
Can self-assembled hydrogels composed of aromatic amino acid derivatives function as drug delivery carriers?†
Priyanka
Tiwari
a,
Ramanathan
Rajagopalan
b,
Mohammad
Moin
b,
Rohit
Soni
b,
Piyush
Trivedi
*b and
Anita
DuttKonar
*ab
aDepartment of Chemistry, Rajiv Gandhi Technological University, Bhopal, Airport Bypass Road, Gandhinagar, Bhopal-462033, India. E-mail: anitaduttkonar@rgtu.net
bSchool of Pharmaceutical Sciences, Rajiv Gandhi Technological University, Bhopal, Airport Bypass Road, Gandhinagar, Bhopal-462033, India. E-mail: piyush.trivedi@rgtu.net
First published on 16th November 2016
Abstract
Low molecular weight hydrogelators (LMOHGs) have attracted recent attention due to their diversified applications. In an attempt to artificially imitate their importance in the design of drug delivery carriers, we have synthesized two simple N-terminally protected aromatic amino-acid derivatives that form efficient stable hydrogels at room temperature. The gelation properties of the hydrogels have been thoroughly investigated using various techniques and their strength has been determined by rheological studies. In order to explore the efficacy of the hydrogels as tools for drug delivery, we have developed hydrogel nanoparticles (HNPs) using a surfactant and high-speed homogenization approach. Interestingly, our hydrogel nanoparticles display good entrapment efficiency and release kinetics of the model drug 5-fluoro uracil from the hydrogel matrix. Our experimental results reveal that hydrogel II displays slightly higher efficiency as a drug delivery carrier, which may be due to the presence of an aromatic ring in the backbone in comparison to hydrogel I. This increased strength may be attributed to the increase in π–π interactions when the aromatic residue is present in the backbone. Therefore the nanoparticles generated from hydrogel II may have better hydrogen bonding abilities with drugs in comparison to hydrogel I; thus resulting in a slightly slower release of drug from the hydrogel matrix. This fact may shed some light on the candidature of our hydrogels as future carriers for drug delivery. However, further studies to evaluate the candidature of these novel types of aromatic amino acid hydrogel nanoparticles for nano-medical applications are under investigation.
Introduction
The principle of self-assembly involves the construction of complex architectures from biological building blocks which exhibit diversified applications in drug delivery, tissue engineering, electronic devices etc.1–4 The building blocks that have been extensively known for the generation of novel structures are nucleic acids, phospholipids and large peptidyl building blocks.5–8 Like large polypeptides, short amino acid derivatives and peptides can also self-assemble into various nanostructures as well as nanoscale ordered hydrogels.9–18Low molecular weight hydrogelators (LMOHGs) composed of amino acids and short peptides represent a promising approach to drug delivery pathways.19 This is because of the fact that these molecules are of biological origin and possess non-toxic behavior. They can form specific secondary, tertiary and quaternary structures which provide unique opportunities for the design of nanomaterials that does not exist with traditional organic molecules.20
Peptide based hydrogel nanoparticles (HNPs) have gained momentum in recent years as a promising nanoparticulate drug delivery system.21–24 This is due to a combination of two different characteristics: (a) the hydrophilicity and extremely high water content of the hydrogels and (b) the nanoparticle which is exclusively small in size. The amino acid/peptide hydrogel nanoparticles (HNPs) can be modulated rationally by controlling the hierarchical self-assembly process which includes the non-involvement of any potentially hazardous chemicals such as cross-linkers that may affect their biocompatibility.24–28 Moreover, their synthesis procedure is very simple and in vivo degradability is non-toxic due to the fact that they are composed of simple ecofriendly amino acids. To date, although significant examples have been documented in the literature, the majority of them either involve synthetic and natural polymers or longer peptides/peptide amphiphiles as building blocks for drug delivery systems.29–41 The exploration of hydrogel nanoparticles from short peptides/simple aromatic amino acid derivatives, where the concept of self-assembly has been explicitly exploited, remains in the rudimentary stage.42
As a part of the investigation, in this report our effort lies in modulating the excellent hydrogelating ability of two N-terminally protected aromatic amino acid derivatives, benzoyloxy carbonyl phenylalanine (Z-Phe-OH) (hydrogelator I) and fluorenylmethoxy carbonyl meta amino benzoic acid (Fmoc-m-ABA-OH) (hydrogelator II) (Fig. 1), that form an efficient, stable hydrogel in water, at room temperature with a minimum gelator concentration of 0.02% and 0.05% w/v, respectively. The gelation properties of the hydrogel have been investigated by using scanning electron microscopy, FT-IR, and rheological studies. In order to explore the efficacy of the hydrogels as tools for drug delivery, we have developed hydrogel nanoparticles (HNPs) using a surfactant and high-speed homogenization approach. Our hydrogel nanoparticles display good entrapment efficiency and release kinetics of the model drug 5-fluoro uracil from the hydrogel matrix. This fact may shed some light on the candidature of our hydrogels as future carriers for drug delivery.
