
Novel DNA sensor system for highly sensitive and quantitative retrovirus detection using virus encoded integrase as a biomarker†
Jing
Wang
a,
Jiangnan
Liu
a,
Jonas
Thomsen
a,
Denis
Selnihhin
a,
Marianne S.
Hede
b,
Freja C. M.
Kirsebom
a,
Oskar
Franch
ac,
Søren
Fjelstrup
ac,
Magnus
Stougaard
d,
Yi-Ping
Ho
ace,
Finn Skou
Pedersen
*ac and
Birgitta R.
Knudsen
*ac
aDepartment of Molecular Biology and Genetics, Aarhus University, Aarhus, Denmark. E-mail: fsp@mbg.au.dk; brk@mbg.au.dk
bZymonostics ApS, Aarhus, Denmark
cInterdisciplinary Nanoscience Center (iNANO), Aarhus University, Aarhus, Denmark
dDepartment of Clinical Medicine, Aarhus University, Denmark
eDivision of Biomedical Engineering, Department of Electronic Engineering, The Chinese University of Hong Kong, Shatin, NT, Hong Kong SAR, China
First published on 2nd December 2016
Abstract
In the current study we describe a novel DNA sensor system that allows the detection of single catalytic DNA integration events mediated by retrovirus encoded integrase (IN) present in viral particles. This is achieved by rolling circle amplification mediated conversion of enzymatic reactions happening within nanometer dimensions to directly detectable micrometer sized DNA products. The system utilizes the unique integration reaction of IN to generate a surface anchored nicked DNA circle that serves as a substrate for rolling circle amplification and allows for specific, quantitative and sensitive detection of purified recombinant IN or virus particles with a detection limit of less than 30 virus particles per μL of sample. Moreover, by modifying the nucleotide sequences of the utilized DNA it was possible to tailor the system to distinguish between the highly pathogenic lentivirus HIV and the gammaretrovirus murine leukemia virus present in a given sample. Infections with HIV remain a major threat to global health with more than 2 million new infections and 1 million deaths each year. The sensitive and specific detection of HIV particles based on IN activity holds promise for the development of a new type of diagnostic tools suitable for early (within hours of infection) detection of HIV, which would be valuable for prevention strategies as well as for efficient treatment.
Introduction
Recent developments demonstrate that DNA nano-sensor systems targeting pathogen specific enzyme activities for highly sensitive and specific molecular diagnosis of infectious diseases may present an attractive alternative to conventional diagnostic tools. By definition, enzymes convert vast numbers of substrate molecules into distinct products without themselves being consumed in the process. Consequently, highly sensitive detection of biomolecules can be achieved by enzymatic signal enhancement. This is exploited in e.g. polymerase driven PCR amplification, which is among the popular tools for current and future diagnosis of infectious diseases.1–4 For applications where need of quantifiability, limited resources or time-to-readout are major concerns, a drawback of PCR is the requirement of thermal cycling. This hampers quantification and requires specialized equipment unavailable to most physicians in rural areas despite efforts to develop portable and user-friendly PCR machines.5 Therefore, alternative means of sensitive detection have been investigated. Such methods include measuring directly the activity of microbe expressed enzymes using various DNA nano-sensors. Examples of real-time optical or electrochemical systems for a variety of enzymes relevant for detection of human diseases such as tuberculosis and cancer,6–8 have been presented. However, common for most real-time sensors is inadequate sensitivity for real-life diagnosis and/or that their performance is hampered in crude biological sample.9 For diagnostic applications additional amplification of enzymatic products without hampering quantitative readout, robustness of the assay, or ease of handling is desirable. One technique for such amplification is Rolling-Circle-Amplification (RCA), by which a circular DNA template is converted to a long tandem repeat Rolling-Circle-amplification-Product (RCP) that can be visualized at the single-molecule level by e.g. fluorescent staining or in bulk using various colorimetric readouts.10,11 Taking advantage of RCA of single stranded DNA circle products generated by the cleavage-ligation activity of the Plasmodium expressed enzyme, topoisomerase IB, we previously demonstrated specific and quantitative detection of malaria parasites.12 This principle for detection, termed Rolling-circle-Enhanced-Enzyme-Activity-Detection (REEAD) allowed pathogen detection in small volumes of crude blood samples with a detection limit of 0.06 parasites per μL, which is comparable to or even better than most PCR protocols.12 Among DNA cleaving/ligation enzymes, topoisomerase IB, is rare in requiring no co-factors for activity and, hence, specificity towards the Plasmodium enzyme was relatively easily achieved by a combination of buffer- and REEAD substrate design.12,13 Although it was envisioned that REEAD detection in principle is adaptable for detection of a broad variety of DNA cleaving/ligating enzymes, it was not obvious how specificity towards target enzymes with more common requirements for co-factors could be achieved. De facto, specific detection of every new enzyme target requires specific design of the DNA substrate system to match the unique properties of the enzyme reaction to-be-detected.In the present study we describe a REEAD assay for highly sensitive, specific and quantitative detection of active retrovirus particles. Members of the retrovirus family, including the well-known human immunodeficiency virus type 1 (HIV-1) (the cause of AIDS), are RNA viruses that replicate through an obligatory DNA-intermediate, which becomes stably integrated into the host genome in a reaction carried out by the virus-encoded enzyme integrase (IN). IN is translated as part of a polyprotein (Gag-Pol), which is present in around 100 copies per viral particle14 and cleaved into its constituents by the retroviral protease during maturation of the virus. Active IN can be extracted from retrovirus particles, whereas virus-producing cells contain only the inactive polyprotein form. Hence, IN activity is a marker of active virus particles and may be well suited for early diagnosis of infection by retroviruses or specific subgroups thereof. Indeed early diagnosis of HIV-1, which currently is difficult to diagnose within the first weeks after infection, may allow early onset of treatment and add considerably to eliminating spreading of disease.15 Hence, aiming for the development of a novel enzyme based assay for early diagnosis of retroviral infections, including HIV we have chosen IN activity as a biomarker. Since IN is unique to retroviruses and present in the virus particle this choice of biomarker allows the specific detection of this group of viruses. The activity of another retroviral enzyme, namely reverse transcriptase, which copies the virus RNA genome into DNA shortly after infection could potentially also be used as a target for early diagnosis. However, this would require a different substrate design that might not be as easily accommodated into the REEAD setup as a substrate system recognized by IN (see below). It is of note, that other virus types may be detected by the REEAD setup provided that they express unique DNA interacting enzymes.
