
Elucidating the influence of materials-binding peptide sequence on Au surface interactions and colloidal stability of Au nanoparticles†
Zak E.
Hughes‡
a,
Michelle A.
Nguyen‡
b,
Yue
Li
c,
Mark T.
Swihart
c,
Tiffany R.
Walsh
 *a and
Marc R.
Knecht
*b
*a and
Marc R.
Knecht
*b
aInstitute for Frontier Materials, Deakin University, Geelong, Victoria 3216, Australia. E-mail: tiffany.walsh@deakin.edu.au
bDepartment of Chemistry, University of Miami, 1301 Memorial Drive, Coral Gables, Florida 33146, USA. E-mail: knecht@miami.edu
cDepartment of Chemical and Biological Engineering, University of Buffalo, The State University of New York, Buffalo, New York 14260, USA
First published on 1st December 2016
Abstract
Peptide-mediated synthesis and assembly of nanostructures opens new routes to functional inorganic/organic hybrid materials. However, understanding of the many factors that influence the interaction of biomolecules, specifically peptides, with metal surfaces remains limited. Understanding of the relationship between peptide sequence and resulting binding affinity and configurations would allow predictive design of peptides to achieve desired peptide/metal interface characteristics. Here, we measured the kinetics and thermodynamics of binding on a Au surface for a series of peptide sequences designed to probe specific sequence and context effects. For example, context effects were explored by making the same mutation at different positions in the peptide and by rearranging the peptide sequence without changing the amino acid content. The degree of peptide-surface contact, predicted from advanced molecular simulations of the surface-adsorbed structures, was consistent with the measured binding constants. In simulations, the ensemble of peptide backbone conformations showed little change with point mutations of the anchor residues that dominate interaction with the surface. Peptide-capped Au nanoparticles were produced using each sequence. Comparison of simulations with nanoparticle synthesis results revealed a correlation between the colloidal stability of the Au nanoparticles and the degree of structural disorder in the surface-adsorbed peptide structures for this family of sequences. These findings suggest new directions in the optimization and design of biomolecules for in situ peptide-based nanoparticle growth, binding, and dispersion in aqueous media.
Introduction
Growing interest in biomimetic materials has led to the use of both traditional (e.g. proteins and peptides)1,2 and unconventional (e.g. peptoids and biopolymers)3,4 biomolecules to synthesize and assemble inorganic materials. Exploiting materials recognition properties of biomolecules could allow the creation of functional nanoassemblies that incorporate both inorganic and organic components.5–7 In particular, bionanocombinatorics is an emerging field that aims to control the assembly of nanomaterials using biological interactions. Peptides provide a unique pathway to achieving this objective. For peptides that bind to inorganic surfaces through multiple noncovalent interactions, both enthalpic and entropic components of binding affinity must be considered when predicting and interpreting binding behavior.8 Biocombinatorial selection techniques used to identify material-binding peptides generally select for overall binding affinity and do not provide information on the components of binding affinity separately. Based on their binding affinity, such peptides have been used to cap and direct the growth of various inorganic nanoparticles under mild conditions.9–11 Specifically, Au nanoparticles are of high interest due to their potential applications in catalysis,12–14 energy harvesting and storage,15,16 sensors,17,18 optics/plasmonics,19,20 and as components in nanoassemblies.21–23 Additionally, Au nanoparticles exhibit a localized surface plasmon resonance (LSPR), which provides a simple means of detecting changes in the size, shape, and aggregation state of the nanostructures via changes in the LSPR optical extinction spectrum.While experimental measurements of peptide binding affinity for a target surface are possible, experimentally characterizing the peptide–metal interface at the molecular level is difficult, if not impossible. As a result, molecular simulation approaches, along with complementary experimental data, are essential for separating out enthalpic and entropic contributions of peptide binding, elucidating configurations of the peptide–metal interface, and linking these to the peptide-surface binding affinity. We previously demonstrated that material-binding peptides can be categorized as either enthalpically- or entropically-driven binders.8 Enthalpically driven material-binding peptides tend to have several anchor residues (those residues with strong and persistent non-covalent interactions with the surface) spaced throughout the sequence. These anchor the biomolecule to the surface, resulting in a limited number of surface-binding configurations. However, strong materials-binding affinity can also arise from the presence of a large number of relatively-weakly-bound adsorbed peptide configurations. Such entropically-driven binding peptides have fewer anchor residues, allowing them to adopt many different binding conformations on the inorganic surface.8 The conformational entropic contribution to binding associated with the large number of possible configurations contributes to higher binding affinity with fewer strong anchor residues compared to enthalpically driven binders.
We previously classified the Pd4 peptide (TSNAVHPTLRHL), originally isolated based on its affinity for Pd,24 as an entropically driven binder on Au.8 This sequence contains only two anchor residues (His6 and His11), resulting in many distinct surface-adsorbed conformations. In comparison, other sequences such as AuBP1 (WAGAKRLVLRRE) and AuBP2 (WALRRSIRRQSY), originally isolated as Au-binding peptides,25 were classified as enthalpic binders. They have five and four anchor residues, respectively, with a significantly smaller number of thermally-accessible conformations on Au compared to Pd4.25 While these broad classifications advanced the understanding of how peptides bind to inorganic surfaces, the effects of the global and local sequence on binding remain poorly understood. In this regard, the importance of sequence, and not just overall residue content, is clearly important; however, how the anchor residues work in concert with neighboring residues and how altering the global sequence of the peptide changes materials binding affinity remain unknown.
Here, Pd4 was employed as a parent peptide to identify the effects of amino acid sequence and the local context of the anchor residues, and to explore how these effects act synergistically to mediate binding affinity for the Au surface. Our modifications included point mutation of histidine anchor residues with alanine (with the aim to diminish localized binding affinity), scrambling of the peptide sequence, and clustering of the anchor residues at the N-terminus (Table 1). The Pd4 peptide was initially studied due to the limited number of anchor residues (two in total)8 and its known affinity for Au. Peptide binding affinities on Au were quantified using Quartz Crystal Microbalance (QCM) measurements, which revealed significant changes in binding affinity between sequences. This information was combined with findings from advanced molecular simulation studies, in which the Boltzmann-weighted ensemble of adsorbed conformations was predicted for each of the sequences. Taken together, the experimental and computational results reveal underlying causes for the observed variation in peptide-surface binding. We also prepared peptide-capped Au nanoparticles using each sequence and characterized them by UV-vis spectroscopy and transmission electron microscopy (TEM). These results demonstrated that the local context of the anchor residues, as well as the global peptide sequence, plays a key role in controlling material affinity. Understanding of these sequence effects can also be exploited to enhance the quality and colloidal stability of peptide-capped Au nanoparticles. Thus, these studies advance efforts to identify heuristics for the design of peptides with affinity and potential selectivity for inorganic surfaces. The ability to rationally tune the binding strength and binding mode on specific surfaces would be extremely valuable for creating peptide-organized structures for optical, catalytic, and nanoparticle self-assembly applications.