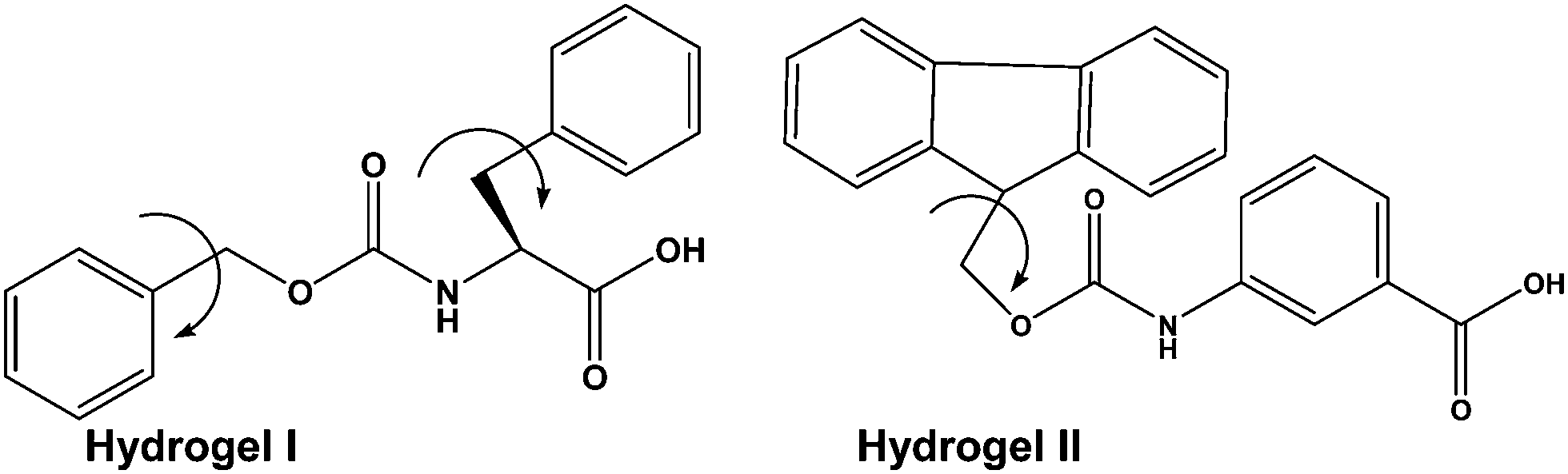 | ||
| Fig. 1 Chemical structures of hydrogels I and II. | ||
Experimental
Materials and methods
Benzoyloxy carbonyl (Z) and fluorenylmethoxy carbonyl (Fmoc) chlorides, 5-fluoro-uracil (5-FU) and all other chemicals were purchased from Spectrochem chemicals. The surfactant vitamin E-TPGS was purchased from Sigma-Aldrich Chemicals Company Pvt. Ltd. All solvents used in the synthesis were purified, dried, or distilled, as required. 1H NMR spectra were recorded using a Bruker Ultra shield (400 MHz) spectrometer. Mass spectra were recorded in the ESI-MS mode on a MicroTOF-Q-II instrument manufactured by Bruker Daltonics; IR spectra were recorded using a Shimadzu, Prestige 21 FT-IR spectrometer.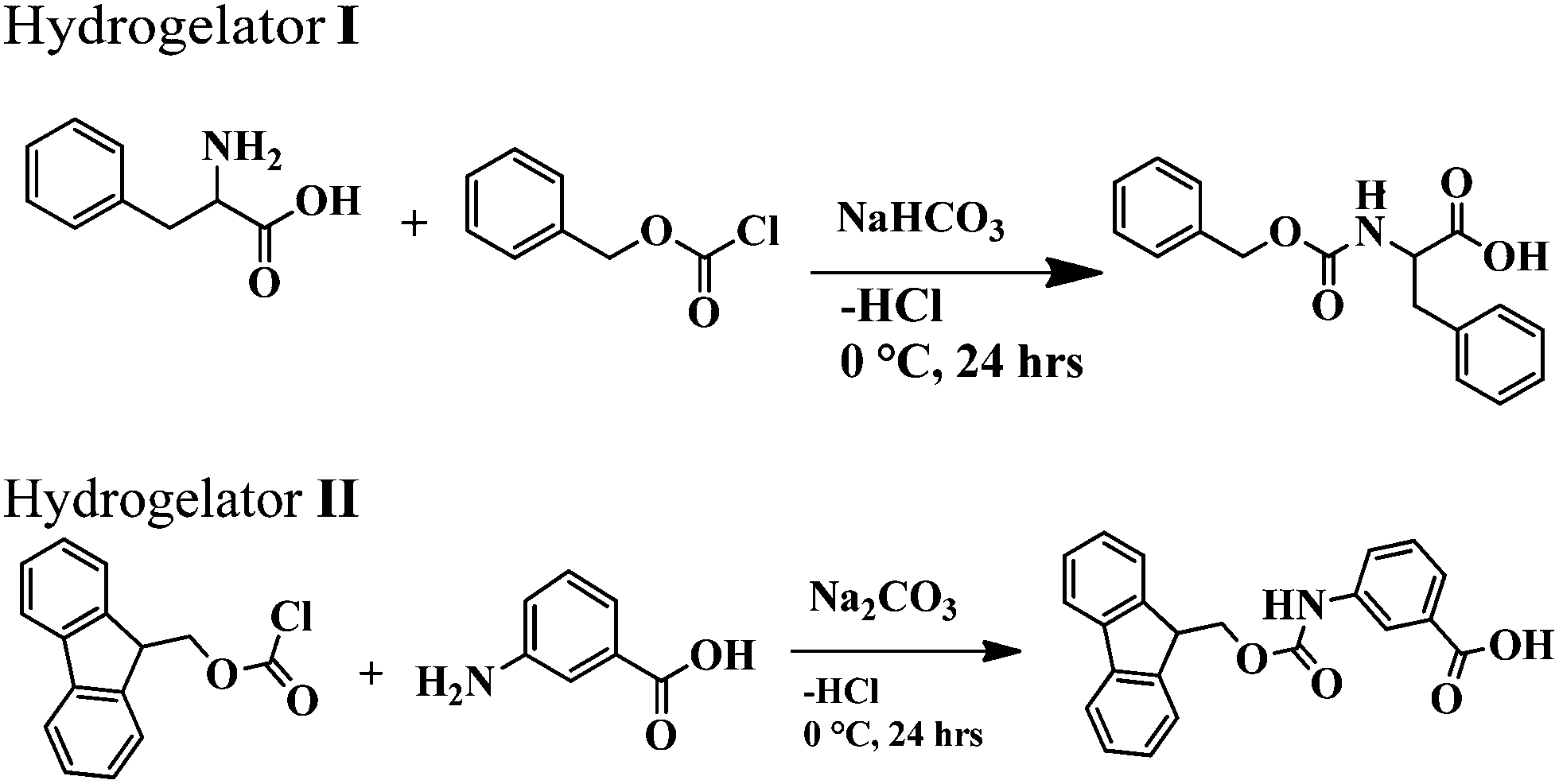
Synthesis of amino acid derivatives I and II
The amino acid derivatives I and II were synthesized using a conventional solution phase methodology, with racemization free techniques. All the intermediates obtained were checked for purity using thin layer chromatography (TLC) on silica gel. The final derivatives were purified using column chromatography using silica gel (100–200 mesh) as the stationary phase and an ethyl acetate and petroleum ether mixture as the eluent. The reported derivatives I and II were fully characterised using NMR, IR spectroscopy and mass spectrometry.Yield: 3 g (83%); m.p. 343–344 °C. FTIR (KBr pellet, cm−1): 1538, 1643, 1696, 3329. 1H-NMR (CDCl3, ppm): 2.8 (1H, d, CβH of Phe), 3.08 (1H, d, CβH of Phe), 4.3 (2H, m, CβHs of benzyloxycarbonyl), 5.0 (1H, m, CαH of Phe), 7–7.4 (10H, m, aromatic H's of Phe and benzoyloxycarbonyl), 7.6 (1H, d, NH of Phe), 12.8 (1H, br, COOH of Phe).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 HCl and extracted with ethylacetate as the organic phase. The organic layer was evaporated to get a solid brownish product of Fmoc-mABA-OH.
