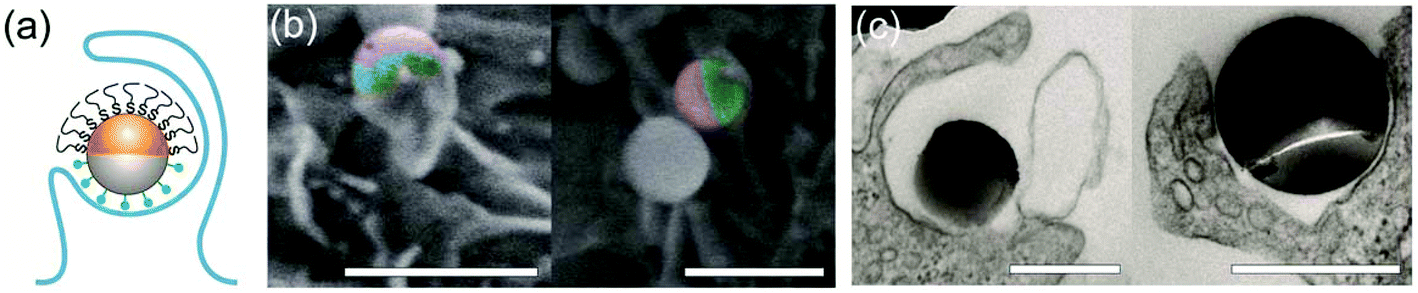 | ||
| Fig. 3 Asymmetric membrane protrusion around IgG–PEG Janus particles. A schematic illustration (a), pseudo-colored SEM images (b), and TEM images (c) demonstrate the different membrane morphology on the two hemispheres of IgG–PEG Janus particles. In the pseudo-colored SEM images, the PEGylated hemispheres are shown in purple and the IgG-coated hemispheres are shown in green. Scale bars: 1 μm in SEM images (b) and 500 nm in TEM images (c). | ||
We carried out a series of experiments to confirm the involvement of macropinocytosis in the uptake of IgG–PEG Janus particles. The first one was to measure the tightness of phagosomes, because phagosomes formed via macropinocytosis are expected to be more spacious than ones formed via ligand-guided phagocytosis. The fraction of phagosome area occupied by the internalized particles was quantified in transmission electron microscopy (TEM) images and used to indicate the tightness of phagosomes. Phagosomes containing IgG–PEG Janus particles are more spacious than ones containing all-IgG particles, as the fraction of particle-occupied area per phagosome is 0.60 ± 0.03 for the former and 0.87 ± 0.02 for the latter (Fig. 4a and b). We then treated macrophage cells using a macropinocytosis inhibitor 5-(N-ethyl-N-isopropyl)-amiloride (EIPA). EIPA inhibits cell membrane ruffling and thus macropinocytosis by blocking the Na+/H+ exchangers.59–63 EIPA treatment significantly reduced the formation of spacious phagosomes containing the IgG–PEG Janus particles, but had no effect on the phagosomes for all-IgG particles. The fraction of particle-occupied area per phagosome after inhibition of macropinocytosis is 0.84 ± 0.02, similar to that for all-IgG particles after the same drug treatment (0.80 ± 0.03). This clearly demonstrates that macropinocytosis in involved in the internalization of IgG–PEG particles (Fig. 4c and d). The EIPA treatment also significantly reduced the overall internalization probability of IgG–PEG Janus particles of all three sizes, 500 nm, 1.2 μm and 1.6 μm (Fig. 5). It did not completely diminish the particle phagocytosis, probably due a heterogeneous response of cells to EIPA inhibition, because previous studies have also shown that macropinocytosis of macrophages and cancer cells is not inhibited completely even at EIPA concentration as high as 75 μM.64,65 The involvement of macropinocytosis in the uptake of IgG–PEG Janus particles was also confirmed in a dextran-uptake assay.43,46 Because dextran of large molecular weight (70 kDa is commonly used) enters macrophages via macropinocytosis, its colocalization with engulfed particles inside phagosomes is typically used as an indicator for macropinocytosis.43,66,67 In our experiments, fluorescently labeled dextran of 70 kDa was incubated with Janus particles during macrophage internalization. Dextran was observed to colocalize with IgG–PEG Janus particles in some phagosomes, providing additional evidence that these spacious phagosomes are the result of macropinocytosis (Fig. S5, ESI†).