| Peptide | Sequence | ΔG (kJ mol−1) | θ (%) |
|---|---|---|---|
| a From QCM experiments: adsorption Gibbs free energy (ΔG) values, as well as surface coverage at the highest peptide concentration studied (15 μg mL−1 peptide). Values are given as mean ± one standard deviation from at least three independent experiments. | |||
| Pd4 |

|
−31.4 ± 0.1 | 79.8 ± 0.5 |
| Pd4-1 |

|
−27.2 ± 0.2 | 41.2 ± 1.6 |
| Pd4-2 |

|
−30.7 ± 0.1 | 75.0 ± 0.9 |
| Pd4-3 |

|
−32.0 ± 0.8 | 82.8 ± 5.0 |
| Pd4-4 |

|
−33.1 ± 0.2 | 88.8 ± 0.8 |
| Pd4-5 |

|
−35.5 ± 0.2 | 95.3 ± 0.4 |
Experimental
Chemicals
All Fmoc-protected Wang resins, Fmoc-protected amino acids, N,N′-diisopropylethylamine (DIPEA), piperidine, 1-hydroxybenzotriazole hydrate (HOBt hydrate), and N-[(1H-benzotriazol-1-yl)(dimethylamino)methylene]-N-methylmethanaminium hexafluoro-phosphate N-oxide (HBTU) were purchased from Advanced ChemTech. N,N′-dimethylformamide, methanol, diethyl ether, ammonium hydroxide (20%), and hydrogen peroxide (30%) were obtained from BDH Chemicals, while trifluoroacetic acid (TFA) and triisopropylsilane (TIS) were acquired from Alfa Aesar. HAuCl4 and NaBH4 were purchased from Sigma-Aldrich and Acros Organics, respectively. Finally, acetonitrile was purchased from EMD Millipore. All chemicals were used as received without further purification. Milli-Q water (18 MΩ cm) was used for all experiments.Peptide synthesis
Peptides were generated using a TETRAS peptide synthesizer (CreoSalus) following standard solid-phase Fmoc protocols.26 A cleavage cocktail of 95% TFA, 2.5% TIS, and 2.5% water was employed to cleave the peptides from the resins. The crude peptides were purified via reversed-phase high performance liquid chromatography (HPLC) with a 0.1% TFA aqueous solution and acetonitrile gradient. Finally, the purified peptides were confirmed by matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry. All peptide solutions were lyophilized and stored at −80 °C prior to use.QCM analysis
All QCM measurements, including those for dissipation energy, were conducted on a Q-Sense E4 instrument (Biolin Scientific) following previously reported procedures.27 Standard polycrystalline gold crystal sensors were employed and cleaned according to manufacturer protocols prior to use. In brief, the gold QCM sensors were first subjected to UV-ozone exposure, followed by immersion in a 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 (v/v/v) water/ammonium hydroxide/hydrogen peroxide solution and an additional UV/ozone treatment. Afterward, an aqueous peptide solution at a concentration between 2.5 to 15.0 μg mL−1 was flowed over the sensor surface at a rate of 150 μL min−1. The frequency change and dissipation energy were recorded for 30 min to ensure saturation. This frequency change is directly related, via the Sauerbrey equation, to the mass of peptide adsorbed, from which binding kinetic parameters can be determined.28 All measurements were performed at 22.5 °C, and the pH of the peptide solutions ranged from 3.9–4.2.
:1:1 (v/v/v) water/ammonium hydroxide/hydrogen peroxide solution and an additional UV/ozone treatment. Afterward, an aqueous peptide solution at a concentration between 2.5 to 15.0 μg mL−1 was flowed over the sensor surface at a rate of 150 μL min−1. The frequency change and dissipation energy were recorded for 30 min to ensure saturation. This frequency change is directly related, via the Sauerbrey equation, to the mass of peptide adsorbed, from which binding kinetic parameters can be determined.28 All measurements were performed at 22.5 °C, and the pH of the peptide solutions ranged from 3.9–4.2.
Molecular simulations
To investigate the effect of the variations of the parent Pd4 peptide sequence on the peptide-surface adsorption, the conformational ensemble of the adsorbed peptides was predicted using Replica Exchange with Solute Tempering (REST)29,30 molecular dynamics (MD) simulations. Each simulation comprised a single peptide chain adsorbed at the Au(111) interface (represented by a five-layer Au slab) in the presence of liquid water. Three-dimensional periodic boundary conditions were applied throughout. The polarizable GolP-CHARMM force-field,31,32 the CHARMM22* force-field,33,34 and the modified TIP3P potential35,36 were used to describe the interactions involving the Au surface, the peptide, and water, respectively. All REST simulations were performed at a thermal temperature of 300 K. Additional details about the simulation procedures and analysis of the simulation trajectories are provided in the ‘Additional methodology’ section in the ESI.† Note that previous studies37 have shown that the binding modes of peptide sequences at the described planar surface are very similar to the binding modes on Au NPs dispersed in solution. Thus REST simulations of peptides at the aqueous Au (111) interface are able to provide useful insight into the principal contact modes and conformational entropic contributions of the same peptide adsorbed onto the surface of Au NPs, which is in turn related to colloidal stability of the Au NPs.At present, capturing the true complexity of the polycrystalline Au substrate used in the QCM experiments using atomistic MD simulation is not possible. A viable means of addressing this challenge is to focus on the dominant Au crystal orientations present on the polycrystalline Au surface. Based on previously-published analyses,38 the Au(111) surface is expected to be the dominant facet at the aqueous interface of polycrystalline Au. In particular, because our study encompasses seventeen different peptide sequences (see below), for pragmatic reasons we focused our REST MD simulations solely on the aqueous Au(111) interface. Probing the binding of these peptides at other Au surfaces, such as the Au(100) interface that is predicted to be the next most dominant facet on the polycrystalline Au surface, would be interesting. However, for flat planar Au surfaces (i.e. not those present on nanoparticle surfaces), previous studies suggest that the Au(100) native surface relaxes to the Au(100) (5 × 1) reconstruction,38 which features a lateral hexagonal arrangement of Au atoms in the surface plane that is very similar to that of Au(111). Due to this lateral hexagonal arrangement, our previous studies indicate that peptide adsorption to the aqueous Au(100) (5 × 1) surface is similar to that corresponding to Au(111). In particular, the anchor residues identified on Au(111) were generally found to be strong-binding residues on Au(100) (5 × 1).38 We therefore suggest that the Au(111) surface is an appropriate structural model for comparison with experimental data obtained from polycrystalline Au QCM sensor surfaces.
Special attention was paid to the protonation state of the histidine residues (see Results and discussion). For the original sequence and each mutated sequence (see Table 1), we separately considered the deprotonated (charge-neutral) and protonated (positively-charged) states of each histidine present in that sequence. For each sequence that contained only one histidine, this resulted in two different REST simulations: one with the histidine modeled as protonated (denoted PS1) and one with the histidine deprotonated (denoted PS0). For sequences with two histidines, this resulted in four different REST simulations: (1) no histidines protonated (denoted PS0), (2) only the first histidine in the sequence protonated (denoted PS1A), (3) only the second histidine in the sequence protonated (denoted PS1B), and (4) both histidines protonated (denoted PS2). In summary, we performed a total of seventeen REST simulations to cover all possibilities for the mutant sequences.
Au nanoparticle synthesis
Peptide-capped Au nanoparticles were prepared according to synthetic methods established by Li et al.12 The fabrication of peptide-capped Au nanoparticles with a Au:peptide ratio of 2 is described; however, changes to the reagent volumes (to equal a total volume of 5.0 mL) were employed to reach the appropriate metal loading. In detail, 500 μL of an aqueous 1.0 mM peptide solution was added to 4.460 mL of water in a vial. Next, 10 μL of an aqueous 100 mM HAuCl4 solution was added. The solution was briefly agitated and allowed to stand for 10 min. This was followed by the addition of 30 μL of a freshly prepared aqueous 100 mM NaBH4 solution. A NaBH4:Au ratio of 3 was used for all syntheses. The solution was briefly agitated, and the reaction was allowed to proceed undisturbed at room temperature for 1.0 h to ensure complete reduction. Identical synthetic protocols were used to prepare Au nanoparticles at a Au:peptide ratio of 1:1; however, the amount of the stock HAuCl4 solution was decreased to 5 μL and the water volume was increased to reach a final reaction volume of 5.0 mL.