:3 HCl and extracted with ethylacetate as the organic phase. The organic layer was evaporated to get a solid brownish product of Fmoc-mABA-OH.
Yield: 4.19 g (80%); m.p. 220–225 °C. FTIR (KBr pellet, cm−1): 1536, 1694, 3306. 1H NMR (CDCl3, ppm): 4.28–4.30 (1H, m, CH of fluoren), 4.48 (1H, d, J = 6.4 Hz, CH2 of fluoren), 7.30–7.41 (8H, m, aromatic protons of fluoren), 7.41 (2H, br, mABAHb and Hd), 7.56–7.66 (3H, m, mABAHb, Hd&Hc), 7.72–7.88 (8H, m, aromatic protons of fluoren), 8.13 (1H, s, mABA Ha), 9.89 (NH of mABA), 12.91 (COOH of mABA).
Gel-melting temperature (Tm)
The melting temperature of the resultant gel in water was determined using an ‘inverse flow method’. Samples were gelled in 10 mm diameter glass vials and attached inverted to a thermometer near the bulb end. This assembly was immersed in a stirred water bath, the temperature of which was raised at a rate of 5 °C min−1 using a hot plate. The temperature at which the solid mass falls from the top was recorded as Tm. This procedure was repeated on three independent samples, and the average values are reported.Field emission scanning electron microscopy (FESEM) study
The morphology of the xerogels obtained from amino acid derivatives I and II was investigated using a FESEM microscope (JEOL JSM – 6700F) and were gold coated.Synthesis of HNPs
The amino acid derivative based hydrogel nanoparticles (HNPs) were prepared using a self-assembly and modified inverse emulsion technique in which vitamin E-TPGS was used as an emulsion stabilizer. In short, HNPs were formulated by the dilution of stock solution (100 mg mL−1) in pure water to a final peptide concentration of 10 mg mL−1. The resulting solution was added drop-wise into 50 mL of slightly warmed mineral oil containing vitamin E-TPGS at a concentration of 0.4% wt/v and homogenized using a high-speed homogenizer. Next, nanoparticles were allowed to self-assemble for 2 h with continuous stirring at 4 °C to allow surfactant monolayer accumulation on the surface of the nanoparticle. Upon completion of the self-assembly process, the resulting suspension was mixed with a non-polar solvent, and centrifuged to obtain phase separation. The supernatant was removed and HNPs were washed again to remove the remaining residues of mineral oil and were finally vacuum dried. The obtained HNPs were used immediately, or stored at 4 °C for later use.Nanoparticle characterization
Nanoparticle size diameter, polydispersity, and surface charge were measured using a Malvern Zetasizer, with a 4 mW 633 He–Ne Laser (DTS version 4.10, Malvern, UK), with appropriate viscosity and refractive index settings. The temperature was maintained at 25 °C during the measurement.Transmission electron microscopy (TEM)
A total of 500 μL of HNPs was prepared and suspended in PBS and placed on a 400-mesh copper grid. After 2 min, the excess fluid was removed. Negative staining was obtained by covering the grid with 10 μL of 2% uranyl acetate in water. After 2 min, excess uranyl acetate solution was removed. Samples were viewed using a FEI-TECNAI G2 (Netherlands) TEM operating at an accelerating voltage of 200 kV. Images were acquired digitally using a Gatan CCD camera.Entrapment efficiency of the hydrogel HNPs
The entrapment efficiency of the hydrogel nanoparticles was determined by the separation of nanoparticles from the aqueous medium containing non-associated drug via centrifugation at 15000 rpm at 4 °C for 40 min. The amount of free drug was measured using a UV spectrophotometer at 267 nm. The entrapment efficiency of the nanoparticles was calculated as per the equation given below: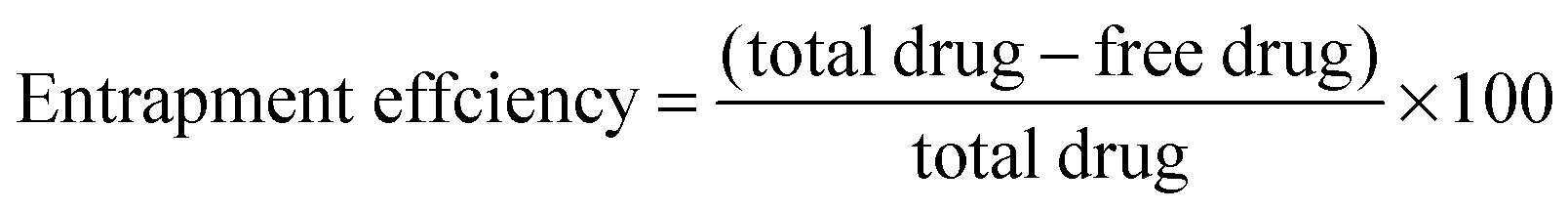
In vitro drug release of 5-fluorouracil from the HNPs
The in vitro release profile of the model drug 5FU from the drug loaded HNPs was determined using a dialysis membrane previously soaked for 24 h in the dissolution membrane and stretched around at one end of the tube. The drug-loaded formulations were carried out using pretreated membranes which were immersed into 30 mL of phosphate buffer solution at pH 7.4 at room temperature and were magnetically stirred at 50 rpm. At selected time intervals aliquots were withdrawn from the release medium and replaced with the same amount of phosphate buffer (1 mL). The samples were analyzed thrice using a UV-spectrophotometer at 267 nm. The percentage of cumulative drug release was plotted against time to obtain the release curves.Rheology
Rheological measurements were carried out on a Rheoplus MCR302 (Anton paar) rotational rheometer with parallel plate geometry and the obtained data were processed using start rheometer software. For the oscillatory shear measurements, parallel top plates with a 25 mm diameter and 1.0 mm gap distance were used. Gels (6 mg mL−1) for rheological experiments were prepared on the bottom plate of the rheometer. The shear modulus (storage modulus G′ and loss modulus G′′) was measured against % strain from 0.001% to 1%. A frequency sweep experiment was performed from 0.11 to 100 rad s−1 at a constant strain of 1%.Results and discussion
Preparation and characterization of the hydrogels
From the literature documentation, it is evident that π–π interactions play a vital role in gelation. Therefore the presence of aromatic groups in the molecule is an obvious choice. So our first design consists of Z-F-OH (hydrogel I) (Fig. 1). This molecule consists of a single aromatic unit at the N-terminus and another aromatic unit in the sidechain. In addition, the insertion of two methylene units in between the two aromatic groups provides two rotational degrees of freedom to the aromatic units which was expected to accelerate the gelation process.43 In order to decipher whether the position and number of aromatic groups present in the conjugate plays any role in the gelation process, we have designed Fmoc-mABA-OH (hydrogel II) (Fig. 1). In this conjugate the aromatic group in the sidechain has been shifted to the backbone (Phe to mABA-OH) and the Z-group (1 aromatic unit) in the N-terminus has been replaced by a Fmoc group (2 aromatic units).In this study, the aromatic amino acid derivatives I and II undergo hydrogelation with a minimum gelation concentration of 0.02% w/v and 0.05% w/v, respectively, without any sonication at room temperature. At first the hydrogelators were dissolved in a drop of DMSO under mild heating conditions and allowed to cool. To this, the required amount of water was added to make the volume up to 1 mL. It was allowed to stand at room temperature for 8–10 min to obtain an opaque gel. The formation of the gel was confirmed using an inverted test tube method (Fig. S1, ESI†).