IN catalyzes the integration of viral DNA into the host genome by two sequential reactions. In the first reaction the enzyme cleaves off two to three 3′-terminal nucleotides at the ends of the long terminal repeats (LTRs) flanking the linear double stranded viral DNA. Key motifs for IN recognition are found as inverted repeats at the two termini in the U5 and U3 regions, respectively (see Fig. 1). These inverted repeat sequences differ among retroviral genera or subgroups, although variations in substrate specificity preferences among IN from different species are observed. In the second reaction, termed strand transfer, the recessed 3′-OH ends generated during cleavage are inserted in the phosphodiester backbone in opposing strands of the host genome. This step of the reaction occurs without any sequence preferences.16,17
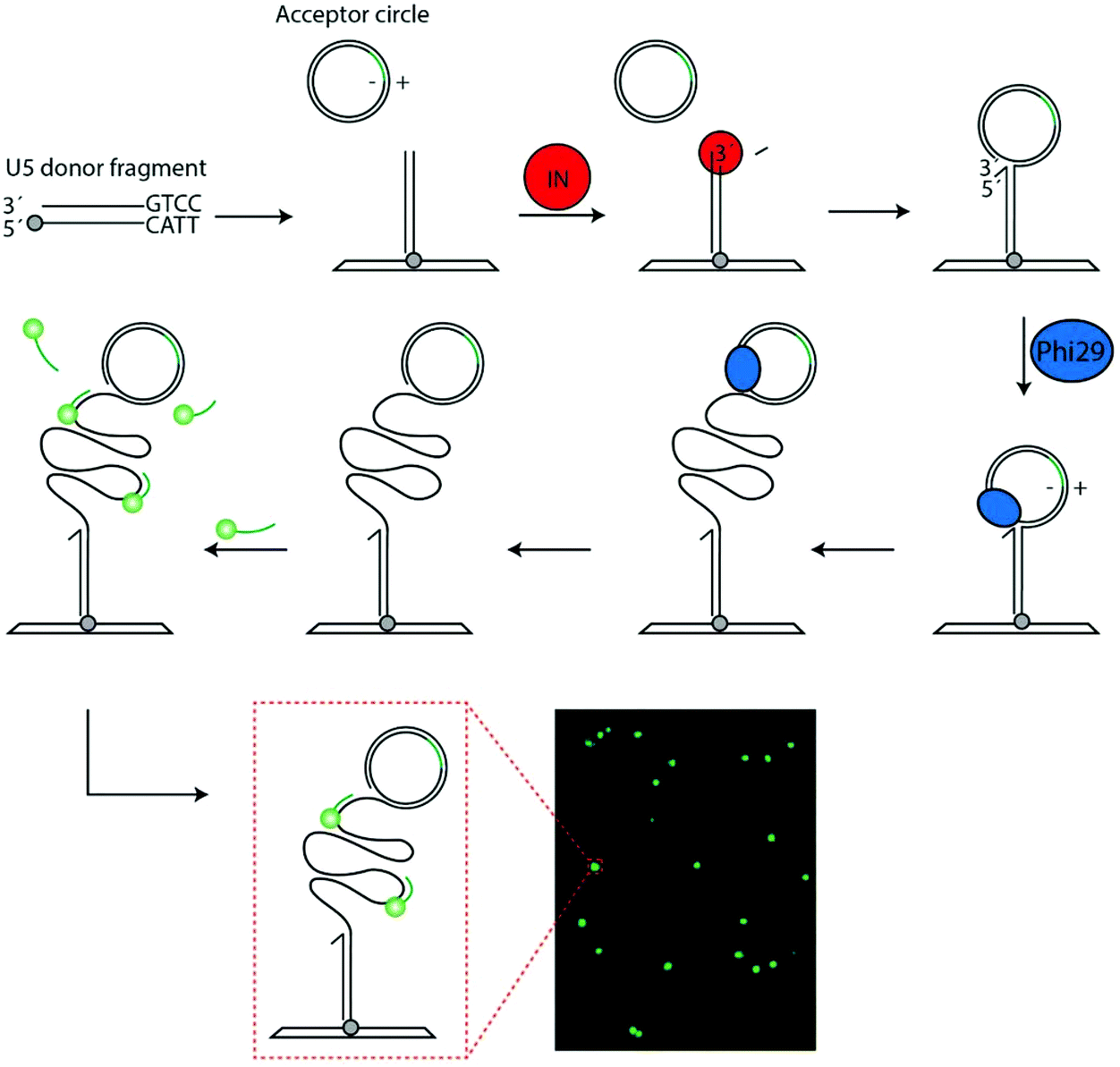 | ||
| Fig. 1 Schematic illustration of the assay. The U5 donor fragment is coupled to a glass slide via the primary amine (grey circle). The acceptor circle (containing a probe annealing site marked in green) and IN (red circle) containing sample is added. The reaction is initiated by IN mediated cleavage of the donor fragment. This generates a recessed 3′-end, which is integrated by IN into the acceptor circle. Upon integration the acceptor circle becomes covalently attached to glass slide and a free 3′-OH end is generated. Phi-29 polymerase (blue circle) is added to support RCA of the uncut strand of the acceptor circle generating a long tandem repeat product that can be visualized in a fluorescence microscope upon hybridization of fluorescently labeled probes. An example of a typical microscopic image obtained using this method is shown at the bottom of the figure (lower, right panel). The green fluorescent spots represent one RCP generated upon amplification of the product generated an integration event mediated by HIV IN. The orientation of the strands in the acceptor circle is indicated using plus-minus nomenclature. Only integration in the +strand will result in signals. | ||
Each of the IN mediated reactions have been reconstituted in vitro. A common assay for investigating cleavage and strand transfer utilizes short synthetic “integration” DNA fragments with ends that mimic the viral DNA end i.e. contains various lengths of the LTR typically including U5. Such donor fragments with a U5-terminus can be cleaved and integrated in an “acceptor” DNA fragment by purified IN defining the minimal part of the LTR to support specific cleavage and integration reactions.11 In the present study we utilize this basic reaction to generate an efficient substrate for RCA allowing specific and sensitive detection of two members of the retrovirus family, namely the human pathogenic lentivirus HIV and the gammaretrovirus murine leukemia virus (MLV). This is the first report of a REEAD setup that allows specific detection of a non-type IB topoisomerase target enzyme, which depends on commonly used divalent cations for reaction. Although IN differs from type IB topoisomerases with respect to the reaction it catalyzes and the requirements for cofactors the obtained results demonstrate a detection limit below 30 virus particles per μL of serum (using viruses that all expressed IN without necessarily being infectious). This points to the same advantages in terms of sensitive and specific detection as was previously observed for malaria specific REEAD. Hence, these findings support REEAD not only as a new important tool for early diagnosis of retrovirus inflicted diseases e.g. HIV but also a putative diagnostic tool in broader terms.
Results and discussion
Design of REEAD setup for measurements of single catalytic events mediated by IN
As mentioned in the introduction the natural reaction of IN is to catalyze integration of a double stranded viral DNA genome into the host chromosome in a two-step reaction involving first cleavage of the viral DNA ends, and secondly integration of the recessed 3′-OH ends generated during cleavage into opposing strands of the host genome. This basic reaction was utilized to generate an IN specific REEAD substrate system composed of (1) a donor DNA fragment with a U5 end mimicking the viral DNA end and, hence, serving as a substrate for cleavage and subsequent as a donor for integration mediated by IN and (2) a closed double stranded DNA circle that served as an acceptor for integration.In the experimental design (see Fig. 1), the donor fragment was modified with a 5′-amine in the non-U5 containing end to allow anchoring of the fragment on a functionalized glass surface. Addition of IN and acceptor circle to the surface attached donor fragment was anticipated to allow the U5 end of the donor to be processed and the generated recessed 3′-OH end to be integrated in one of the strands of the acceptor circle. This would result in the generation of a strand interruption in the acceptor circle and a free 3′-OH end that could prime RCA upon addition of phi-polymerase. The acceptor circle was designed with an identifier sequence identical to the sequence of a fluorescently labeled visualization probe on one strand (termed the (−) strand) and complementary to the sequence of the probe on the other (+) strand. Hence, integration into the (+) strand of the acceptor circle followed by addition of phi-polymerase generated RCA products that could subsequently be visualized at the single molecule level upon hybridization to the fluorescently labeled visualization probes essentially as described previously for topoisomerase IB specific REEAD assays.12,13,18 Since the described assay relies only on isothermal amplification each RCA product corresponded to one catalytic event mediated by IN. Consequently, at optimal conditions the described IN specific REEAD was anticipated to allow for direct quantification of IN activity by converting integration reactions happening within a few hundred nanometer dimensions to micrometer-sized product that could be directly visualized. Note, however, that not all IN mediated reactions may result in a visible RCA product, since the site of integration by retroviral IN is random.19 One example of an un-detectable reaction event is integration in the (−) strand of the acceptor circle, which would lead to the generation of RCA products with identifier sequences identical and not complementary to the visualization probe (see Fig. 1). Another example is integration of two integration fragments on each their strand of the acceptor circle, which would lead to linearization of the circle and, hence, prevent RCA. Indeed, the latter reaction resembles the natural reaction of IN, in which a tetramer of the enzyme catalyzes the concerted integration of the two ends of the viral genome in each their strand of the host genome20 and, as will be described below, care needed to be taken to avoid this type of integration that compromised the quantitative nature of the assay.
