
Cooperation and competition between halogen bonding and van der Waals forces in supramolecular engineering at the aliphatic hydrocarbon/graphite interface: position and number of bromine group effects†
Bao
Zha
,
Meiqiu
Dong
,
Xinrui
Miao
 *,
Shan
Peng
,
Yican
Wu
,
Kai
Miao
,
Yi
Hu
and
Wenli
Deng
*
*,
Shan
Peng
,
Yican
Wu
,
Kai
Miao
,
Yi
Hu
and
Wenli
Deng
*
College of Materials Science and Engineering, South China University of Technology, Wushan Road, Tianhe District, Guangzhou 510640, P. R. China. E-mail: msxrmiao@scut.edu.cn; wldeng@scut.edu.cn; Tel: (+86)020-22236708
First published on 17th November 2016
Abstract
Herein, the photophysical properties of two π-conjugated thienophenanthrene derivatives (6,9- and 5,10-DBTD) are reported. Their self-assembled monolayers in aliphatic hydrocarbon solvents under different concentrations were investigated by scanning tunneling microscopy on a graphite surface. The STM results revealed that the self-assembled structures of the two geometrical isomers exhibited absolutely different behaviors. At the aliphatic solvent/graphite interface, 6,9-DBTD produced almost a single stable coassembled linear structure, except for that with n-tridecane as the solvent. However, the self-assembly of 5,10-DBTD showed structural diversity, and it presented a gradient variety through increasing the chain length of the aliphatic solvents as well as the solution concentration. All ordered self-assembled adlayers critically depend on not only the interchain van der Waals (vdW) interactions, but also on multiple intermolecular interactions, including Br⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and Br⋯S hetero-halogen bonds, homo-Br⋯Br interactions, and H⋯Br and H⋯O hydrogen bonds. We proposed that the cooperation and competition of the intermolecular interactions involving a Br atom and interchain vdW forces induce this structural variety. Density functional theory calculations support to unravel the different elementary structural units based on halogen bonds and hydrogen bonds and were useful tools to dissect and explain the formation mechanism.
C and Br⋯S hetero-halogen bonds, homo-Br⋯Br interactions, and H⋯Br and H⋯O hydrogen bonds. We proposed that the cooperation and competition of the intermolecular interactions involving a Br atom and interchain vdW forces induce this structural variety. Density functional theory calculations support to unravel the different elementary structural units based on halogen bonds and hydrogen bonds and were useful tools to dissect and explain the formation mechanism.
Introduction
π-Conjugated thiophene-based oligomers have generated a great deal of attention due to their improved solubility, well-defined chemical structure, and excellent electronic properties, which make them promising candidates for applications in fabricating organic electronic devices, such as light-emitting diodes, field-effect transistors, and organic photovoltaic cells.1–3 Understanding the molecular arrangement and the intermolecular interactions is an important issue in fabricating organic electronic devices. Scanning tunneling microscopy (STM) is especially well suited to exploring the role of the molecule in assembling processes and surface engineering by self-assembly as well as in molecular device fabrication. Self-assembly is the spontaneous organization of molecular components into larger supramolecular architectures and is one of the most powerful methods to fabricate molecular devices by a bottom-up approach.4,5 The assembly process is sensitive to a delicate balance among the intermolecular non-covalent interactions, involving the van der Waals (vdW) forces,6–8 hydrogen bonding,9–13 dipolar interaction,14–16 π–π stacking,17,18 and so on.As a high directional analog to hydrogen bonding, halogen bonding is an attractive tool for developing the construction of ordered surface-bound assemblies, and its binding geometry can adopt a few configurations,19,20 which have been intensely investigated in 3D crystal engineering21,22 and biological systems.23 Besides, these obvious advantages of halogen bonding have been utilized for the design and effective control of various supramolecular structures for rigid molecules with halogen substituents.24–30 For example, Gutzler et al. reported the first case of halogen bonding in monolayers at the liquid/solid interface using STM to study the self-assembly of tetrabrominated derivatives.26 However, few reports describe the effects of the alkyl chain and bromine substituents in rigid molecules on two-dimensional (2D) surfaces. On the surface, the adsorption behavior depends on the chemical substituent and is of special interest. Having a substituent with a slight difference in the position can produce a variety of morphologies and vastly different extents of polymorphism.31,32 For example, Stöhr et al. demonstrated the formation of highly-ordered self-assembled structures of pyrene derivatives on the Au (111) surface under vacuum conditions, which was influenced by the position and number of bromine substituents.25 The relationship between the position of the bromine substituents and the types of halogen bonds is still not fully understood. Therefore, it is necessary and also important to understand for certain how the position of bromine substituents affect the surface-supported self-assembly process. In addition to the intermolecular interactions, the molecule–solvent interaction is also a key factor that can drastically modify the intermolecular binding.28 In the present study, we focused on understanding the competition in the delicate interplay between interchain vdW interactions and intermolecular halogen bonding, as well as the solvent and solution concentration effects on the formation of self-assembly polymorphism.
Previously, we investigated the self-assembly of a thienophenanthrene derivative (6,9-DBTD) at the aliphatic acid/graphite interface. Apart from the intermolecular vdW interaction, halogen bonding consisting of a triangular binding motif based on Br⋯Br⋯H bond dominated the diversity of the studied self-assembled nanostructures. We found that the thienophenanthrene derivative could be switched among dissimilar polymorphs with different halogen-bond densities formed by adjusting the solution concentration.33 Then, we further investigated the effect of different halogen substituents of thienophenanthrene derivatives (5,10-DBTD and 5,10-DITD) on the self-assembly at the liquid/graphite interface and found that the orientation of the ester substituent for thienophenanthrene derivatives affects the positive charge distribution of halogens, thus determining the formation of an intermolecular halogen bond and different self-assembled patterns.34 Besides, such molecules were chosen because π-conjugated thienophenanthrene cores composed of a fused-ring compound with one more ring than phenanthrene have been applied in organic photovoltaic cells.35–37
Herein, we investigated the photophysical properties of two brominated π-conjugated geometrical isomers (depending on the position of bromine in the molecule), 1,3-dicarboxylic acid dihexadecyl ester-6,9-dibromo-phenanthro[9,10-c]thiophene (6,9-DBTD) and 1,3-dicarboxylic acid dihexadecyl ester-5,10-dibromo-phenanthro[9,10-c]thiophene (5,10-DBTD) (Scheme 1a and b), and the influence of the position of the bromine substituents on their 2D self-assembly at the aliphatic hydrocarbon (n-tridecane, n-tetradecane, n-pentadecane and n-hexadecane)/graphite interface. The STM results revealed that the 6,9- and 5,10-DBTD molecules can construct polymorph nanostructures via the cooperation and competition between vdW interactions of molecule–molecule, intermolecular interactions including Br⋯OC and Br⋯S hetero-halogen bonds, and homo-Br⋯Br interactions, H⋯Br and H⋯O hydrogen bonds (HBs) along with varying the solvent and solution concentration. With the help of density functional theory (DFT) calculations, we clarified how slightly tuning the molecular structure can define the geometry of the 2D self-assembled nanoarchitecture through different halogen bonding ways. It would be reasonable to believe that our current work could provide a new object of study toward controlling and fabricating 2D patterns based on halogen bonds.
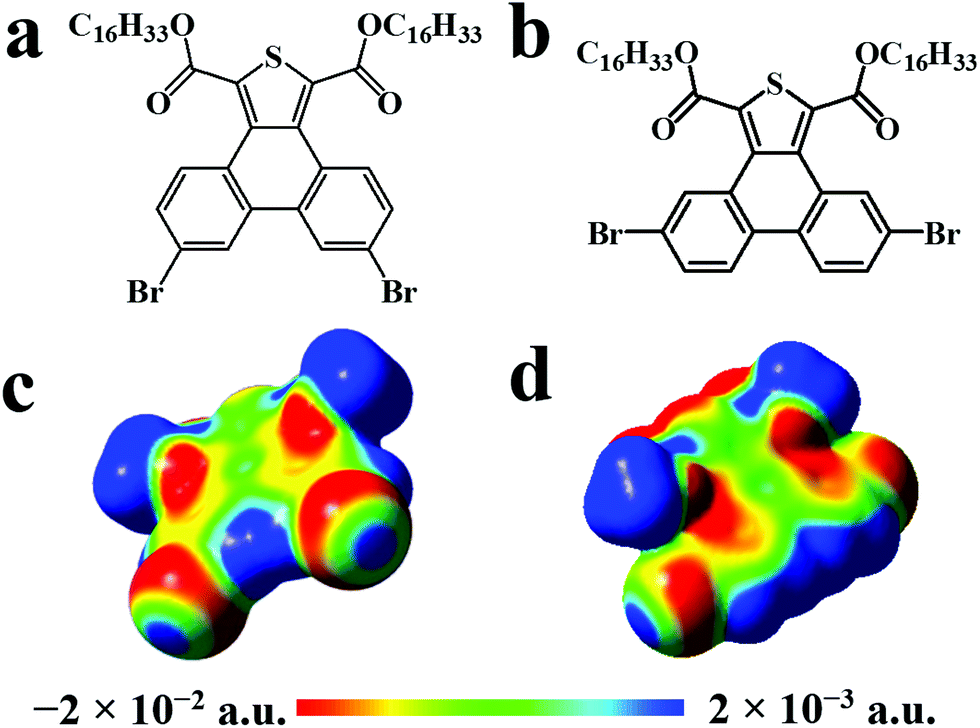 | ||
| Scheme 1 Chemical structures of the thienophenanthrene derivatives: (a) 6,9-DBTD and (b) 5,10-DBTD. (c, d) Calculated electrostatic potential maps of 6,9-DBTD and 5,10-DBTD under vacuum, presented by blue (positive) and red (negative) regions. The hexadecyloxy side chains were replaced by the methoxy groups. | ||
Experiment section
Synthesis
6,9-DBTD and 5,10-DBTD were synthesized as described in previous reports.33,34 The synthesis of 6-DBTD is described in the ESI.†Scanning tunneling microscopy
n-Tridecane, n-tetradecane, n-pentadecane, and n-hexadecane were purchased from TCI. The samples were prepared by dropping a droplet (∼1 μL) of solution containing the DBTDs (5,10-DBTD, 6,9-DBTD, and 6-DBTD, concentration: 10−3–10−6 M) onto a freshly cleaved atomically flat surface of HOPG (quality ZYB, Bruker, USA). Most the images obtained at the liquid/solid interface were recorded within 3 h after dropping the solution. STM measurements were performed on a Nanoscope IIIa Multimode SPM (Bruker, USA) at ambient conditions with the tip immersed in the supernatant liquid. The tips were mechanically cut from Pt/Ir wires (80/20). All the images were recorded with the constant current mode and are shown herein without further processing. The tunneling parameters are given in the corresponding figure captions. Different tips and samples were used to check the reproducibility, and to exclude any image artifacts caused by the tips or the samples. The HOPG lattice was used as an internal standard to modify each STM image and to determine the average lattice parameters of the DBTDs monolayer by examining at least 4 images.Computational simulation
Structural models of the assembled structures were built by using Materials Studio 4.4. The models of the monolayers were constructed by placing the molecules according to the intermolecular distances and angles that were obtained from the analysis of the STM images.DFT calculations were performed with the Gaussian 09 software package. The geometry full-optimizations of those structures were done through the M06-2X method together with the split-valence polarized 6-31+g(d) basis set. The M06-2X functional is known to be more accurate for systems involving non-covalent interactions, and hence was chosen for the description of the halogen-bonded complexes.31,38 The 6-31+g(d) basis set was found to provide the lowest total energy and to be more suitable for modeling hydrogen-bonded species.39 Halogen bonds, which are similar to hydrogen bonds, are known to be a kind of non-covalent intermolecular interaction. The topological properties of the electron densities in all the complexes were assessed using the Multiwfn program.40
The geometry optimization for the ground state (S0) was conducted by DFT using the M06-2X functional along with the 6-31+g(d) basis set. The electronic absorption and fluorescence spectra of the two thienophenanthrene derivatives were assessed with the time-dependent density functional theory (TD-DFT) at the CAM-B3LYP/6-31+g(d) level based on the optimized structures of the ground state.