Nanoparticle characterization
TEM studies were performed using a JEOL JEM-2010 microscope operating at 200 kV. The samples were prepared by drop casting 5 μL of the colloidal nanoparticle dispersion onto a carbon-coated 200 mesh Cu grid, which was allowed to dry overnight. UV-vis monitoring of particle formation and aggregation employed an Agilent 8453 photodiode array spectrophotometer. All samples were analyzed using a quartz cuvette with a 1 cm optical path length.Results and discussion
QCM analysis of peptide binding to Au
Pd4, originally isolated as a Pd-binding peptide,24 also binds to Au.8,12 Our previously-published molecular simulations predicted that the native Pd4 peptide has two anchor residues for Au binding, specifically two histidines at the 6 and 11 positions.8 To elucidate effects of sequence and anchor residue context on peptide binding, a set of six different Pd4 variant peptide sequences was prepared (Table 1). More specifically, to understand how the binding thermodynamics change when one or both of the anchor residues is removed, two peptides, Pd4-1 and Pd4-2, were prepared in which a single histidine residue was replaced with alanine at the 6 or 11 position, respectively. A doubly mutated peptide, Pd4-3, was generated with both histidines replaced by alanines. Previous predictions of amino acid binding free energies on aqueous Au(111) suggested that alanine is not a strong binder.21 Thus, minimal Au surface binding at the alanine sites was anticipated. However, these sequences were designed to probe how the local context of anchor residues affected the surface conformation and Au-binding affinity of the peptide. To probe the effects of the global sequence for Au binding behavior (i.e. to test the possibility that overall binding affinity is just a summation of the individual residues’ affinities, in which details of the sequence play no role in binding), two additional peptides were designed: the Pd4-4 sequence (a random rearrangement of the residues present in the native Pd4 peptide) and the Pd4-5 peptide (which has the two histidine anchors grouped together at the N-terminus).QCM analysis was used to determine peptide binding kinetics and thermodynamics for each sequence. Fig. 1a presents the observed frequency changes for the Pd4-5 peptide at five different concentrations as an example. The full QCM analysis for all of the peptides is presented in the ESI, Fig. S1.† Higher peptide concentrations produced faster frequency changes, reflecting increased binding rates. The dissipation energy, which is an indicator of the viscoelasticity of the bound peptide layer, is shown for the highest peptide concentration (15 μg mL−1). For all of the peptides studied here, the dissipation energy was <5% of the total frequency change. This indicates that the absorbed peptide layer was rigid, and that multiple layers were not formed. Note that for all QCM data, inverted plots are displayed for more intuitive data interpretation.
 | ||
| Fig. 1 QCM analysis of the Pd4-5 peptide to obtain ka and kd values. Part (a) shows the inverted frequency change vs. time for five concentrations, as well as the dissipation energy plot for the highest concentration studied. Part (b) shows kobs values vs. peptide concentration, obtained from the data in part (a) by fitting using Langmuir kinetics. Note that no error bars are presented as they are smaller than the points in the graph. The slope and y-intercept in part (b) provide the rate constants for adsorption and desorption, respectively. | ||
Fitting each QCM-derived binding curve to Langmuir kinetics provided pseudo-first order adsorption rate constants (kobs) as in prior studies.8,27,28 Plotting the kobs values as a function of the peptide concentration gives the binding constants for adsorption (ka) and desorption (kd) as the slope and y-intercept of the best fit line of the plot, respectively (Fig. 1b). Using these values, the binding equilibrium constant, Keq, can then be calculated as ka/kd. The binding affinity, defined as the Gibbs free energy change (ΔG) for peptide binding, is then obtained as ΔG = −RTln(Keq). Using this approach, the ΔG of binding for the Pd4-5 peptide on Au was determined to be −35.5 ± 0.2 kJ mol−1. This analysis was carried out for each of the peptides in the designed library to yield the ΔG values presented in Table 1. These ranged from −27.2 to −35.5 kJ mol−1 for the weakest (Pd4-1) and strongest (Pd4-5) binders, respectively. Note that the pH of the peptide solutions for the QCM analysis ranged from 3.9 to 4.2. A full summary of the ka, kd, and Keq values for each peptide is provided in Table S1 of the ESI.† The native Pd4 peptide has a ΔG value of −31.4 ± 0.1 kJ mol−1. This value is similar to values reported in previous studies of Pd4 binding on Au.4 When a single histidine was replaced by alanine, as in Pd4-1 and Pd4-2, lower ΔG values of −27.2 ± 0.2 kJ mol−1 and −30.7 ± 0.1 kJ mol−1, respectively, were measured. This indicates that mutation of the individual histidine anchor residues does substantially affect the Au affinity; however, the effect was larger for the residue at the 6 position. When histidines at both the 6 and 11 positions were replaced with alanines (Pd4-3), we observed an unexpected increase in the Au binding affinity (ΔG = −32.0 ± 0.8 kJ mol−1). Note that while the average ΔG values for Pd4 and Pd4-3 are different, the error limits for the measured values overlap. The relatively strong affinity of Pd4-3 was surprising; mutation of a second anchor residue (histidine) would be expected to further decrease the binding affinity, given the decreases observed upon replacement of either of the anchor residues individually. Finally, an increase in binding affinity was noted for the remaining two peptide variants, Pd4-4 and Pd4-5. A ΔG value of −33.1 ± 0.2 kJ mol−1 was determined for the Pd4-4 peptide, while the strongest binding of the set was observed from the Pd4-5 peptide. The weakest binding was observed from the Pd4-1 peptide.
From Langmuir fitting of the QCM data, the steady-state fractional surface coverage, θ, of the sensor by the peptide can also be directly determined.8 For the Pd4-5 peptide at the highest concentration studied (15 μg mL−1 peptide), the surface coverage reached a maximum value of 95.3 ± 0.4%, consistent with its strong binding affinity. At the other extreme of binding affinity, Pd4-1 at the same concentration had a maximum surface coverage of 41.2 ± 1.6%.
Molecular dynamics simulations of peptide binding to Au
The conformational ensemble of adsorbed peptide states provided by our molecular simulation data allows for conceptual links to be established between the primary sequence of the peptide, the observed experimental binding data, and the predicted structure(s) of the peptide in the adsorbed state. One additional challenge in predicting the conformational ensemble of the Pd4 sequence and its variants is associated with the presence of His residues. In the amino acid form, the side-chain in histidine is likely to be protonated (i.e. carry an overall +1 charge) in aqueous solution at the pH values investigated experimentally (pH 3.9–4.2); the relevant pKa value is ∼6.2 for the imidazole side-chain. However, the pKa corresponding to protonation of the side-chain of a His residue in a peptide sequence in solution may differ substantially from that of the corresponding amino acid.39 This is due to the modification in the immediate dielectric environment of the relevant His in a protein/peptide, which can depend on the conformational state (or ensemble of states) of the protein/peptide. For example, the degree to which the residue is shielded from water, based upon the biomolecular structure, can substantially affect the localized dielectric environment around the histidine, thus potentially altering the protonation state of the imidazole side chain.40,41 Moreover, the relevant pKa of a His residue in the surface-adsorbed state at the aqueous Au interface might also differ from that of a free (i.e. un-adsorbed) peptide in solution.At present, the prediction of pKa values for residues such as His for peptides/proteins in aqueous solution, which can account for environment/conformation effects, is extremely challenging.42 Going further, to the authors’ knowledge, information regarding the influence exerted by the presence of the aqueous Au interface on the pKa of His residues in surface-bound peptide is also scarce. Therefore, the protonation state of each His residue in the surface-adsorbed state of the peptide may not match expectations based on the known pKa values generated for the His amino acid.