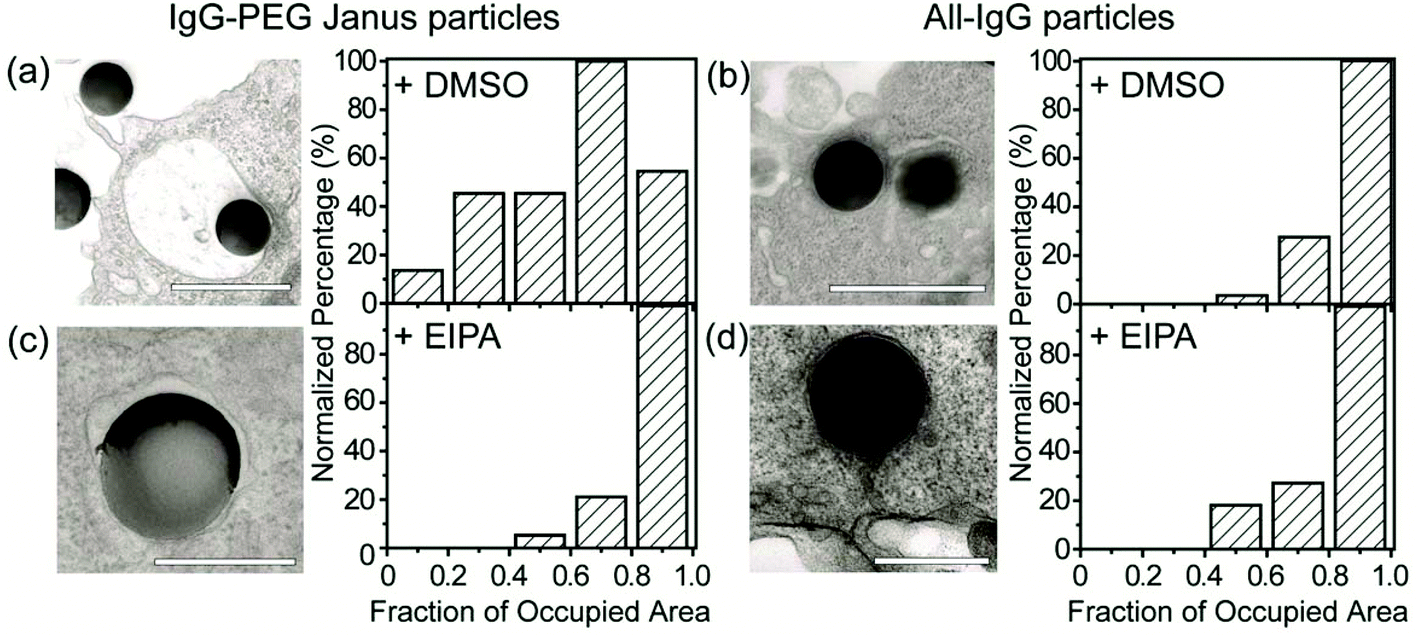 | ||
| Fig. 4 Quantification of tightness of phagosomes. (a) and (c) are for 500 nm IgG–PEG Janus particles in DMSO-treated (a) and EIPA-treated macrophages (c). (b) and (d) are for 500 nm all-IgG control particles in DMSO-treated (b) and EIPA-treated macrophages (d). All bar graphs show the distribution of phagosome areas occupied by single particles. The fraction of occupied area is 0.60 ± 0.03 (average ± SEM) for (a), 0.84 ± 0.02 for (b), 0.87 ± 0.02 for (c) and 0.80 ± 0.03 for (d). A total of N = 57 particles were analyzed for (a), N = 38 for (b), N = 24 for (c) and N = 16 for (d). Scale bars: 1 μm in (a) and (b), and 500 nm in (c) and (d). | ||
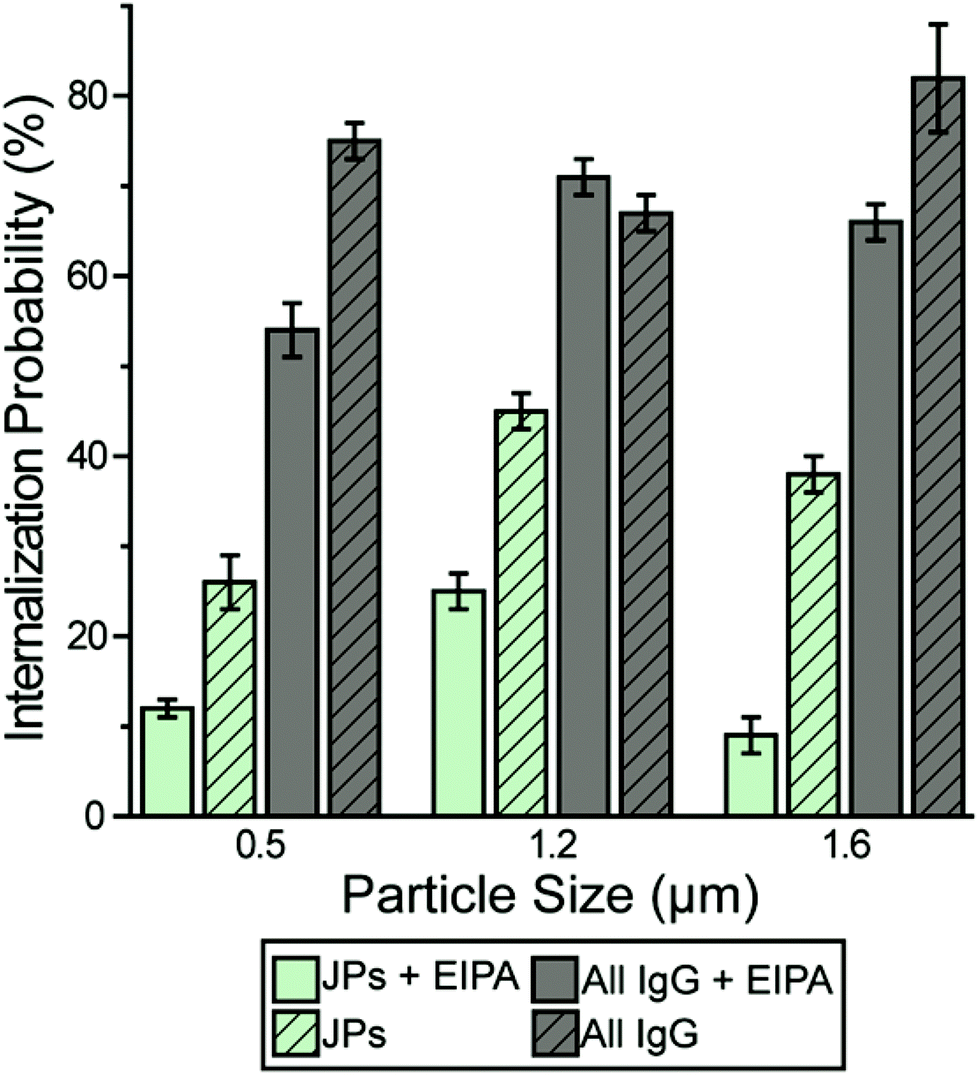 | ||
| Fig. 5 Macrophage internalization probability of particles following 10 μM EIPA treatment. The bar graph compares the internalization probability of IgG–PEG and all-IgG particles with or without EIPA treatment. Each data set was obtained from 18–54 images in 3–6 independent samples. | ||
All the results combined demonstrate that the phagocytosis of IgG–PEG Janus particles involves two distinct mechanisms. The IgG-coated side is first engulfed via ligand-guided zipper-like phagocytosis, and subsequently the PEGylated side is enveloped by macropinocytosis. Because most macropinocytosis events do not lead to successful engulfment of particles as observed in both our previous work and this study,32,33 a majority of half-PEG particles are not internalized, as shown in our results (Fig. 1c). We have shown in a previous study that particles half-coated with T cell-binding ligands are internalized by T cells via a combination of ligand-guided membrane protrusion and membrane ruffling.32 The previous work and this present study were done with Janus particles of different surface functionalities and with different cell systems. The observation of the same particle internalization mechanism in the two different systems suggests that the combined mechanism of both ligand-guided phagocytosis and macropinocytosis in the internalization of Janus particles is a general phenomenon caused by the asymmetric presentation of ligands.