Results
Photophysical properties of the thienophenanthrene derivatives
The UV-vis absorption spectra of the two thienophenanthrene derivatives were recorded in dichloromethane (DCM) solution, as shown in Fig. 1a. In this figure both derivatives show several adsorption bands; however, these are the same with the longest wavelength absorption peak due to a π → π* charge-transfer transition appearing in the 355–370 nm spectral range, with a peak position centered at 360 nm. The adsorption band shows moderate sensitivity toward the substituent, indicating the characteristic of the thienophenanthrene core, while the position of the bromine substituents has no significant effect on the photoabsorption performance.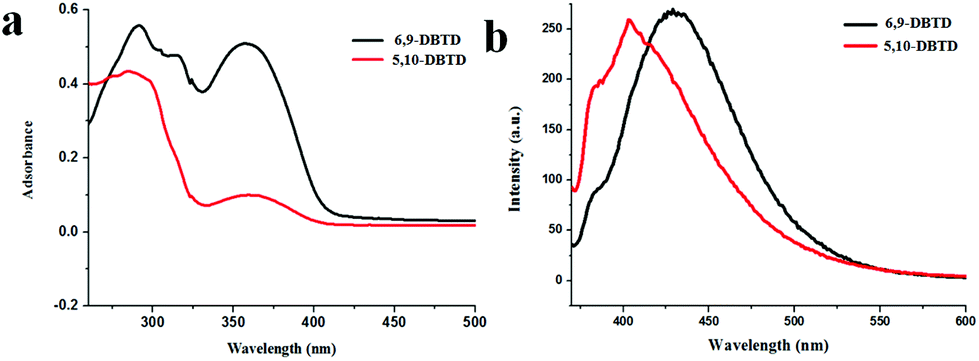 | ||
| Fig. 1 (a) UV-vis absorption spectra of 6,9-DBTD and 5,10-DBTD in DCM solution. (b) Fluorescence spectra of 6,9-DBTD and 5,10-DBTD in DCM solution at room temperature. λEX = 370 nm. | ||
In order to gain a detailed insight into the nature of the UV-vis absorption of the two thienophenanthrene (TP) derivatives, we calculated the singlet–singlet electronic transition in DCM, based on the optimized geometries of the ground state of the two compounds using the TD-DFT method at the CAM-B3LYP/6-31+g(d) level. From Table 1, we can see that an intense S0 → S1 transition for 6,9-DBTD was predicted at about 345 nm with an oscillator strength of 0.1296, which was assigned as a dominant π → π* charge-transfer transition from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO) (Table S1†), with smaller components of HOMO−1 → LUMO+1 and HOMO−3 → LUMO. The S0 → S1 transition for 5,10-DBTD, which was also mainly donated by a HOMO → LUMO transition with an oscillator strength of 0.2247, lies at about 336 nm.
| UV–vis | Fluorescence | |||
|---|---|---|---|---|
| 6,9-DBTD | 5,10-DBTD | 6,9-DBTD | 5,10-DBTD | |
| a The numbers in parentheses are the excitation energy in wavelength. b Oscillator strength. c H stands for HOMO and L stands for LUMO. d CI coefficients are absolute values. | ||||
| Transition | S0 → S1 | S0 → S1 | S1 → S0 | S1 → S0 |
| Energya (eV) | 3.59 (345 nm) | 3.69 (336 nm) | 2.89 (428 nm) | 3.01 (411 nm) |
| Oscillator strength (f)b | 0.1296 | 0.2247 | 0.3608 | 0.3265 |
| Compositionc | H → L | H → L | L → H | L → H |
| H → L+1 | H−2 → L | L → H−2 | ||
| H−3 → L | H−1 → L+2 | |||
| CId (%) | 67.3 | 65.8 | 69.0 | 67.7 |
| 13.1 | 18.3 | 16.2 | ||
| 10.4 | 11.2 | |||
For 6,9-DBTD and 5,10-DBTD, in which an electron withdrawing group (–Br) is appended, the electronic transitions for HOMO → LUMO are of the π → π* type from the local transition assigned to the electron density delocalized over the TP π-core moiety. These expectations based on the TD-DFT calculations were verified by the experimental observations (Fig. 1a).
The fluorescence spectra of the two compounds were explored in the 1 × 10−5 M region. Upon excitation at 370 nm, the two TP derivatives showed fluorescence properties in DCM, as presented in Fig. 1b. In order to study the nature of the fluorescence emission observed for these two compounds, the geometries of the first excited singlet state (S1) were optimized for the TP derivatives in DCM using the TD-DFT method at the CAM-B3LYP/6-31+g(d) level. It can be seen from Table 1 that 6,9-DBTD (428 nm, f = 0.3068) belongs to a LUMO → HOMO transition, as well as the type of 5,10-DBTD (411 nm, f = 0.3265). These S1 → S0 emissions with a π → π* transition character are donated from the local transition assigned to the electron density delocalized over the TP π-core moiety. These values based on DFT/TD-DFT calculations were also verified by the experimental observations (Fig. 1b). On the basis of the electronic transitions, we can see that the Br groups act as electron donors and play an indispensable role in the different orbits involved in the excitation. Furthermore, the S1 → S0 transition is a fully allowed reverse transition, as indicated by the overlapping between the HOMO and LUMO (Table S1†), as well as the transition oscillator strength (f).41,42 Thus, this TP π-core is potentially fluorescent. It is believed that these findings will be important for the future design of new TP-based fluorophores. In general, different packing arrangements and molecular conformations result in different intermolecular interactions, which play the most significant role in tailoring fluorescence.
Self-assembled structures of 6,9-DBTD at the aliphatic solvent/graphite interface
To evaluate the solution concentration effect on the geometry of the 2D self-assembled networks, the monolayer formation of 6,9-DBTD was investigated at the n-tridecane/HOPG interface with different solution concentrations (from 10−3 to 10−6 M). After a drop of a saturated solution of 6,9-DBTD was drop-casted on the basal plane of a freshly cleaved graphite surface at ambient conditions, a nanostructured adlayer was spontaneously formed at the liquid/solid interface. The well-ordered lamellar structure of 6,9-DBTD (Fig. S1a and b†) was obtained. From the high-resolution STM image in Fig. 2a, the conjugated TP π-cores of 6,9-DBTD appear as bright triangular features, corresponding to high tunneling current, whereas the dark areas are attributed to the side alkyl chains, because of their lower electron density.43 It can be seen that two TP π-cores form a dimer and arrange in a head-to-head fashion via two pairs of Br⋯Br and H⋯Br bonds, while adjacent dimers are connected through a pair of C–H⋯OC and H⋯Br hydrogen bonds. The enlarged inset in Fig. 2c provides a model view of the possible halogen and hydrogen bonds, which are marked by dotted lines formed by two neighboring molecules. The side chains are stacked in a tail-to-tail fashion, locating at the same side of the molecules and arranged orthogonally with respect to the rows. The suggested assembled model is shown in Fig. 2c. The unit cell parameters were determined to be: a = 1.0 ± 0.1 nm, b = 6.2 ± 0.1 nm, and β = 74 ± 1°.
 | ||
| Fig. 2 High-resolution (scan area: 20 × 20 nm2) STM images of 6,9-DBTD self-assembly in n-tridecane on the HOPG surface. (a) Lamellar structure (concentration: 1.2 × 10−4 M; tunneling parameters: It = 426 pA and Vbias = 669 mV). (b) Coadsorbed linear pattern (concentration: 6.2 × 10−5 M; It = 421 pA and Vbias = 671 mV). (c, d) Structural models correspond to the observed structures in (a–b). | ||
Upon decreasing the solution concentration (7.5 × 10−4–1.6 × 10−5 M), a coadsorbed linear pattern (Fig. S1c and d†), coexisting with the lamellar structure, arose at the n-tridecane/graphite interface. When the concentration was lower than 1.6 × 10−5 M, only the coadsorbed linear pattern covered the whole scan area (Fig. S1e and f†). The equal distant bright lines were distributed orderly on the HOPG surface. Fig. 2b is the high-resolution STM image, showing the detailed coadsorbed linear pattern of 6,9-DBTD. In one row, two TP π-cores construct a dimer through double C–H⋯OC hydrogen bonds with an antiparallel configuration, while adjacent dimers are arranged in a tail-to-tail fashion, which leads to the formation of Br⋯Br interactions. The possible halogen and hydrogen bonds are marked by dotted lines formed by two neighboring molecules, as shown in the enlarged inset of Fig. 2d. The side chains of 6,9-DBTD keep their interdigitated arrangement and lie flat on the surface. By counting the number of chains and the side chains of 6,9-DBTD, we concluded that the n-tridecane molecules can occupy the space between the alkoxy chains of 6,9-DBTD, and coadsorb on the HOPG surface due to the steric effect and molecule–solvent vdW interactions. On the basis of the above analyses, a structural model for the coadsorbed pattern is proposed in Fig. 2d. The unit cell parameters were measured to be a = 2.6 ± 0.1 nm, b = 2.9 ± 0.2 nm, and β = 71 ± 1°.