Detection of purified HIV-1 IN by REEAD
To test out the ability of the IN specific REEAD to detect enzyme activity we aimed for measuring integration by recombinant HIV-1 IN expressed and purified to approximately 50% purity from E. coli (see ESI S1†). Hence, for the first set of experiments the IN specific REEAD system was designed for detection of HIV-1 IN (HIV-1 REEAD system in the following) by inserting the HIV-1 U5 sequence that is preferentially cleaved by HIV-1 IN, into the donor fragment. An example of fluorescent signals in the microscopic views obtained upon incubation of the HIV-1 donor fragment and acceptor circle with or without added HIV-1 IN followed by RCA and hybridization of visualization probes is shown in Fig. 1. As expected the addition of purified enzyme resulted in the appearance of fluorescent spots each representing one integration event, while no signals were observed in samples without added HIV-1 IN. Quantification of the number of signals obtained upon assaying three different concentrations of HIV-1 IN indicated a dose dependent measurement of IN activity by the IN specific REEAD setup (Fig. 2A) with a significant difference between the samples with or without IN. Assaying a broader range of different HIV-1 IN concentrations, however, revealed that the dose–response of the assay depended on the concentration of donor fragments printed on the glass slides. Hence, high concentrations of printed donor fragment appeared to compromise the expected quantitative relationship between enzyme concentrations to signal numbers (see ESI S2†). As described in ESI S2† this is consistent with the mechanism of IN action and can most easily be explained by the integration of two or more integration fragments, when high concentrations of IN are incubated with a large excess of donor fragment relative to acceptor circle. This indeed would lead to fragmentation of the circle and result in loss-of-signals. In agreement with this theory, the HIV-1 donor fragment was diluted to 5 fmol (final amount in the printed volume) before printing, which resulted in a direct correlation between the number of signals observed and the HIV-1 IN concentration added to the assay within a final amount of IN ranging from 1.5 fmol–1500 fmol (ESI S2†). We therefore decided to use 5 fmol of printed donor fragment in combination with 50 fmol acceptor circle for all the following experiments. This resulted in quantitative detection of virus particles (see later) in the range of 30 to 4000, which is the range anticipated relevant for diagnosis of patients in the acute phase.21 In the experiments using purified enzymes, however, the absolute IN protein amount is in large surplus relative to the REEAD substrate concentration stated above (see ESI S2†). This is in conflict with normal assay setups aiming for quantitative enzyme detection. However, it is likely that the IN fraction that was expressed in and purified from E. coli, contained a relatively large percentage of inactive enzyme, leaving the effective IN concentration much lower than suggested by the protein amount.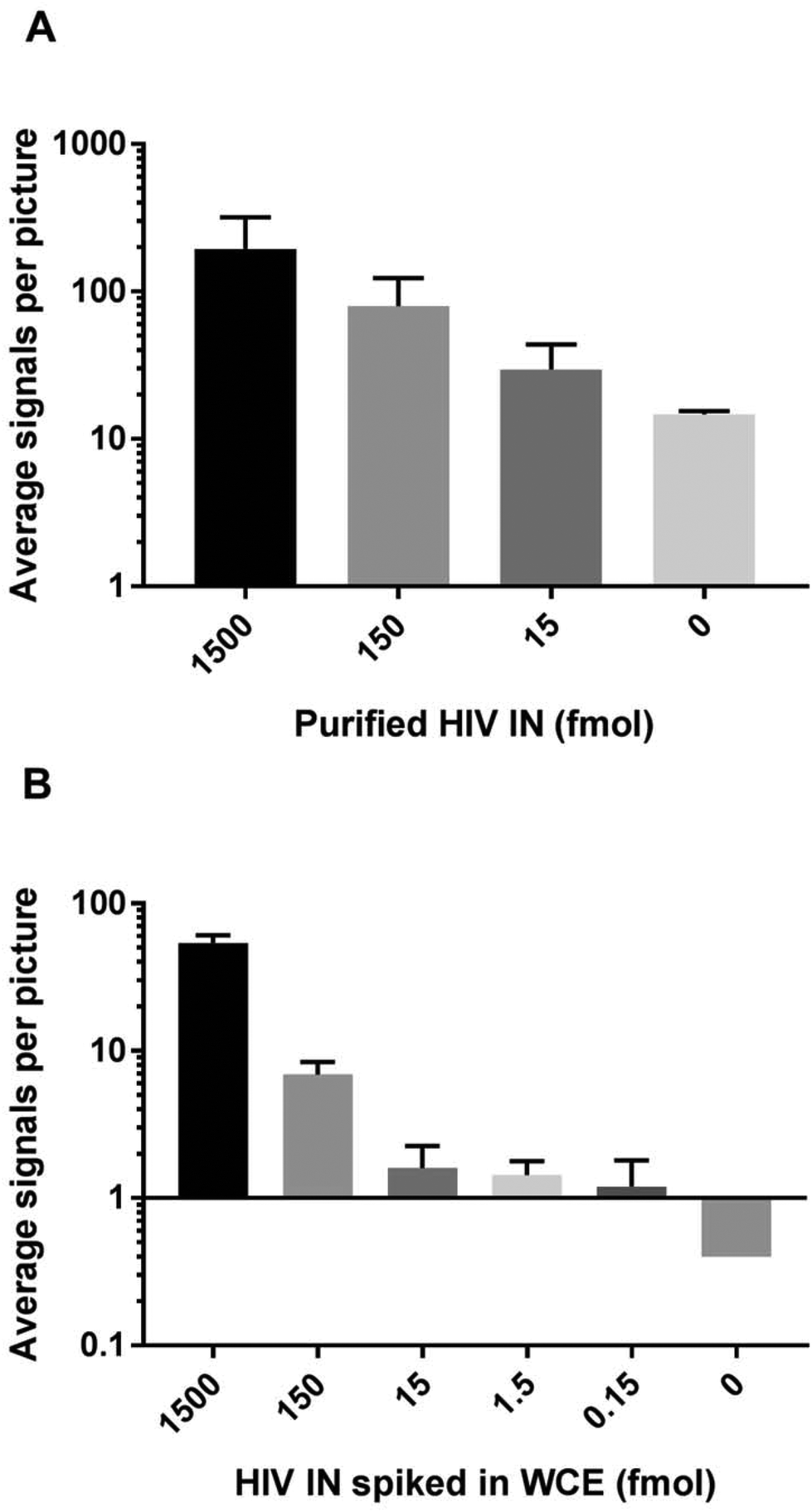 | ||
| Fig. 2 Bar charts showing the results obtained when measuring the activity of (A) purified HIV IN alone or (B) spiked in whole cell extract (WCE) from human cells grown in culture using the HIV REEAD setup. Notice that the vertical axes are logarithmically scaled. Error bars are standard deviations calculated from three individual experiments. | ||
Besides research purposes, quantitative detection of retrovirus IN will be highly relevant for diagnostic purposes, where IN activity may be a biomarker for early detection (within the first hours post-infection) of infection.15 Since patient samples (typically serum) intended for diagnosis of HIV might contain at least a small number of human cells, the potential use of IN specific REEAD for diagnostic purposes strongly depend on the assay being specific towards IN when present in crude extracts from human cells. The detection range and specificity of the HIV-1 REEAD was, therefore, addressed by assaying the activity of decreasing concentrations of purified HIV-1 IN ranging from 1.5 amol to 1500 fmol (lowest concentrations not shown) spiked in human whole cell extract. As evident from Fig. 2B, it was possible to detect HIV IN activity by the REEAD setup in a dose dependent manner and as observed for IN alone there was a significant difference between the samples with or without IN. The approximate detection limit was 0.150 fmol of purified HIV IN spiked in extract from 250 HEK293T cells. The lack of signals observed upon incubation of crude cell extract without spike in HIV IN demonstrated that the assay was specific for HIV IN in a background of the human cell content when using a relatively low number of cells (250 cells). This is anticipated appropriate to mimic worst case scenario (in terms of cell contaminations) in human serum.