The gels are stable over a range of pHs, from pH 5.5 to 7 (water), and also over a period of six months. Sol–gel transition temperatures (Tgel) for both the hydrogelators I and II (% w/v) were plotted against different concentrations of hydrogelators (Fig. 2). These plots show that the Tgel values of the aromatic amino acid derivatives increase with an increase in the concentration (% w/v) of the hydrogelator until the plateau region is reached. This plateau region indicates that the formation of the hydrogel network has reached the saturation limit. No further addition of any molecule is capable of increasing the gel melting temperature (Tgel value).44
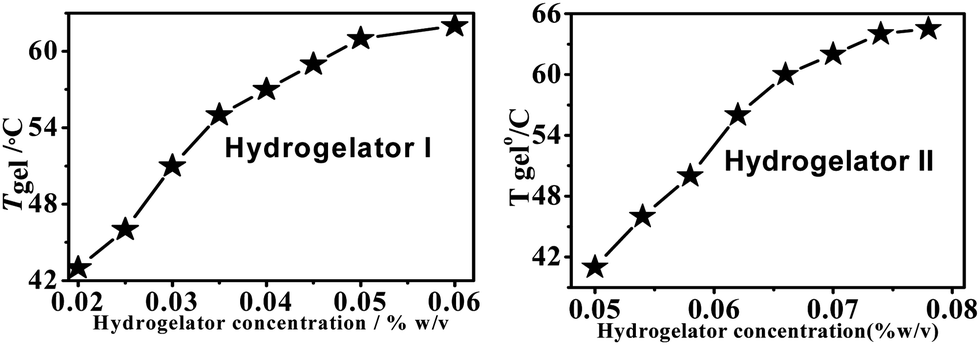 | ||
| Fig. 2 The change in the Tgel profile of hydrogelators I (left) and II (right) with respect to concentration. | ||
To understand the mechanism governing the self-assembly propensities of the aromatic amino acid derivatives I and II, FTIR measurements were carried out (Fig. S2, ESI†). The FTIR spectra of the xerogels obtained from the amino acid derivatives show a major band at 3300–3340 cm−1, a characteristic feature of hydrogen bonded NH stretching. No band was observed around 3400 cm−1 for either of the derivatives, which indicates the absence of any free NH group. Both xerogels of the derivatives I and II further exhibited peaks in the region 1696, 1694 cm−1 (amide I) and 1538, 1536 cm−1 (amide II), respectively, characteristic features of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching and NH bending frequencies, respectively. These data therefore suggest the presence of β-sheet conformations for both conjugates in their corresponding dried gel state.45,46
O stretching and NH bending frequencies, respectively. These data therefore suggest the presence of β-sheet conformations for both conjugates in their corresponding dried gel state.45,46
In order to gain insight into the morphology of the hydrogels, a field emission scanning electron microscopy (FESEM) investigation was carried out. The FESEM images (Fig. 3) of the xerogels of hydrogelators I and II exhibit flat ribbon-like morphology with widths ranging from 600–800 nm and 100–120 nm respectively. The fibers were found to be several micrometers long. Interestingly, hydrogelator II exhibits dense mesh-like aggregates (Fig. 3d at higher magnification) formed due to overlapping of the flat ribbon-like structures in comparison to hydrogelator I. This difference in morphology may be attributed to the lower number of π-surfaces as well as fewer hydrogen bonding elements in hydrogelator I with respect to hydrogelator II.
 | ||
| Fig. 3 FESEM images of the xerogels I and II showing flat ribbon-like morphology of the fibrils ((a and c): lower magnification; (b and d): higher magnification). | ||
Rheology characterisation
In order to address the mechanical strength and stability of hydrogelators I and II, rheological studies were performed where the storage modulus G′ (elastic response) and loss modulus G′′ (viscous response) were measured in frequency sweep experiments.47As is evident from Fig. 4 (left), in the lower viscoelastic region (LVR) the storage modulus G′ is higher than the loss modulus (G′′) for both of the amino acid derivatives. This observation suggests a soft ‘solid-like’ gel phase formation.48 Moreover a crossover point is noticed in the experimental frequency regions, where the value of loss modulus G′′ slightly exceeds the value of the storage modulus G′ and transformed into the solution state (Fig. 4 right). This particular point where the crossover occurs is called yield stress (σy). These gelators showed a G′ (storage modulus) value that was sufficiently larger than the G′′ (loss modulus); this difference remained almost constant up to about 0.5% and 0.05% strain, respectively, for both the amino acid derivatives I and II respectively (Fig. 4 right). As the strain was increased beyond this point, both the storage modulus (G′) and the loss modulus (G′′) started to fall because intermolecular forces that held the gel together started being overcome by the applied strain and the fibrils were unable to withstand large deformations. The frequency-sweep experiments further showed that both the G′ and G′′ values were weakly dependent on the frequency (Fig. 4 left), which was indicative of an entangled network-like system.