3.4 Spatial segregation of PEGylation and cell-targeting ligands on particles
To demonstrate the cell-targeting capability of half-PEG Janus particles, we functionalized their silica hemisphere with biotinylated anti-CD3 antibodies, which bind to T cell receptors. The gold hemisphere was coated with PEGs. We observed that the Janus particles bound to T cells uni-directionally on the anti-CD3 coated side (Fig. 6a). Even when the particles were initially oriented with the PEGylated side facing the cells, the T cells actively rotated the particles to eventually bind the anti-CD3 coated side. This was similar to the observed rotation of IgG–PEG particles by macrophages (Fig. S6, ESI†). Among all PEG-anti-CD3 Janus particles added to T cells, 26.4 ± 0.8% of them bound to the cell membrane after a 30 min incubation period. In contrast, only 4.2 ± 0.7% of particles uniformly coated with PEGs and anti-CD3 (referred to as “PEG-anti-CD3 uniform particles”) were determined to be membrane-bound (Fig. 6c). The sharp difference indicates that the spatial segregation of PEGs and anti-CD3 on particles effectively enhances particle binding to cells by reducing the hindrance effect from PEGs on the targeting side.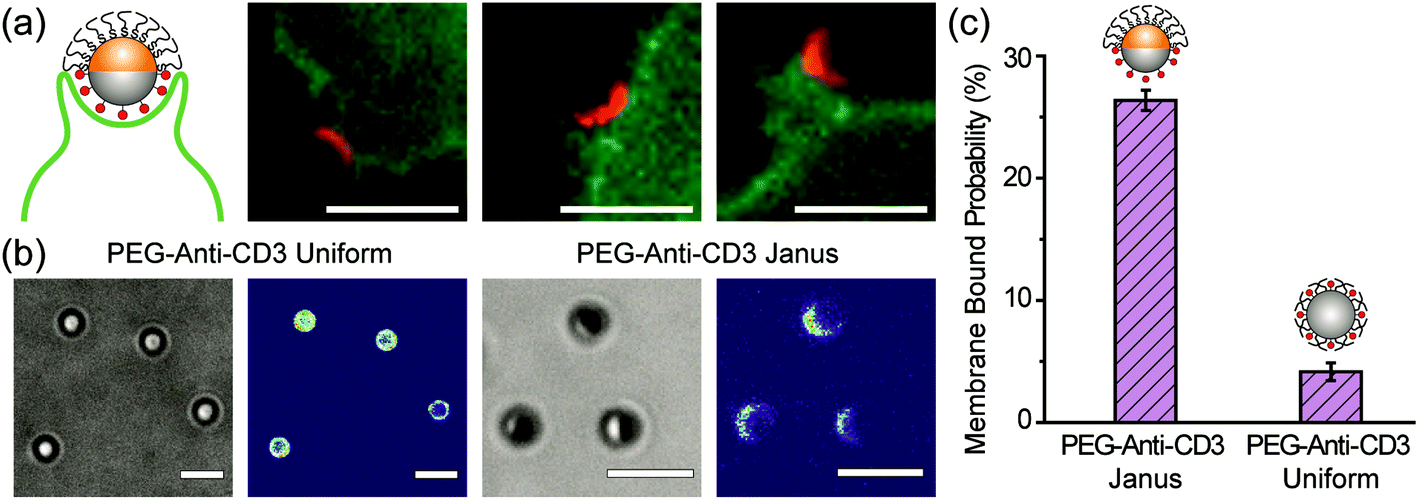 | ||
| Fig. 6 (a) Uni-directional binding of the anti-CD3 coated hemisphere (shown in red) of 1.6 μm Janus particles to Jurkat T cells (shown in green). The other hemisphere of the Janus particles is PEGylated. The images are representative of the entire population of 16 particles and 12 cells. (b) Bright-field and fluorescence images show the conjugation of anti-CD3 labeled with Alexa 568 on the PEG-anti-CD3 Janus and uniform particles. Fluorescence intensity of anti-CD3 Alexa 568 is shown on the same scale in both images for direct comparison. (c) The fraction of membrane-bound particles is 26.4 ± 0.8% for PEG-anti-CD3 Janus particles and 4.2 ± 0.7% for particles uniformly coated with PEGs and anti-CD3 (PEG-anti-CD3 uniform). A total of N = 258 T cells were analyzed for the PEG-anti-CD3 Janus particles and N = 82 cells for the uniformly coated particles. Scale bars: 3 μm. | ||
4. Conclusions
In this study, we report that PEGylation on only one hemisphere of particles reduces macrophage internalization of particles as effectively as a full coating of PEGs. We also reveal that the partial PEG coating changes the conventional internalization mechanism of particles. For Janus particles that are coated with PEGs on one side and IgG (antibodies for triggering phagocytosis) on the other, two mechanisms are involved in one cell entry event. The initial engulfment of the IgG-coated side occurs through zipper-like membrane protrusion that is guided by ligands on the particles. The PEGylated hemisphere is subsequently enveloped by large membrane ruffles through a