
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Use of apomyoglobin to gently remove heme from a H2O2-dependent cytochrome P450 and allow its reconstitution†
Shih-Cheng
Chien
a,
Osami
Shoji
*ab,
Yoshiko
Morimoto
c and
Yoshihito
Watanabe
*c
aDepartment of Chemistry, Graduate School of Science, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8602, Japan. E-mail: shoji.osami@a.mbox.nagoya-u.ac.jp
bCore Research for Evolutional Science and Technology (CREST), Japan Science and Technology Agency, 5 Sanbancho, Chiyoda-ku, Tokyo 102-0075, Japan
cResearch Center for Materials Science, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8602, Japan. E-mail: p47297a@nucc.cc.nagoya-u.ac.jp
First published on 22nd November 2016
Abstract
The heme of hydrogen peroxide-dependent cytochrome P450BSβ (P450BSβ) was removed by apomyoglobin under mild conditions to give apo-P450BSβ without the need for acidic conditions and organic solvents. The circular dichroism spectrum of the apo-P450BSβ was essentially identical to that of holo-P450BSβ, showing a small structural change resulting from the removal of heme using apomyoglobin. The apo-P450BSβ was reconstituted with hemin or manganese protoporphyrin IX (MnPPIX), and the resulting reconstituted P450BSβ catalyzed the one-electron oxidation of guaiacol using hydrogen peroxide as an oxidant. A higher catalytic activity was observed for P450BSβ reconstituted with MnPPIX when meta-chloroperoxybenzoic acid (mCPBA) was used as the oxidant.
Introduction
Heme, Fe-protoporphyrin IX, is one of the most important metal complexes in nature, serving as the prosthetic group for hemoproteins that perform diverse functions, including oxygen storage and transport, gas sensing, electron transfer, and catalysis.1,2 The properties of hemoproteins largely depend on the nature of heme; therefore, replacement of the heme with heme analogues are expected to drastically change the properties of hemoproteins.3 A variety of reconstituted hemoproteins containing heme analogues, and heme derivatives including different metal ions, chemically modified hemes, and synthetic metal complexes have been constructed with the aim of developing synthetic proteins with (improved) catalytic activity or entirely different functions.4,5 To allow the reconstitution of hemoproteins with synthetic metal complexes, the tightly bound heme must be removed to give apoproteins. Noncovalently bound hemes (b-type hemes) can be extracted from hemoproteins using organic solvents such as butan-2-one and acetone under acidic conditions (pH 2–4). Teale et al. first reported the removal of heme from hemoproteins utilizing acid–butanol (pH = 2) to prepare apo forms of horse heart and skeletal muscle myoglobin (Mb).6 Several methods based on a slight modification of this acid–butanol method have been reported for the preparation of apo forms of hemoproteins such as HRP and P450cam (see Fig. S1 and S2 for these different heme enzyme structures, ESI†).7,8 However, these methods are not suitable if the hemoproteins as well as their apo-forms are not sufficiently stable in organic solvents and/or under acidic conditions. Although the preparation of apoproteins in bacterial cells has been developed as an alternative, their reconstitution was limited to metal complexes possessing a protoporphyrin IX framework. To avoid the use of harsh chemical treatments in the preparation of apoproteins, and to expand the range of applicable metal complexes to include various synthetic metal complexes, it is important to develop a simple strategy for removing the heme of hemoproteins without the need for organic solvents and acidic conditions.Cytochrome P450BSβ (P450BSβ) is a hemoprotein that is irreversibly denatured during the preparation of apoprotein under the methods described above. Therefore, it has been believed to be impossible to prepare reconstituted P450BSβ. However, P450BSβ is regarded as a promising candidate for the construction of a biocatalyst because P450BSβ and its unique family, the hydrogen peroxide-dependent P450s, efficiently utilize hydrogen peroxide to catalyze various C–H bond hydroxylations and some other types of oxidation reactions.9,10 If the heme of P450BSβ could be replaced with synthetic metal complexes, the properties of the enzymatic reaction catalyzed by P450BSβ could be altered to allow the development of a versatile biocatalyst.
Here we report a simple and useful method for the preparation of apo-P450BSβ using apo-Mb under very mild conditions (Scheme 1). We decided to employ apo-Mb as a heme scavenger because it binds strongly to heme and can be readily prepared by the aforementioned acid–organic solvent-methods; the dissociation constant of apo-Mb for heme is reported to be 10−14 M−1.11 We also demonstrate that the resulting apo-P450BSβ can be reconstituted with manganese protoporphyrin IX (MnPPIX) and that the resulting reconstituted P450BSβ exhibits improved one-electron oxidation activity when mCPBA was employed as an oxidant.