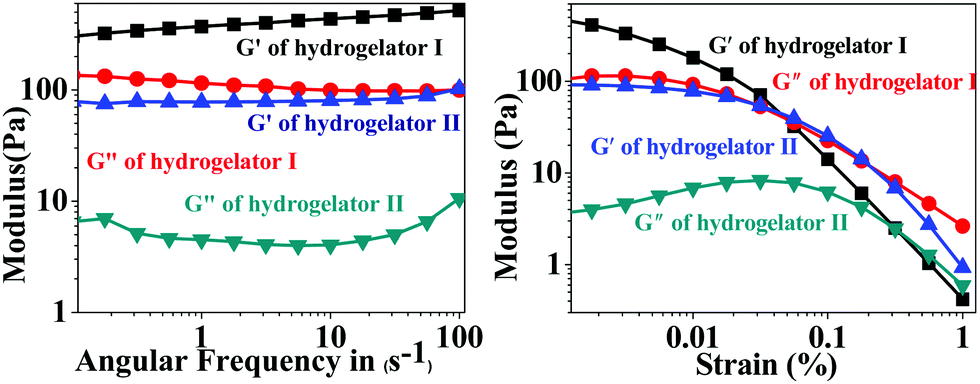 | ||
| Fig. 4 Rheological study of the hydrogels I and II showing the frequency sweep study (in the left) and amplitude sweep study (in the right). | ||
The principle guidelines of gelation involve a balance between enthalpy and entropy, the nature of the weak interactions involved, such as hydrogen bonds and π–π resulting in thermodynamic minimum conditions. In hydrogelator I, two methylene units are being inserted in between the aromatic moieties, in comparison to hydrogelator II (that contains one methylene unit). Insertion of methylene units between aromatic groups are already known to accelerate the gelation process.43 Although more π–π interactions are observed in hydrogelator I, due to the presence of an aromatic residue in the backbone, we observe lower melting points of hydrogelator II than hydrogelator I, at the same concentration. This may be attributed to the more entropic contribution from the two methylene units of hydrogelator I, which might overrule the extra stability gained due to π–π interaction, due to the presence of an aromatic moiety in the backbone, resulting in thermodynamic minimum conditions. Interestingly, the ratio between the storage modulus and loss modulus of both the aromatic amino acid derivatives I and II is approximately one order of magnitude higher which indicates significant mechanical stability of the hydrogels. These data prompted us to explore the candidature of our hydrogels in drug delivery applications. A similar observation where gels of similar strength have been utilized for various applications has been reported by other groups.49–53
Preparation and characterization of HNPs
Literature documentation reveals that hydrogel nanoparticles (HNPs) provide a promising alternative approach towards drug delivery applications.16–18,34 But most of the report demonstrates the use of extremely complex techniques.Our aromatic amino acid derivative I and II based hydrogels are prepared using the concept of self-assembly involving weak interactions. Henceforth they are ecofriendly in nature. Therefore we became interested in exploring whether our hydrogels could be utilized for the synthesis of hydrogel nanoparticles. As illustrated in Fig. 5 a modified inverse emulsion technique (water-in-oil) was used for HNP formulation. In order to synthesize the hydrogel nanoparticles, the peptide was first dissolved in its monomeric state in an organic solvent (DMSO), followed by dilution in water. This solution is considered to be the aqueous phase. This aqueous phase was drop-wise added into the organic phase containing the surfactant vitamin E-TPGS dissolved in light paraffin oil. Vitamin E-TPGS was chosen as a surfactant due to its enormous advantage.54,55 Moreover, the hydroxylic groups in vitamin E-TPGS may provide sites for the attachment of targeting units.
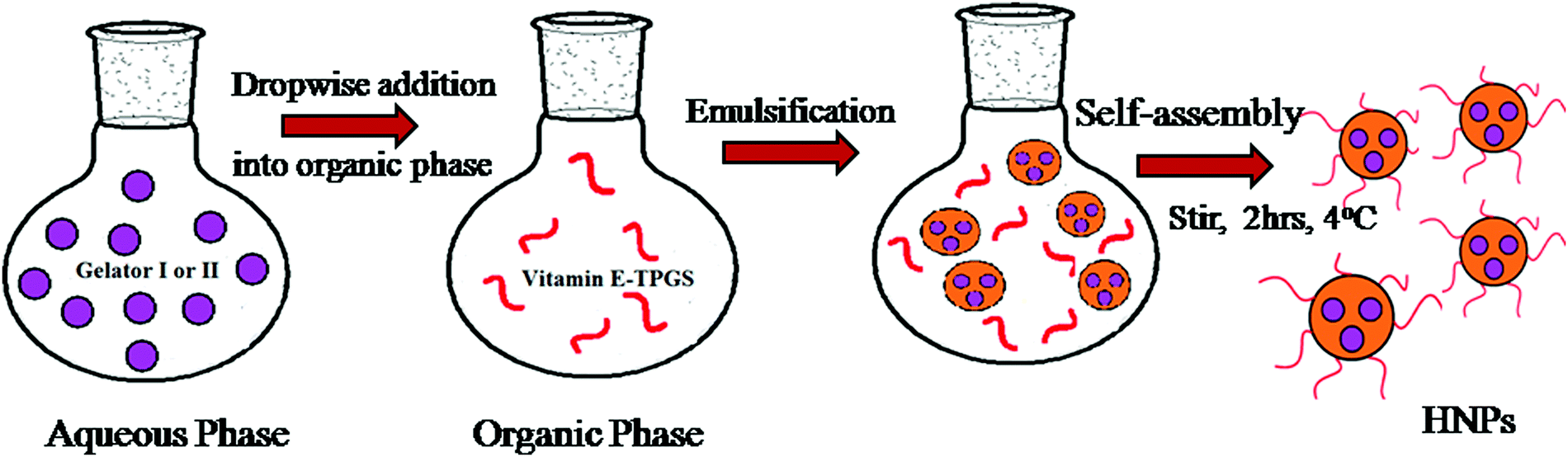 | ||
| Fig. 5 Schematic representation of hydrogel nanoparticle formulation (HNP) preparation using a modified inverse emulsion method. | ||
The heterogeneous mixture was then subjected to homogenization using a high-speed homogenizer operating at 25000 rpm for ten minutes. During this process the hydrogelator I and II mixture was dispersed well and allowed to self-assemble into particle aggregates.56
The mixture was then stirred at 4 °C for 2 h to allow the attachment of the surfactant along the surface of the hydrophilic core.