macropinocytosis process. Even though we reported previously how a partial coating of cell-binding ligands changes phagocytosis,32 this study provides the first evidence showing the effectiveness of half-PEGylation on reducing the macrophage phagocytosis of particles, and reveals the mechanism underlying this phenomenon. Moreover, our results suggest that spatially separating PEGs and cell targeting ligands on two hemispheres of single particles may overcome the challenge that PEGs sterically hinder the binding of particles to targeted cells. Of course, further studies on the PEGylated Janus particle system will be necessary to fully evaluate the effectiveness of this spatial decoupling strategy. First, an important question to explore is how the internalization of half-PEGylated particles is affected by the surface grafting density of PEGs. Macrophage uptake efficiency of particles is known to depend on the conformation of PEG chains at different grafting density.23,48 It is possible that a half coating of PEGs becomes inefficient in preventing macrophage uptake below a threshold surface density. Second, this study uses IgG as the opsonin, one of the four major corona-forming proteins, but it alone may not fully mimic the complex composition of protein corona formed in vivo.68 Third, macrophage cells, the cell system used in this study, also represent only one type of phagocytic cells that remove drug delivery particles in the blood stream.69 In future studies, phagocytosis of the half-PEGylated Janus particles will be investigated in blood serums and with other phagocytic immune cell systems. Systematic studies that vary the length and surface density of PEGs will also be necessary. These results altogether will evaluate whether or not a complete coating of PEGs is necessary for preventing the immune clearance of synthetic particles in vivo.Acknowledgements
The authors gratefully acknowledge Dr. B. Stein and Dr. D. Morgan of the IUB Electron Microscopy Imaging Center and Dr. J. Powers at the IUB Light Microscopy Imaging Center, and Dr. J. Karty at the IUB Mass Spectrometry Facility for assistance with instrument use and helpful discussions. The idea of MALDI-TOF MS measurement was conceived based on helpful discussions with Mr. M. Kappler in the Yu group. Fabrication and characterization of Janus particles were performed at the Nanoscale Characterization Facility at Indiana University. This work was supported by the National Science Foundation under the Award no. CBET-1554078 and Indiana University. L. S. was supported by the Graduate Training Program in Quantitative and Chemical Biology under Award no. T32 GM109825 and Indiana University. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.Notes and references
- D. E. Owens and N. A. Peppas, Int. J. Pharm., 2006, 307, 93–102 CrossRef CAS PubMed.