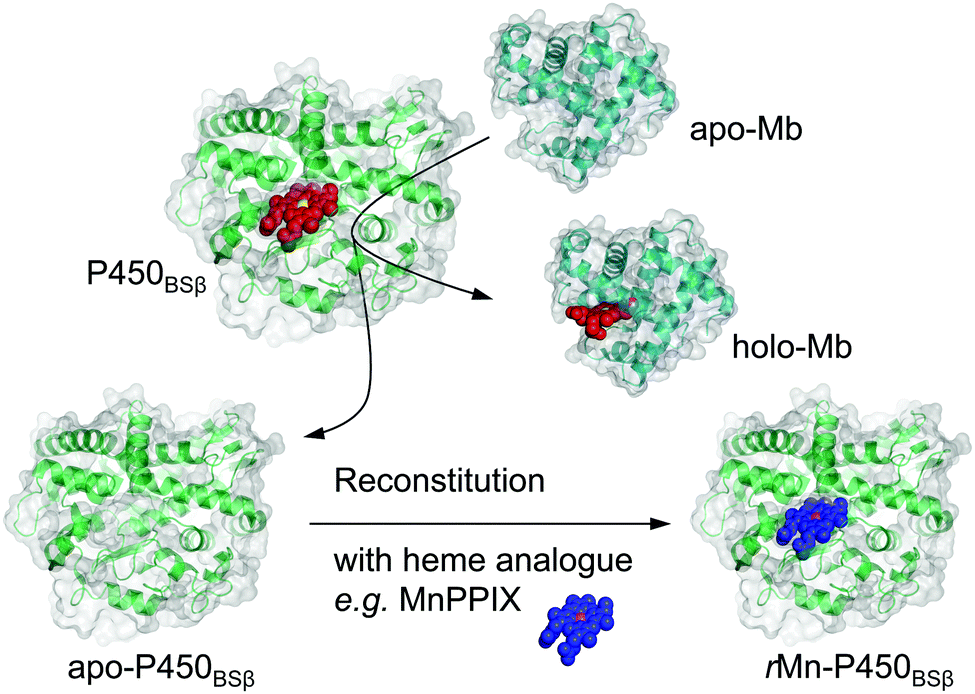 | ||
| Scheme 1 Schematic illustration of apo-Mb capturing the native cofactor heme (red with the iron atom in yellow) and subsequently reconstituted with an artificial cofactor (blue with the manganese atom in magenta) to generate a functional enzyme from holo-form H2O2-dependent P450BSβ. | ||
Experimental methods
Materials
All chemicals were purchased from commercial sources and used without further purification unless otherwise indicated. Mn(III) protoporphyrin IX chloride was obtained from Frontier Scientific, Inc. (Logan, UT, USA) and used after exchanging its Cl− for BF4− by treating with AgBF4. Hydrogen peroxide, dithiothreitol (DTT), ethylene glycol, sodium dodecyl sulfate (SDS), isopropyl-β-D-1-thiogalactopyranoside (IPTG), and kanamycin sulfate were obtained from WAKO Pure Chemical Industries, Ltd (Osaka, Japan). Glycerol, potassium chloride, urea, hydrochloric acid, potassium phosphate, ampicillin sodium salt and phenylmethylsulfonyl fluoride were purchased from Nacalai Tesque Inc. (Kyoto, Japan). Horse skeletal muscle Mb was obtained from Sigma-Aldrich Co. (St. Louis, MO, USA). Concentrations of chemicals described in procedures are the final concentrations.Measurements
UV-visible (UV-Vis) absorption spectra were recorded on a UV-2600 PC spectrophotometer (Shimadzu Corporation) or an Agilent 8453 equipped with an 89090A thermal controller (Agilent Technologies, Inc.), and the data were collected using screw-capped quartz cuvettes of 1 cm path length. Circular dichroism (CD) spectra were recorded with quartz cuvettes of 0.1 cm path length using a J-720WI spectropolarimeter (Jasco Corporation) equipped with a temperature controller, and the data were recorded from 200 to 260 nm at 24 °C and 20 nm min−1 scan speed, and an average spectrum was produced from four independent consecutive scans. Size distributions were recorded by dynamic light scattering (DLS) using a Nano ZS (Malvern Instruments Ltd) with disposable plastic cuvettes and caps. Inductively coupled plasma-optical emission spectrometer (ICP-OES) measurements were performed on a Varian Vista-Pro ICP Spectrometer (Varian Inc.). The purification procedures described below were carried out using a Bioassist eZ system (Tosoh Corporation). The protein figures were generated using the PyMOL Molecular Graphics System (version 1.8; DeLano Schrödinger, LLC.).Protein expression and purification
For the expression of recombinant P450BSβ, 6× histidine-tagged (6× His-tagged) P450BSβ was expressed in the transformed Escherichia coli strain M15. Cells were cultivated at 27 °C in Luria–Bertani medium supplemented with 100 μg mL−1 of ampicillin and 25 μg mL−1 of kanamycin. Once the culture reached an optical density at 600 nm (OD600) of 0.7–0.8, heme precursor δ-aminolevulinic acid (0.5 mM) was added, and the culture was incubated for an additional 30 min. At OD600 = 1.0–1.2, gene expression was induced by addition of IPTG (0.1 mM), and cultivation was followed by culturing at 20 °C for 20 h rotating at 80 rpm. Cells were then harvested by centrifugation and lysed in 0.1 M potassium phosphate buffer (pH 7.0) with 0.3 M potassium chloride, 50 mM imidazole, and 20% (v/v) glycerol. Purification of 6× His-tagged P450BSβ was performed by Ni-chelate affinity chromatography (GE Healthcare, Little Chalfont, UK). The protein was further purified using S-200 Sephacryl (GE Healthcare) size exclusion chromatography. An ASoret/A280 absorbance ratio ≥1 indicated the high purity of the protein, which was also confirmed by SDS-polyacrylamide gel electrophoresis (SDS-PAGE). Reduced CO-difference spectra were used to determine the concentration of P450 by subjecting the purified recombinant protein to sodium dithionite and CO gas.12 Protein samples were stored at −80 °C for further use.Preparation of apo-Mb