REEAD allows quantitative and specific measurements of virus particles
To evaluate the potential of IN specific REEAD for HIV diagnostic purposes, the ability of the setup to detect the presence of virus particles in crude samples was investigated. For these experiments we chose to use engineered defective HIV particles or non-human-pathogenic murine leukemia virus (MLV) cultivated in standard cell culture medium with 10% fetal calf serum as mimics of patient samples. This was done due to safety issues concerned with handling of infectious HIV virus.Extracts from serial dilutions of the defective HIV particles were prepared and subjected to analyses in the HIV-1 REEAD setup as described above for purified HIV-1 IN. As expected the number of REEAD signals decreased significantly with decreasing numbers of HIV particles in the sample showing a detection limit of less than 1 colony forming unit (cfu) (corresponding to 30 virus particles per μL) when compared to the number of background signals generated by the negative sample containing 10% fetal calf serum without any virus particles (see Fig. 3A). Note that the sample type for HIV diagnosis is typically serum isolated from blood samples taken from suspected cases. Hence we believe that the positive and negative samples assayed in the described experiment represent a realistic mimic of patient samples. Also note, that although REEAD proved capable of detecting the presence of HIV particles in the sample in a dose dependent manner, we did not observe a clear linear relationship between the virus concentrations and the number of REEAD signals. While this was true in some experiments, others showed a clear linear relationship between IN or virus concentrations and number of REEAD signals. The reason for these differences is unclear, but may relate to a different extent of aggregation between IN monomers in the individual experiments.22 In all cases, however, it is important to note that we do observe a dose dependence between IN/virus concentrations and REEAD signals.
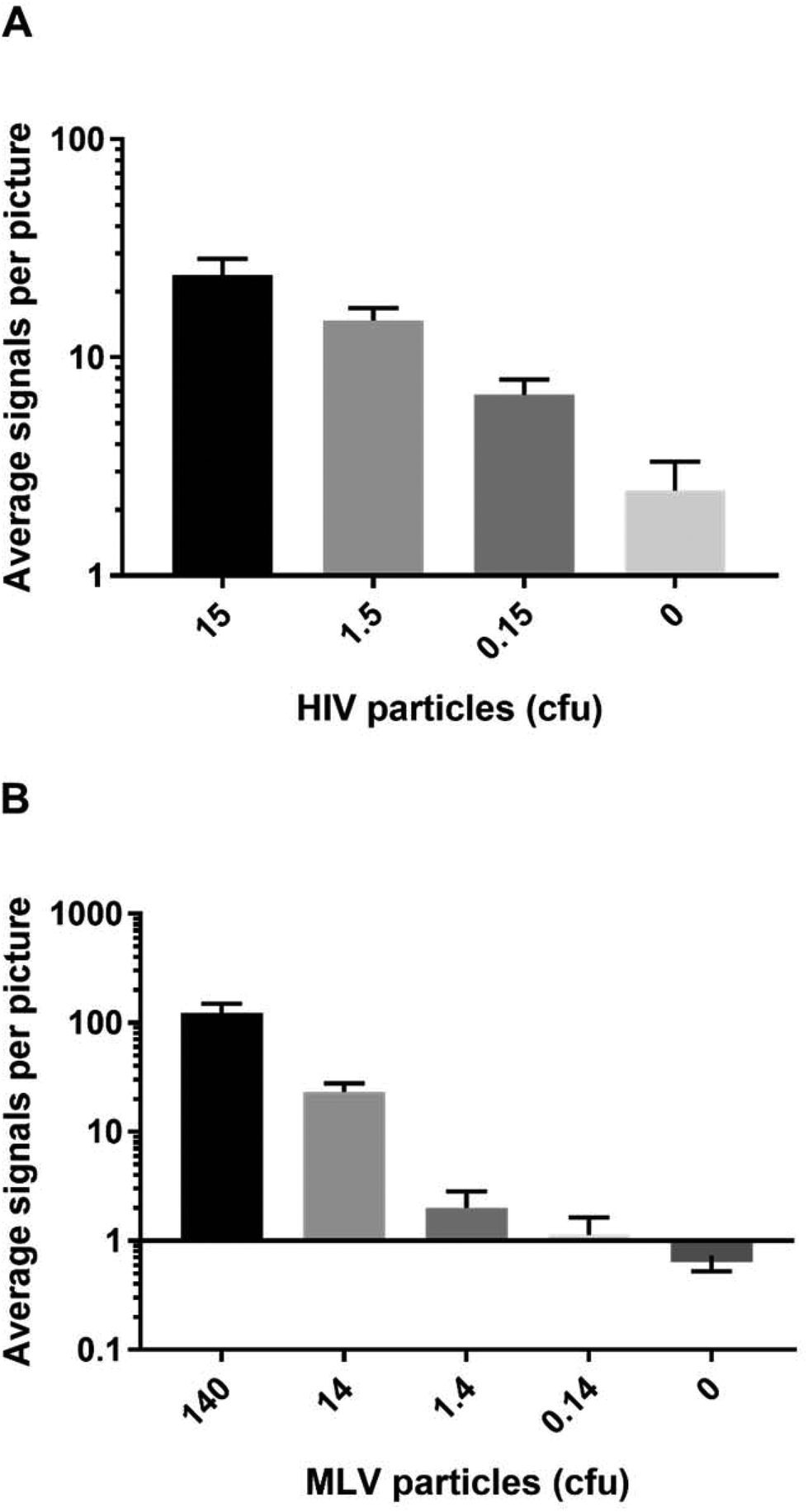 | ||
| Fig. 3 Bar chart representing the results of analyzing serial dilutions of extracts from gene-modified HIV particles in HIV REEAD (A) or from MLV in MLV REEAD (B), respectively. The amount of virus particles is stated in cfu (colony forming units) corresponding to 30 virus particles per μL in the initial sample. Notice that the vertical axis in (B) is logarithmically scaled. Error bars represent standard deviations calculated from the results of three individual experiments. | ||
To expand the investigations to natural retrovirus particles we used extracts from MLV as an additional model for patient samples. Like HIV, MLV belongs to the retrovirus family and can be regarded as a model of HIV, since the two viruses share all important aspects of life cycle and protein expression.23 However, since members of the retrovirus family differ with respect to the terminal sequences of their genomes, which are integrated in the host chromosome by IN, we used a modified REEAD setup (termed MLV REEAD in the following) for these experiments. The MLV REEAD substrate system was, as HIV REEAD, composed of the acceptor circle and a donor fragment. However, to allow processing by MLV IN the donor fragment contained a MLV U5 sequence instead of the HIV-1 U5 sequence that was included in the donor fragment of HIV-1 REEAD. Cultivated MLV was serially diluted in 10% fetal calf serum, extracted and subjected to analyses using the MLV REEAD system. The results are depicted as average number of signals observed on each microscopic image as a function of MLV concentration indicated as cfu. Consistent with the results observed for defective HIV particles the MLV REEAD allowed dose depended detection of MLV virus with a significant difference between number of signals in samples with or without virus particles, a linear signal-to-particle-number relationship and a detection limit of approximately 1 cfu (Fig. 3B).