- R. Gref, Y. Minamitake, M. T. Peracchia, V. Trubetskoy, V. Torchilin and R. Langer, Science, 1994, 263, 1600–1603 CAS.
- P. Pacheco, D. White and T. Sulchek, PLoS One, 2013, 8, 1–9 Search PubMed.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS PubMed.
- C.-M. J. Hu, R. H. Fang, B. T. Luk and L. Zhang, Nanoscale, 2014, 6, 65–75 RSC.
- J. A. Champion and S. Mitragotri, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 4930–4934 CrossRef CAS PubMed.
- J. A. Champion and S. Mitragotri, Pharm. Res., 2009, 26, 244–249 CrossRef CAS PubMed.
- N. Doshi, A. S. Zahr, S. Bhaskar, J. Lahann and S. Mitragotri, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21495–21499 CrossRef CAS PubMed.
- I. García, A. Sánchez-Iglesias, M. Henriksen-Lacey, M. Grzelczak, S. Penadés and L. M. Liz-Marzán, J. Am. Chem. Soc., 2015, 137, 3686–3692 CrossRef PubMed.
- P. L. Rodriguez, T. Harada, D. A. Christian, D. A. Pantano, R. K. Tsai and D. E. Discher, Science, 2013, 339, 971–975 CrossRef CAS PubMed.
- B. T. Luk and L. Zhang, J. Controlled Release, 2015, 220, 600–607 CrossRef CAS PubMed.
- C.-M. J. Hu, L. Zhang, S. Aryal, C. Cheung, R. H. Fang and L. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 10980–10985 CrossRef CAS PubMed.
- S. Faraasen, J. Vörös, G. Csúcs, M. Textor, H. P. Merkle and E. Walter, Pharm. Res., 2003, 20, 237–246 CrossRef CAS.
- B. T. Luk, C.-M. Jack Hu, R. H. Fang, D. Dehaini, C. Carpenter, W. Gao and L. Zhang, Nanoscale, 2014, 6, 2730–2737 RSC.
- C. D. Walkey, J. B. Olsen, H. Guo, A. Emili and W. C. W. Chan, J. Am. Chem. Soc., 2012, 134, 2139–2147 CrossRef CAS PubMed.
- K. P. Garcia, K. Zarschler, L. Barbaro, J. A. Barreto, W. O'Malley, L. Spiccia, H. Stephan and B. Graham, Small, 2014, 10, 2516–2529 CrossRef CAS PubMed.
- M. Yoshida, K. H. Roh, S. Mandal, S. Bhaskar, D. W. Lim, H. Nandivada, X. P. Deng and J. Lahann, Adv. Mater., 2009, 21, 4920–4925 CrossRef CAS PubMed.
- M. Q. Zhang, T. Desai and M. Ferrari, Biomaterials, 1998, 19, 953–960 CrossRef CAS PubMed.
- E. Wischerhoff, K. Uhlig, A. Lankenau, H. G. Boerner, A. Laschewsky, C. Duschl and J.-F. Lutz, Angew. Chem., Int. Ed., 2008, 47, 5666–5668 CrossRef CAS PubMed.
- C. D. Walkey and W. C. W. Chan, Chem. Soc. Rev., 2012, 41, 2780–2799 RSC.
- P. Kingshott, H. Thissen and H. J. Griesser, Biomaterials, 2002, 23, 2043–2056 CrossRef CAS PubMed.
- T. Arakawa and S. N. Timasheff, Biochemistry, 1985, 24, 6756–6762 CrossRef CAS PubMed.
- Q. Dai, C. Walkey and W. C. W. Chan, Angew. Chem., Int. Ed., 2014, 53, 5093–5096 CrossRef CAS PubMed.