A heme moiety was extracted from horse skeletal muscle Mb by the butan-2-one extraction according to a published procedure6 with slight modifications. The pH of a Mb solution (100 mg Mb in 40 mL Milli-Q water) was adjusted to 2.5 in an ice-water bath using chilled 0.1 M HCl(aq). The solution was transferred to a separation funnel, and an equal volume of 2-butanone was added. The mixture was gently shaken for 30 s and then allowed to stand for 10 min at 4 °C. The colorless aqueous phase was separated and dialyzed for 24 h at 4 °C against 5 L of Milli-Q water, and then for 24 h against PBS buffer (pH 7.3). Next, the solution was passed through a 0.22 μm filter and stored at 4 °C for further use. The concentration of prepared apo-Mb was spectrophotometrically determined using its extinction coefficient of 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 M−1 cm−1 at 280 nm.13
700 M−1 cm−1 at 280 nm.13
Preparation of apo-P450BSβ
The removal of heme from P450BSβ to prepare apo-P450BSβ was performed by treating P450BSβ with apo-Mb. The reaction was carried out in 0.1 M potassium phosphate buffer (pH 7.0) containing 0.3 M potassium chloride and 20% (v/v) glycerol with P450BSβ (25 mL, 8.5 μM). Solid Na2S2O4 was dissolved into the solution (1 mg mL−1) at 25 °C to generate reduced P450BSβ. Five equivalents of apo-Mb (25 mL, 42.5 μM) was added to the reaction buffer, and the final concentration of glycerol was adjusted to 5% using additional PBS buffer. The reaction was maintained in a gas-tight flask for 24 h without stirring. Before purification, 0.02% of Triton X-100 was added to aid the separation of P450BSβ and Mb. Purification of 6× His-tagged apo-P450BSβ was performed by Ni-chelate affinity chromatography to remove holo-Mb and excess apo-Mb. This is the same purification procedure used for recombinant P450BSβ. Sample purity was checked by SDS-PAGE. Pure apo-P450BSβ was stored at 4 °C for further use or stored at −80 °C for long-term preservation.Reconstitution of apo-P450BSβ
Reconstitution of apo-P450BSβ was performed by adapting the procedure for apo-P450cam14 at 25 °C as well as subsequent protein purification at 4 °C. After the preparation of protoporphyrin complex solution, it should be added to apo-P450BSβ buffer immediately. Any delay in the addition of protoporphyrin complex solution to apo-P450BSβ led to incomplete recombination due to its low solubility in water solution.13000 M−1 cm−1),15 diluted tenfold with Milli-Q water, was added dropwise to the solution of apoprotein. The whole reaction was then incubated in a gas-tight flask wrapped in aluminum foil for 24 h without stirring. Before purification, an Amicon ultracentrifugal filter was used to remove excess MnPPIX and other chemicals. Purification of 6× His-tagged rMn–P450BSβ was performed by Ni-chelate affinity chromatography to remove free MnPPIX by the same procedure as that used for recombinant P450BSβ. The final purified rMn–P450BS gave a single band on SDS-PAGE, and the ASoret(381)/A280 absorbance ratio was 0.7.