The self-assembled aromatic conjugate occupies the hydrophilic core of the HNPs, and a vitamin E-TPGS monolayer occupies the outer hydrophobic periphery as in micelles (Fig. 6). Finally, the HNPs were purified via centrifugation and a series of washings using a non-polar solvent were undertaken. The particle dimensions were then recorded using a zetasizer. Regarding pharmaceutical applications, the physical dimensions of the nanoparticles (like particle size, polydispersity index, zeta potential) influence biodistribution and in vivo efficacy. We were able to control the size of the HNPs by standardizing the parameters of the emulsion process, such as the stirring speed, stirring time, concentration of gelators and concentration of the surfactant. Our optimized formulation results reflect that the hydrogelators I and II exhibit an average particle size of 254 nm and 482 nm respectively. Furthermore, we analyzed the HNPs using transmission electron microscopy (TEM) analysis. The TEM images obtained demonstrate the presence of well-discrete spherical structure nanoparticles of various sizes (Fig. 7 and 8). The zeta potential indicates the degree of repulsion between adjacent, similarly charged particles in dispersion. For molecules and particles that are small enough, a high zeta potential will confer stability, i.e. the solution or dispersion will resist aggregation. When the potential is low, attraction exceeds repulsion and the dispersion will break and flocculate. The zeta potential value of the hydrogel nanoparticles for both the hydrogelators was found to be −25.5 mV for the optimized formulation, which indicates significant stability of the nanoparticles. The pKa value of 5-FU is 8.1. Under experimental conditions, the pH of the medium remains neutral (since water has been used for gelation) or slightly acidic (which may occur due to ionization of the organic acids that has been used as hydrogelators). Henceforth the drug is expected to remain un-ionized under these conditions as the pH of the medium is much lower than the pKa value. The negative charge of the zeta potential could be attributed to the presence of carboxyl groups in the amino acid derivatives as well as the emulsion stabilizer, the vitamin E-TPGS coating around the hydrophobic core.57
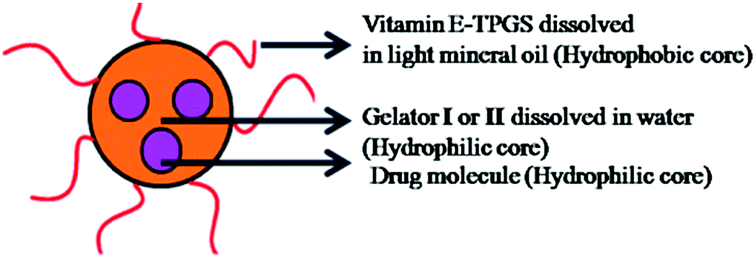 | ||
| Fig. 6 Cartoon diagram of drug encapsulated hydrogel nanoparticles formed after self-assembly. | ||
 | ||
| Fig. 7 TEM images of the hydrogel nanoparticles (HNPs) from optimized formulation: (a) hydrogelator I and (b) hydrogelator II. | ||
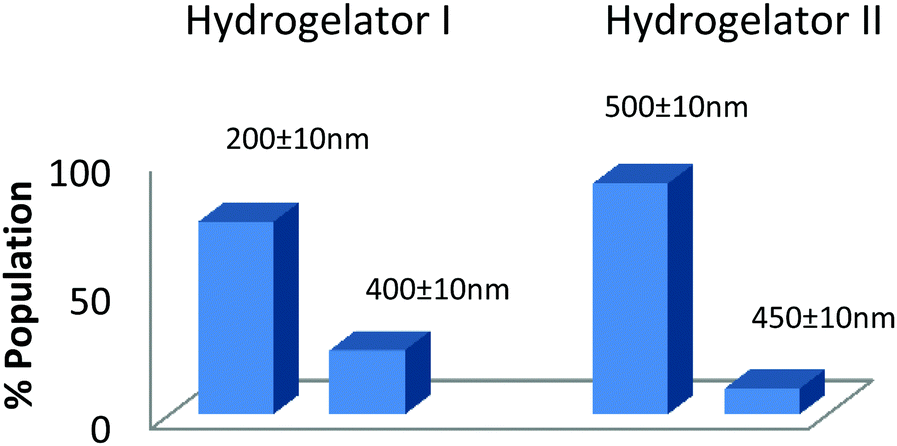 | ||
| Fig. 8 Particle size distribution of the hydrogel nanoparticles (HNPs) obtained from the optimized formulation. | ||
Generally, stresses caused by lyophilization may cause fusion of the HNPs.58 Therefore, before subjecting to lyophilization, cryoprotectants are added to protect the fragile structure of the nanoparticles. In our case we have used vitamin E-TPGS, which not only serves as a surfactant but also as a cryoprotectant.59 After lyophilisation we determined the particle size distribution after three months (Fig. 9a and b). The results show that the particles have similar diameters and morphology even after three months, thus emphasizing the stability of the formulation. These results further indicate that after formulation, the particles could be readily turned into the dry form, and re-suspended prior to use.
 | ||
| Fig. 9 TEM images of the stored hydrogel nanoparticles (HNPs) after three months: (a) hydrogelator I and (b) hydrogelator II. | ||
Drug encapsulation and in vitro release studies
As a part of the investigation, thereafter our idea was to explore the efficacy of our nanoparticles for the encapsulation and release studies of a model drug 5-fluorouracil (5-Fu) from the hydrogel matrix. Therefore, the drug loading efficiency and release kinetics were determined using a dialysis membrane in HBS at 37 °C under gentle stirring, to mimic physiological conditions. The encapsulation of the model drug 5-flourouracil (5-Fu) from the HNP matrix showed an entrapment efficiency of 77.57% and 84.52% for hydrogelators I and II respectively. The concentration of the released drug was measured in solution over time, and quantified using UV spectroscopy. The resulting release profiles of 5-Fu from both the derivatives are presented in Fig. 10. It has been noticed that the release profile of 5-Fu loaded HNPs slightly differ for both the derivatives. The release of 50% of 5-Fu from the HNPs was within 3 h/4 h for hydrogelator I and II, respectively, and after 7 h/9 h the release kinetics reached a plateau. This difference in release kinetics between 5-Fu for both the derivatives might be due to their dissimilar chemical structures. We propose that aromatic interactions and hydrogen bond formation between the amino acid derivative and drug may play a crucial role in its efficient encapsulation. The slightly higher efficiency of hydrogelator II as a carrier may be attributed to the presence of an aromatic ring in the backbone which may increase the extent of hydrogen bonding and π–π interactions to a greater extent with the drug molecule (in comparison to hydrogel I), thereby resulting in slightly slower release of the drug from the hydrogel matrix.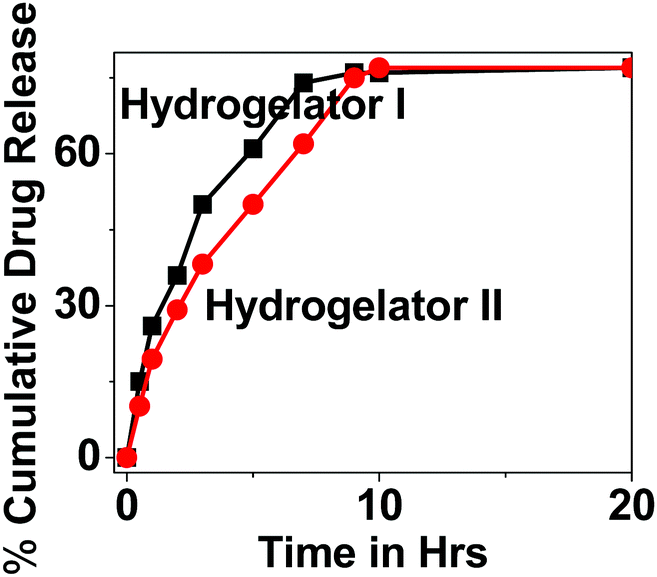 | ||