For diagnostic purposes it may be important not only to detect HIV or other pathogenic retroviruses in a background of human cells but also to distinguish between infections by different, yet related, viruses. To evaluate the potential of the IN specific REEAD setup to discriminate between different members of the retrovirus family we investigated the ability of the HIV- and MLV-REEAD, respectively, to detect the activity of their non-target IN enzyme. In these experiments we used extracts from MLV particles as a source of MLV IN, while HIV IN was available in purified form. For both the HIV- and the MLV-REEAD setup, incubation with the matching IN enzyme in either purified (for HIV IN) or crude form (MLV IN) resulted in a high number of signals that significantly decreased with decreasing concentrations of added enzyme/virus until background levels (the number of signals observed when no enzyme was added) were reached (Fig. 4A and B). In comparison, as evident by comparing the light grey columns with dark grey columns of Fig. 4A and B, the activity of either IN enzyme was approximately 10 times reduced when assayed using their non-matching REEAD setups (e.g. when incubating MLV extracts with the HIV REEAD setup or purified HIV IN with the MLV REEAD setup). These results point towards discrimination by the described REEAD setup between the target virus and other related viruses. Note, however, that the current REEAD setup measures one end integration by IN. Available evidence demonstrate that the natural two ended integration mediated by IN during integration of the viral genome into the host genome is associated with a higher degree of species specific sequence recognition of the viral DNA than a one ended integration.11 Hence, if necessary a more efficient species differentiation may be obtained by designing an IN specific REEAD substrate that depend on two end integration for RCA and detection.
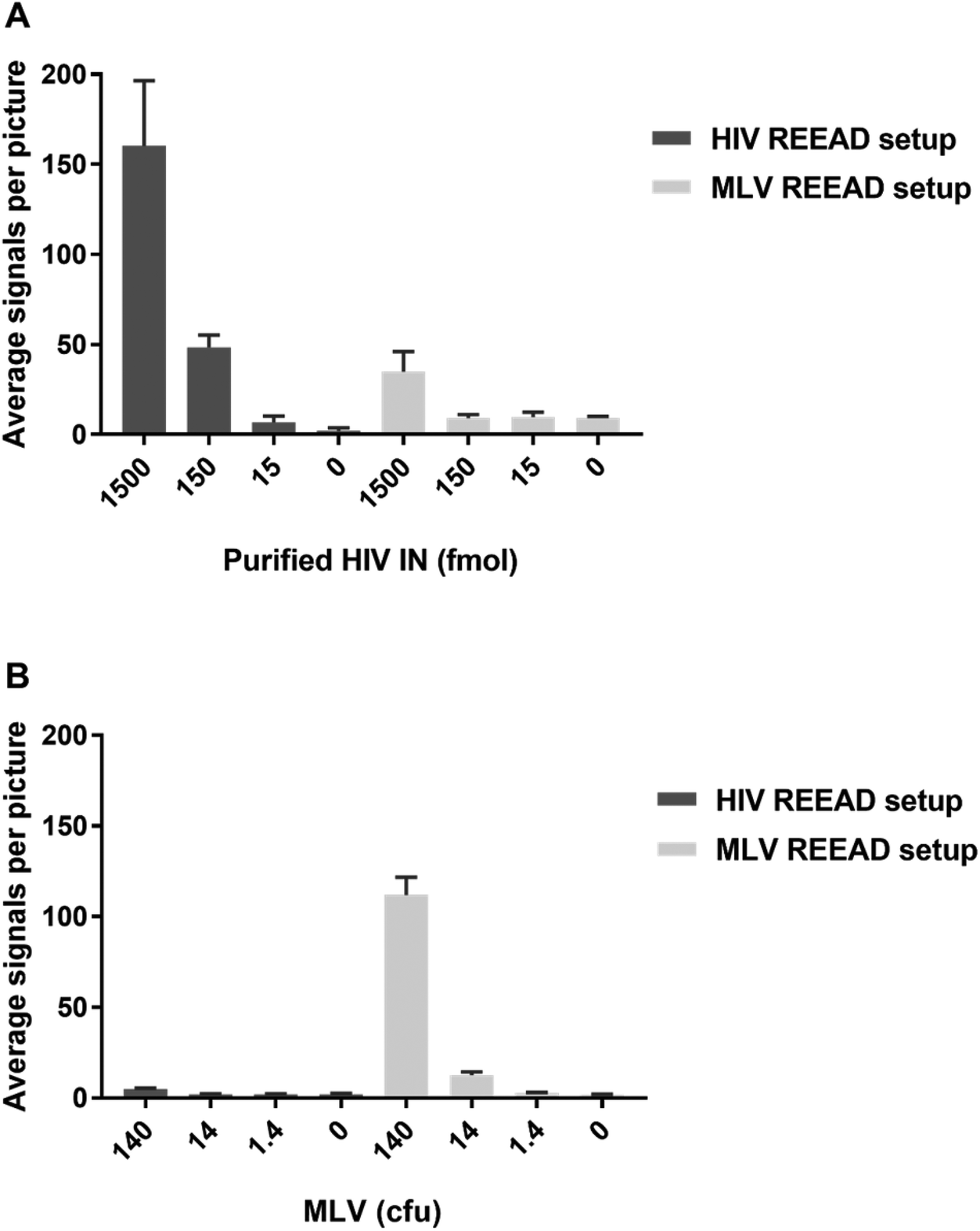 | ||
| Fig. 4 Bar chart representing the results of analyzing serial dilutions of purified HIV IN (A) or MLV particles (B) with the HIV or the MLV specific REEAD setup as indicated. The concentrations of virus particles are stated in cfu (colony forming units) corresponding to 30 virus particles per μL. Error bars represent standard deviations calculated from the results of three individual experiments. | ||
Conclusion
In conclusion the presented REEAD setup allowed for sensitive, dose dependent and specific detection of HIV-1 and MLV in crude biological samples and even demonstrated the ability to discriminate between different viruses. Current FDA-approved state-of-the-art diagnostic test of HIV are based on host-antibody detection in a Western-blotting or ELISA-based setup.24–26 Unlike tests, which suffer from the disadvantages of yielding false negative results due to the time window of several weeks from infection to seroconversion, the REEAD setup is likely to allow early diagnosis by detecting IN activity, which is present in mature virus particles at the stage of infection. The risk of false negatives posed by antibody detection can currently be circumvented by PCR based tests. However, such tests require a level of laboratory standard not available at the general practitioners and a regional infrastructure lacking in the parts of the World, where the HIV burden is largest.15 In contrast, the presented REEAD setup is easy-to-operate, requires no thermal cycling, and can be adapted to simple colorimetric readout systems by incorporation of biotinylated nucleotides in the RCA product followed by coupling to streptavidin tagged horseradish peroxidase or alkaline phosphatase essentially as described in.10,11 This will enable detection by a simple color reaction that will be directly visible by the naked eye making the method an appealing possibility for Point-of-Care (POC) testing even at low resource settings. Alternatively electrochemical readout principles can be tested out. Further highlighting such application even in the current proof-of-principle setup time-to-readout (∼2 hours) and prize (less than two USD per test) of the retrovirus specific REEAD is acceptable for routine use and is expected considerably reduced, once a POC test is fully developed. Moreover, the presented REEAD setup presents the first example of a REEAD substrate system that allows specific detection of an enzyme, which requires a divalent cation for activity and for which specificity cannot be ensured simply by depleting reaction buffers for divalent cations as it was previously done for detection of human or Plasmodium expressed topoisomerase I. Hence, the presented setup points towards the future possibilities of designing novel REEAD substrate systems for detection of vast number of target enzymes, with more complicated requirements, for diagnosis of disease in broader terms or for other nano-medical applications.Materials and methods
Materials