Determination of P450 concentration
The concentration of the recombinant P450BSβ was measured by reduced CO-difference spectroscopy.12 In the case of rFe–P450BSβ with a higher content of P420 isoform (Fig. S4, ESI†), the concentration was measured by reduced pyridine-hemochrome spectroscopy.16 The concentrations of holo- and apo-P450 for CD spectroscopy measurements were determined by the Bradford protein assay.17 rMn–P450BSβ was quantitated from the Bradford protein assay and multiplied by values of ASoret/A280.Guaiacol oxidation
600 M−1 cm−1.18 All initial turnover numbers (TONs) are the average of at least three measurements, and are expressed as nmol product per nmol P450 per min ± standard deviation.
ICP-OES
The iron and manganese contents in rP450BSβ were determined by ICP-OES. Concentrated samples of purified rFe–P450BSβ and rMn–P450BSβ protein were diluted with 0.2 M HNO3 to 2 mL of 1.5, 3.0, 4.5 and 6.0 μM for the determinations. The solution was placed in a new glass vial and subjected to ICP-OES via a peristaltic pump. The sample was aspirated with argon, passed over plasma, and analyzed for iron and manganese. A control experiment was performed using Milli-Q water as a sample. Commercial calibration standards and multi-element standards (Wako Pure Chemical Industries, Ltd) were used.Results and discussion
The removal of the heme from P450BSβ to prepare apo-P450BSβ was performed by treating P450BSβ with apo-Mb. When five equivalents of apo-Mb were added to a solution of histidine-tagged P450BSβ at 25 °C, apparent shifts of the Soret absorption band and Q-band were observed.‡ The absorption peak of P450BSβ at 417 nm shifted to 409 nm, which is readily attributed to the formation of holo-Mb (Fig. 1A). After 24 h incubation, the resulting apo-P450BSβ was purified by Ni-chelate affinity chromatography, but a small amount of holo-P450BSβ was still observed. To bring the heme transportation to completion, P450BSβ was reduced using Na2S2O4 because the dissociation rate constant of reduced heme (FeII) coordinated with cysteine is rapid compared with that of heme (FeIII).19 DFT calculations also showed a weaker binding of cysteine to ferroheme than to ferriheme. A clear shift of the absorption maxima of heme was again observed after the addition of apo-Mb, and no remarkable change was observed after 24 h (Fig. 1B).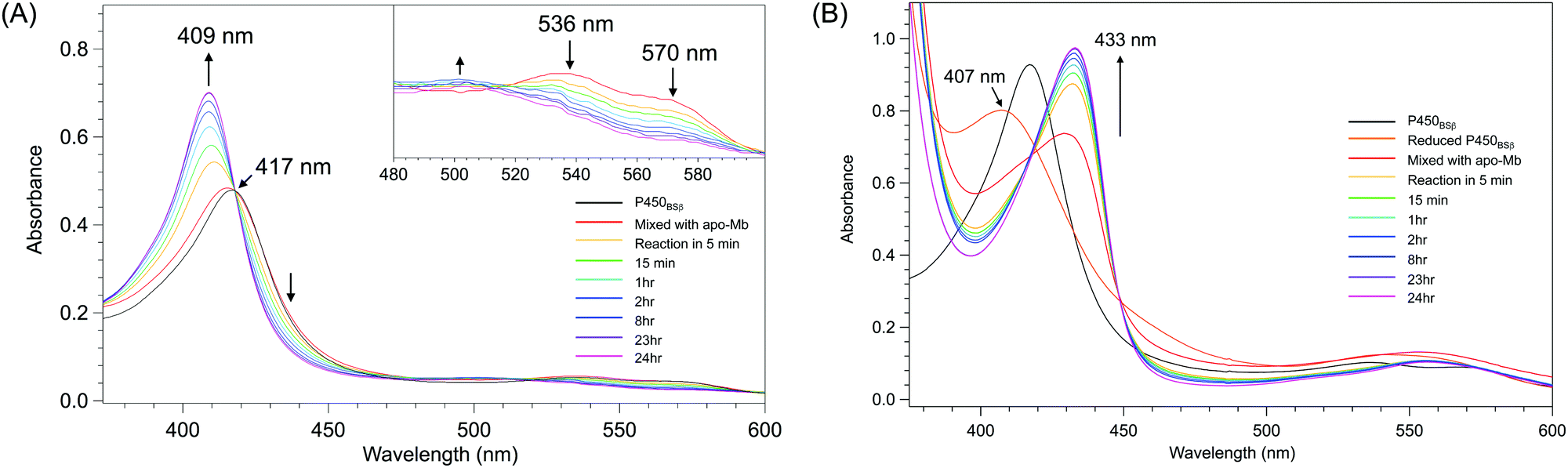 | ||