- P. E. Saw, J. Park, E. Lee, S. Ahn, J. Lee, H. Kim, J. Kim, M. Choi, O. C. Farokhzad and S. Jon, Theranostics, 2015, 5, 746–754 CrossRef CAS PubMed.
- T. Wang, V. A. Petrenko and V. P. Torchilin, J. Nanomed. Nanotechnol., 2012, S4, 1–6 Search PubMed.
- A. A. Kale and V. P. Torchilin, J. Liposome Res., 2007, 17, 197–203 CrossRef CAS PubMed.
- H. Hatakeyama, H. Akita and H. Harashima, Adv. Drug Delivery Rev., 2011, 63, 152–160 CrossRef CAS PubMed.
- W. Gao, R. Langer and O. C. Farokhzad, Angew. Chem., Int. Ed., 2010, 49, 6567–6571 CrossRef CAS PubMed.
- C. Clawson, L. Ton, S. Aryal, V. Fu, S. Esener and L. Zhang, Langmuir, 2011, 27, 10556–10561 CrossRef CAS PubMed.
- Z. Poon, D. Chang, X. Zhao and P. T. Hammond, ACS Nano, 2011, 5, 4284–4292 CrossRef CAS PubMed.
- T. J. Harris, G. von Maltzahn, M. E. Lord, J.-H. Park, A. Agrawal, D.-H. Min, M. J. Sailor and S. N. Bhatia, Small, 2008, 4, 1307–1312 CrossRef CAS PubMed.
- Y. Gao and Y. Yu, J. Am. Chem. Soc., 2013, 135, 19091–19094 CrossRef CAS PubMed.
- Y. Gao and Y. Yu, Langmuir, 2015, 31, 2833–2838 CrossRef CAS PubMed.
- S. Reculusa and S. Ravaine, Chem. Mater., 2003, 15, 598–605 CrossRef CAS.
- B. van Duffel, R. H. A. Ras, F. C. De Schryver and R. A. Schoonheydt, J. Mater. Chem., 2001, 11, 3333–3336 RSC.
- S. M. Ansar, F. S. Ameer, W. Hu, S. Zou, C. U. Pittman and D. Zhang, Nano Lett., 2013, 13, 1226–1229 CrossRef CAS PubMed.
- M. Yuan, S. Zhan, X. Zhou, Y. Liu, L. Feng, Y. Lin, Z. Zhang and J. Hu, Langmuir, 2008, 24, 8707–8710 CrossRef CAS PubMed.
- H. Chen and M. He, J. Am. Soc. Mass Spectrom., 2005, 16, 100–106 CrossRef CAS PubMed.
- S. E. A. Gratton, P. A. Ropp, P. D. Pohlhaus, J. C. Luft, V. J. Madden, M. E. Napier and J. M. DeSimone, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11613–11618 CrossRef CAS PubMed.
- T. Patino, J. Soriano, L. Barrios, E. Ibanez and C. Nogues, Sci. Rep., 2015, 5, 1–12 Search PubMed.
- L. Sanchez, P. Patton, S. M. Anthony, Y. Yi and Y. Yu, Soft Matter, 2015, 11, 5346–5352 RSC.
- S. Falcone, E. Cocucci, P. Podini, T. Kirchhausen, E. Clementi and J. Meldolesi, J. Cell Sci., 2006, 119, 4758–4769 CrossRef CAS PubMed.
- C. Commisso, R. J. Flinn and D. Bar-Sagi, Nat. Protoc., 2014, 9, 182–192 CrossRef CAS PubMed.
- I. Rasmussen and F. Vilhardt, J. Virol., 2015, 89, 1851–1866 CrossRef PubMed.
- N. Araki, T. Hatae, T. Yamada and S. Hirohashi, J. Cell Sci., 2000, 113, 3329–3340 CAS.
- J. Mercer and A. Helenius, Science, 2008, 320, 531–535 CrossRef CAS PubMed.
- M. E. Handley, G. Pollara, B. M. Chain and D. R. Katz, J. Immunol. Methods, 2005, 297, 27–38 CrossRef CAS PubMed.