DNA oligonucleotides HIV-IN-fw and HIV-IN-rv (see Table 1) were purchased from DNA-Technology. All other oligonucleotides were purchased from Sigma-Aldrich.| U5MLVB | AATGAAAGACCCCCGCTGACGGGTAGTCAA |
| U5HIVB | ACTGCTAGAGATTTTCCACACTGACTAAA |
| 5′-Amine-U5HIVA | Am-TTTAGTCAGTGTGGAAAACTCTAGCAGT |
| 5′-Amine-U5MLVA | Am-TTGACTACCCGTCAGCGGGGGTCTTTCATT |
| Acceptor-circle-fw | CCGCCCTGCAGCCTCAATGCACATGTTTGGCTCCC |
| Acceptor-circle-rv | TAATTCTGCAGACGATAGCGGTACATCTCGG |
| HIV-IN-fw | AACTGGCGCGCCATGGCTTCTGAC |
| HIV-IN-rv | TTAATCTTCGTCCTGACGAGAAGCAACG |
| FAM-ID33 | FAM-CCTCAATGCACATGTTTGGCTCC |
| FAM-IDA1 | FAM-CCGAGATGTACCGCTATCGT |
pTrcHis-TOPO TA Expression Kits, TOPO® TA Cloning® Kit of the expression vector pTrcHis-TOPO, was purchased from Invitrogen. The synthetic gene MLV_IN of the expression vector pET303_CT-His_A118 was purchased from GeneArt AG. The pEGFP-PK-IN vector was purchased from Addgene. E.Z.N.A.®Gel Extraction Kit and E.Z.N.A.®Plasmid Mini Kit were purchased from Omega Bio-tek. NucleoBond®Xtra Maxi Kit was purchased from Macherey-Nagel. Pst I restriction endonuclease and T4 DNA ligase were purchased from New England Biolabs. The phi29 DNA polymerase was purchased from Thermo Scientific. Ni2+NTA Superflow was purchased from Qiagen. Protease inhibitor EDTA-free tablet was purchased form Roche. Primary antibody – Anti-His Antibody (mouse IgG1) purchased from Qiagen. Secondary Antibody – Polyclonal Rabbit Anti-Mouse Immunoglobulin purchased from Dako. ECL Plus Western Blotting Detection Reagents were purchased from GE Healthcare Life Sciences. E. coli XL1-Blue, E. coli K12 and E. coli BL21 were purchased from Thermo Fisher. CodeLink Activated Slides were from Surmodics, and Vectashield was from Vector Laboratories.
Recombinant plasmid DNA description
The synthetic gene MLV_IN was cloned into pET303_CT-His_A118 using Xba I and Xho I cloning sites. The generated plasmid, 13AASNMC_MLV_IN_ pET303_CT-His, was purified from transformed bacteria E. coli K12.The HIV IN gene was amplified from the vector pEGFP-PK-IN (Addgene: pPS2986) by a HotStart PCR system. The PCR program was set to 5 min at 95 °C, 30 cycles of 30 s at 95 °C, 30 s at 57 °C, 1 min at 72 °C and a terminal step of 10 min at 72 °C. The PCR product was cloned into expression vector pTrcHis-TOPO using the pTrcHis TOPO®TA Expression Kit. The resulting plasmid, pTrcHis-TOPO-HIV_IN, was amplified in E. coli XL1-Blue cells and purified.
Expression and purification of MLV and HIV IN
E coli BL21 was transformed with either 13AASNMC_MLV_IN_ pET303_CT-His or pTrcHis-TOPO-HIV_IN and grown in LB medium. Expression of IN was induced at OD 0.6 with IPTG (0.3 mM) for 3 h at 30 °C. The cells were then pelleted and either lysed immediately or stored at −80 °C. The pellets were resuspended in 40 ml of ice-cold solubilization buffer (50 mM sodium phosphate buffer, pH 8.0, 300 mM NaCl), 10 mM imidazole, 10 mM Chaps (3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulfonate), 1 protease inhibitor EDTA-free tablet (Roche) per 50 ml of buffer. The cells were kept on ice, added 100 mg of lysozyme, and inverted 5–10 times to insure sufficient mixing. The cells were incubated on ice for 15 min with gentle agitation. Samples were then sonicated for 15 s and placed on ice for 1 min. Sonication was repeated 5 times until samples appeared clear (not viscous), then the extracts were centrifuged at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 1 h at 4 °C. The supernatants containing soluble MLV IN and HIV IN proteins, respectively, were transferred to a new 50 ml Nunc tube and IN was purified using fast protein liquid chromatography (FPLC) on Ni2+-NTA columns. A column with 3 mL Ni-NTA Superflow was packed, system pumps filled with 80% ethanol, and the column washed with 10 column volumes double deionized water by gravitational flow. Then the column was attached to the pump outlet without introducing air into the system. The column outlet was attached to collecting system tubing. The column was equilibrated with 10 column volumes of equilibration buffer (10 mM Tris-HCl, pH 7.5, 200 mM NaCl, 5 mM MgCl2, 10% glycerol, 1 mM PMSF) at 0.5 mL min−1. Solubilized integrase (IN) protein extract was diluted using an equal volume dilution buffer (10 mM Tris-HCl, pH 7.5, 10% glycerol, 1 mM PMSF) with extra 1 protease inhibitor EDTA-free tablet (Roche) per 50 mL of buffer before loading. Soluble protein extract was loaded onto the column using the system pump at a flowrate of 0.5 mL min−1. Bound proteins were washed with 10 column volumes of wash buffer (10 mM Tris-HCl, pH 7.5, 200 mM NaCl, 20 mM imidazole, 10% glycerol, 1 mM PMSF) and then eluted with 25 ml elution buffer (10 mM Tris-HCl, pH 7.5, 200 mM NaCl, 150 mM imidazole, 5 mM MgCl2, 10% glycerol, 1 mM PMSF) collecting 0.5 mL fractions of 0.5 mL. PMSF was added to the relevant buffers immediately prior to use. Purified fractions were pooled and diluted in 200 mM KCl, 10 μM ZnCl2, 50% glycerol. Aliquots of fractions were stored at −20 °C or −80 °C. The protein fractions were analyzed by SDS-PAGE.