| Fig. 1 Time course of UV-Vis absorption spectra changes during the reactions at 25 °C for 24 h. (A) P450BSβ with apo-Mb. Inset: spectral expansion of 480 ∼ 600 nm; (B) Reduced P450BSβ with apo-Mb. | ||
Apo-P450BSβ purified by Ni-chelate affinity chromatography did not show any absorption assignable to heme, indicating that the heme of P450BSβ can be removed completely (Fig. S5, ESI†). Interestingly, the CD spectrum of apo-P450BSβ was essentially identical to that of the holo form, indicating a small structural change upon the removal of the heme (Fig. 2B), while the CD spectra of apoproteins produced by acid–organic solvent methods were generally significantly different from those of the holo form.20 For example, apo-P450cam prepared by the acid–butanone method gave a weaker CD signal, indicative of partial denaturation. The hydrodynamic diameters of holo- and apo-P450BSβ determined by DLS analysis were 4.36 and 5.05 nm, respectively (Fig. 2C and Fig. S6, ESI†), which are consistent with the size of holo-P450BSβ (PDB ID: 3WSP), indicating that there is no appreciable denaturation or aggregation. These results clearly showed that apo-P450BSβ could be prepared using apo-Mb and the resulting apo-P450BSβ retained almost the same conformation as its holo form without denaturation.
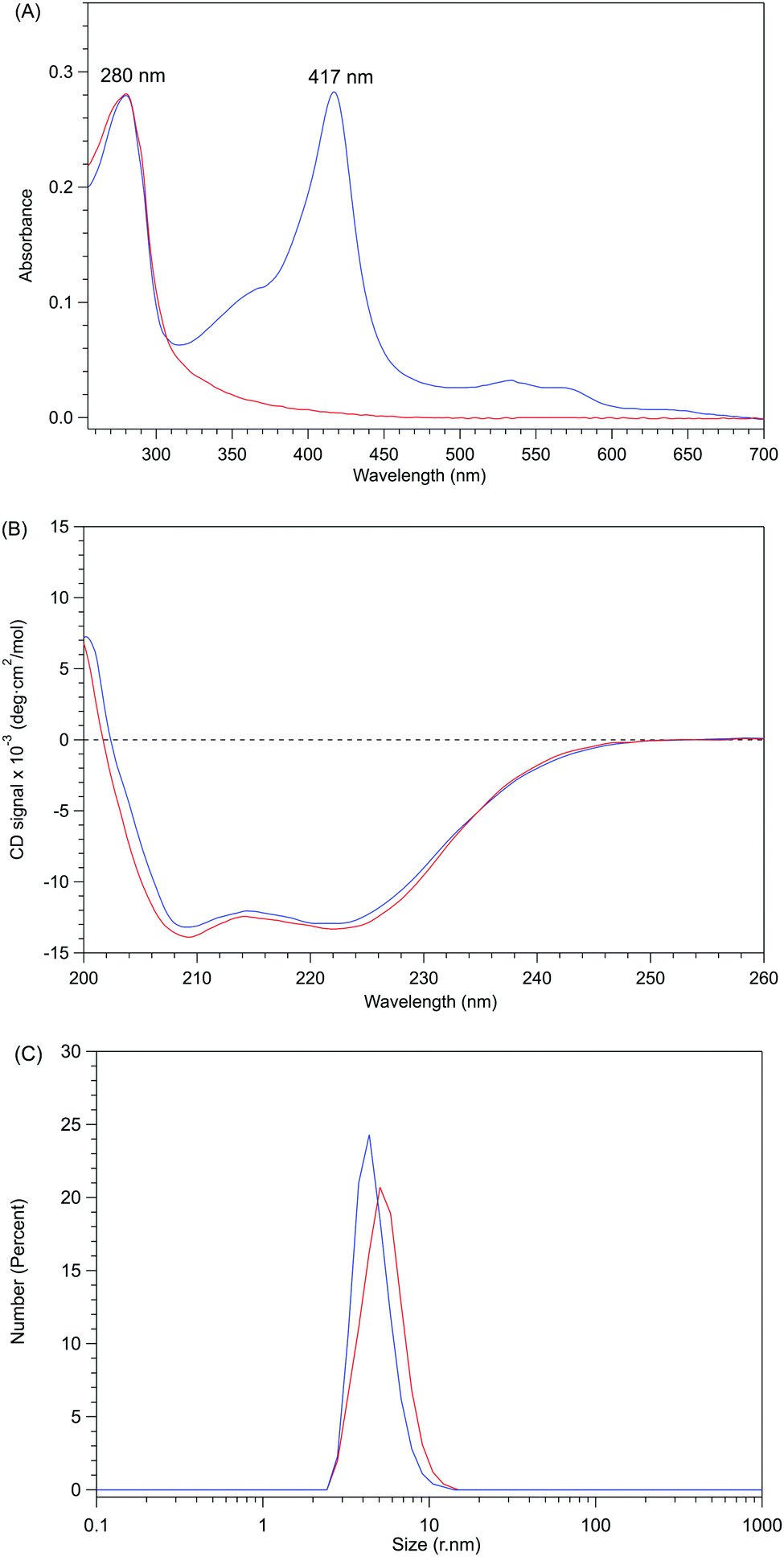 | ||
| Fig. 2 Comparisons between holo- and apo-form P450BSβ. Holo-P450BSβ is shown as a blue solid line and apo-P450BSβ as a red solid line. (A) UV-Vis absorption spectra of holo- and apo-P450BSβ. (B) CD spectra (0.0025 M) of holo- and apo-P450BSβ; the concentrations of protein were calculated using the Bradford method. (C) DLS spectra of holo- and apo-P450BSβ. | ||