When the solvent was replaced by n-tetradecane, n-pentadecane, and n-hexadecane, only the coadsorbed linear pattern of 6,9-DBTD could be observed (Fig. S2–S4†), and the solution concentration had no effect on the formation of the self-assembled configuration. The coadsorbed linear patterns were similar to the molecular packing observed in the n-tridecane solvent at low concentrations, which indicated that the coadsorbed linear pattern was a more stable structure. The results further demonstrate that, in addition to the steric hindrance effect, the molecule–solvent vdW interactions play a dominant role in the coadsorption of solvent molecules on the HOPG surface.
Self-assembled structures of 5,10-DBTD at the aliphatic solvent/graphite interface
C and Br⋯S halogen bonds. The neighboring 5,10-DBTD units were connected through C–H⋯Br and C–H⋯OC hydrogen bonds, which further strengthened this ribbon motif. Two rows of molecules were packed in a reverse orientation and formed the vertebra-like pattern, resulting in the minimized polarity of the adlayer.16 Thus, 5,10-DBTD exhibited distinct and nonidentical halogen bonds, which could vary as a function of the halogen identity. The unit cell parameters were measured to be a = 2.3 ± 0.1 nm, b = 5.8 ± 0.1 nm, and β = 84 ± 1°.
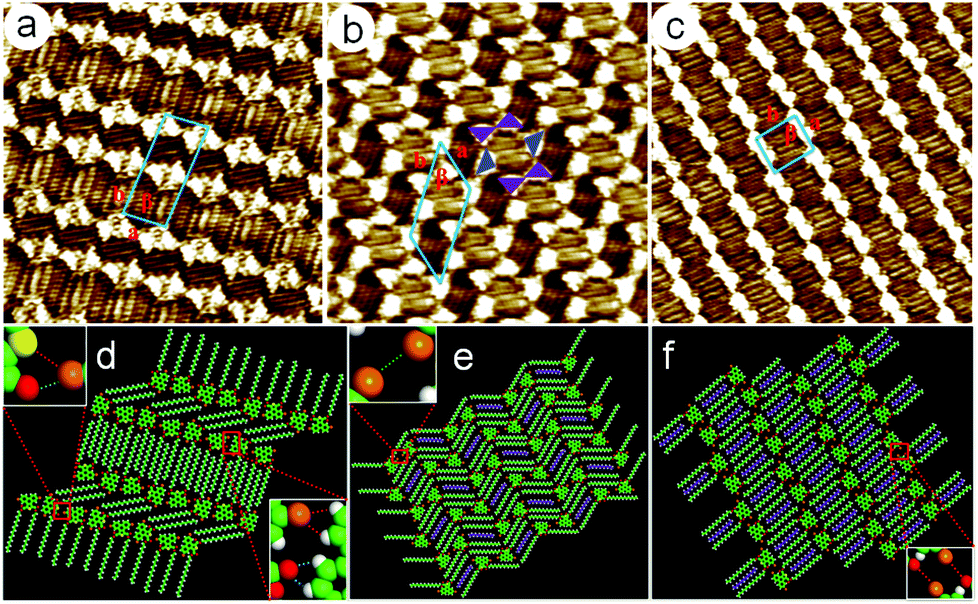 | ||
| Fig. 3 High-resolution STM image of the 5,10-DBTD self-assembly in n-tridecane. (a) Vertebra-like pattern at a high concentration (concentration: 2.8 × 10−4 M; 20 × 20 nm2; It = 430 pA and Vbias = 655 mV). (b) Hexagonal network pattern with a mediate concentration (5.7 × 10−5 M; 17 × 17 nm2; It = 432 pA and Vbias = 658 mV). (c) Linear pattern under a low concentration (5.3 × 10−6 M; 20 × 20 nm2; It = 431 pA and Vbias = 651 mV). (d–f) Structural models correspond to (a–c). | ||
A hexagonal network pattern (Fig. S5c and d†) could also be discerned with decreasing the concentration (4.6 × 10−4–6.8 × 10−5 M). A typical high-resolution STM image is given in Fig. 3b, which depicts the details of the hexagonal network pattern of the 5,10-DBTD molecule. Here, the hexagonal network with a 6-fold ring elementary structural motif is enclosed by two pairs of molecules and two single molecules (indicated by triangles). Two TP π-cores appear in pairs with reverse orientations through Br⋯Br interactions, indicated by triangles with the same color. At the same time, the TP π-cores labeled by blue triangles form Br⋯Br interactions with the molecules in the adjacent hexagonal ring, respectively, as shown in Fig. 3b. Two neighboring hexagonal units in each lamella have two TP π-cores in common. The angle of the side chains in the neighboring hexagonal units is 120°, indicating that the arrangements of alkyl chains are consistent with the lattice symmetry of HOPG. By counting the number of chains and the side chain of 5,10-DBTD, we found that one n-tridecane molecule is coadsorbed with the 5,10-DBTD molecules in each hexagonal unit due to space matching and the interchain vdW interactions, just as the suggested model shows in Fig. 3e. The unit cell parameters of the hexagonal network pattern were measured to be a = 2.9 ± 0.1 nm, b = 5.2 ± 0.1 nm, and β = 52 ± 1°.
When the concentration was further decreased (5.7 × 10−5–8.2 × 10−5 M), the typical STM image (Fig. S5e†) of 5,10-DBTD self-assembly in n-tridecane on the graphite surface shows the coexistence of the linear pattern and hexagonal network pattern. With the decrease in the solution concentration (8.2 × 10−5 M), the assembling structure undergoes a variation from the hexagonal network pattern to the linear pattern (Fig. S5f†). As shown in Fig. 3c, all the 5,10-DBTD molecules are anti-aligned, and the side chains located on the same side of the molecules are arranged orthogonally with respect to the rows. Thus, we estimated that all the carboxyl groups orient toward the conjugated TP cores. Except for the interchain vdW interactions, the Br⋯OC XBs play a decisive role in the structural formation. The adjacent TP cores arrange through a single Br⋯Br interaction and form a kinked lamella. Similarly, we found that the n-tridecane molecules filled in the arrangement space between each side chain lamella of 5,10-DBTD due to the spatial matching. The unit cell parameters were determined to be: a = 2.4 ± 0.1 nm, b = 2.8 ± 0.1 nm, and β = 83 ± 1°. A molecular model was constructed and is shown in Fig. 3f.
 | ||
| Fig. 4 (a, b) Large-scale (70 × 70 nm2) and high-resolution (20 × 20 nm2) STM images of the 5,10-DBTD self-assembly showing the hexagonal network pattern in n-tetradecane with a high concentration (2.1 × 10−4 M) on HOPG surfaces. It = 422 pA and Vbias = 676 mV. (c) Structural model corresponding to the hexagonal network pattern. (d) Coexisting pattern (100 × 100 nm2) with a medium concentration (3.3 × 10−5 M) on the HOPG surface. It = 429 pA and Vbias = 686 mV. (e) Loosely packed double linear pattern (17 × 17 nm2) with a low concentration (7.9 × 10−5 M). It = 417 pA and Vbias = 682 mV. (f) Structural model corresponding to the loosely packed double linear pattern. | ||
When the solution concentration decreased to about 5.3 × 10−5 M, the densely packed double linear pattern disappeared, and the whole scanning area was then covered by a regular loosely packed double linear pattern, as shown in Fig. 4e and S6d.† As the green arrows indicate in Fig. 4e, in adjacent one-row molecular lamellae, the conjugated TP π-cores arrange in reverse orientations. The high-resolution STM image clearly displays that more alkyl chains can be identified in the side chain lamellae. This phenomenon demonstrates that the n-tetradecane molecules fill the gap between adjacent rows, and take part in the assembly process, as the purple alkyl chain models indicate in Fig. 4f. The unit cell parameters were determined to be: a = 1.7 ± 0.1 nm, b = 6.2 ± 0.1 nm, and β = 61 ± 1°. Moreover, it was found that the interchain vdW interactions were enhanced distinctly due to the coadsorption of more n-tetradecane molecules, which resulted in the absence of intermolecular halogen bonding.