| Fig. 10 Release profiles of the model drug from the hydrogel matrix. | ||
Conclusions
In summary, we have demonstrated the formation of two simple amino acid derivatives that display excellent hydrogelating abilities under mild conditions. The gelation properties of the hydrogel were investigated using various microscopic techniques and its strength was determined by rheological measurements. Interestingly, these amino acid conjugate based hydrogels have been nicely tuned to prepare and stabilize hydrogel nanoparticles (HNPs) utilizing an inverse emulsion procedure. The method proposed here is environmentally benign, as simple amino acid conjugates have been utilized. The approach adopted is the concept of self-assembly utilizing weak interactions, unlike other polymers that require harsh conditions and complex techniques for their fabrication. To date, significant examples have been documented in the literature for synthetic and natural polymers and longer peptides/peptide amphiphiles where they have been used as building blocks for the drug delivery systems. However, to the best of our knowledge, these derivatives represent one of the very few reports of HNP formation resulting from the self-assembly of simple amino acid derivatives, solely driven by an environmentally benign approach i.e. weak interactions, unlike other polymers that require drastic conditions and complex techniques for their fabrication. The obtained HNPs were characterized and evaluated for their size, zeta-potential and post-formulation stability. Our gelators display good entrapment efficiency and in vitro release kinetics of the model drug 5-FU. The obtained results clearly indicate that these amino acid derivative based HNPs may be suitable for use as a potential drug delivery system. However, further studies to evaluate the candidature of this novel type of amino acid conjugate based HNPs for nano-medical applications are under investigation.Acknowledgements
ADK wishes to thank UGC (F.4-(55)/2014(BSR)/FRP) and DST, New Delhi, India (Project no. SR/FT/CS-118/2013), for financial support. All the authors sincerely acknowledge Dr Sanjit Konar, IISER Bhopal, and Dr C. Karthikeyan, RGPV, for their valuable suggestions. ADK is very grateful to Prof. N. K. Jain and Dr Anindya Basu, RGPV, Bhopal, for their very valuable input during manuscript preparation. The authors would like to thank Mr Digambar Waiker and Mrs Vandana Gupta (SOPS, RGPV) and Mr Amit Kumar Mondal (IISER Bhopal) for various help during manuscript preparation. We thank Mr Manoj Sharma and Mr Maharsi Gajjar, Antonn Parr New Delhi for rheological measurements and the TEM facilities at AIIMS New Delhi for recording TEM images.Notes and references
- S. Zhang, Nat. Biotechnol., 2003, 21, 1171 CrossRef CAS PubMed.
- M. Reches and E. Gazit, Curr. Nanosci., 2006, 2, 105 CrossRef CAS.
- C. Mao, D. J. Solis, B. D. Reiss, S. T. Kottmann, R. Y. Sweeney, A. Hayhurst, G. Georgiou, B. Iverson and A. M. Belcher, Science, 2004, 303, 213 CrossRef CAS PubMed.
- A. B. Sanghvi, K. P. Miller, A. M. Belcher and C. E. Schmidt, Nat. Mater., 2005, 4, 496 CrossRef CAS PubMed.
- M. R. Ghadiri, J. R. Granja, R. A. Milligan, D. E. Mcree and N. Khazanovich, Nature, 1993, 366, 324 CrossRef CAS PubMed.
- Y. V. Zastavker, N. Asherie, A. Lomakin, J. Pande, J. M. Donovan, J. M. Schnur and G. B. Benedek, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 7883 CrossRef CAS.
- S. Vauthey, S. Santoso, H. Y. Gong, N. Watson and S. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5355 CrossRef CAS.
- M. Hamidi, A. Azadi and P. Raife, Adv. Drug Delivery Rev., 2008, 60, 1638–1649 CrossRef CAS PubMed.
- A. Dutt, M. G. B. Drew and A. Pramanik, Org. Biomol. Chem., 2005, 2250 CAS.
- P. Koley and A. Pramanik, Soft Matter, 2012, 8, 5364 RSC.
- P. Koley and A. Pramanik, Adv. Funct. Mater., 2011, 21, 4126 CrossRef CAS.
- A. Sharma Gangele, S. Goswami, R. Rajagopalan and A. DuttKonar, Supramol. Chem., 2015, 27(10), 669 CrossRef.
- A. Sharma Gangele, S. Goswami, A. Bar, P. Tiwari, S. Konar and A. DuttKonar, Cryst. Growth Des., 2016, 16, 2130 CAS.
- A. DuttKonar, CrystEngComm, 2013, 15, 10569 RSC.
- A. DuttKonar, CrystEngComm, 2012, 14, 6689 RSC.
- A. DuttKonar, CrystEngComm, 2013, 15, 2466 RSC.
- A. Mahler, M. Reches, M. Rechter, S. Cohen and E. Gazit, Adv. Mater., 2006, 18, 1365 CrossRef CAS.
- R. Orbach, L. A. Abramovich, S. Zigerson, I. M. Harpaz, D. Seliktar and E. Gazit, Biomacromolecules, 2009, 10, 2646 CrossRef CAS PubMed.
- A. M. Jonker, D. W. P. M. Löwik and J. C. M. van Hest, Chem. Mater., 2012, 24, 759–773 CrossRef CAS.
- M. Zelzer and R. V. Ulijn, Chem. Soc. Rev., 2010, 39, 3351–3357 RSC.
- M. Hamidi, A. Azadi and P. Raife, Adv. Drug Delivery Rev., 2008, 60, 1638–1649 CrossRef CAS PubMed.
- Z. Yu, Q. Xu, C. Dong, S. S. Lee, L. Gao, Y. Li, M. D. Ortenzio and J. Wu, Curr. Pharm. Des., 2015, 21, 4342–4354 CrossRef CAS PubMed.