With pure apo-P450BSβ in hand, we reconstituted it according to the method reported for preparing reconstituted P450cam.14 Initially, apo-P450BSβ was reconstituted with heme to confirm that it is possible to reinsert heme into an appropriate position in P450BSβ and to fully recover its catalytic activity. A solution of apo-P450BSβ was added with 2.5 equivalents of hemin in the presence of 10 mM DTT and 10 mM histidine, and then incubated for 24 h under argon. Reconstituted P450BSβ (rFe–P450BSβ) could be purified by Ni-chelate affinity chromatography. The UV-Vis absorption spectrum showed a Soret peak at 417 nm. The CD spectrum of reconstituted P450BSβ remained similar to those of recombinant P450BSβ and α-helical proteins (Fig. S7, ESI†).
rFe–P450BSβ exhibited catalytic activity similar to that of native P450BSβ for the one-electron oxidation of guaiacol with the assistance of n-heptanoic acid, which accelerates the generation of active species of P450BSβ because the carboxylate of n-heptanoic acid serves as an acid–base catalyst.21 The initial TON (nmol product per nmol P450 per min) of one-electron oxidation of guaiacol by rFe–P450BSβ with heptanoic acid was estimated to be 3150, which is almost the same as that of P450BSβ under similar conditions (Table 1).22 These results indicate that apo-P450BSβ prepared using apo-Mb can be reconstituted with heme and that the reconstituted rFe–P450BSβ has a catalytic activity comparable to that of recombinant P450BSβ.
|
|
||
|---|---|---|
| Enzyme protein | TON (min−1) | |
| H2O2a | mCPBA | |
| a Heptanoic acid assisted these catalytic experiments. b The value of TON is adapted from Angew. Chem., Int. Ed., 2007, 46, 3656–3659. c For the proposed oxidation mechanism of guaiacol with P450, see Scheme S1, ESI. | ||
| P450BSβ | 3750b | 113 ± 5 |
| rFe–P450BSβ | 3150 ± 50 | — |
| rMn–P450BSβ | 40 ± 8 | 2860 ± 40 |

Given that apo-P450BSβ could be reconstituted with heme, we have attempted to reconstitute apo-P450BSβ with MnPPIX in a similar manner, and obtained P450BSβ reconstituted with MnPPIX (rMn–P450BSβ). The CD spectrum of rMn–P450BSβ confirmed that rMn–P450BSβ had the same protein structure as P450BSβ (Fig. 3 and Fig. S7, ESI†). The Soret absorption band of rMn–P450BSβ was observed at 381 nm, which corresponded to that of P450cam reconstituted with MnPPIX (Fig. 3).23,24 ICP-OES showed that 70% of the apo-P450BSβ was reconstituted with MnPPIX. The UV-Vis absorbance ratio (ASoret(381)/A280) of rMn–P450BSβ was consistent with 70% reconstitution.