 | ||
| Fig. 5 (a) High-resolution (20 × 20 nm2) STM image of 5,10-DBTD self-assembly at the n-pentadecane/graphite interface. It = 422 pA and Vbias = 667 mV. (b) Molecular model corresponding to the self-assembled structure. | ||
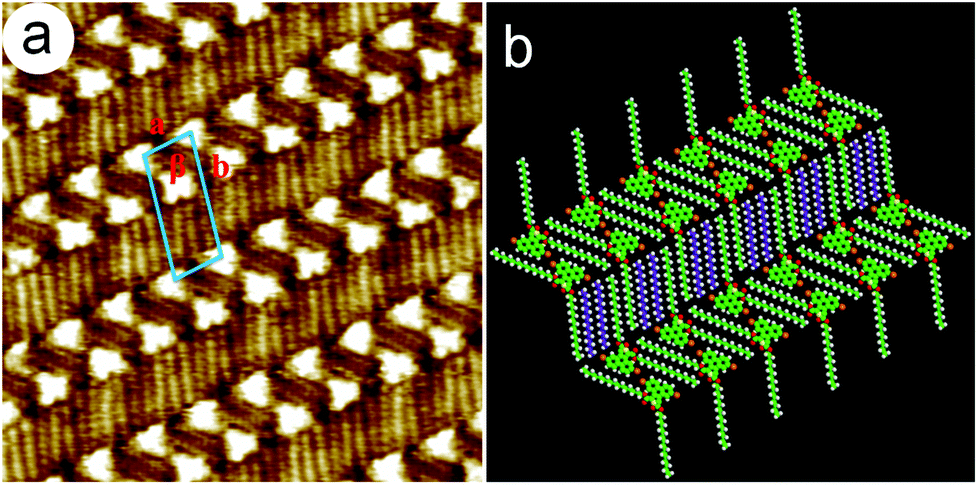 | ||
| Fig. 6 (a) High-resolution (16 × 16 nm2) STM image of the 5,10-DBTD self-assembly at the n-hexadecane/HOPG interface. It = 422 pA and Vbias = 667 mV. (b) Molecular model corresponding to the self-assembled structure. | ||
Discussion
Table 2 summarizes the geometric characteristics, molecular self-assembled models, and unit cell parameters of the physisorbed adlayers of DBTDs (6,9- and 5,10-DBTD) at the aliphatic solvents (n-tridecane, n-tetradecane, n-pentadecane, and n-hexadecane)/graphite interfaces with different concentrations.| Molecule | Solvent | Structural model | Concentration (M) | a (nm) | b (nm) | α (°) | Density, S (nm2 per molecule) |
|---|---|---|---|---|---|---|---|
| 6,9-DBTD | n-Tridecane |
|
>7.5 × 10−4, saturated | 1.0 ± 0.1 | 6.2 ± 0.1 | 74 ± 1 | 3.97 |
|
<7.5 × 10−4 | 2.4 ± 0.1 | 2.8 ± 0.1 | 76 ± 1 | 4.34 | ||
| n-Tetradecane |
|
>10−6, saturated | 2.6 ± 0.1 | 2.9 ± 0.2 | 71 ± 1 | 3.56 | |
| n-Pentadecane |
|
>10−6, saturated | 2.5 ± 0.1 | 3.3 ± 0.1 | 80 ± 1 | 4.06 | |
| n-Hexadecane |
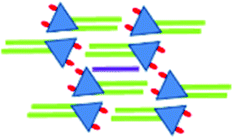
|
>10−6, saturated | 2.3 ± 0.1 | 2.9 ± 0.1 | 76 ± 1 | 4.41 | |
| 5,10-DBTD | n-Tridecane |
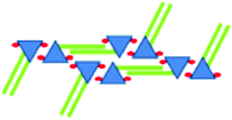
|
>4.6 × 10−4, saturated | 2.3 ± 0.1 | 5.8 ± 0.1 | 84 ± 1 | 3.31 |
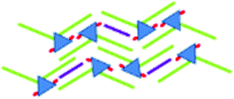
|
4.6 × 10−4–6.8 ×10−5 | 2.9 ± 0.1 | 4.9 ± 0.1 | 56 ± 1 | 3.96 | ||
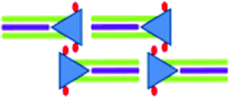
|
<6.8 × 10−5 | 2.4 ± 0.1 | 2.8 ± 0.1 | 83 ± 1 | 4.45 | ||
| n-Tetradecane |

|
>7.1 × 10−4, saturated | 2.9 ± 0.1 | 5.2 ± 0.1 | 52 ± 1 | 3.96 | |
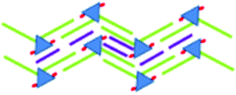
|
<7.1 × 10−5 | 1.7 ± 0.1 | 6.2 ± 0.1 | 61 ± 1 | 4.61 | ||
| n-Pentadecane |
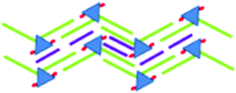
|
<4.1 × 10−4 | 2.0 ± 0.1 | 5.6 ± 0.1 | 61 ± 1 | 4.89 | |
| n-Hexadecane |
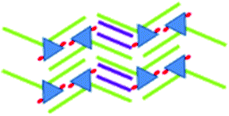
|
>10−6, saturated | 1.9 ± 0.1 | 4.3 ± 0.1 | 75 ± 1 | 4.51 |
The number of bromine substituents induces the diversity of the self-assembled structures
6,9-DBTD and 5,10-DBTD are geometrical isomers, so the only difference between them lies in the position of the Br substituents. The dipole moment of 5,10-DBTD (3.97 D) is larger than that of 6,9-DBTD (1.75 D). In our system, at the n-tridecane/graphite interface with a higher concentration without solvent adsorption, most Br atoms of 6,9-DBTD and 5,10-DBTD take part in the formation of halogen bonding, although these two molecules exhibit completely different self-assembled patterns. We suggest that in order to coordinate the halogen bond direction efficiently and to form stable halogen bonds, the conjugated TP π-cores adopt different orientations, which results in the different adlayers.In order to demonstrate the influence of the number of Br substituents on the halogen bonding formation in the 2D self-assembled structure, the 6-DBTD molecule with a single bromine atom was home-synthesized (ESI†) (Fig. 7a). The self-assembly of 6-DBTD was then extensively investigated at the aliphatic solvents/HOPG interfaces under different solution concentrations. No solvent and concentration effects on the self-assembly were observed. STM images (Fig. 7a and S9†) showed that 6-DBTD forms a linear structure. From the molecular model based on the STM image (Fig. 7b), it can be observed that two 6-DBTD molecules form a dimer with a head-to-head fashion via a pair of H⋯Br bonds. The linear phase had a density of 2.58 nm2 per molecule. The unit cell parameters of the 2D assembly were measured to be a = 2.0 ± 0.1 nm, b = 2.9 ± 0.1 nm, and β = 63 ± 1°. Besides the vdW interaction between the side chains, the dimer was stabilized by the H⋯Br hydrogen bonds formed with the neighboring molecules. Interestingly, the H⋯Br bonds were formed by the opposite charge distribution areas of the Br (δ−) and H (δ+) atoms within the dimer nodes. The absence of Br⋯Br bonds in the self-assembly of 6-DBTD could be explained by taking a weaker intermolecular interaction and thermodynamic considerations into account. First, besides the intermolecular vdW interaction and H⋯Br bonds, the molecules arrange in pairs by antiparallel dipole–dipole interactions extinguishing the polarity of a single 6-DBTD molecule, such that the overall dipole moment around the area is actually zero. Second, compared with the non-covalent bonding, the interplay of entropy and enthalpy also plays an important role in the nanostructure formation.25,44 The decrease in entropy during self-assembly must be compensated by a gain in enthalpy via the formation of favorable intermolecular interactions. Since 6-DBTD has one terminal Br substituent, two adjacent molecules will preferably bind via H⋯Br bonds to maximize the attractive interactions and to minimize the repulsive Br⋯Br interactions. In this way, a minimum in the Gibbs free energy is obtained, corresponding to a stable state for the overall system.25
 | ||
| Fig. 7 (a) High-resolution (20 × 20 nm2) STM image of the 6-DBTD self-assembly at the liquid/solid interface. It = 427 pA and Vbias = 665 mV. (b) Molecular model corresponding to the self-assembled structure. | ||
Self-assembly mechanism: computing simulation for halogen bonding and hydrogen bonding
Except for the intermolecular vdW interaction and dipole–dipole interaction, two other decisive factors affecting the adlayers of the DBTDs were the bromine substituents and their position. The Br atoms are electron withdrawing groups and are highly polarizable; i.e., these atoms have strong electronegativity, giving rise to a rearrangement in the electronic density distribution of the molecules, and further causing distinct types of Br bonds, which affects the self-assembled structures of the monolayers.38 The electrostatic potential (ESP) maps (Schemes 1c and d) reveal the charge distribution, which is particularly noteworthy with the Br atoms. A region of positive electrostatic potential (blue) was evident on the outermost portion of the Br atom surface, where it intersected the C–Br axis, while a negative potential (red) encompasses the bromine atom.45–47 Such polarization is given the name of a “σ-hole” and is responsible for the halogen bonding.20,26 As we know, in our study, there were two different types of Br⋯Br bonds, which were classified based on the C–Br⋯Br angles.48 When the C–Br⋯Br angles α1 and α2 tend to be close to 180° and 90°, respectively, they are known as type-II halogen bonding (Scheme S1†). Type-II contacts were interpreted as an attractive Br+⋯Br− interaction and conformed to the Williams model.19 However, the same angle for α1 and α2 implies that the Br⋯Br (type-I) interaction is in accordance with the vdW type.49In order to gain deeper insights into the intermolecular interactions mechanisms within the adlayers, we carried out DFT calculations by using the M06-2X functional and applying the 6-31+g(d) basis set. For the self-assembly of 6,9-DBTD at the liquid/HOPG interfaces, besides the vdW interactions between the side chains, the halogen bonds and hydrogen bonds were the most dominant interactions in stabilizing the 2D self-assembled structures. A careful observation revealed that the self-assembled patterns with four elementary structural units (dimer-I to dimer-IV) could be classified in categories, whereby dimer-I and dimer-II and then dimer-III and dimer-IV are the basic units of the lamellar structure and coadsorbed linear pattern, respectively. The geometry optimizations were closely consistent with the four elementary structural units of the observed 6,9-DBTD system, as shown in Fig. 8 and Table 3. The formation of dimer-I was mainly driven by the Br⋯Br⋯H triangular binding motifs formed by H⋯Br and Br⋯Br (type-II) bonds. The molecules in dimer-II were connected through a pair of C–H⋯OC and H⋯Br hydrogen bonds, which have been observed for different molecules in 2D and 3D supramolecular assembles.26,50 The most significant close-contacts were the Br⋯Br and H⋯Br (see Table 3), where the distances of the two atoms are smaller than the sum of the vdW radii (ΣvdW = 3.70 Å for Br⋯Br, 3.05 Å for H⋯Br).27 The same angle for α1 and α2 in dimer-III indicates that the Br⋯Br bond is in accordance with a type-I repulsive interaction. For dimer-IV, the TP π-cores arrange in an antiparallel mode, concatenated through a pair of C–H⋯OC hydrogen bonds. The calculated binding energy densities per unit area obtained from the DFT calculations were 4.73 and 1.42 kcal mol−1 nm−2 for the lamellar structure and coadsorbed linear pattern, respectively. These results illustrate that the lamellar structure is a more stable arrangement. Although the binding energy density per unit area of the lamellar structure was higher, the intermolecular vdW interactions were enhanced due to the solvent coadsorption and the interdigitation of the side chains, which gave rise to the stable coadsorbed linear pattern. The results also display that the coadsorption of aliphatic solvents can change the contact modes of the Br atom, in which the type of Br⋯Br interactions change from type-II halogen bonds to type-I contact bonds.