000 rpm for 1 h at 4 °C. The supernatants containing soluble MLV IN and HIV IN proteins, respectively, were transferred to a new 50 ml Nunc tube and IN was purified using fast protein liquid chromatography (FPLC) on Ni2+-NTA columns. A column with 3 mL Ni-NTA Superflow was packed, system pumps filled with 80% ethanol, and the column washed with 10 column volumes double deionized water by gravitational flow. Then the column was attached to the pump outlet without introducing air into the system. The column outlet was attached to collecting system tubing. The column was equilibrated with 10 column volumes of equilibration buffer (10 mM Tris-HCl, pH 7.5, 200 mM NaCl, 5 mM MgCl2, 10% glycerol, 1 mM PMSF) at 0.5 mL min−1. Solubilized integrase (IN) protein extract was diluted using an equal volume dilution buffer (10 mM Tris-HCl, pH 7.5, 10% glycerol, 1 mM PMSF) with extra 1 protease inhibitor EDTA-free tablet (Roche) per 50 mL of buffer before loading. Soluble protein extract was loaded onto the column using the system pump at a flowrate of 0.5 mL min−1. Bound proteins were washed with 10 column volumes of wash buffer (10 mM Tris-HCl, pH 7.5, 200 mM NaCl, 20 mM imidazole, 10% glycerol, 1 mM PMSF) and then eluted with 25 ml elution buffer (10 mM Tris-HCl, pH 7.5, 200 mM NaCl, 150 mM imidazole, 5 mM MgCl2, 10% glycerol, 1 mM PMSF) collecting 0.5 mL fractions of 0.5 mL. PMSF was added to the relevant buffers immediately prior to use. Purified fractions were pooled and diluted in 200 mM KCl, 10 μM ZnCl2, 50% glycerol. Aliquots of fractions were stored at −20 °C or −80 °C. The protein fractions were analyzed by SDS-PAGE.
Generation of acceptor DNA circles
An approximately 500 bp circle gene fragment was amplified from the vector pTrcHis-TOPO by a HotStart PCR system using the primers Acceptor-circle-fw and Acceptor-circle-rv (see Table 1). The PCR program was set to 5 min at 95 °C, 30 cycles of 45 s at 95 °C, 30 s at 57 °C and 30 s at 72 °C followed by a terminal step of 10 min at 72 °C. The PCR product was cloned into the vector pTrcHis-TOPO by TOPO®TA Cloning®Kit and the plasmid DNA was transformed into E. coli DH5αcells. A large amount of the 500 bp gene fragment was excised from the plasmid by Pst I and purified using the E.Z.N.A.®Gel Extraction Kit. The cohesive end 500 bp DNA fragment was circularized by T4 DNA ligase. The resulting covalently closed double stranded DNA circles were referred to as DNA acceptor circles. The circles were purified using Illustra GFX PCR DNA and Gel Band Purification Kits. Circle concentration was determined using a Nanodrop spectrophotometer (Thermo Scientific, USA).Rolling circle amplification-based retrovirus integrase detection
Five fmol of the 5′-amine-U5MLVA oligonucleotide and the 5′-amine-U5HIVA nucleotide respectively (see Table 1) were linked to CodeLink activated slides as described by the supplier. The U5MLV LTR and U5HIV LTR substrates were completed by hybridization of 5 fmol of the complementary U5MLVB and U5HIVB substrate respectively for 30 min at 37 °C in a humidity chamber on a hot plate (hybridization buffer containing 40% formamide, 4× SSC, and 10% glycerol). Integration of the 5 fmol LTR substrate on the slides into the acceptor DNA circle was done in a reaction buffer consisting of 20 mM 2-(N-morpholino)ethanesulfonic acid (MES, pH 6.2), 200 mM KCl, 10 mM MnCl2, 10 mM MgCl2, 10 mM DTT (dithiothreitol), and 10% glycerol. The reaction was performed in a 5 μL reaction volume containing 0.5 μL IN and 50 fmol of the acceptor circle. The reaction was incubated for 5 min on ice, after which incubation was continued for 2 h at 37 °C. The reaction was stopped by washing in wash buffer 2 (0.1 M Tris-HCl, pH 7.5, 150 mM NaCl, and 0.3% SDS) for 1 min at room temperature and subsequently wash buffer 3(0.1 M Tris-HCl, pH 7.5, 150 mM NaCl, 0.05% Tween-20) for another 1 min. Finally, the slides were dehydrated in 99.9% ethanol for 1 min and air dried. Rolling circle DNA synthesis was performed in 1× phi 29 buffer supplemented with 1 μg μL−1 BSA, 250 μM dNTP, and 1 unit per μL phi29 DNA polymerase for 1 h at 37 °C in a humidity chamber on a hot plate. The reaction was stopped by washing in wash buffers 2 and 3, followed by ethanol mediated dehydration as described above. The RCPs were detected by hybridization to 0.2 μM of detection probes FAM-ID33 and FAM-IDA1 (see Table 1) in a buffer containing 20% formamide, 2× SSC, and 5% glycerol for 30 min at 37 °C. The slides were washed in wash buffer 2 and 3, dehydrated, and mounted with Vectashield without DAPI, and put on coverglass. Epifluorescent and bright-field images were captured with a Leica fluorescence microscope by Leica LAS AF Lite. Monocolor emission from each fluorophore was collected and filtered through appropriate filters. Image processing and analysis were performed with LAS-AF-lite.Virus production and titer assay
Human embryonic kidney HEK 293 cells were cultured in Dulbecco's modified Eagle medium (Gibco) supplemented with 10% fetal bovine serum (FBS) (Gibco), 100 units per mL penicillin, and 100 mg per mL streptomycin (Invitrogen). Cells were incubated in a humidified incubator (5% CO2/95% air atmosphere at 37 °C). Cells were harvested with 0.5% Trypsin-EDTA (Gibco). The medium was discarded and the cells were washed in phosphate-buffered saline (1× PBS) prior to nuclear extraction. Cell pellets were incubated on ice for 5 min in low salt integration reaction buffer (20 mM 2-(N-morpholino)ethanesulfonic acid (MES, pH 6.2), 10 mM DTT, and 10% glycerol). The cell extracts were used for REEAD directly or spiked with purified IN prior to REEAD. Retrovirus was produced in Plat E packaging cells. Retroviral vectors were transfected into Plat E packaging cells. The growth medium of Plat E cells was renewed one day after transfection, and the cells were incubated for one more day before harvesting viral particles. Virus supernatants were harvested 48 hours post-transfection and filtered through a sterile 0.45 μm filter. To determine the virons infectious titer, NIH3T3 cells were plated on a six well plates (2.5 × 104 cells per well) 24 h before transduction in Dulbecco's modified Eagle medium (Gibco) supplemented with 10% new born bovine serum (Gibco), the virus supernatant was diluted 10−1 to 10−6 with 6 μg ml−1 polybrene of medium. The 3 ml of mixture was added to each well. After 24 h infection, the medium was replaced with fresh medium containing 0.6 mg of G418 per ml (Gibco) and cultured for two weeks. The resistant colonies were counted after staining with 1% methylene blue in methanol. The viral titer was calculated by multiplying the number of colonies by dilution factor and volume of virus added to the well, defined as CFU per milliliter, Virions were lysed on ice for 5 min in 0.03 M NaCl, 25 mM HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) pH 7.6, 0.1% Triton X-100, and 1 mM DTT. 0.5 μL viral extract was used with 5 fmol 5′ amine MLV/HIV U5 substrate and 50 fmol 500 bp-circle in a 5 μL integration reaction buffer as described previously.Statement of contribution
All authors contributed extensively to the work presented in this paper.BRK and FSP conceived the study, designed or co-designed all experiments presented and wrote the major part of the manuscript assisted by MS, MSH, OF, Y-PH, JW.