- J. L. Perry, K. G. Reuter, M. P. Kai, K. P. Herlihy, S. W. Jones, J. C. Luft, M. Napier, J. E. Bear and J. M. DeSimone, Nano Lett., 2012, 12, 5304–5310 CrossRef CAS PubMed.
- T. Waku, M. Matsusaki, T. Kaneko and M. Akashi, Macromolecules, 2007, 40, 6385–6392 CrossRef CAS.
- R. Gref, M. Lück, P. Quellec, M. Marchand, E. Dellacherie, S. Harnisch, T. Blunk and R. H. Müller, Colloids Surf., B, 2000, 18, 301–313 CrossRef CAS PubMed.
- Y. Zhang, A. D. Hoppe and J. A. Swanson, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 19332–19337 CrossRef CAS PubMed.
- J. A. Swanson and A. D. Hoppe, J. Leukocyte Biol., 2004, 76, 1093–1103 CrossRef CAS PubMed.
- J. A. Swanson and S. C. Baer, Trends Cell Biol., 1995, 5, 89–93 CrossRef CAS PubMed.
- G. Kaplan, Scand. J. Immunol., 1977, 6, 797–807 CrossRef CAS PubMed.
- P. R. Hoffmann, A. M. deCathelineau, C. A. Ogden, Y. Leverrier, D. L. Bratton, D. L. Daleke, A. J. Ridley, V. A. Fadok and P. M. Henson, J. Cell Biol., 2001, 155, 649–660 CrossRef CAS PubMed.
- J. A. Swanson, Nat. Rev. Mol. Cell Biol., 2008, 9, 639–649 CrossRef CAS PubMed.
- D. R. Shaw and F. M. Griffin, Nature, 1981, 289, 409–411 CrossRef CAS PubMed.
- F. M. Griffin, J. A. Griffin and S. C. Silverstein, J. Exp. Med., 1976, 144, 788–809 CrossRef PubMed.
- J. Mercer and A. Helenius, Nat. Cell Biol., 2009, 11, 510–520 CrossRef CAS PubMed.
- M. C. Freeman, C. T. Peek, M. M. Becker, E. C. Smith and M. R. Denison, mBio, 2014, 5, 1–10 CrossRef PubMed.
- M. Koivusalo, C. Welch, H. Hayashi, C. C. Scott, M. Kim, T. Alexander, N. Touret, K. M. Hahn and S. Grinstein, J. Cell Biol., 2010, 188, 547–563 CrossRef CAS PubMed.
- B. Masereel, L. Pochet and D. Laeckmann, Eur. J. Med. Chem., 2003, 38, 547–554 CrossRef CAS PubMed.
- J. Mercer and A. Helenius, Curr. Opin. Microbiol., 2012, 15, 490–499 CrossRef PubMed.
- C. Commisso, S. M. Davidson, R. G. Soydaner-Azeloglu, S. J. Parker, J. J. Kamphorst, S. Hackett, E. Grabocka, M. Nofal, J. A. Drebin, C. B. Thompson, J. D. Rabinowitz, C. M. Metallo, M. G. Vander Heiden and D. Bar-Sagi, Nature, 2013, 497, 633–637 CrossRef CAS PubMed.
- S. BoseDasgupta and J. Pieters, PLoS Pathog., 2014, 10, 1–18 Search PubMed.
- M. C. Kerr and R. D. Teasdale, Traffic, 2009, 10, 364–371 CrossRef CAS PubMed.
- J. P. Lim and P. A. Gleeson, Immunol. Cell Biol., 2011, 89, 836–843 CrossRef CAS PubMed.
- M. P. Monopoli, C. Aberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 779–786 CrossRef CAS PubMed.
- S. Pooyan, B. Qiu, M. M. Chan, D. Fong, P. J. Sinko, M. J. Leibowitz and S. Stein, Bioconjugate Chem., 2002, 13, 216–223 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6nr07353k |
| This journal is © The Royal Society of Chemistry 2017 |