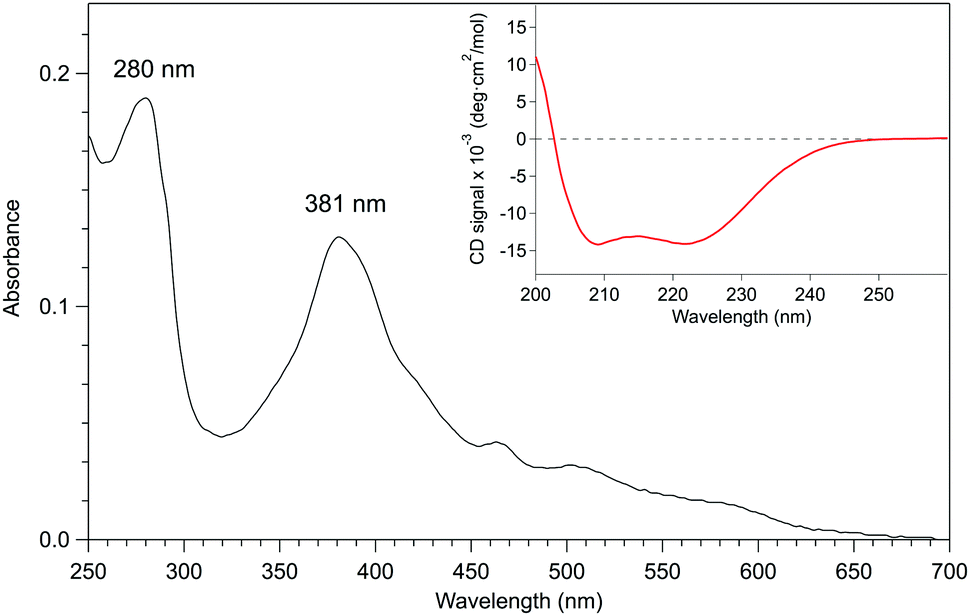 | ||
| Fig. 3 UV-Vis absorption spectrum (black line) of rMn–P450BSβ in 0.1 M potassium phosphate buffer (pH 7.0) containing 0.3 M KCl and 20% (v/v) glycerol. The Soret band from MnPPIX in the deep pocket of the active site of P450BSβ was at 381 nm, and there was no signal of free MnPPIX or adherent (noncovalently bound) MnPPIX at the surface of P450BSβ in the UV-Vis spectrum. The ASoret(381)/A280 absorbance ratio was 70%. The CD spectrum showed that rMn–P450BSβ remained in the same α-helix structure as the recombinant P450BSβ. Inset: CD spectrum (red line) of rMn–P450BSβ in the same potassium phosphate buffer. | ||
The catalytic activity of rMn–P450BSβ for one-electron oxidation of guaiacol using hydrogen peroxide as an oxidant was examined, but the TON was very low at 40 min−1. In contrast, when mCPBA was used as the oxidant, the catalytic activity was improved (2850 min−1) compared with that of recombinant P450BSβ (Table 1). On the other hand, hydroxylation of cyclohexane was not catalyzed by neither recombinant P450BSβ nor rFe–P450BSβ. We also examined the hydroxylation of cyclohexane by rMn–P450BSβ with mCPBA, but no product was detected, suggesting either the MnIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, not the MnVO, species generated by homolytic cleavage of MnIII–OOR or very rapid reduction of MnVO under the conditions and eventually MnIVO is responsible for one-electron oxidation by rMn–P450BSβ in the presence of mCPBA (Scheme S2, ESI†).25
O, not the MnVO, species generated by homolytic cleavage of MnIII–OOR or very rapid reduction of MnVO under the conditions and eventually MnIVO is responsible for one-electron oxidation by rMn–P450BSβ in the presence of mCPBA (Scheme S2, ESI†).25
Conclusions
We have demonstrated that apo-P450BSβ can be prepared using apo-Mb as a heme scavenger without subjecting the proteins to harsh and denaturing conditions. Although the preparation of apo-P450s and their reconstitution is generally difficult, the heme of P450BSβ was completely removed by apo-Mb to give pure apo-P450BSβ that could be reconstituted with MnPPIX. The holo-, apo-, rFe– and rMn–P450BSβ remained as analogous α-helical structures, in contrast to other reported reconstituted P450s and their apo-P450s prepared using the acid–butanone method20 or incubation with hydrogen peroxide.26 Although we focused on the preparation of apo-P450BSβ, other P450s whose apo-forms are difficult to prepare by conventional acid–organic-solvent methods may also be potent targets for our methodology. Furthermore, we believe that this method is useful to selectively remove the heme of hemoproteins containing other cofactors such as flavin derivatives or iron–sulfur clusters.Acknowledgements
This work was supported by Grants-in-Aid for Scientific Research (S) to Y. W. (24225004) from the Ministry of Education, Culture, Sports, Science, and Technology (Japan) and CREST, Japan Science and Technology Agency.Notes and references
- K. K. Kadish, L. M. Smith and R. Guilard, The Porphyrin Handbook, Academic Press, San Diego, USA, 2000 Search PubMed.