These questions discussed above represent a grand challenge in molecular simulation and are not the focus of the present study. Herein, we only consider how the conformational ensemble of the surface-adsorbed peptide sequences can be modulated by the protonation state of each His residue. As will be demonstrated, the position and local environment of each His residue, in addition to the protonation state of the His side-chain, can all influence this conformational ensemble. Therefore, in the REST simulations, each protonation state of each His residue for every peptide sequence that contained any His residues (i.e. Pd4, Pd4-1, Pd4-2, Pd4-4 and Pd4-5) was considered. These sequences comprise a special case in the simulation dataset; however, unless specified otherwise, the results discussed below will refer to the case where all the His in the sequence were protonated. Snapshots of representative structures of the Pd4-1 and Pd4-5 peptides, which are the two peptides that feature the greatest difference in terms of the QCM-measured binding affinity, are shown in Fig. 2. Corresponding snapshots of the other peptides are shown in Fig. S4–S7 in the ESI.†
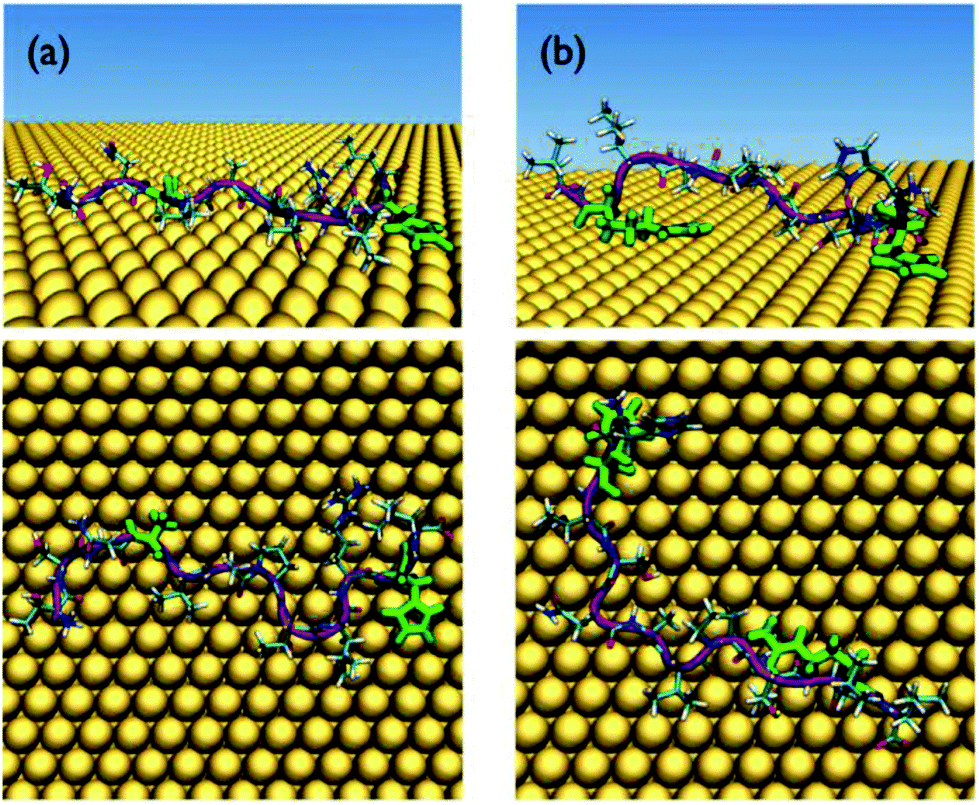 | ||
| Fig. 2 Representative snapshots of the peptides (a) Pd4-1 and (b) Pd4-5 adsorbed at the Au(111) interface, shown in side (top) and plan (bottom) views. The anchor residues are colored bright green, while the peptide backbone is colored purple. Water molecules are not shown for clarity. | ||
As mentioned above, our previously-published simulation studies on the adsorption of Pd4 at the aqueous Au interface have identified Pd4 as an entropically-driven binder,8i.e. that Pd4 supports a large number of distinct conformations in the surface-adsorbed state. The number of distinct surface-adsorbed conformations of a peptide can be determined from a clustering analysis,43 where like structures are grouped together on the basis of peptide backbone conformational similarity (denoted herein as clusters). From this cluster analysis, both the total number of distinct adsorbed conformations, n, and their relative population, pi, can be determined. As introduced in earlier studies,21,44 the conformational entropic contribution, Sconf, associated with the surface-adsorbed peptide can then be estimated from:

These Sconf values are summarized in Table S5 in the ESI;† the actual values of total number of distinct conformations (referred to as clusters), n, and the populations, pi for the top-ten most populated clusters, are also provided in Table S3 of the ESI.† In terms of a relative Sconf score, the native Pd4 sequence was previously ranked as high.8 The current data show that almost all of the variants of the Pd4 sequence could also be classified as high in terms of Sconf score. In the case of three of the variants, Pd4-1, Pd4-2, and Pd4-4, the conformational entropy contribution was substantially enhanced relative to the parent sequence (see Table S5 of the ESI†).
In addition, the adsorbed configurations of each variant sequence were compared in terms of structural similarity against those obtained for the parent peptide sequence. If the root mean-squared deviation (RMSD) between the relative positions of the peptide backbone atoms inherent to the representative structures of the two clusters was less than or equal to 2 Å (the same spatial cut-off used for the clustering analysis, see ‘Additional methodology’ in the ESI†) then the two conformations were classified as a match, i.e. the peptide backbone conformations were similar. The top-ten most populated clusters of the parent peptide have thus been compared against the ten most populated clusters of each of the variants (see ESI, Table S4†). Both the Pd4-1 and Pd4-3 sequences shared reasonable structural similarity with the original Pd4 sequence (i.e. both Pd4-1 and Pd4-3 possess several matched structures with Pd4). In particular, the most populated cluster of Pd4-1 and Pd4-3 both have a structural match with the most populated cluster of Pd4. Pd4-2 had fewer matching conformations with Pd4 but still featured one match between clusters with a non-negligible population. These findings suggest that the Pd4 sequence possesses conformational stability. In other words, Pd4 shows a lack of conformational susceptibility with regards to the mutation of individual residues, even if those residues are anchors. These results do not necessarily suggest a general finding; for example, this may only hold for entropically-driven materials-binding peptides. Further comprehensive and systematic investigations are needed to fully elucidate the rules governing this phenomenon. The more substantial changes to peptide sequence inherent to the other two mutants (Pd4-4 and Pd4-5) resulted in few or no matches with Pd4, respectively. Overall, the extensive conformational ensemble identified for all six peptide sequences, combined with the conformational recalcitrance of the sequences corresponding with point mutations, indicates that the underlying explanation for the differences in the experimentally-measured binding affinities is related to the anchor residues, i.e. the histidines.
Fig. 3 shows the predicted degree of direct Au surface contact calculated for each residue in each of the six peptides, in the instance where all His residues are protonated. Fig. S8 in the ESI† shows the residue-surface contact averaged over different configurations of the protonation states, while the numerical contact data (as percentages) are provided in Table S2 of the ESI† for all possible combinations of histidine protonation state for every sequence. From these data, the anchor residues can be identified and the degree to which the mutations of the peptide chain alter the binding of residues within the peptide can be assessed. For the parent Pd4 sequence, H6 and H11 are anchor residues in all protonation state scenarios (Table S2 in the ESI†). At pH 4, R10 is also an anchor residue. Overall, the peptide is predicted to be adsorbed to the Au surface at two locations in the sequence: in the middle of the chain and close to the C-terminus of the chain. In Pd4-1 the H6A mutation generates a change in the residue-surface contact of the peptide (Fig. 3 and Table S2 in the ESI†). In this case, the A6 residue showed only weak contact with the Au(111) surface, while the A4 residue became an anchor. Interestingly, this is the only non-His/Arg residue that acted as an anchor in all the systems considered. At the C-terminal end of Pd4-1, H11 remained an anchor residue but R10 did not. However, the enthalpic contribution of A4 to the overall peptide binding strength is predicted to be considerably less than that of a His or Arg residue.21,31,32,45,46 In summary, when compared to Pd4, Pd4-1 suffered a reduction from two strong anchor residues (in Pd4) to one strong anchor (H11) and one weak anchor (A4), which may explain why the measured binding affinity of Pd4-1 is weaker than Pd4.