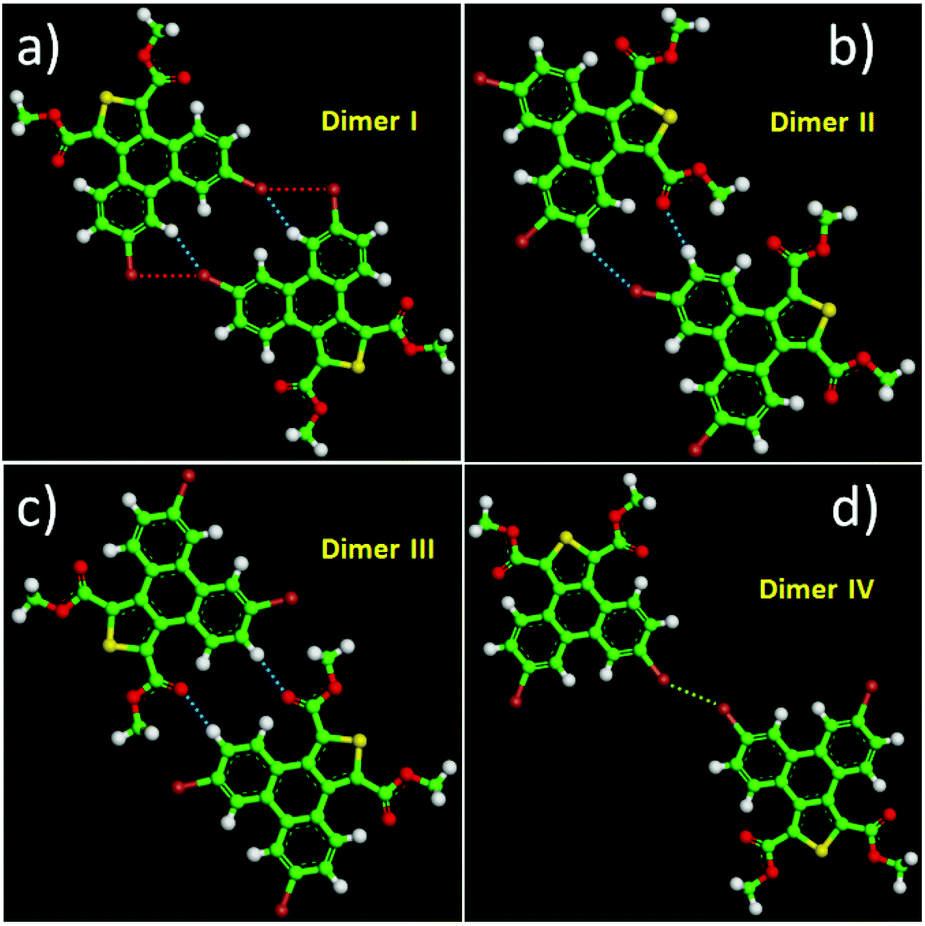 | ||
| Fig. 8 (a–d) DFT optimized intermolecular interactions for four different dimers formed by 6,9-DBTD. The intermolecular bonds are labelled by red (halogen bond) and blue (hydrogen bond) dashed lines, respectively. | ||
| Molecular block | Dimer I | Dimer II | Dimer III | Dimer IV | ||
|---|---|---|---|---|---|---|
|
|
|
|
|
|||
| Bond | H⋯Br–C | C–Br⋯Br | H⋯Br–C | C–H⋯O | C–Br⋯Br | C–H⋯O |
| Distance (Å) | 2.68 | 3.53 | 2.99 | 2.47 | 3.40 | 2.46 |
| Angle (°) | 151.7 | 95.0 | 146.7 | 173.6 | 153.7 | 175.5 |
| ΔE/(kcal mol−1) | −8.40 | −5.70 | −2.44 | −6.80 | ||
| ρ(CP)/(e Å−3) | 0.0690 | 0.0619 | 0.0364 | 0.0585 | 0.0635 | 0.0562 |
| ∇2ρ(CP)/(e Å−5) | 0.8639 | 0.6926 | 0.4492 | 0.7714 | 0.8160 | 0.7620 |
| G(CP)/(kJ mol−1 bohr−3) | 19.21 | 15.10 | 9.29 | 18.64 | 17.30 | 18.27 |
| V(CP)/(kJ mol−1 bohr−3) | −14.91 | −11.33 | −6.34 | −16.48 | −12.37 | −15.79 |
| H(CP)/(kJ mol−1 bohr−3) | 4.30 | 3.77 | 2.95 | 2.16 | 4.93 | 2.48 |
| |V(CP)|/G(CP) | 0.78 | 0.75 | 0.68 | 0.88 | 0.72 | 0.86 |
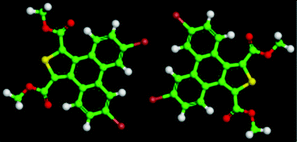

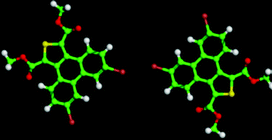
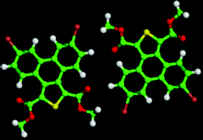
The 5,10-DBTD molecules formed several self-assembled structures via interchain vdW interactions and intermolecular interactions between the TP cores. A detailed analysis of the well-resolved STM images showed four styles of dimers (dimer-I to dimer-IV), which were classified based on the Br atoms forming Br⋯X (O, S, H and Br) bonds between adjacent TP π-cores, as shown in Fig. 9 (see Table 4). In dimer-I, the Br atom forms the bifurcated hetero-halogen Br⋯OC and Br⋯S bonds, while in dimer-II, the Br atom forms a H⋯Br bond with the β-H atom, along with the formation of two C–H⋯OC hydrogen bonds. Dimer-I and dimer-II are the basic units of the vertebra-like pattern, which was formed by 5,10-DBTD in n-tridecane at high concentrations. For dimer-III, the TP π-cores arrange in an antiparallel mode, concatenated through a pair of C–Br⋯OC halogen bonds with two centers. The formation of a C–Br⋯OC halogen bond should be attributed to the bromine atom acting as a Lewis acid and the carbonyl oxygen atom (CO) as a Lewis base. This result was consistent with the previous literature.51–53 This type of halogen bond is only available in n-tridecane. At the aliphatic solvent/HOPG interfaces, dimer-IV features frequently appear in the self-assembly of 5,10-DBTD, such as the hexagonal network pattern, the linear pattern, and the densely packed double linear pattern. In dimer-IV, the C–Br⋯Br angles α1 and α2 are the same, indicating that the Br⋯Br bond is of the vdW type (type-I).
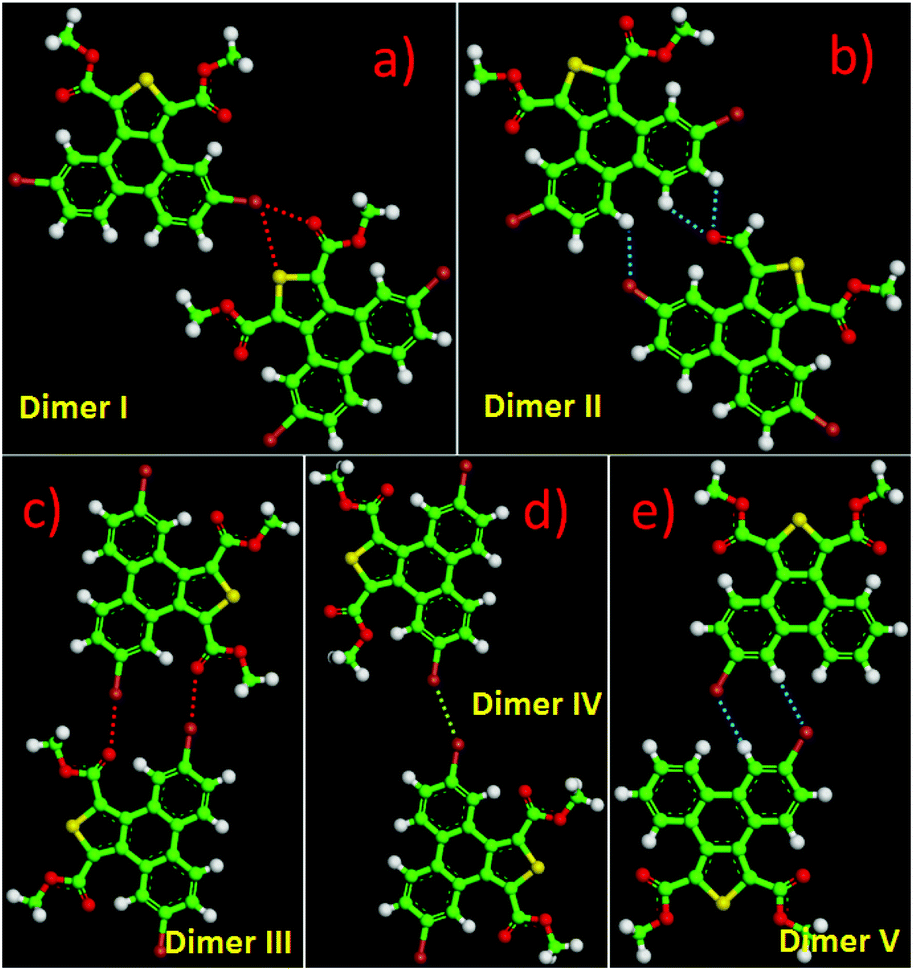 | ||
| Fig. 9 (a–d) DFT optimized intermolecular interactions for four different dimers formed by 5,10-DBTD. (e) DFT optimized intermolecular interactions for the dimer formed by 6-DBTD. The intermolecular bonds are labelled by red (halogen bond) and blue (hydrogen bond) dashed lines, respectively. | ||
| Molecular block | Dimer I | Dimer II | Dimer III | Dimer IV | Dimer V | ||
|---|---|---|---|---|---|---|---|
|
|
|
|
|
|
|||
| Bond | C–Br⋯O | C–Br⋯S | C–H⋯Br | C–H⋯O | C–Br⋯O | C–Br⋯Br | H⋯Br–C |
| Distance (Å) | 2.84 | 3.56 | 2.54 | 2.83 | 2.93 | 3.44 | 2.90 |
| 2.93 | |||||||
| Angle (°) | 166.8 | 114.9 | 96.2 | 127.8 | 178.4 | 150.2 | 124.1 |
| 177.7 | |||||||
| ΔE/(kcal mol−1) | −3.85 | −6.20 | −9.43 | −2.36 | −6.96 | ||
| ρ(CP)/(e Å−3) | 0.1086 | 0.0553 | 0.0594 | 0.0607 | 0.0864 | 0.0616 | 0.0518 |
| 0.0493 | |||||||
| ∇2ρ(CP)/(e Å−5) | 1.5327 | 0.6362 | 0.1759 | 0.7157 | 1.2234 | 0.7703 | 0.5974 |
| 0.7181 | |||||||
| G(CP)/(kJ mol−1 bohr−3) | 36.49 | 13.92 | 19.17 | 16.28 | 28.88 | 16.27 | 12.78 |
| 15.49 | |||||||
| V(CP)/(kJ mol−1 bohr−3) | −31.51 | −10.24 | −16.54 | −13.39 | −24.42 | −11.72 | −9.82 |
| −11.55 | |||||||
| H(CP)/(kJ mol−1 bohr−3) | 4.98 | 3.68 | 2.63 | 2.89 | 4.46 | 4.55 | 2.96 |
| 3.94 | |||||||
| |V(CP)|/G(CP) | 0.86 | 0.74 | 0.86 | 0.82 | 0.85 | 0.72 | 0.77 |
| 0.75 | |||||||
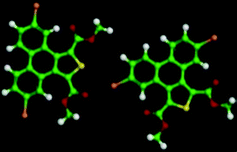
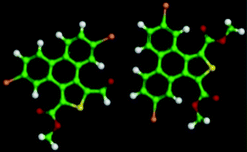

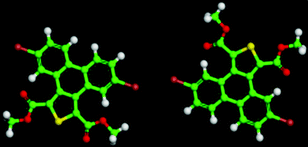
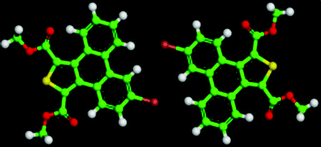
The bond distances and angles, as well as the binding energies, were obtained from the DFT calculations, as summarized in Table 4. The distances of these bonds were shorter than the sum of the vdW radii, thereby implying that these intermolecular interactions were weak bonds. The computational results show that dimer-III with the lowest energy (−9.43 kcal mol−1) is the most stable packing. It is now well documented in studies of halogen bonding that the ideal C–Br–O (OC) angle is 180°.54,55 However, the observed C–Br–O (OC) angle for 5,10-DBTD in dimer-I was 166.8°. The obvious different geometry was formed because of the enhanced positive charge distribution of the halogen atom and the coexistence of weak C–Br⋯S halogen bonds, which could induce a little bit of deflection for the angle of the C–Br–O (OC).34