- T. R. Hoare and D. S. Kohane, Polymer, 2008, 5, 1993–2007 CrossRef.
- A. Dasgupta, J. Hassan Mondal and D. Das, RSC Adv., 2013, 3, 9117–9149 RSC.
- X. Gong, Nano LIFE, 2015, 5, 1542002 CrossRef CAS.
- D. N. Woolfson and M. G. Raydnov, Curr. Opin. Chem. Biol., 2006, 10, 559–567 CrossRef CAS PubMed.
- L. Zhang, J. Zhong, L. Huang, L. Wang, Y. Hong and Y. Sha, J. Phys. Chem., 2008, 112, 805–816 Search PubMed.
- M. Ma, L. Zhong and W. Li, Soft Matter, 2013, 9, 11325–11333 RSC.
- M. Suzuki and K. Hanabusa, Chem. Soc. Rev., 2010, 39, 455–466 RSC.
- M. Suzuki, C. Setoguchi, H. Shirai and K. Hanabusa, Chem. – Eur. J., 2007, 13, 8193–8200 CrossRef CAS PubMed.
- Y. Osada and J. P. Gong, Adv. Mater., 1998, 10, 827–837 CrossRef CAS.
- S. Nayak and L. A. Lyon, Angew. Chem., Int. Ed., 2005, 44, 7686–7708 CrossRef CAS PubMed.
- J. M. Chan, L. Zhang, K. P. Yuet, G. Lio, J. W. Rhee, R. Langer and O. C. Farokhzad, Biomaterials, 2009, 30, 1627–1634 CrossRef CAS PubMed.
- Y. Nagai, L. D. Unsworth, S. Koutsopoulos and S. Zhang, J. Controlled Release, 2006, 115, 18–25 CrossRef CAS PubMed.
- R. V. Rughani and J. P. Schneider, MRS Bull., 2011, 33, 530–535 CrossRef.
- C. Yan, M. E. Mackay, K. Czymmek, R. P. Nagarkar, J. P. Schneider and D. J. Pochan, Langmuir, 2012, 28, 6076–6087 CrossRef CAS PubMed.
- C. Yan, A. Altunbas, T. Yucel, R. P. Nagarkar, J. P. Schneider and D. J. Pochan, Soft Matter, 2010, 6, 5143–5156 RSC.
- S. Zarhitsky and H. Rapaport, J. Colloid Interface Sci., 2011, 360, 525–531 CrossRef PubMed.
- L. Mao, H. Wang, M. Tan, L. Ou, D. Kong and Z. Yang, Chem. Commun., 2012, 48, 395–397 RSC.
- Y. Zhao, T. Tan, H. Yokoi, M. Tanaka and T. Kinoshita, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 4927–4933 CrossRef CAS.
- C. Y. Yang, B. Song, Y. Ao, A. P. Nowak, R. B. Abelowitz, R. A. Korsak, L. A. Havton, T. J. Deming and M. V. Sofroniew, Biomaterials, 2009, 30, 2881–2898 CrossRef CAS PubMed.
- R. Ischakov, L. A. Abramovich, L. Buzhansky, T. Shekhter and E. Gazit, Bioorg. Med. Chem. Lett., 2013, 21, 3517–3522 CrossRef CAS PubMed.
- A. Reddy, A. Sharma and A. Srivastava, Chem. – Eur. J., 2012, 18, 7575–7581 CrossRef CAS PubMed.
- C. S. Love, A. R. Hirst, V. Chechik, D. K. Smith, I. Ashworth and C. Brennan, Langmuir, 2004, 20, 6580–6585 CrossRef CAS PubMed.
- M. Mutter, F. Maser, K. H. Altmann, C. Toniolo and G. M. Bonora, Biopolymers, 1985, 24, 1057–1074 CrossRef CAS PubMed.
- V. Moretto, M. Crisma, G. M. Bonora, C. Toniolo, H. Balaram and P. Balaram, Macromolecules, 1989, 22, 2939–2944 CrossRef CAS.
- A. Reddy, A. Sharma, Q. Maqbool and A. Srivastava, RSC Adv., 2013, 3, 18900–18907 RSC.
- A. K. Das, R. Collins and R. V. Ulijn, Small, 2008, 4, 279–287 CrossRef CAS PubMed.
- R. Avalos-Diaz, C. Long, E. Fontano, M. Balbirnie, R. Grothe, D. Eisenberg and D. L. D. Caspar, Fiber Diffrac. Rev., 2003, 11, 79–86 Search PubMed.
- J. J. Balbach, Y. Ishii, O. N. Antzutkin, R. D. Leapman, N. W. Rizzo, F. Dyda, J. Reed and R. Tycko, Biochemistry, 2000, 39, 13748–13759 CrossRef CAS PubMed.
- B. Adhikari, J. Nanda and A. Banerjee, Soft Matter, 2011, 7, 8913–8922 RSC.
- G. Palui, A. Garai, J. Nanda, A. K. Nandi and A. Banerjee, J. Phys. Chem. B, 2010, 114, 1249–1256 CrossRef CAS PubMed.
- B. Adhikari, J. Nanda and A. Banerjee, Chem. – Eur. J., 2011, 17, 11488–11496 CrossRef CAS.
- L. Mu and S. S. Feng, J. Controlled Release, 2002, 80, 129–144 CrossRef CAS PubMed.
- S. S. Feng, W. T. Zeng, Y. T. Lim, L. Y. Zhao, K. Y. Win, R. Oakley, S. H. Teoh, R. C. H. Lee and S. R. Pan, Nanomedicine, 2007, 2, 333–344 CrossRef CAS PubMed.
- B. Abismaïl, J. P. Canselier, A. M. Wilhelm, H. Delmas and C. Gourdon, Ultrason. Sonochem., 1999, 6, 75–83 CrossRef.
- W. Abdelwahed, G. Degobert, S. Stainmesse and H. Fessi, Adv. Drug Delivery Rev., 2006, 58, 1688–1713 CrossRef CAS PubMed.
- Y. Fu and W. Kao, Expert Opin. Drug Delivery, 2010, 7, 429–444 CrossRef CAS PubMed.
- M. Rehman, A. Madni, A. Ihsan, W. S. Khan, I. Khan, M. Ahmad, M. Mahmood, S. Z. Ashfaq and I. Shakir, Int. J. Nanomed., 2015, 10, 2805–2814 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Figures of gel, 1H NMR, IR, and mass spectra. See DOI: 10.1039/c6nj02125e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2017 |