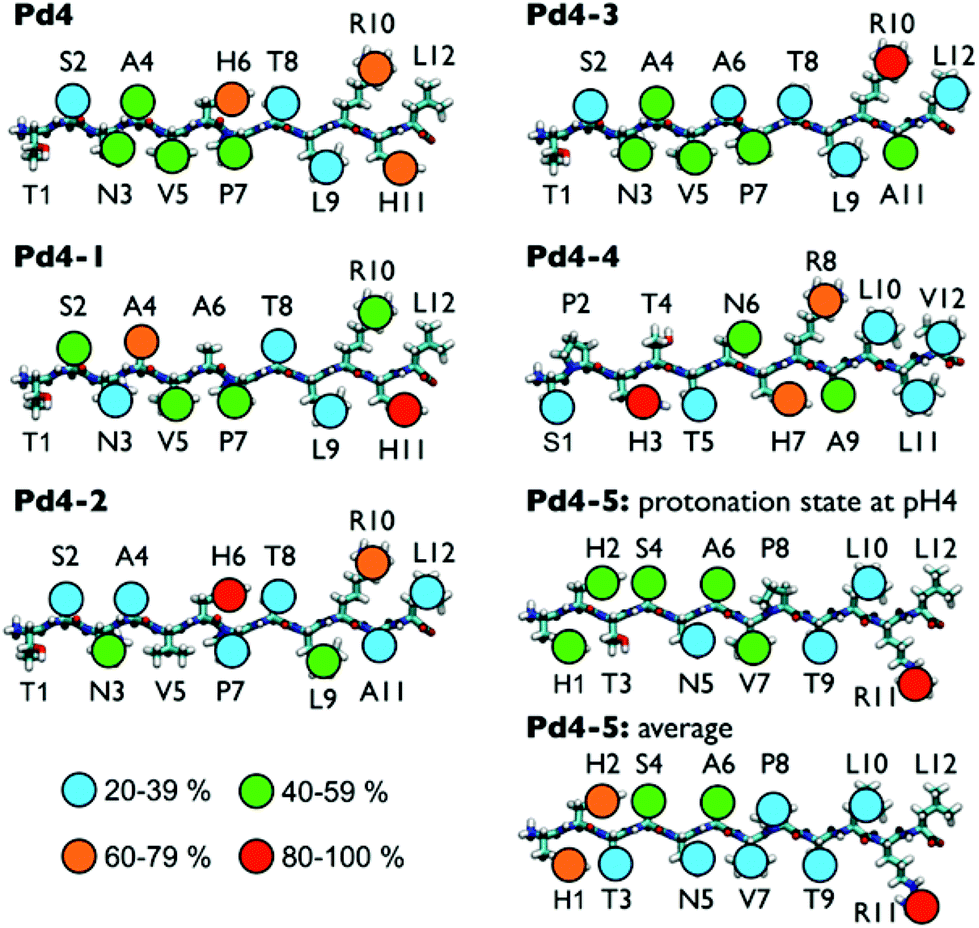 | ||
| Fig. 3 Degree of residue-surface contact for each of the six peptide molecules, adsorbed at the aqueous Au interface, predicted from the REST simulations. All data shown for peptides simulated at pH 4 and for the average over different protonation states for Pd4-5. | ||
In the case of Pd4-2, the H11A mutation is seen to exert less effect on the surface contact points along the peptide chain. H6 remained an anchor (regardless of protonation state, see Table S2 in the ESI†); in addition, despite the (expectedly) weak binding of A11, R10 assumed the role of the sole strong surface contact point in the C-terminal region of the chain. Both His and Arg are thought to be strong binding residues at the aqueous Au interface.8 We propose that for a dyad sequence motif where His and Arg are adjacent in the sequence (i.e. either HR or RH), this can lead to a competition between the two residues in determining which residue acts as the surface contact point. However, this competition to act as an anchor appears to also be influenced by the His protonation states (see data for Pd4 and Pd4-4 in Table S2 of the ESI†).
The mutation of both His residues in Pd4-3 removed two anchor points. Intuitively, this would suggest that Pd4-3 should bind more weakly than Pd4; however, as seen for Pd4-2, R10 again assumed the role as anchor residue in the absence of H11. Moreover, while Pd4-3 did not feature any additional anchor residues, this sequence did however exhibit a substantial contiguous segment (N3–P7) that featured moderate binding. For the Pd4 parent, the surface contact for residues in the N3–P7 segment varies with the protonation state of the His residues (Table S2†); if the contact is averaged over all the possible protonation states (Fig. S8†), the contact of the N3–P7 segment in Pd4-3 is comparable to that of Pd4. In particular, the contact of the N3–A4–V5 triad is higher in Pd4-3 than the parent peptide. The probability of at least two of these three residues being adsorbed simultaneously is 51% and 35% for Pd4-3 and Pd4, respectively. This, in partnership with the anchor at R10, could in part account for the similarity in binding strength of Pd4-3 compared with Pd4. Despite the scrambling of the sequence in Pd4-4, both of the His residues and the Arg residue all remained as anchors for three out of the four His protonation configurations considered here. While we cannot determine the relative weighting of these four protonation configurations in the overall ensemble of Pd4-4, a naïve averaging of the number of anchors over the four states suggests a greater number of anchor points compared with the corresponding analysis for Pd4. This assumption is reasonable given the lack of certainty regarding the pKa of each His residue when the peptide is adsorbed at the aqueous Au interface. These results are consistent with the experimentally-observed binding constants.
The data from the QCM experiments indicate that Pd4-5 was the strongest binder to Au. For the doubly-protonated state (where both His are protonated), at first glance it may seem counter-intuitive that Pd4-5 featured only one single strong anchor residue, R11. However, further analysis (see a summary of conditional residue-surface binding probabilities in Table S6 of the ESI†) demonstrated that the majority (92%) of the simulation trajectory had at least one of the His residues also in contact. In other words, a competitive binding scenario, similar to that seen for RH/HR, was also noted for the HH dyad motif, where the binding of the two His residues was seen to toggle. Our analysis confirmed that only 8% of the trajectory featured neither of the His residues in a surface-bound state. Therefore, any given adsorbed conformation of doubly-protonated Pd4-5 was likely to have at least two strong surface contact points (R11 and either H1 or H2, or all three residues), as indicated in Fig. 2b. As with Pd4-4, the relative proportion of each protonation configuration is unknown, but again, an unweighted average over states suggests that Pd4-5 had a greater average number of anchors than did Pd4. This could in part explain the stronger binding affinity observed for Pd4-5.
Au nanoparticle synthesis and characterization
With a clearer understanding of the binding between the Au surface and the peptides of Table 1, Au nanoparticles were fabricated using the biomolecules as capping ligands. For nanoparticle synthesis, each peptide was mixed with Au3+ ions at Au:peptide molar ratios of 1:1 and 2:1. After allowing time for complexation of the metal ions to the peptide, each solution was reduced with a 3-fold molar excess of NaBH4 relative to Au3+. In all cases, a dramatic color change was observed upon reduction. The absorbance of the nanoparticle dispersions was characterized using UV-vis spectroscopy (Fig. 4). Fig. 4a specifically presents the parent Pd4 system. The spectrum of the Pd4 peptide solution was featureless, while those of the Au3+/peptide complex showed a weak absorbance peak at 300 nm, at both Au:peptide ratios. After reduction, an absorbance peak associated with the Au nanoparticle LSPR was observed. For a Au:peptide ratio of 2, the LSPR absorbance was quite broad and red-shifted relative to the typical position for the LSPR of Au particles. However, when a Au:peptide ratio of 1 was employed, a distinct LSPR band was evident at 525 nm. This suggests that the Au nanoparticles prepared at a ratio of 2 are larger, more polydisperse, and/or more aggregated in solution compared to particles prepared at the lower ratio.47 Identical analyses were conducted for each of the peptides at both ratios. Overall, for all of the peptide-capped Au nanoparticles produced at a Au:peptide ratio of 2, the LSPR absorbance was broad and red-shifted, beginning near 580 nm, while at a ratio of 1, the materials typically had a distinct LSPR band that was positioned between 525 and 550 nm.
 | ||
| Fig. 4 UV-vis analysis of Au nanoparticles capped with (a) Pd4, (b) Pd4-1, (c) Pd4-2, (d) Pd4-3, (e) Pd4-4, and (f) Pd4-5 peptide. Ratios represent molar ratio of Au:peptide. | ||
TEM analysis was conducted to examine the effect of peptide sequence/mutations and the Au:peptide ratio on particle size, shape, and stability. Fig. 5 presents TEM images of the Au nanoparticles capped with each of the peptides after 1 h of reduction. The top and bottom rows display the materials prepared at ratios of 2 and 1, respectively. For the Pd4-capped materials fabricated at a ratio of 2, polydisperse particles with an average size of 7.7 ± 3.6 nm were observed, consistent with previous studies;12,47 however, the particles generated with the alanine-substituted peptides decreased in size to 5.2 ± 2.0 nm (Pd4-1), 3.9 ± 1.1 nm (Pd4-2), and 5.3 ± 1.7 nm (Pd4-3). For the materials capped with the Pd4-4 peptide that randomized the peptide sequence and the Pd4-5 sequence that grouped the histidine anchor residues at the N-terminus, particles of 5.3 ± 2.8 nm and 4.1 ± 1.6 nm were noted, respectively. Overall, all of the modified or mutated peptides generated smaller Au nanoparticles than the parent Pd4 sequence; however, no clear trends with respect to biomolecule binding strength (ΔG) and average particle size were evident. Furthermore, all of the particles were polydisperse in size and overall morphology.