At the n-tridecane/HOPG interface, the calculation of the binding energy density per unit area for the vertebra-like pattern, the hexagonal network pattern, and the linear pattern were 1.52, 0.80, and 1.77 kcal mol−1 nm−2, respectively. The intermolecular binding energy density of the linear pattern was higher than those of the vertebra-like pattern and hexagonal network pattern due to the formation of C–Br⋯OC halogen bonds. In the DFT calculations, however, the interchain vdW interactions were not taken into account. For the vertebra-like and the hexagonal network patterns, the side chains in all of the molecules stretch in different directions along the lattice of HOPG and interdigitate with the adjacent molecules to maximize their interactions. While for the linear pattern, all the side chains are located on the same side of the molecules to arrange orthogonally with respect to the rows. Thus, the vdW interactions in the vertebra-like and in the hexagonal network patterns were larger than those in the linear pattern. At the aliphatic solvent/HOPG interfaces, the intermolecular interactions were enhanced by increasing vdW interactions due to the solvent coadsorption, leading to the stable packing patterns. We propose that the coadsorption of aliphatic solvents changes the contact modes of the Br atom, whereby the type of Br⋯X interactions change from Br⋯OC and Br⋯S to Br⋯Br (type-I) interactions, or even disappear. At the aliphatic solvent/HOPG interfaces, compared with 6,9-DBTD, the 5,10-DBTD molecule presents more dispersed distribution,34 which further results in the formation of multiple intermolecular halogen bonds, leading to structural diversity.
While for 6-DBTD, the dimer is stabilized only by the H⋯Br bond, instead of the Br⋯Br bond. We realized by DFT calculations that the optimal configuration has two H⋯Br bonds. In the optimal configuration, the distance of H⋯Br (2.90 Å) was significantly smaller than the sum of the vdW radii (ΣvdW = 3.05 Å),27,56 as shown in Fig. 9e. The direction of the H⋯Br bond was determined by the negative potential region of a Br atom,48 which has also been observed for different molecules in 2D and 3D self-assembled structures.25,57 Although the 6-DBTD had one Br atom, the calculated binding energy for the linear structure was −6.96 kcal mol−1 (Fig. 8d), which is getting close to that of dimer-II for 5,10-DBTD (−6.20 kcal mol−1) and that of dimer-IV for 6,9-DBTD (−6.80 kcal mol−1). These results reveal that the hydrogen bonding is the primary driving force (HB > XB) to stabilize the 2D self-assembled adlayer, which is the reason why there is no solvent and concentration effects in the self-assembly of 6-DBTD.
Deformation density and topological features of the intermolecular interactions
The topological values for all the intermolecular interactions based on the quantum theory of atoms in molecules (QTAIM) at their bond critical points (BCPs) are given in Tables 3 and 4. Several important features, such as the electron density ρ(CP) and the Laplacian ∇2ρ(CP), are associated with the BCP (Fig. S10 and S11†), which can be used to characterize an intermolecular interaction.58 The value of ρ(CP) represents the accumulation of the electron density at the BCP, and is also a good indicator of the bond strength.59 In the common situation, the larger the ρ(CP) value, the stronger the bond is.60 The ρ(CP) values for the hetero-halogen bonds (Br⋯OC and Br⋯S) and homo-halogen Br⋯Br interactions in the present study varied in a wide range between 0.0553 and 0.1086 e Å−3, indicating that the Br⋯OC halogen bond was the strongest, while the Br⋯S halogen bond was the weakest. The ρ(CP) values for the H⋯Br and C–H⋯OC hydrogen bonds ranged from 0.0364 to 0.0690 e Å−3, which demonstrate that the magnitude of both types of hydrogen bonds was only approximate. Further, the Laplacian value [∇2ρ(CP)] is a measure of the local curvature of ρ(CP) and can indicate whether the electron density is locally concentrated or depleted at the given points.51 In the case of a closed-shell interaction ∇2ρ(CP) > 0, which indicates that the density is locally depleted at the BCP.58 The topological values provide strong evidence that these interactions in the complexes are characteristic of closed-shell interactions.
For better understanding and visualizing the halogen bonding and hydrogen bonding features for the studied system, 2D (Fig. 10 and 11) and 3D (Fig. S12 and S13†) deformation electron density maps were developed. These maps provide detailed information about the nonspherical distribution of electron density around the bromine atom. The bromine atom can therefore interact with nucleophiles, such as O, S, Br atoms, along the C–Br bond (Br⋯OC, Br⋯S and type-II Br⋯Br), and with electrophiles, such as an H atom (H⋯Br), in the directions perpendicular to the C–Br bond. The character of these weak interactions agrees well with the 2D and 3D maps of the deformation electron density. The charge rearrangement due to the bond formation was represented in terms of standard deformation density maps with the promolecular reference density.61 Moreover, according to the “hole–lump” concept, the formation of halogen bonding can be regard as an interaction between a region of charge depletion (CD), referred to as a “hole”, on the bromine atom and a region of charge concentration (CC), referred to as a “lump”, on an electron donor atom (Br, O and S);62 for instance, the 2D deformation electron density features were observed in C–Br⋯OC intermolecular space, where a “hole”, corresponding to the CD region at the Br atom (shown in red), is seen facing a “lump”, corresponding to the CC region on the carbonyl oxygen atom (shown in blue color) (Fig. 11a and c). At the same time, 3D deformation electron density maps (Fig. S13a and c†) can also be obtained, which can be better depicted in the intermolecular regions.