- S. Schneider, J. Marles-Wright, K. H. Sharp and M. Paoli, Nat. Prod. Rep., 2007, 24, 621–630 RSC.
- L. Fruk, C. H. Kuo, E. Torres and C. M. Niemeyer, Angew. Chem., Int. Ed., 2009, 48, 1550–1574 CrossRef CAS PubMed.
- H. Sato, T. Hayashi, T. Ando, Y. Hisaeda, T. Ueno and Y. Watanabe, J. Am. Chem. Soc., 2004, 126, 436–437 CrossRef CAS PubMed.
- T. Hayashi, Y. Hitomi, T. Ando, T. Mizutani, Y. Hisaeda, S. Kitagawa and H. Ogoshi, J. Am. Chem. Soc., 1999, 121, 7747–7750 CrossRef CAS.
- F. W. J. Teale, Biochim. Biophys. Acta, 1959, 35, 543 CrossRef CAS.
- C. A. Yu and I. C. Gunsalus, J. Biol. Chem., 1974, 249, 107–110 CAS.
- G. C. Wagner, I. C. Gunsalus, M. Y. R. Wang and B. M. Hoffman, J. Biol. Chem., 1981, 256, 6266–6273 CAS.
- D. S. Lee, A. Yamada, H. Sugimoto, I. Matsunaga, H. Ogura, K. Ichihara, S. Adachi, S. Y. Park and Y. Shiro, J. Biol. Chem., 2003, 278, 9761–9767 CrossRef CAS PubMed.
- O. Shoji, C. Wiese, T. Fujishiro, C. Shirataki, B. Wünsch and Y. Watanabe, JBIC, J. Biol. Inorg. Chem., 2010, 15, 1109–1115 CrossRef CAS PubMed.
- M. S. Hargrove, D. Barrick and J. S. Olson, Biochemistry, 1996, 35, 11293–11299 CrossRef CAS PubMed.
- I. C. Gunsalus and G. C. Wagner, Methods Enzymol., 1978, 52, 166–188 CAS.
- S. C. Harrison and E. R. Blout, J. Biol. Chem., 1965, 240, 299–303 CAS.
- G. C. Wagner, M. Perez, W. A. Toscano and I. C. Gunsalus, J. Biol. Chem., 1981, 256, 6262–6265 CAS.
- E. M. Harvat, O. Daltrop, F. Sobott, M. Moreau, P. D. Barker, J. M. Stevens and S. J. Ferguson, Metallomics, 2011, 3, 363–368 RSC.
- E. A. Berry and B. L. Trumpower, Anal. Biochem., 1987, 161, 1–15 CrossRef CAS PubMed.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS PubMed.
- G. D. Depillis, B. P. Sishta, A. G. Mauk and P. R. O. Demontellano, J. Biol. Chem., 1991, 266, 19334–19341 CAS.
- K. Kitanishi, J. Igarashi, K. Hayasaka, N. Hikage, I. Saiful, S. Yamauchi, T. Uchida, K. Ishimori and T. Shimizu, Biochemistry, 2008, 47, 6157–6168 CrossRef CAS PubMed.
- W. Pfeil, B. O. Nolting and C. Jung, Biochemistry, 1993, 32, 8856–8862 CrossRef CAS PubMed.
- O. Shoji, T. Fujishiro, H. Nakajima, M. Kim, S. Nagano, Y. Shiro and Y. Watanabe, Angew. Chem., Int. Ed., 2007, 46, 3656–3659 CrossRef CAS PubMed.
- O. Shoji and Y. Watanabe, Metallomics, 2011, 3, 379–388 RSC.
- M. H. Gelb, W. A. Toscano and S. G. Sligar, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 5758–5762 CrossRef CAS.
- O. Daltrop and S. J. Ferguson, J. Biol. Chem., 2004, 279, 45347–45353 CrossRef CAS PubMed.
- T. Matsui, S. Nagano, K. Ishimori, Y. Watanabe and I. Morishima, Biochemistry, 1996, 35, 13118–13124 CrossRef CAS PubMed.
- V. Y. Uvarov, V. E. Tretiakov and A. I. Archakov, FEBS Lett., 1990, 260, 309–312 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary figures including crystal structures, UV-Vis and CD spectra, the spectrum of purification and the proposed mechanisms of Mn–P450BSβ with mCPBA. See DOI: 10.1039/c6nj02882a |
| ‡ The absorption spectral change was very slow when the reaction of preparing apo-P450BSβ was under 4 °C after 24 h incubation (Fig. S3, ESI†). |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2017 |