 | ||
| Fig. 5 TEM analysis of the Au nanoparticles capped with the indicated mutant peptides. TEM grids were prepared 1 h after reduction. The top row displays the materials fabricated at a Au:peptide ratio of 2, while the bottom row shows the structures generated at a ratio of 1. All scale bars are 50 nm. | ||
Different results were obtained when the synthesis reactions were carried out at a Au:peptide ratio of 1. Spherical particles with an average diameter of 3.0 ± 0.6 nm (Pd4), 3.6 ± 0.9 nm (Pd4-1), 2.6 ± 0.5 nm (Pd4-2), 3.5 ± 1.1 nm (Pd4-3), 3.2 ± 1.1 nm (Pd4-4), and 2.7 ± 0.5 nm (Pd4-5) were formed at this lower Au:peptide ratio. For all particle sizing, at least 100 particles were measured over multiple TEM images. Size distribution histograms are available in the ESI, Fig. S2 and S3.† The effect of decreasing the Au:peptide ratio on particle size and shape is illustrated in these samples. In general, after 1 h of reduction, using a 2:1 ratio to prepare the materials gave larger, more polydisperse, and somewhat aggregated particles, while employing a 1:1 ratio produced smaller, more spherical particles. Such effects likely arise from insufficient peptide in solution at the higher ratio (2:1) to passivate the Au nanoparticle surfaces at smaller particle sizes, leading to coalescence of the materials to generate the observed sample. At the smaller ratio (1:1), enough peptide is present in the reaction to cap the growing nanoparticles at the smaller sizes. These results are consistent with the UV-vis spectra of the materials, in which all the structures generated at a ratio of 2, independent of which peptide was used to cap them, displayed a broad and red shifted Au LSPR absorbance relative to the distinct LSPR peak at 525–550 nm for the particles prepared at a ratio of 1.
Fig. 6 presents UV-vis and TEM analyses of samples 24 h after reduction. The aggregation process was monitored by measuring the absorbance at the LSPR peak of the Au nanoparticles over 24 h, taking one spectrum every 10 min for 24 h. For the reactions using a Au:peptide ratio of 2, as presented in Fig. 6a, aggregation of the particles prepared with Pd4-1, Pd4-2, and Pd4-4 was observed in less than 24 h. Although a significant increase in absorbance was evident initially for the Pd4-3 capped materials, arising from the nucleation and initial growth of the particles, a sharp decrease in the absorbance was noted beginning at 16 h, indicating precipitation was occurring. The Pd4-5 reaction products were stable for the duration of the experiment as seen in the plateau of its LSPR absorbance. Interestingly, the absorbance for the Pd4 particles slowly decreased over 24 h and ended at the same absorbance as the Pd4-3 reaction when it had precipitated; however, the Pd4-capped materials remained stable and dispersed in water. This suggests that the Pd4-capped particles were somewhat aggregated in solution, even though they do not aggregate to a sufficient extent to show visible precipitation. TEM imaging of the Pd4 and Pd4-5 capped materials 24 h post reduction revealed particle sizes of 5.5 ± 2.4 nm and 3.7 ± 2.1 nm, respectively, which are relatively similar to the particle sizes after 1 h of reduction. When a Au:peptide ratio of 1 was employed, a similar trend was also found in that materials produced using Pd4 and Pd4-5 remained stable over 24 h; however, bulk precipitation was evident in the particles capped with Pd4-1, Pd4-2, Pd4-3, and Pd4-4 peptides (Fig. 6b). TEM imaging 24 h after reduction gave identical particle sizes of 2.7 ± 0.5 nm (Pd4) and 2.7 ± 0.5 nm (Pd4-5), which are again similar to the particle sizes after 1 h of reduction.
 | ||
| Fig. 6 UV-vis and TEM analysis for the stability of the Au nanoparticle dispersions 24 h after reduction. Part (a) presents the data for the materials prepared at a Au:peptide ratio of 2, while part (b) shows the data for the particles produced at a ratio of 1. | ||
Overall, no clear trend was evident between the binding strengths of the peptides and the size of the particles produced. For example, Pd4-4 and Pd4-5 have the strongest affinity for Au based upon their ΔG values; however, these sequences generated Au nanoparticles with neither the largest or smallest sizes among all the peptides studied. This suggests that additional binding parameters beyond surface affinity govern particle formation and growth using peptides. Interestingly, the Pd4 and Pd4-5 peptides generated nanoparticles that remained stable for more than 24 h at both ratios of 2 and 1, even though their ΔG values were very different. This indicates that particle stability depends upon other factors such as the conformation in which the peptide binds to the metal surface, rather than simply the affinity of the peptide for the NP material.
To date, there has been no systematic rationale to understand or predict why a given peptide sequence may or may not produce a stable dispersion of peptide-capped Au nanoparticles in aqueous solution. Aggregation is likely if patches of exposed Au surface are present on two neighboring nanoparticles in solution. This condition is more probable for a peptide/nanoparticle interface that features a relatively high degree of conformational entropic contribution to the binding (Sconf), i.e., a large number of distinct adsorbed structures and few strong anchor points. In terms of the underlying potential energy landscape that corresponds to a high value of Sconf, two scenarios are plausible. In the first, the large number of distinct surface-adsorbed conformations could be separated by very low (less than or equal to kBT) potential energy barriers. This would result in a highly dynamic, disordered system where the adsorbed peptide conformations could readily interconvert at room temperature, producing a high degree of mobility of the surface-adsorbed peptides. The second scenario would correspond to the presence of high (much greater than kBT) potential energy barriers with respect to interconversion between the large number of distinct conformations. In this case, the peptide conformations would be frozen into a wide range of adsorbed structures, and would be essentially immobile on the surface, presumably from their first instance of contact on the Au nanoparticle surface. In the former scenario, a high Sconf value, corresponding to high mobility of surface-adsorbed peptides, could be conducive to the thermally-activated exposure of patches on the Au nanoparticle surface. Our predicted averaged Sconf values, provided in Fig. 7 (see Table S5 in the ESI† for the numerical values), indicate that the sequences Pd4-1, Pd4-2, Pd4-3, and Pd4-4, which correspond to unstable nanoparticle dispersions, all have very high Sconf values. This contrasts with Pd4 and Pd4-5, which have the lowest Sconf values in the set and also yield the only nanoparticle dispersions that remained stable after 24 h. These data suggest a connection between the Sconf value and nanoparticle stability in solution. However, because multiple factors are likely to influence nanoparticle stability, extension of this hypothesis to other peptide systems requires further investigation.
 | ||
| Fig. 7 Values of the conformational contribution to the entropy of peptide-surface binding (Sconf) for the parent Pd4 peptide and all variants on this sequence. Numerical values are provided in Table S5 of the ESI.† | ||
In a previous study of peptide-mediated Au NP formation using a diverse set of sequences,12 we also found that there was no direct correlation between binding affinity and the size of the resulting NPs. Consistent with the present study, this shows that simply selecting sequences for their overall binding affinity for a particular inorganic material may not guarantee identification of sequences that will produce stable peptide-capped NPs. This earlier study also showed that the catalytic activity of the NPs correlated with the number of strong anchor residues. Peptides characterized as enthalpic binders, with a relatively large number of anchor residues exhibited high activation energies for a prototypical surface catalysed reaction. This picture was refined in the study by Bedford et al.37 With the present study, a coherent picture begins to emerge. Synthesis of small, colloidally stable peptide-capped NPs seem to be favored by use of enthalpically-driven binders with relatively low conformational entropy. Sequences with fewer anchor residues and high conformational entropy provide greater exposure of the inorganic surface, which may be desirable, e.g. for improved catalytic activity, but may also promote aggregation.