 | ||
| Fig. 10 (a–d) Deformation charge density maps in the plane for four different dimers formed by 6,9-DBTD. Positive contours are shown with blue lines, whereas negative contours are shown with red lines. | ||
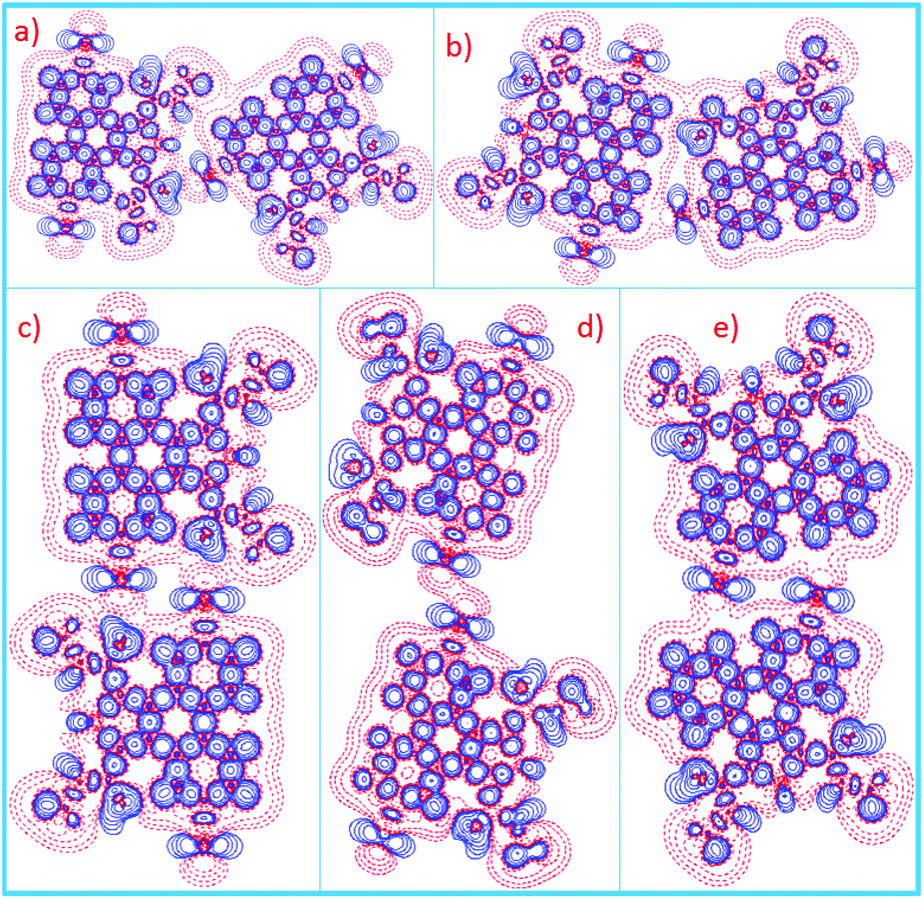 | ||
| Fig. 11 (a–d) Deformation charge density maps in the plane for four different dimers formed by 5,10-DBTD. (e) Deformation charge density maps in the plane dimer formed by 6-DBTD. Positive contours are shown with blue lines, whereas negative contours are shown with red lines. | ||
Further, Cremer and Kraka proposed the use of the kinetic electron energy density G(CP), the potential electron energy density V(CP), and the electron energy density H(CP) to characterize intermolecular interactions, as tabulated in Tables 3 and 4.63 When |V(CP)|/G(CP) < 1 and H(CP) > 0, and is therefore positive, the interaction can be classified as a closed-shell. In Tables 3 and 4, it can be seen, in general, that the kinetic energy density G(CP) is only slightly larger than the potential energy density |V(CP)|, and the ratio of |V(CP)|/G(CP) < 1. Such an observation indicates that the interactions in this complex belong to closed-shell non-covalent interactions. This work provides a new development to obtain a deeper insight into the bonding features of these weak interactions in a 2D self-assembled system.
Morphology change: concentration and coadsorption of solvent
It is generally known that the solute concentration is directly related to the different stabilities and molecular densities of different polymorphs.64 Molecular adsorption onto a surface from a solution is a kinetic process, and all the assembled structures could develop simultaneously. The assembly process depends on the adsorption–desorption equilibrium toward a minimum of the overall free energy.65,66 Systematic studies have found that the molecular density tends to increase with the decrease in concentration, as shown in Table 2.At the aliphatic solvent/HOPG interfaces, only in n-tridecane at high concentrations did 6,9-DBTD and 5,10-DBTD form the lamellar structure and double linear pattern without solvent coadsorption, respectively. This can be explained from the point of view of solubility. The solubility of these two molecules in n-tridecane (v = 1.61 mN s m2) is the best compared with those in n-tetradecane (v = 2.18 mN s m2), n-pentadecane (v = 2.88 mN s m2), and n-hexadecane (v = 3.34 mN s m2). Only the number of dissolved molecules in n-tridecane is enough to form close packing structures.
At the aliphatic solvent/HOPG interfaces under low concentrations, to maximize the substrate coverage and to be favored for enthalpic reasons, the aliphatic solvent molecules coadsorb on the side chain lamellae, giving rise to a closely packed structure and favorable vdW interactions between the alkyl chains. This observation can be interpreted from geometric considerations, in which the aliphatic solvent molecules could fit into the voids left by the side chains of the solute molecules. For 6,9-DBTD, a similar coadsorbed linear pattern was formed in the aliphatic solvent, in which only one Br atom in each molecule formed a Br⋯Br type-I contact bond with the adjacent molecule in each dimer. The alkyl chain length of the solvents had no effect on the self-assembly formation. The enhanced molecule–solvent vdW forces were strong enough to change the type of Br⋯Br interaction from type-II to type-I.
Taking steric hindrance into account, aliphatic molecules were coadsorbed on the HOPG surface with the 6,9-DBTD molecules, which could better stabilize the coadsorbed linear pattern. Whereas the self-assembly of 5,10-DBTD in n-tridecane, n-tetradecane, and n-pentadecane showed structural diversity and exhibited concentration-induced self-assembled structures, resulting from the coadsorption of the solvent and from steric constraints. For 5,10-DBTD in n-hexadecane, no concentration-dependent effect was observed and only a kind of densely packed double linear pattern was formed. The reason for this was that the side chain length of the 5,10-DBTD molecule was almost equal to that of n-hexadecane, which resulted in good structural size matching for the solvent molecules to coadsorb. This arrangement of coadsorption can maximize the molecule–solvent interactions. However, the packing patterns of 5,10-DBTD at the aliphatic solvent/HOPG interfaces under low concentrations were completely different, and they presented a gradient change by varying the chain length of aliphatic solvents as well as the concentration. The underlying reasons for the gradient change were: (1) the solubility of these solvents; (2) the molecular self-assembly of 5,10-DBTD on the surface under liquid conditions is a balance of the cooperation and competition of halogen bond, hydrogen bond, and vdW interactions, and where the longer the alkyl chain length of the solvents, the greater the molecule–solvent vdW interactions. Compared with the intermolecular vdW interactions, the intermolecular halogen bonding is weaker, in which the bonds change from a C–Br⋯OC halogen bond to type-I Br⋯Br interactions, and even no intermolecular interactions involved in Br atom are formed. Thus, the 5,10-DBTD molecules in n-tridecane form a linear pattern by continuous intermolecular C–Br⋯OC halogen bond and Br⋯Br interactions in each stripe. The loosely packed double linear pattern in n-tetradecane and n-pentadecane without Br⋯Br contacts were stabilized by the increasing molecule–solvent vdW interactions, because more solvent molecules adsorb in the side chain lamellae. Especially, in the loosely packed double linear pattern, the intermolecular Br⋯Br interactions were completely destroyed by the coadsorption of n-tetradecane or n-pentadecane molecules at low concentrations. In n-hexadecane, the 5,10-DBTD molecules in the densely packed double linear pattern formed a lateral dimer by Br⋯Br (type-I) interactions and no halogen bonds exist between the adjacent dimers in each lamella. Such an arrangement is formed from the strongest molecule–solvent vdW interactions based on the alkyl chains matching. The results also display that the coadsorption of aliphatic solvents change the Br atom contact modes, and result in different types of halogen bonding.
Conclusion
In summary, we accomplished assessing the photophysical properties, and self-assembly of two thienophenanthrene derivatives (6,9- and 5,10-DBTD) at liquid/solid interfaces using aliphatic hydrocarbons with different chain lengths as the solvents at different concentrations by using STM. The STM experiments demonstrated that the different self-assembly behaviors of the two isomers could be explained by the position of the bromine substituents, which could construct different types of halogen bonds and hydrogen bonds, which could further affect the self-assembled structures on HOPG surfaces. The aliphatic solvent can act as a coadsorbed component via vdWs interactions rather than simply as a dispersant, which further changes the Br atom contact modes in the 5,10-DBTD system. The type of Br⋯X interactions varied from Br⋯OC and Br⋯S to Br⋯Br (type-I) interactions, or even disappeared due to the solvent adsorption. The cooperation and completion between the interchain vdW interactions and the intermolecular halogen bonding and hydrogen bonding had a significant effect on the formation of self-assembly polymorphism. Moreover, combining the experimental results with DFT calculations helped to explore the effect of the position of bromine groups on the molecular self-assembly. Our results open up a new avenue for tailoring molecular self-assembly by changing the position and number of halogen substituents, the solvent, and the solution concentration. There is no doubt that XB has the potential to become as ubiquitous an interaction as HB in supramolecular chemistry. In future work, we will further synthesize a multitude of molecules with X atoms and other donor groups, which will be designed to investigate the intermolecular XB in molecular self-assembly and functional organic devices.
Acknowledgements
Financial supports from the National Natural Science Foundation of China (21573077, 21103053, 51373055), the National Program on Key Basic Research Project (2012CB932900), and the Fundamental Research Funds for the Central Universities (SCUT) are gratefully acknowledged.References
- L. Zalewski, S. Brovelli, M. Bonini, J. M. Mativetsky, M. Wykes, E. Orgiu, T. Breiner, M. Kastler, F. Dotz, F. Meinardi, H. L. Anderson, D. Beljonne, F. Cacialli and P. Samori, Adv. Funct. Mater., 2011, 21, 834–844 CrossRef CAS.
- F. Zhang, D. Q. Wu, Y. Y. Xu and X. L. Feng, J. Mater. Chem., 2011, 21, 17590–17600 RSC.
- E. K. Schillinger, E. Mena-Osteritz, J. Hentschel, H. G. Borner and P. Bauerle, Adv. Mater., 2009, 21, 1562–1567 CrossRef CAS.
- A. R. Alemozafar and R. J. Madix, Surf. Sci., 2004, 557, 231–242 CrossRef CAS.
- A. Ciesielski, P. J. Szabelski, W. Rzysko, A. Cadeddu, T. R. Cook, P. J. Stang and P. Samori, J. Am. Chem. Soc., 2013, 135, 6942–6950 CrossRef CAS PubMed.
- A. Mourran, U. Ziener, M. Moller, M. Suarez and J. M. Lehn, Langmuir, 2006, 22, 7579–7586 CrossRef CAS PubMed.
- K. Tahara, S. B. Lei, J. Adisoejoso, S. De Feyter and Y. Tobe, Chem. Commun., 2010, 46, 8507–8525 RSC.
- L. Y. Liao, Y. B. Li, J. Xu, Y. F. Geng, J. Y. Zhang, J. L. Xie, Q. D. Zeng and C. Wang, J. Phys. Chem. C, 2014, 118, 28625–28630 CrossRef CAS.
- K. S. Mali, J. Adisoejoso, E. Ghijsens, I. De Cat and S. De Feyter, Acc. Chem. Res., 2012, 45, 1309–1320 CrossRef CAS PubMed.
- L. Xu, X. R. Miao, B. Zha and W. L. Deng, Chem. – Asian J., 2013, 8, 926–933 CrossRef CAS PubMed.
- F. Silly, J. Phys. Chem. C, 2014, 118, 11975–11979 CrossRef CAS.
- C. Y. Fu, H. P. Lin, J. M. Macleod, A. Krayev, F. Rosei and D. F. Perepichka, Chem. Mater., 2016, 28, 951–961 CrossRef CAS.
- J. Zhang, P. C. Chen, B. K. Yuan, W. Ji, Z. H. Cheng and X. H. Qiu, Science, 2013, 342, 611–614 CrossRef CAS PubMed.
- L. Xu, X. R. Miao, B. Zha, K. Miao and W. L. Deng, J. Phys. Chem. C, 2013, 117, 12707–12714 CrossRef CAS.
- M. Viciano-Chumillas, D. Z. Li, A. Smogunov, S. Latil, Y. J. Dappe, C. Barreteau, T. Mallah and F. Silly, Chem. – Eur. J., 2014, 20, 13566–13575 CrossRef CAS PubMed.
- Y. Zhang, Y. Luo, Y. Zhang, Y.-J. Yu, Y.-M. Kuang, L. Zhang, Q.-S. Meng, Y. Luo, J.-L. Yang, Z.-C. Dong and J. G. Hou, Nature, 2016, 531, 623–627 CrossRef CAS PubMed.
- R. Gutzler, S. Lappe, K. Mahata, M. Schmittel, W. M. Heckl and M. Lackinger, Chem. Commun., 2009, 680–682 RSC.
- S. L. Lee, Z. Yuan, L. Chen, K. S. Mali, K. Mullen and S. De Feyter, J. Am. Chem. Soc., 2014, 136, 4117–4120 CrossRef CAS PubMed.
- P. Metrangolo, F. Meyer, T. Pilati, G. Resnati and G. Terraneo, Angew. Chem., Int. Ed., 2008, 47, 6114–6127 CrossRef CAS PubMed.
- H. Wang, W. Z. Wang and W. J. Jin, Chem. Rev., 2016, 116, 5072–5104 CrossRef CAS PubMed.
- J. Marti-Rujas, L. Colombo, J. Lu, A. Dey, G. Terraneo, P. Metrangolo, T. Pilati and G. Resnati, Chem. Commun., 2012, 48, 8207–8209 RSC.
- X. Pang, X. R. Zhao, H. Wang, H. L. Sun and W. J. Jin, Cryst. Growth Des., 2013, 13, 3739–3745 Search PubMed.
- E. Parisini, P. Metrangolo, T. Pilati, G. Resnati and G. Terraneo, Chem. Soc. Rev., 2011, 40, 2267–2278 RSC.
- J. K. Yoon, W.-J. Son, K.-H. Chung, H. Kim, S. Han and S.-J. Kahng, J. Phys. Chem. C, 2011, 115, 2297–2301 CrossRef CAS.
- T. A. Pham, F. Song, M. T. Nguyen and M. Stöhr, Chem. Commun., 2014, 50, 14089–14092 RSC.
- R. Gutzler, O. Ivasenko, C. Y. Fu, J. L. Brusso, F. Rosei and D. F. Perepichka, Chem. Commun., 2011, 47, 9453–9455 RSC.
- R. Gutzler, C. Fu, A. Dadvand, Y. Hua, J. M. MacLeod, F. Rosei and D. F. Perepichka, Nanoscale, 2012, 4, 5965–5971 RSC.
- F. Silly, J. Phys. Chem. C, 2013, 117, 20244–20249 CrossRef CAS.
- B. Zha, X. R. Miao, Y. J. Li, P. Liu, K. Miao and W. L. Deng, J. Nanomater., 2013, 624927 Search PubMed.
- D. Peyrot and F. Silly, ACS Nano, 2016, 10, 5490–5498 CrossRef CAS PubMed.
- R. Gatti, J. M. MacLeod, J. A. Lipton-Duffin, A. G. Moiseev, D. F. Perepichka and F. Rosei, J. Phys. Chem. C, 2014, 118, 25505–25516 CrossRef CAS.
- Z. C. Mu, L. J. Shu, H. Fuchs, M. Mayor and L. F. Chi, Langmuir, 2011, 27, 1359–1363 CrossRef CAS PubMed.
- B. Zha, X. R. Miao, P. Liu, Y. M. Wu and W. L. Deng, Chem. Commun., 2014, 50, 9003–9006 RSC.
- B. Zha, M. Dong, X. Miao, K. Miao, Y. Hu, Y. Wu, L. Xu and W. Deng, J. Phys. Chem. Lett., 2016, 7, 3164–3170 CrossRef CAS PubMed.
- J. Hachmann, R. Olivares-Amaya, A. Jinich, A. L. Appleton, M. A. Blood-Forsythe, L. R. Seress, C. Roman-Salgado, K. Trepte, S. Atahan-Evrenk, S. Er, S. Shrestha, R. Mondal, A. Sokolov, Z. A. Bao and A. Aspuru-Guzik, Energy Environ. Sci., 2014, 7, 698–704 Search PubMed.
- I. Schnapperelle and T. Bach, Chem. – Eur. J., 2014, 20, 9725–9732 CrossRef CAS PubMed.
- J. C. Huang, B. Xiao, J. Wang, Y. F. Wang, X. H. Peng, X. R. Miao, Q. W. Pan, Y. Q. Mo, W. L. Deng, H. B. Wu and Y. Cao, Org. Electron., 2014, 15, 2311–2321 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed.
- A. Chatterjee, L. Zhang and K. T. Leung, Langmuir, 2013, 29, 9369–9377 CrossRef CAS PubMed.
- T. Lu and F. W. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- S. M. Ji, J. Yang, Q. Yang, S. S. Liu, M. D. Chen and J. Z. Zhao, J. Org. Chem., 2009, 74, 4855–4865 CrossRef CAS PubMed.
- X. Zhang, L. N. Chi, S. M. Ji, Y. B. Wu, P. Song, K. L. Han, H. M. Guo, T. D. James and J. Z. Zhao, J. Am. Chem. Soc., 2009, 131, 17452–17463 CrossRef CAS PubMed.
- R. Lazzaroni, A. Calderone, J. L. Bredas and J. P. Rabe, J. Chem. Phys., 1997, 107, 99–105 CrossRef CAS.
- F. Jackel, M. Ai, J. S. Wu, K. Mullen and J. P. Rabe, J. Am. Chem. Soc., 2005, 127, 14580–14581 CrossRef PubMed.
- M. G. Sarwar, B. Dragisic, L. J. Salsberg, C. Gouliaras and M. S. Taylor, J. Am. Chem. Soc., 2010, 132, 1646–1653 CrossRef CAS PubMed.
- T. M. Beale, M. G. Chudzinski, M. G. Sarwar and M. S. Taylor, Chem. Soc. Rev., 2013, 42, 1667–1680 RSC.
- P. Politzer, P. Lane, M. C. Concha, Y. G. Ma and J. S. Murray, J. Mol. Model., 2007, 13, 305–311 CrossRef CAS PubMed.
- T. T. T. Bui, S. Dahaoui, C. Lecomte, G. R. Desiraju and E. Espinosa, Angew. Chem., Int. Ed., 2009, 48, 3838–3841 CrossRef CAS PubMed.
- M. Fourmigue, Curr. Opin. Solid State Mater. Sci., 2009, 13, 36–45 CrossRef CAS.
- H. F. Lieberman, R. J. Davey and D. M. T. Newsham, Chem. Mater., 2000, 12, 490–494 CrossRef CAS.
- V. R. Hathwar, R. G. Gonnade, P. Munshi, M. M. Bhadbhade and T. N. G. Row, Cryst. Growth Des., 2011, 11, 1855–1862 Search PubMed.
- K. E. Riley, J. S. Murray, J. Fanfrlik, J. Rezac, R. J. Sola, M. C. Concha, F. M. Ramos and P. Politzer, J. Mol. Model., 2013, 19, 4651–4659 CrossRef CAS PubMed.
- K. E. Riley, J. S. Murray, J. Fanfrlik, J. Rezac, R. J. Sola, M. C. Concha, F. M. Ramos and P. Politzer, J. Mol. Model., 2011, 17, 3309–3318 CrossRef CAS PubMed.
- F. C. Pigge, V. R. Vangala, D. C. Swenson and N. P. Rath, Cryst. Growth Des., 2010, 10, 224–231 Search PubMed.
- K. E. Riley, J. S. Murray, P. Politzer, M. C. Concha and P. Hobza, J. Chem. Theory Comput., 2009, 5, 155–163 CrossRef CAS PubMed.
- O. Navon, J. Bernstein and V. Khodorkovsky, Angew. Chem., Int. Ed. Engl., 1997, 36, 601–603 CrossRef CAS.
- M. Yamada and F. Hamada, Cryst. Growth Des., 2015, 15, 1889–1897 Search PubMed.
- D. J. Wolstenholme, K. N. Robertson, E. M. Gonzalez and T. S. Cameron, J. Phys. Chem. A, 2006, 110, 12636–12643 CrossRef CAS PubMed.
- P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2010, 12, 7748–7757 RSC.
- W. D. Arnold and E. Oldfield, J. Am. Chem. Soc., 2000, 122, 12835–12841 CrossRef CAS.
- F. F. Zhou, J. Han, R. R. Liu, P. Li and H. Y. Zhang, Comput. Theor. Chem., 2014, 1044, 80–86 CrossRef CAS.
- K. Eskandari and H. Zariny, Chem. Phys. Lett., 2010, 492, 9–13 CrossRef CAS.
- D. Cremer and E. Kraka, Angew. Chem., Int. Ed. Engl., 1984, 23, 627–628 CrossRef.
- T. Kudernac, S. B. Lei, J. A. A. W. Elemans and S. De Feyter, Chem. Soc. Rev., 2009, 38, 402–421 RSC.
- D. F. Padowitz, D. M. Sada, E. L. Kemer, M. L. Dougan and W. A. Xue, J. Phys. Chem. B, 2002, 106, 593–598 CrossRef CAS.
- M. Lackinger, S. Griessl, L. Kampschulte, F. Jamitzky and W. M. Heckl, Small, 2005, 1, 532–539 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional STM images of 6,9- and 5,10-DBTD in different solvents, synthesis and characterization of 6-DBTD. See DOI: 10.1039/c6nr07693a |
| This journal is © The Royal Society of Chemistry 2017 |