JW, FCMK cloned the retroviral integrase genes and performed initial characterizations.
JW, JL, and DS expressed, purified, and characterized the HIV-IN and MLV-IN enzymes as well as performed the presented REEAD experiments.
JT, SF, and JW produced virus particles and performed and analyzed the REEAD experiments on these particles.
Acknowledgements
This work was supported by Marie & M. B. Richters Foundation, Minister Erna Hamiltons Legat for Science and Art, Civilingeniør Frode V. Nyegaard og Hustrus Foundation, Augustinus Foundation, Danish AIDS Foundation and “Fonden til Lægevidenskabens Fremme”. Jing Wang and Jiangnan Liu were supported by fellowships from China Scholarship Council.References
- N. Mehta, S. Trzmielina, B. A. Nonyane, M. N. Eliot, R. Lin, A. S. Foulkes, K. McNeal, A. Ammann, V. Eulalievyolo, J. L. Sullivan, K. Luzuriaga and M. Somasundaran, PLoS One, 2009, 4, e5819 Search PubMed.
- Y. N. Kim, K. M. Kim, H. N. Choi, J. H. Lee, H. S. Park, K. Y. Jang, W. S. Moon, M. J. Kang, D. G. Lee and M. J. Chung, J. Mol. Diagn., 2015, 17, 597–604 CrossRef CAS PubMed.
- Y. Yamamoto, Clin. Diagn. Lab. Immunol., 2002, 9, 508–514 CAS.
- E. Kamau, S. Alemayehu, K. C. Feghali, D. W. Juma, G. M. Blackstone, W. R. Marion, P. Obare, B. Ogutu and C. F. Ockenhouse, Malar. J., 2014, 13, 158 CrossRef PubMed.
- D. R. Almassian, L. M. Cockrell and W. M. Nelson, Chem. Soc. Rev., 2013, 42, 8769–8798 RSC.
- M. L. Jepsen, C. Harmsen, A. A. Godbole, V. Nagaraja, B. R. Knudsen and Y. P. Ho, Nanoscale, 2016, 8, 358–364 RSC.
- X. Li, D. Deng, J. Xue, L. Qu, S. Achilefu and Y. Gu, Biosens. Bioelectron., 2014, 61, 512–518 CrossRef CAS PubMed.
- H. Liu, Y. Lou, F. Zhou, H. Zhu, E. S. Abdel-Halim and J. J. Zhu, Biosens. Bioelectron., 2015, 71, 249–255 CrossRef CAS PubMed.
- W. Abu al-Soud and P. Radstrom, J. Clin. Microbiol., 2001, 39, 485–493 CrossRef PubMed.
- T. Takahashi, T. Mitsuda and K. Okuda, Anal. Biochem., 1989, 179, 77–85 CrossRef CAS PubMed.
- W. Cao, M. M. Su and S. S. Zhang, Electrophoresis, 2010, 31, 659–665 CrossRef CAS PubMed.
- S. Juul, C. J. Nielsen, R. Labouriau, A. Roy, C. Tesauro, P. W. Jensen, C. Harmsen, E. L. Kristoffersen, Y. L. Chiu, R. Frohlich, P. Fiorani, J. Cox-Singh, D. Tordrup, J. Koch, A. L. Bienvenu, A. Desideri, S. Picot, E. Petersen, K. W. Leong, Y. P. Ho, M. Stougaard and B. R. Knudsen, ACS Nano, 2012, 6, 10676–10683 CrossRef CAS PubMed.
- C. Tesauro, S. Juul, B. Arno, C. J. Nielsen, P. Fiorani, R. F. Frohlich, F. F. Andersen, A. Desideri, M. Stougaard, E. Petersen and B. R. Knudsen, Conference proceedings: Annual International Conference of the IEEE Engineering in Medicine and Biology Society. IEEE Engineering in Medicine and Biology Society. Annual Conference, 2012, vol. 2012, pp. 2416–2419.
- E. Yung, M. Sorin, E. J. Wang, S. Perumal, D. Ott and G. V. Kalpana, J. Virol., 2004, 78, 2222–2231 CrossRef CAS PubMed.
- J. Ambrosioni, A. Calmy and B. Hirschel, J. Int. AIDS Soc., 2011, 14, 28 CrossRef PubMed.
- T. Fujiwara and K. Mizuuchi, Cell, 1988, 54, 497–504 CrossRef CAS PubMed.
- M. J. Roth, P. L. Schwartzberg and S. P. Goff, Cell, 1989, 58, 47–54 CrossRef CAS PubMed.
- M. Stougaard, J. S. Lohmann, A. Mancino, S. Celik, F. F. Andersen, J. Koch and B. R. Knudsen, ACS Nano, 2009, 3, 223–233 CrossRef CAS PubMed.
- S. Desfarges and A. Ciuffi, Viruses, 2010, 2, 111–130 CrossRef CAS PubMed.
- F. S. Pedersen, M. Pyrz and M. Duch, Encyclopedia Of Life Sciences, 2011, DOI:10.1002/9780470015902.a0000430.pub3.
- P. An and C. A. Winkler, Trends Genet., 2010, 26, 119–131 CrossRef CAS PubMed.
- E. Guiot, K. Carayon, O. Delelis, F. Simon, P. Tauc, E. Zubin, M. Gottikh, J. F. Mouscadet, J. C. Brochon and E. Deprez, J. Biol. Chem., 2006, 281, 22707–22719 CrossRef CAS PubMed.
- A. Rein, Adv. Virol., 2011, 2011, 403419 Search PubMed.
- K. A. Curtis, D. L. Rudolph, I. Nejad, J. Singleton, A. Beddoe, B. Weigl, P. LaBarre and S. M. Owen, PLoS One, 2012, 7, e31432 CAS.
- C. D. Chin, T. Laksanasopin, Y. K. Cheung, D. Steinmiller, V. Linder, H. Parsa, J. Wang, H. Moore, R. Rouse, G. Umviligihozo, E. Karita, L. Mwambarangwe, S. L. Braunstein, J. van de Wijgert, R. Sahabo, J. E. Justman, W. El-Sadr and S. K. Sia, Nat. Med., 2011, 17, 1015–1019 CrossRef CAS PubMed.
- http://www.fda.gov/ .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6nr07428f |
| This journal is © The Royal Society of Chemistry 2017 |