Conclusions
In conclusion, we have presented a peptide synthetic mutation analysis that advances the understanding of the role of peptide sequence in controlling and modulating the affinity of the biomolecule for its target inorganic surface. In this regard, mutation of anchor points demonstrated that the local affinity can be modulated based upon the neighboring residues in the sequence. Thus, sequence context effects can influence the materials-binding ability of peptides. This observation was corroborated by both experimental and computational findings that pointed to important thermodynamic considerations for isolating the underlying basis of binding affinity. Further Au nanoparticle synthesis results demonstrated that the overall particle morphology was not directly associated with the ΔG of binding to the material surface, suggesting that other biomolecular factors govern the size, shape, and stability of peptide-capped materials. A feature that is suggested to govern the stability of the peptide-capped nanoparticle dispersion was identified, namely the number of distinct conformations of the peptide in the surface-adsorbed state. Such results are important for the creation of rules for the a priori design of new biomolecules that can tune peptide affinity and specificity, as well as nanoparticle capping and stabilization ability for target inorganic structures. Such capabilities would be of significant use in the one-pot, aqueous synthetic design of functional materials, for applications wherein the ligand overlayer structure/morphology is critical to the intended functionality. For instance, controlling the bioligand surface display could be important for applications in biosensing, especially for Au nanorings48,49 that are sensitive to the biomolecule orientation, and catalysis where selective peptide surface structures could be designed to enhance the particle functionality.Acknowledgements
This material is based upon work supported by the Air Office of Scientific Research, grant number FA9550-12-1-0226. We gratefully acknowledge the Victorian Life Sciences Computation Facility (VLSCI) for allocation of computational resources, and TRW thanks veski for an Innovation Fellowship.References
- J. S. Evans, “Tuning in” to Mollusk Shell Nacre- and Prismatic-Associated Protein Terminal Sequences. Implications for Biomineralization and the Construction of High Performance Inorganic−Organic Composites, Chem. Rev., 2008, 108, 4455–4462 CrossRef CAS PubMed.
- C. J. Munro, Z. E. Hughes, T. R. Walsh and M. R. Knecht, Peptide Sequence Effects Control the Single Pot Reduction, Nucleation, and Growth of Au Nanoparticles, J. Phys. Chem. C, 2016, 120, 18917–18924 CAS.
- C.-L. Chen, R. N. Zuckermann and J. J. DeYoreo, Surface-Directed Assembly of Sequence-Defined Synthetic Polymers into Networks of Hexagonally Patterned Nanoribbons with Controlled Functionalities, ACS Nano, 2016, 10, 5314–5320 CrossRef CAS PubMed.
- C.-L. Chen, J. Qi, J. Tao, R. N. Zuckermann and J. J. DeYoreo, Tuning Calcite Morphology and Growth Acceleration by a Rational Design of Highly Stable Protein-Mimetics, Sci. Rep., 2014, 4, 6266 CrossRef CAS PubMed.
- M. Sarikaya, C. Tamerler, A. K. Y. Jen, K. Schulten and F. Baneyx, Molecular Biomimetics: Nanotechnology through Biology, Nat. Mater., 2003, 2, 577–585 CrossRef CAS PubMed.
- S. Y. Park, A. K. R. Lytton-Jean, B. Lee, S. Weigand, G. C. Schatz and C. A. Mirkin, DNA-Programmable Nanoparticle Crystallization, Nature, 2008, 451, 553–556 CrossRef CAS PubMed.
- C.-L. Chen and N. L. Rosi, Preparation of Unique 1-D Nanoparticle Superstructures and Tailoring their Structural Features, J. Am. Chem. Soc., 2010, 132, 6902–6903 CrossRef CAS PubMed.
- Z. Tang, J. P. Palafox-Hernandez, W.-C. Law, Z. E. Hughes, M. T. Swihart, P. N. Prasad, M. R. Knecht and T. R. Walsh, Biomolecular Recognition Principles for Bionanocombinatorics: An Integrated Approach to Elucidate Enthalpic and Entropic Factors, ACS Nano, 2013, 7, 9632–9646 CrossRef CAS PubMed.
- C.-Y. Chiu, Y. Li, L. Ruan, X. Ye, C. B. Murray and Y. Huang, Platinum Nanocrystals Selectively Shaped Using Facet-Specific Peptide Sequences, Nat. Chem., 2011, 3, 393–399 CrossRef CAS PubMed.
- N. M. Bedford, H. Ramezani-Dakhel, J. M. Slocik, B. D. Briggs, Y. Ren, A. I. Frenkel, V. Petkov, H. Heinz, R. R. Naik and M. R. Knecht, Elucidation of Peptide-Directed Palladium Surface Structure for Biologically Tunable Nanocatalysts, ACS Nano, 2015, 9, 5082–5092 CrossRef CAS PubMed.
- L. Ruan, H. Ramezani-Dakhel, C. Lee, Y. Li, X. Duan, H. Heinz and Y. Huang, A Rational Biomimetic Approach to Structure Defect Generation in Colloidal Nanocrystals, ACS Nano, 2014, 8, 6934–6944 CrossRef CAS PubMed.
- Y. Li, Z. Tang, P. N. Prasad, M. R. Knecht and M. T. Swihart, Peptide-Mediated Synthesis of Gold Nanoparticles: Effects of Peptide Sequence and Nature of Binding on Physicochemical Properties, Nanoscale, 2014, 6, 3165–3172 RSC.
- R. Bhandari, R. Coppage and M. R. Knecht, Mimicking Nature's Strategies for the Design of Nanocatalysts, Catal. Sci. Technol., 2012, 2, 256–266 CAS.
- R. Coppage, J. M. Slocik, M. Sethi, D. B. Pacardo, R. R. Naik and M. R. Knecht, Elucidation of Peptide Effects that Control the Activity of Nanoparticles, Angew. Chem., Int. Ed., 2010, 49, 3767–3770 CrossRef CAS PubMed.
- Y. J. Lee, Y. Lee, D. Oh, T. Chen, G. Ceder and A. M. Belcher, Biologically Activated Noble Metal Alloys at the Nanoscale: For Lithium Ion Battery Anodes, Nano Lett., 2010, 10, 2433–2440 CrossRef CAS PubMed.
- Y. Lee, J. Kim, D. S. Yun, Y. S. Nam, Y. Shao-Horn and A. M. Belcher, Virus-Templated Au and Au-Pt Core-Shell Nanowires and Their Electrocatalytic Activities for Fuel Cell Applications, Energy Environ. Sci., 2012, 5, 8328–8334 CAS.
- K. Saha, S. S. Agasti, C. Kim, X. Li and V. M. Rotello, Gold Nanoparticles in Chemical and Biological Sensing, Chem. Rev., 2012, 112, 2739–2779 CrossRef CAS PubMed.
- N. J. Halas, Plasmonics: An Emerging Field Fostered by Nano Letters, Nano Lett., 2010, 10, 3816–3822 CrossRef CAS PubMed.
- S. Liu, G. Chen, P. N. Prasad and M. T. Swihart, Synthesis of Monodisperse Au, Ag, and Au–Ag Alloy Nanoparticles with Tunable Size and Surface Plasmon Resonance Frequency, Chem. Mater., 2011, 23, 4098–4101 CrossRef CAS.
- K.-S. Lee and M. A. El-Sayed, Gold and Silver Nanoparticles in Sensing and Imaging: Sensitivity of Plasmon Response to Size, Shape, and Metal Composition, J. Phys. Chem. B, 2006, 110, 19220–19225 CrossRef CAS PubMed.
- J. P. Palafox-Hernandez, Z. Tang, Z. E. Hughes, Y. Li, M. T. Swihart, P. N. Prasad, T. R. Walsh and M. R. Knecht, Comparative Study of Materials-Binding Peptide Interactions with Gold and Silver Surfaces and Nanostructures: A Thermodynamic Basis for Biological Selectivity of Inorganic Materials, Chem. Mater., 2014, 26, 4960–4969 CrossRef CAS.
- Y. Kim, R. J. Macfarlane and C. A. Mirkin, Dynamically Interchangeable Nanoparticle Superlattices through the Use of Nucleic Acid-Based Allosteric Effectors, J. Am. Chem. Soc., 2013, 135, 10342–10345 CrossRef CAS PubMed.
- R. J. Macfarlane, M. R. Jones, B. Lee, E. Auyeung and C. A. Mirkin, Topotactic Interconversion of Nanoparticle Superlattices, Science, 2013, 341, 1222–1225 CrossRef CAS PubMed.
- D. B. Pacardo, M. Sethi, S. E. Jones, R. R. Naik and M. R. Knecht, Biomimetic Synthesis of Pd Nanocatalysts for the Stille Coupling Reaction, ACS Nano, 2009, 3, 1288–1296 CrossRef CAS PubMed.
- M. Hnilova, E. E. Oren, U. O. S. Seker, B. R. Wilson, S. Collino, J. S. Evans, C. Tamerler and M. Sarikaya, Effect of Molecular Conformations on the Adsorption Behavior of Gold-Binding Peptides, Langmuir, 2008, 24, 12440–12445 CrossRef CAS PubMed.
- W. C. Chan and P. D. White, Fmoc Solid Phase Peptide Synthesis: A Practical Approach, Oxford University Press, New York, 2000 Search PubMed.
- C. Tamerler, E. E. Oren, M. Duman, E. Venkatasubramanian and M. Sarikaya, Adsorption Kinetics of an Engineered Gold Binding Peptide by Surface Plasmon Resonance Spectroscopy and a Quartz Crystal Microbalance, Langmuir, 2006, 22, 7712–7718 CrossRef CAS PubMed.
- C. Tamerler, M. Duman, E. E. Oren, M. Gungormus, X. Xiong, T. Kacar, B. A. Parviz and M. Sarikaya, Materials Specificity and Directed Assembly of a Gold-Binding Peptide, Small, 2006, 2, 1372–1378 CrossRef CAS PubMed.
- T. Terakawa, T. Kameda and S. Takada, On Easy Implementation of a Variant of the Replica Exchange with Solute Tempering in GROMACS, J. Comput. Chem., 2011, 32, 1228–1234 CrossRef CAS PubMed.
- L. B. Wright and T. R. Walsh, Efficient Conformational Sampling of Peptides Adsorbed onto Inorganic Surfaces: Insights from a Quartz Binding Peptide, Phys. Chem. Chem. Phys., 2013, 15, 4715–4726 RSC.
- L. B. Wright, P. M. Rodger, S. Corni and T. R. Walsh, GolP-CHARMM: First-Principles Based Force Fields for the Interaction of Proteins with Au(111) and Au(100), J. Chem. Theory Comput., 2013, 9, 1616–1630 CrossRef CAS PubMed.
- L. B. Wright, P. M. Rodger, T. R. Walsh and S. Corni, First-Principles-Based Force Field for the Interaction of Proteins with Au(100)(5 × 1): An Extension of GolP-CHARMM, J. Phys. Chem. C, 2013, 117, 24292–24306 CAS.
- A. D. MacKerell, D. Bashford, M. Bellott, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiórkiewicz-Kuczera, D. Yin and M. Karplus, All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins, J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef CAS PubMed.
- S. Piana, K. Lindorff-Larsen and D. E. Shaw, How Robust Are Protein Folding Simulations with Respect to Force Field Parameterization?, Biophys. J., 2011, 100, L47–L49 CrossRef CAS.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, Comparison of Simple Potential Functions for Simulating Liquid Water, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- E. Neria, S. Fischer and M. Karplus, Simulation of Activation Free Energies in Molecular Systems, J. Chem. Phys., 1996, 105, 1902–1921 CrossRef CAS.
- N. M. Bedford, Z. E. Hughes, Z. Tang, Y. Li, B. D. Briggs, Y. Ren, M. T. Swihart, V. G. Petkov, R. R. Naik, M. R. Knecht and T. R. Walsh, Sequence-Dependent Structure/Function Relationships of Catalytic Peptide-Enabled Gold Nanoparticles Generated under Ambient Synthetic Conditions, J. Am. Chem. Soc., 2016, 138, 540–548 CrossRef CAS PubMed.
- L. B. Wright, J. P. Palafox-Hernandez, P. M. Rodger, S. Corni and T. R. Walsh, Facet Selectivity in Gold Binding Peptides: Exploiting Interfacial Water Structure, Chem. Sci., 2015, 6, 5204–5214 RSC.
- J. Khandogin and C. L. Brooks, Toward the Accurate First-Principles Prediction of Ionization Equilibria in Proteins, Biochemistry, 2006, 45, 9363–9373 CrossRef CAS PubMed.
- S. P. Edgcomb and K. P. Murphy, Variability in the pKa of Histidine Side-Chains Correlates with Burial within Proteins, Proteins, 2002, 49, 1–6 CrossRef CAS PubMed.
- J. E. Nielsen, M. R. Gunner and B. García-Moreno, The pKa Cooperative: A Collaborative Effort to Advance Structure-Based Calculations of pKa Values and Electrostatic Effects in Proteins, Proteins, 2011, 79, 3249–3259 CrossRef CAS PubMed.
- E. Alexov, E. L. Mehler, N. Baker, A. Baptista, Y. Huang, F. Milletti, J. E. Nielsen, D. Farrell, T. Carstensen, M. H. M. Olsson, J. K. Shen, J. Warwicker, S. Williams and J. M. Word, Progress in The Prediction of pK(a) Values in Proteins, Proteins, 2011, 79, 3260–3275 CrossRef CAS PubMed.
- X. Daura, K. Gademann, B. Jaun, D. Seebach, W. F. van Gunsteren and A. E. Mark, Peptide Folding: When Simulation Meets Experiment, Angew. Chem., Int. Ed., 1999, 38, 236–240 CrossRef CAS.
- J. P. Palafox-Hernandez, C.-K. Lim, Z. Tang, K. L. M. Drew, Z. E. Hughes, Y. Li, M. T. Swihart, P. N. Prasad, M. R. Knecht and T. R. Walsh, Optical Actuation of Inorganic/Organic Interfaces: Comparing Peptide-Azobenzene Ligand Reconfiguration on Gold and Silver Nanoparticles, ACS Appl. Mater. Interfaces, 2016, 8, 1050–1060 CAS.
- M. Hoefling, F. Iori, S. Corni and K.-E. Gottschalk, Interaction of Amino Acids with the Au(111) Surface: Adsorption Free Energies from Molecular Dynamics Simulations, Langmuir, 2010, 26, 8347–8351 CrossRef CAS PubMed.
- J. Feng, R. B. Pandey, R. J. Berry, B. L. Farmer, R. R. Naik and H. Heinz, Adsorption Mechanism of Single Amino Acid and Surfactant Molecules to Au {111} Surfaces in Aqueous Solution: Design Rules for Metal-Binding Molecules, Soft Matter, 2011, 7, 2113–2120 RSC.
- B. D. Briggs, Y. Li, M. T. Swihart and M. R. Knecht, Reductant and Sequence Effects on the Morphology and Catalytic Activity of Peptide-Capped Au Nanoparticles, ACS Appl. Mater. Interfaces, 2015, 7, 8843–8851 CAS.
- J. Ye, M. Shioi, K. Lodewijks, L. Lagae, T. Kawamura and P. Van Dorpe, Tuning Plasmonic Interaction between Gold Nanorings and a Gold Film for Surface Enhanced Raman Scattering, Appl. Phys. Lett., 2010, 97, 163106 CrossRef.
- J. Aizpurua, P. Hanarp, D. S. Sutherland, M. Käll, G. W. Bryant and F. J. García de Abajo, Optical Properties of Gold Nanorings, Phys. Rev. Lett., 2003, 90, 057401 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional simulation methodology, binding constants for the mutant peptides, details of residue-surface contact analysis, comparison of most populated clusters, conformational entropy scores, conditional binding probabilities, QCM analysis, size distribution histograms, and snapshots of simulation results for different protonation states. See DOI: 10.1039/c6nr07890g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |