
An efficient methodology to introduce o-(aminomethyl)phenyl-boronic acids into peptides: alkylation of secondary amines†
Erik T.
Hernandez
,
Igor V.
Kolesnichenko
,
James F.
Reuther
and
Eric V.
Anslyn
*
Department of Chemistry, University of Texas at Austin, Austin, TX 78712-1224, USA. E-mail: anslyn@austin.utexas.edu
First published on 17th November 2016
Abstract
Current approaches for incorporating boronic acids into peptides require one of the following: the synthesis of commercially unavailable pinacol-protected boronate ester amino acid building blocks, amidation of small-molecule amine-containing boronic acids, or reductive amination of amine residues with 2-formylphenyl boronic acid. These methods have drawbacks, such as the use of excess starting materials, the lack of reactive-site specificity, or the inability to add multiple boronic acids in solution. In addition, several of these approaches do not allow for incorporation of the critical o-aminomethyl functionality that allows for binding of saccharides under physiological conditions. In this work, we report three methods to functionalize synthetic peptides with boronic acids using solid-phase and solution-phase chemistries by alkylating a secondary amine with o-(bromomethyl)phenylboronic acid. Solution-phase chemistries afforded the highest yields, and were used to synthesize seven complex biotinylated multi-boronic acid peptides.
Introduction
Boronic acid containing compounds continue to gain interest in the areas of chemosensing, materials chemistry, fluorescence imaging, mass spectrometry, and biomedical engineering.1–24 From the sensing of sugars to the formation of dynamic covalent structures, or serving as probes for the enrichment of analytes, boronic acid compounds are seen as an important organic moiety for developing novel analytical and in vivo applications. Continued growth in the biological sciences has sparked interest for synthesizing boronic acid containing peptides. These are attractive synthetic targets because peptides are versatile platforms for incorporating binding specificity to a target of interest. Boronic acid peptides could also be used in the design of combinatorial libraries. A method for incorporating a boronic acid into the side chains at controllable positions increases the diversity of compounds accessible. We believe these methods should be more modular; thus, providing greater utility when compared to current approaches.25,26Common approaches for synthesizing boronic acid containing peptides include the use of building blocks with a boronate ester side chain, amidation of boronic acid compounds, or reductive amination of residues with 2-formylphenyl boronic acid. The most common approach is incorporating a building block during solid-phase synthesis. However, long synthetic routes to non-commercially available boronic acid reactants are required (Fig. 1a).27–35 To derivatize the C-terminus, amidation of amino boronic acids is usually used (Fig. 1b). Recently, solid-phase approaches for adding boronic acids at the C-terminus has been reported.36 However, peptides with aspartates or glutamates have to be avoided. If unprotected carboxylate side chains are used, non specific amidation can occur. Such a limitation decreases the diversity of peptides available by limiting the natural side chains that can be used. Lastly, reductive amination, performed previously in our group, consisted of modifying only one amine residue (Fig. 1c).30
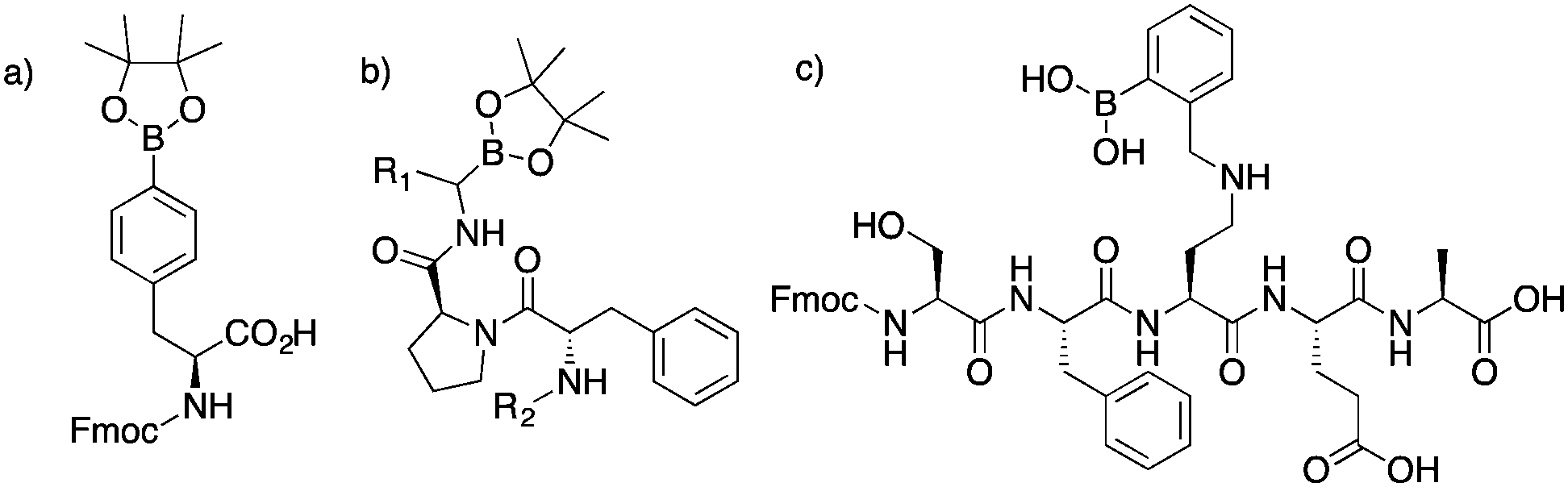 | ||
| Fig. 1 (a) Synthesized building block. Requires a total of five synthetic steps. (b) Boronate ester moieties introduced at the C-terminus via coupling conditions. (c) Polypeptide with ortho-aminomethyl boronic acid, synthesized by reductive amination of 2-formylphenyl boronic acid.27–35 | ||
Most boronic acids do not bind saccharides efficiently at neutral pH. While electron withdrawing groups on the phenyl ring, such as in m-nitrophenylboronic acid, can act to promote boronate ester formation at neutral pH, the most common structural feature that promotes binding is an o-aminomethyl group.37,38 This structural feature is often overlooked when incorporating a boronic acid into a receptor meant to be used in a biological setting. The only previously published strategy that allows for easy incorporation of an o-aminomethyl group is reductive amination. Alternatively, it would be feasible to incorporate the o-aminomethyl moiety into an amino acid building block in advance of peptide synthesis, but this would further increase the number of synthetic steps. The methods we present in this paper were devised to be synthetically simple and afford the critical o-aminomethyl group.
To increase the number of targetable sites on peptides while minimizing the difficulty and time required to synthesize o-(aminomethyl)phenylboronic acid building blocks, we developed chemistries for functionalizing peptides with secondary amines. Primary amines are present in the side chains of natural amino acids such as lysine. However, alkylation of primary amines typically suffers from over-alkylation. If secondary amines residues are present, then alkylation approaches become a viable alternative. The commercial availability of o-(bromomethyl)phenylboronic acid is an additional benefit to an alkylation approach.
Herein, we report three methods that introduce secondary amines and subsequent o-(aminomethyl)phenylboronic acids to peptides. The methods include both solid-phase and solution-phase approaches. The first approach was the selective modification of cysteine residues in solution using an iodoacetamide linker with a Boc-protected secondary amine. In the second approach, a cysteamine resin and an azido lysine building block was used in the solid-phase synthesis of peptides with a secondary amine. The third approach, using the commercially available Nε-methyl lysine, was the most direct way for making our desired peptides. Once a secondary amine was incorporated, a reaction with o-(bromomethyl)phenylboronic acid afforded the o-aminomethyl moiety on all our peptides.
Finally, to demonstrate the usefulness of our chemistry for making complex structures, seven peptides containing Nε-methyl lysine amino acids and derivatized at the N-terminus with a pegylated biotin, were synthesized. This organic moiety was seen to add to the utility of boronic acid peptides for applications that use streptavidin to be reported in future publications.
Results and discussion
Solution-phase incorporation of secondary amines and alkylation
To first test if an alkylation strategy would be successful, N,N′-dimethylethylenediamine (DMEDA) was used as a model (Scheme 1). The alkylation occurred with a purified yield of 16% (1). The boronic acid community, has reported on the difficulty to isolate boronic acid compounds because of the strong interaction with the stationary phase in chromatographic methods.39 Therefore, pinacol-protected boronate esters are more commonly used due to facilitate the isolation of boronic acid compounds. However, the reaction conditions required for deprotection of the boronic ester were considered to be too harsh for peptides, and thus unprotected boronic acids were chosen. Irrespective of the low yield of this model reaction, we pushed forward with peptide derivatization because of the ease of the technique. The desired product was confirmed by mass spectrometry and NMR.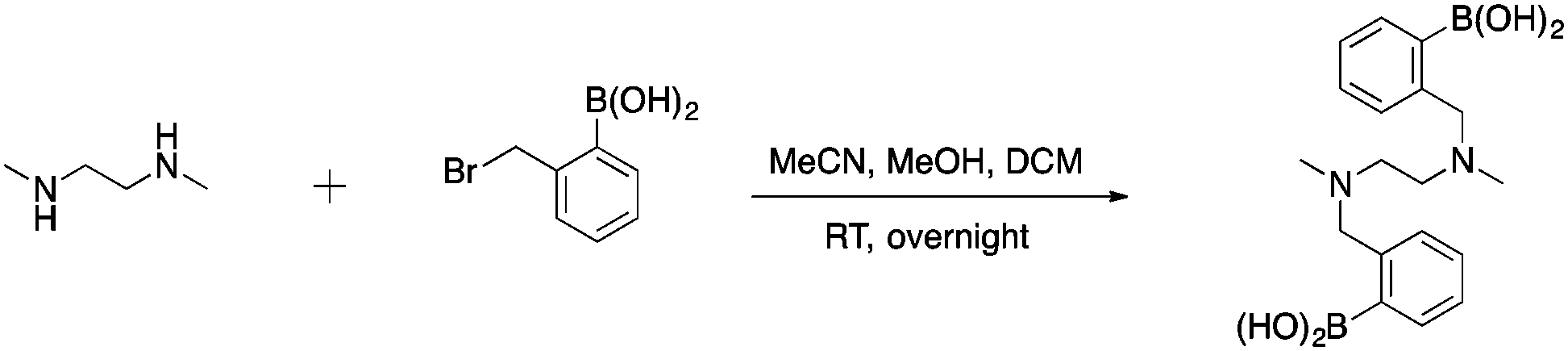 | ||
| Scheme 1 Synthesis of compound 1. | ||
After successful functionalization of DMEDA, as a proof of concept experiment, we set out to devise a linker approach to place secondary amines on peptides. Cysteine residues are reported to be selectively modified with iodoacetamides. Therefore, a linker with this functional tether provided a method for selectively introducing a secondary amine.40 The synthesis of this linker involved a single Boc protection of DMEDA (Scheme 2). Literature accounts regularly describe mono-protection with diamine compounds by using an excess of the amino compound relative to Boc-anhydride.41 A mixture of products was observed: unreacted starting material, singly protected product, and doubly protected product. Aqueous washes removed unreacted starting material. No further purification was performed because the doubly protected product was inert in the following step. Mono-Boc DMEDA was modified with chloroacetyl chloride, and the desired product was isolated. A Finklestein reaction afforded iodoacetamide linker 2. During purification, the doubly Boc product was removed. The cysteine in Fmoc-CysAla-OH was modified with the linker (Scheme 3). Deprotection with TFA solution occurred quantitatively, followed by alkylation with o-(bromomethyl)phenylboronic acid (BMPBA) to give compound 3. Linker 2 should prove widely useful to selectively modify peptides with cysteine residues.31
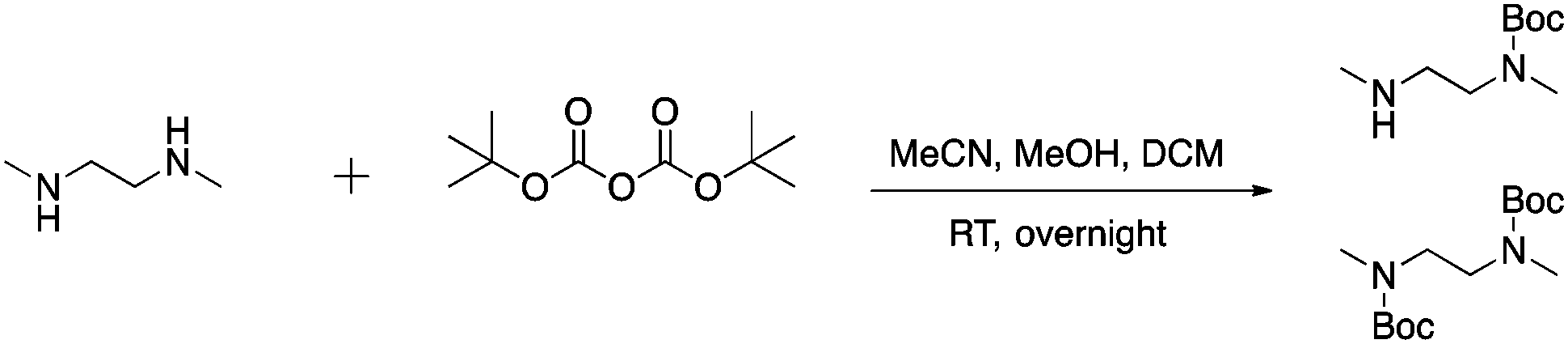 | ||
| Scheme 2 Synthesis of mono-Boc diamine. | ||
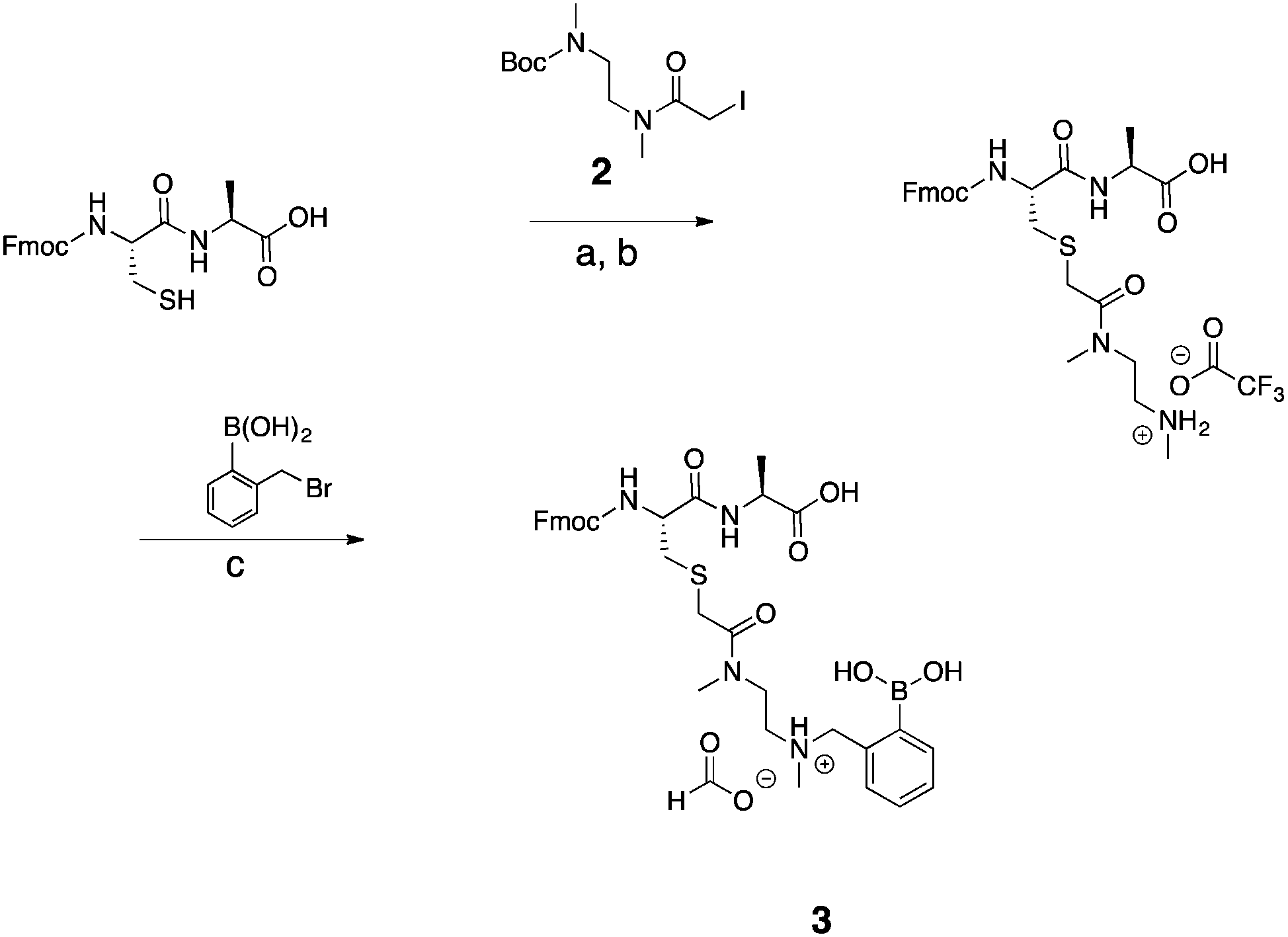 | ||
| Scheme 3 Synthesis of compound 3. (a) MeCN, MeOH, TEA, Pyr, H2O, RT, 4 h. (b) TFA, TIS, H2O, RT, 4 h. (c) MeCN, H2O, RT, overnight. | ||
Solid-phase incorporation of secondary amines and alkylation
To expand the utility of the alkylation approach, we sought to develop solid-phase reactions for the incorporation of secondary amines. Click approaches via Sharpless reactions, generally are not compatible with boronic acids.42 Despite efforts to use fluoride to minimize deborylation,43 copper clicking of boronic acids did not work. A cysteamine resin and an azido lysine building block also allowed incorporation of a secondary amine (Scheme 4). The unnatural azido residue can be modified via click chemistry while immobilized on the solid support.44 We used N-methyl propargylamine (NMPA) to click on a secondary amine. If the Fmoc protecting group is needed in future solution-phase modification, deprotection by the basic amine was avoided by doing repetitions of the click reaction. Three one-hour repetitions were the maximum number before Fmoc deprotection of product became significant.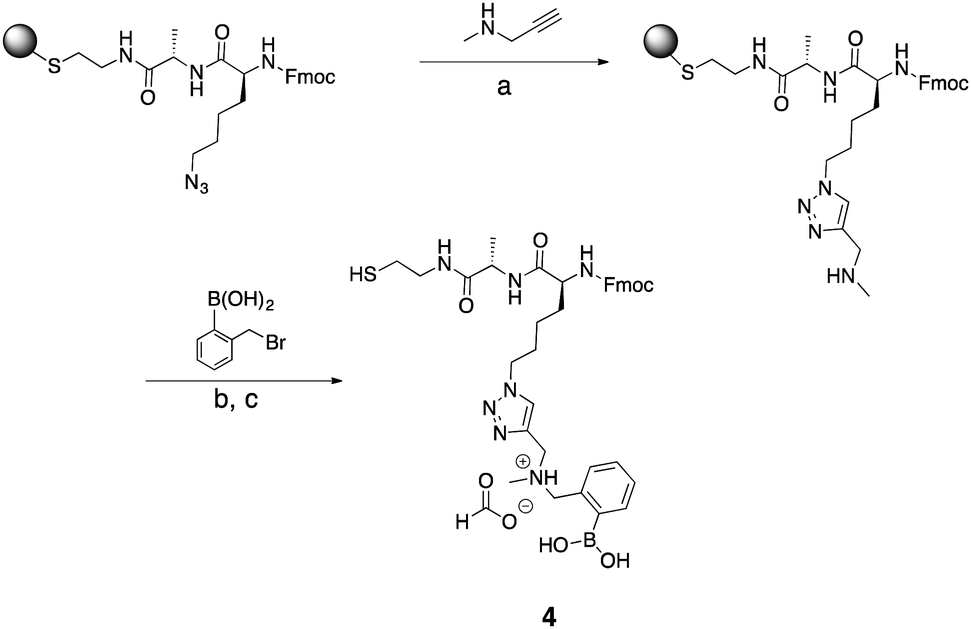 | ||
| Scheme 4 Synthesis of compound 4. (a) DMF, CuI, TBTA, sodium ascorbate, RT, 1 h (3 repetitions). (b) MeCN, H2O, overnight. (c) TFA, TIS, H2O, RT, 4 h. | ||
Interestingly, Wang resin did not yield the desired product. However, “clicking” did occur with a cysteamine resin. Upon cleavage, the product (4) contained a thiol functional group as the C-terminus, which could be used for solution-phase derivatization if desired.
Building-block incorporation of secondary amines and alkylation
Using commercially available and synthetically accessible Nε-methyl lysine as a building block proved to be the most straightforward method for introducing secondary amines. This building block ensured the placement of multiple secondary amines along a sequence with high reliability. Fmoc-Lys(Nε,Me)Ala-OH gave the highest purified yield (60%; Scheme 5). Only a single modification step post-solid phase synthesis was required to make a boronic acid peptide. Thus, it minimized the cumulative inefficiencies at each reaction step in the former two approaches. Using this approach, seven complex biotinylated, using D-biotin, multi-boronic acid peptides were synthesized (Scheme 5 and Fig. 2).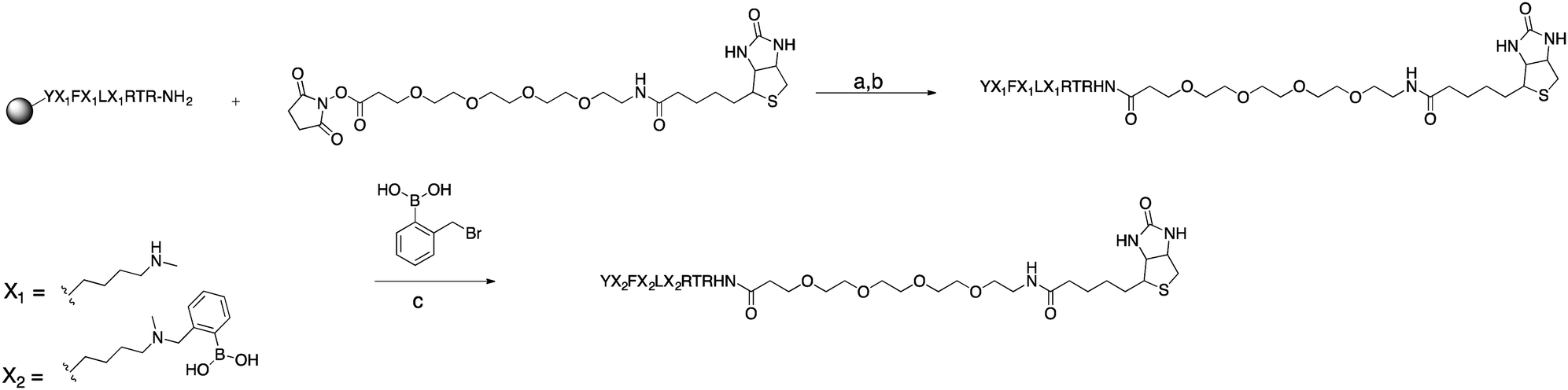 | ||
| Scheme 5 Synthesis of compound 6. (a) DMF, DIPEA, RT, 2 h (3 repetitions). (b) TFA, TIS, H2O, RT, 4 h. (c) MeCN, H2O, RT, overnight. | ||
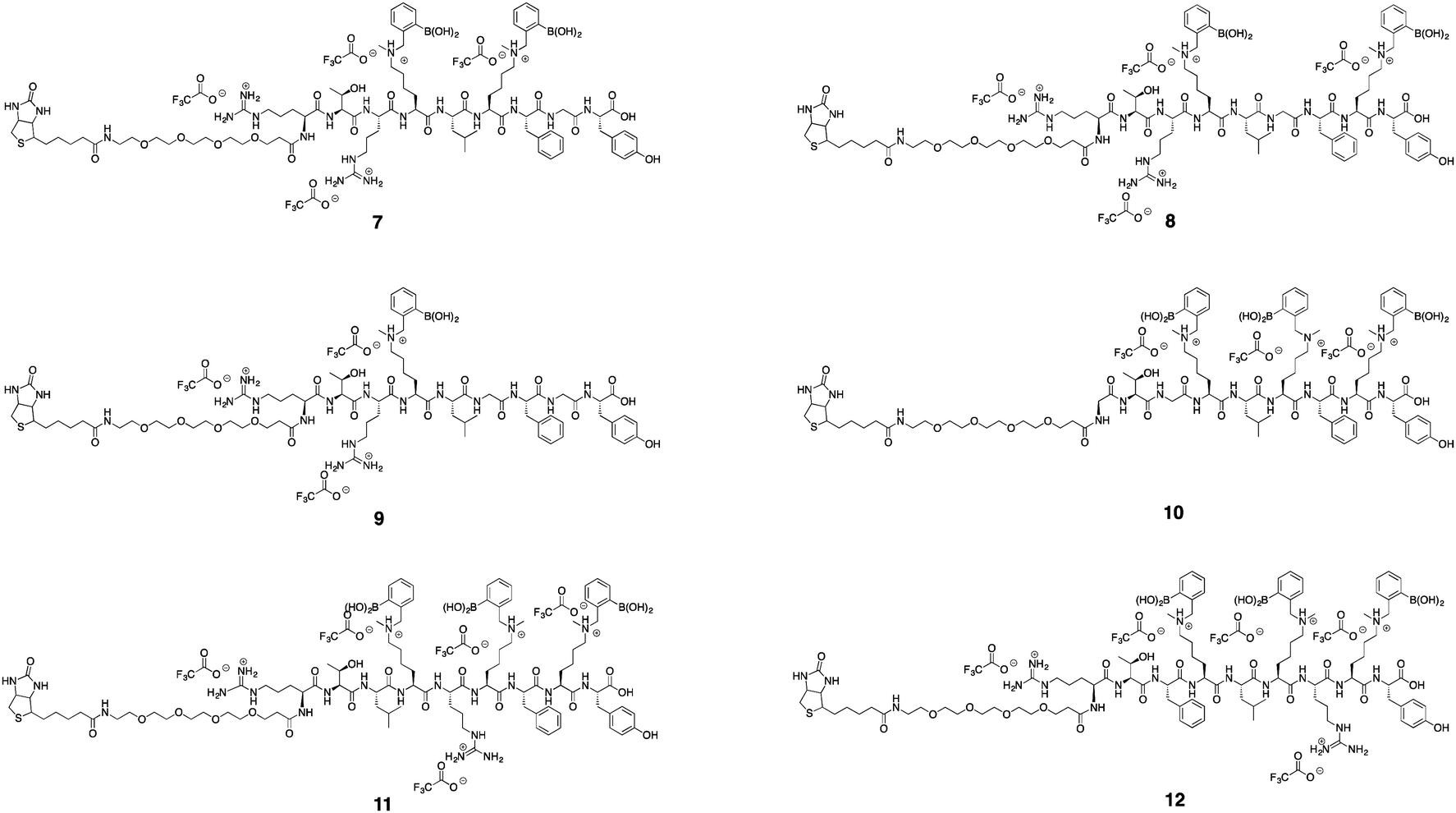 | ||
| Fig. 2 Biotinylated, using D-biotin, boronic acid peptides. | ||
Of the seven biotinylated peptides, 9 had the highest purified yield (45%). The purified yields of peptides with two or three boronic acids ranged from 8–22%. We attributed the lower yields to the increasing difficulty in purification when the number of boronic acids grew. Streaking of product occurred more readily during purification.
The HPLC analytical trace for 9 gave a relatively sharp peak as the product eluted. However, when two or three boronic side chains were present, the elution peak was broadened, suggesting again that the phenyl boronic acid moieties interact with the column. Higher percentages of organic solvent were required to elute the peptide. The broadening of the elution peak led to low signal intensity during HPLC purification. This broadening led to a low signal intensity during HPLC purification, which made identification of the peaks challenging, and hence poor detection of sample elution was seen to contribute to the lower yields for peptides with more than one boronic acid.
Conclusions
We developed three different approaches for derivatizing peptides with secondary amines, followed by modification with o-(bromomethyl)phenylboronic acid. Unlike most previously published methods for incorporation of boronic acids into peptides, this alkylation strategy allows for easy incorporation of an o-aminomethyl functionality on the aryl boronic acid, which has been previously shown to increase binding affinity of boronic acids to saccharides. The first approach involved the synthesis of a linker with an iodoacetamide at one side and Boc protected secondary amine on the other end. Cysteine residues were selectively targeted in a model peptide. After deprotection, the secondary amine was alkylated with a boronic acid. In the second approach, N-methyl propargyl amine was used in click chemistry to introduce secondary amines followed by alkylation with a boronic acid in the solid phase. Post cleavage, a boronic acid peptide with a C-terminal thiol group was isolated. Lastly, the commercially available Nε-methyl lysine building block was used as the most direct method for making boronic acid peptides. It gave the highest yield and was the method of choice to make a set of seven biotinylated peptides. These peptides had up to three boronic acids, although purification became difficult and the associated yields decreased with increasing numbers of boronic acids (Table 1).| Compound | Sequence | Yield (%) |
|---|---|---|
| 6 | RTRX2LX2FX2Y | 7.5 |
| 7 | RTRX2LX2FGY | 11 |
| 8 | RTRX2LGFX2Y | 19 |
| 9 | RTRX2LGFGY | 45 |
| 10 | GTGX2LX2FX2Y | 25 |
| 11 | RTLX2RX2FX2Y | 17 |
| 12 | RTFX2LX2RX2Y | 22 |
Compounds such as 6–12 are currently being investigated as pull-down reagents for cell-surface saccharides. Due to the utility of the synthetic methodologies we report herein, we anticipate that others can adopt them for site-specific incorporation of boronic acids in peptides and proteins.
Experimental
General methods
For automated Fmoc amino solid-phase peptide synthesis, Ala, Pbf(Arg), Trt(Cys), Gly, Leu, Boc(Lys), Phe, and Boc(Thr) were purchased from P3 biosystems. Fmoc-Lys(Nε,Me)-OH was purchased from Chem. Pep. Inc. Fmoc-Lys(N3)-OH was purchased from Chem-Impex, Inc. Cysteamine 2-ClTrt (0.95 mmol g−1) resin and Fmoc-Tyr(tBu)-Wang resin (0.46 mmol g−1) were purchased from AnaSpec, Inc. Fmoc-Ala-Wang (100–200 mesh, 0.72 mmol g−1) was purchased NovaBiochem. DMF, DCM, piperidine used for automated solid-phase peptide synthesis were purchased from Fisher Scientific and Sigma-Aldrich. N,N′-Dimethylethylenediamine, chloroacetyl chloride, and sodium iodide, N-methyl propargyl amine, copper iodide, and sodium ascorbate were purchased from Sigma-Aldrich. o-(Bromomethyl)phenylboronic acid was purchased from Combi-Blocks. EZ-Link™ NHS-PEG4-biotin was purchased from Thermo Scientific.A Prelude peptide synthesizer (Protein Technologies, Inc.) was used for automated-solid phase synthesis of the peptides. For the longer peptides, a Liberty Blue microwave peptide synthesizer was used. Preparative HPLC purification of peptides was performed using an Agilent Zorbax SB-C18 Prep HT column 21.2 × 250 mm; 10 mL min−1, 5–95% MeOH in 90 min. Analytical HPLC characterization of peptides was performed using an Agilent Zorbax column 4.6 × 250 mm; 1 mL min−1, 5–95% MeCN (0.1% TFA) in 35 min (RT). A Gemini C18 3.5 micron 2.1 × 50 mm was used for online separation; 0.7 mL min−1, 5–95% MeCN (0.1% formic acid) in 12 min (RT). An Agilent Technologies 6530 Accurate Mass QTofLC/MS was used for high-resolution mass spectra of purified peptides. Solvents used were HPLC grade. For small molecule organic compounds, reverse-phase combi-flash was used for purification if necessary.
General procedure (A): synthesis of dimeric peptides and Nε-methyl lysine peptides
Fmoc-CysAla-Wang resin, Fmoc-Lys(N3)Ala-S-resin, Fmoc-Lys(Nε,Me)Ala-Wang, and biotinylated Nε-methyl lysine peptides resin (100 μmol) were synthesized by automated sequential coupling of Nα-Fmoc-amino acid (0.1 M) in DMF in the presence of N,N,N,N-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate (HBTU, 0.15 M) and Hünig's base (0.2 M) with gentle nitrogen bubbling for 30 minutes at room temperature. A total of three repetitions were performed for each amino acid building block incorporated, followed by DMF (3 mL, 3 min, 3×) and DCM (3 mL, 3 min, 3×) washes. For peptides longer than three amino acid residues, a 0.8 M LiCl wash (3 mL, 3 min, 3×) was included after swelling with DCM. After the synthesis, resin was washed with glacial AcOH (5 mL, 3×), DCM (5 mL, 3×), and MeOH (5 mL, 3×). Resins were placed under vacuum overnight. Fmoc-Cys Ala was cleaved from the resin using trifluoroacetic acid (TFA), triisopropylsilane, 1,2-ethanedithiol (EDT), and nanopure water (94![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.0:2.5:2.5) (4 h) for Fmoc-CysAla-OH. TFA was evaporated and the remaining oil was precipitated with diethyl ether at 0 °C. No further purification of the crude peptide was performed.
:1.0:2.5:2.5) (4 h) for Fmoc-CysAla-OH. TFA was evaporated and the remaining oil was precipitated with diethyl ether at 0 °C. No further purification of the crude peptide was performed.
General procedure (B): solution-phase alkylation with o-(bromomethyl)phenylboronic acid
Peptides were dissolved in a mixture of H2O and MeCN (0.6 mL, 1:1 v/v). To this solution, 0.1 mL of Hünig's base was added. If the peptide precipitated, 0.15 mL of MeOH was added. The solution was further diluted with 0.2 mL of MeCN, followed by addition of 3.5 equivalents of o-(bromomethyl)phenylboronic acid. The reaction was allowed to stir overnight at RT. The following day, additional boronic acid (3.5 equivalents) was added, followed by addition of 0.05 mL of Hünig's base. The reaction was incubated at room temperature for an additional 2 h. Preparative HPLC was used to purify the peptides. Purified samples were placed on the rotary evaporator to remove MeOH. The aqueous remnants were frozen and lyophilized overnight.
General procedure (C): solid-phase modification of the N-terminus with EZ-Link™ NHS-PEG4-biotin: post automated solid phase synthesis
Fmoc group was removed with 3 mL of piperidine (20% vol in DMF, 3 min, 3×) followed by washes with DMF (3 mL, 3 min, 3×), DCM (3 mL, 3 min, 3×), and 0.8 M LiCl (3 mL, 3 min, 3×). A 1.5 mL solution of EZ-Link™ NHS-PEG4-biotin was introduced with 0.5 mL solution of 1.2 M Hünig's base. Gentle stirring under nitrogen was performed for 1 h at RT, followed by washes with DMF (3 mL, 3 min, 3×), DCM (3 mL, 3 min, 3×), and 0.8 M LiCl (3 mL, 3 min, 3×). Coupling of biotin to peptide was repeated a total of three times. Resins were washed with glacial AcOH (5 mL, 3×), DCM (5 mL, 3×), and MeOH (5 mL, 3×), and resins were placed under vacuum overnight. Peptides were cleaved with trifluoroacetic acid (TFA), triisopropylsilane, and nanopure water (95:2.5:2.5) (4 h), followed by removal of TFA, and precipitated with diethyl ether at 0 °C. No further purification of the crude peptide was performed.
Synthesis of compounds
:TEA:Pyr:H2O (7:1:1:1) (v/v/v). A solution of compound 3 (0.07 mmol) in 0.5 mL MeCN:H2O (69:31) (v/v) was introduced. The Boc protecting group was removed using trifluoroacetic acid (TFA), triisopropylsilane, and nanopure water (95:2.5:2.5) (4 h). TFA was removed, and the product was precipitated with diethyl ether at 0 °C. No further purification of the peptide was performed. The peptide was alkylated with o-(bromomethyl)phenylboronic acid following general procedure B. The peptide was purified via preparative HPLC. MeOH was removed by rotary evaporation, followed by freezing and lyophilization of aqueous remnants. Purified yield: 6%. HRMS-ESI+ (MeOH/H2O): calcd: m/z = 659.2711; found: m/z = 659.2712 [M − 2H2O + H]+. 1H NMR (500 MHz, DMSO-d6) δ 8.30 (s, 1H), 7.82 (d, J = 7.6 Hz, 2H), 7.65 (t, J = 6.8 Hz, 2H), 7.38 (t, J = 7.4 Hz, 2H), 7.33–7.25 (m, 3H), 7.16 (d, J = 7.3 Hz, 1H), 4.25 (t, J = 6.6 Hz, 2H), 4.20–4.11 (m, 1H), 3.94 (d, J = 7.0 Hz, 1H), 3.88 (s, 4H), 3.72 (d, J = 10.9 Hz, 1H), 3.57–3.39 (m, 2H), 3.40–3.30 (m, 1H), 2.91 (dd, J = 13.8, 5.2 Hz, 1H), 2.71 (d, J = 14.0 Hz, 3H), 2.68–2.58 (m, 4H), 2.24 (s, 2H), 2.16 (s, 1H), 1.18 (dd, J = 7.0, 1.7 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 170.31, 156.90, 144.37, 144.36, 144.25, 141.36, 135.41, 135.40, 130.34, 130.14, 128.60, 128.02, 126.00, 120.83, 66.76, 63.25, 63.23, 54.97, 54.97, 52.75, 50.19, 47.26, 47.26, 44.12, 41.44, 40.45, 40.45, 40.06, 40.02, 39.90, 39.87, 39.84, 39.73, 39.70, 39.67, 39.56, 39.53, 39.51, 39.40, 39.37, 39.34, 39.23, 39.20, 39.17, 39.04, 39.01, 35.87, 34.48, 34.12, 18.92, 18.91.
:1.0:2.5:2.5) (4 h). TFA was evaporated and remaining oil was precipitated with diethyl ether at 0 °C. Peptide was purified via preparative HPLC. MeOH was removed by rotary evaporation, followed by freezing and lyophilization of aqueous remnants. Purified yield: 14%. HRMS-ESI+ (MeOH/H2O): calcd: m/z = 750.3222; found: m/z = 750.3228 [M + Na]+. 1H NMR (500 MHz, DMSO-d6) δ 8.30 (s, 1H), 7.97 (d, J = 6.7 Hz, 1H), 7.84 (t, J = 6.8 Hz, 2H), 7.65 (q, J = 6.2 Hz, 2H), 7.58 (d, J = 7.1 Hz, 1H), 7.39 (q, J = 7.0 Hz, 2H), 7.29 (q, J = 7.5, 7.0 Hz, 2H), 7.22 (dd, J = 8.7, 6.6 Hz, 1H), 7.15 (q, J = 6.8 Hz, 1H), 4.30 (t, J = 6.8 Hz, 2H), 4.22 (dd, J = 7.4, 4.0 Hz, 2H), 4.16 (td, J = 6.9, 3.3 Hz, 2H), 3.93 (dd, J = 9.4, 4.9 Hz, 1H), 3.65 (d, J = 4.4 Hz, 2H), 3.47 (d, J = 5.2 Hz, 0H), 3.33 (dt, J = 13.5, 6.8 Hz, 1H), 3.25 (dt, J = 13.4, 6.6 Hz, 1H), 2.78–2.63 (m, 2H), 2.03 (d, J = 4.5 Hz, 3H), 1.77 (d, J = 9.3 Hz, 3H), 1.64 (s, 1H), 1.52 (d, J = 11.8 Hz, 1H), 1.35–1.19 (m, 4H), 1.16 (d, J = 7.1 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 172.78, 172.08, 156.53, 144.13, 144.03, 143.25, 141.86, 141.07, 134.48, 129.44, 129.21, 129.09, 128.16, 127.57, 127.19, 125.65, 124.80, 121.78, 120.51, 72.44, 66.14, 61.71, 60.55, 54.72, 49.83, 49.70, 48.63, 47.02, 42.26, 40.28, 40.11, 40.02, 39.94, 39.86, 39.77, 39.69, 39.60, 39.52, 39.43, 39.35, 39.26, 39.18, 39.07, 39.02, 38.16, 37.41, 35.91, 31.38, 29.67, 22.76, 18.51.
Acknowledgements
We thank our following funding sources: the National Institute of Health (5DP1GM106408), the Defense Advanced Research Projects Agency (DARPA, N66001-14-2-4051), and the Welch Regents Chair (F-0046).References
- S. Abdellaoui, B. C. Corgier, C. A. Mandon, B. Doumeche, C. A. Marquette and L. J. Blum, Electroanalysis, 2013, 25, 671–684 CrossRef CAS.
- R. Amin and S. A. Elfeky, Spectrochim. Acta, Part A, 2013, 108, 338–341 CrossRef CAS PubMed.
- S. Arimori, S. Ushiroda, L. M. Peter, A. T. A. Jenkins and T. D. James, Chem. Commun., 2002, 2368–2369 RSC.
- J. B. Crumpton, W. Zhang and W. L. Santos, Anal. Chem., 2011, 83, 3548–3554 CrossRef CAS PubMed.
- W. W. Bachovchin, W. Y. L. Wong, S. Farr-Jones, A. B. Shenvi and C. A. Kettner, Biochemistry, 1988, 27, 7689–7697 CrossRef CAS PubMed.
- W. L. A. Brooks and B. S. Sumerlin, Chem. Rev., 2016, 116, 1375–1397 CrossRef CAS PubMed.
- J. N. Cambre and B. S. Sumerlin, Polymer, 2011, 52, 4631–4643 CrossRef CAS.
- T. M. El Dine, J. Rouden and J. Blanchet, Chem. Commun., 2015, 51, 16084–16087 RSC.
- T. D. James, K. R. A. S. Sandanayake and S. Shinkai, Angew. Chem., Int. Ed., 1994, 33, 2207 CrossRef.
- S. Kotha, K. Lahiri and D. Kashinath, Tetrahedron, 2002, 58, 9633–9695 CrossRef CAS.
- K. Lacina and P. Skládal, Electrochim. Acta, 2011, 56, 10246 CrossRef CAS.
- K. Lacina, P. Skládal and T. D. James, Chem. Cent. J., 2014, 8, 1–17 CrossRef PubMed.
- J. Langen, U. Fischer, M. Cavalar, C. Coetzee, P. Wegmann-Herr and H.-G. Schmarr, Anal. Bioanal. Chem., 2016, 408, 2425–2439 CrossRef CAS PubMed.
- J. Li, A. S. Grillo and M. D. Burke, Acc. Chem. Res., 2015, 48, 2297–2307 CrossRef CAS PubMed.
- M. Li, W. Zhu, F. Marken and T. D. James, Chem. Commun., 2015, 51, 14562–14573 RSC.
- J. X. Qiao and P. Y. S. Lam, Synthesis, 2011, 315–361 CAS.
- L. Rocard, A. Berezin, F. De Leo and D. Bonifazi, Angew. Chem., Int. Ed., 2015, 54, 15739–15743 CrossRef CAS PubMed.
- G. Springsteen and B. Wang, Chem. Commun., 2001, 1608–1609 RSC.
- X. Sun, W. Zhai, J. S. Fossey and T. D. James, Chem. Commun., 2016, 52, 3456–3469 RSC.
- Q. Zhang, N. Tang, J. W. C. Brock, H. M. Mottaz, J. M. Ames, J. W. Baynes, R. D. Smith and T. O. Metz, J. Proteome Res., 2007, 6, 2323–2330 CrossRef CAS PubMed.
- W. Zhang, D. I. Bryson, J. B. Crumpton, J. Wynn and W. L. Santos, Chem. Commun., 2013, 49, 2436–2438 RSC.
- X. Sun and T. D. James, Chem. Rev., 2015, 115, 8001–8037 CrossRef CAS PubMed.
- M. S. Melicher, J. Chu, A. S. Walker, S. J. Miller, R. H. G. Baxter and A. Schepartz, Org. Lett., 2013, 15, 5048–5051 CrossRef CAS PubMed.
- Y. Zou, D. L. Broughton, K. L. Bicker, P. R. Thompson and J. J. Lavigne, ChemBioChem, 2007, 8, 2048–2051 CrossRef CAS PubMed.
- F. D'Hooge, S. A. Elfeky, S. E. Flower, S. I. Pascu, A. T. A. Jenkins, J. M. H. v. d. Elsen, T. D. James and J. S. Fossey, RSC Adv., 2012, 2, 3274–3280 RSC.
- S. A. Elfeky, F. D'Hooge, L. Poncel, W. Chen, S. P. Perera, J. M. H. van den Elsen, T. D. James, A. T. A. Jenkins, P. J. Cameron and J. S. Fossey, New J. Chem., 2009, 33, 1466–1469 RSC.
- S.-H. Chung, T.-J. Lin, Q.-Y. Hu, C.-H. Tsai and P.-S. Pan, Molecules, 2013, 18, 12346 CrossRef CAS PubMed.
- J. J. Deadman, S. Elgendy, C. A. Goodwin, D. Green, J. A. Baban, G. Patel, E. Skordalakes, N. Chino and G. Claeson, J. Med. Chem., 1995, 38, 1511–1522 CrossRef CAS PubMed.
- P. J. Duggan and D. A. Offermann, Aust. J. Chem., 2007, 60, 829–834 CrossRef CAS.
- N. Y. Edwards, T. W. Sager, J. T. McDevitt and E. V. Anslyn, J. Am. Chem. Soc., 2007, 129, 13575–13583 CrossRef CAS PubMed.
- G. A. Ellis, M. J. Palte and R. T. Raines, J. Am. Chem. Soc., 2012, 134, 3631–3634 CrossRef CAS PubMed.
- O. V. Gozhina, J. S. Svendsen and T. Lejon, J. Pept. Sci., 2014, 20, 20–24 CrossRef CAS PubMed.
- M. Kita, J. Yamamoto, T. Morisaki, C. Komiya, T. Inokuma, L. Miyamoto, K. Tsuchiya, A. Shigenaga and A. Otaka, Tetrahedron Lett., 2015, 56, 4228–4231 CrossRef CAS.
- C.-H. Tsai, C.-H. Lin, C.-T. Hsieh, C.-C. Cai, T.-J. Lin, P.-Y. Liu, M.-H. Lin, M.-J. Wu, C.-C. Fu, Y.-C. Wu, F.-R. Chang and P.-S. Pan, Res. Chem. Intermed., 2014, 40, 2187–2198 CrossRef CAS.
- K. L. Bicker, J. Sun, J. J. Lavigne and P. R. Thompson, ACS Comb. Sci., 2011, 13, 232–243 CrossRef CAS PubMed.
- M. A. M. Behnam, T. R. Sundermann and C. D. Klein, Org. Lett., 2016, 18, 2016–2019 CrossRef CAS PubMed.
- B. E. Collins and E. V. Anslyn, Chem. – Eur. J., 2007, 13, 4700–4708 CrossRef CAS PubMed.
- B. E. Collins, S. Sorey, A. E. Hargrove, S. H. Shabbir, V. M. Lynch and E. V. Anslyn, J. Org. Chem., 2009, 74, 4055–4060 CrossRef CAS PubMed.
- D. G. Hall, Boronic Acids, Wiley-VCH Verlag GmbH & Co. KGaA, 2006, pp. 1–99 Search PubMed.
- J. M. Chalker, G. J. L. Bernardes, Y. A. Lin and B. G. Davis, Chem. – Asian J., 2009, 4, 630–640 CrossRef CAS PubMed.
- N. Fomina, C. McFearin, M. Sermsakdi, O. Edigin and A. Almutairi, J. Am. Chem. Soc., 2010, 132, 9540–9542 CrossRef CAS PubMed.
- D. K. Scrafton, J. E. Taylor, M. F. Mahon, J. S. Fossey and T. D. James, J. Org. Chem., 2008, 73, 2871–2874 CrossRef CAS PubMed.
- S. Jin, G. Choudhary, Y. Cheng, C. Dai, M. Li and B. Wang, Chem. Commun., 2009, 5251–5253 RSC.
- V. Castro, H. Rodríguez and F. Albericio, ACS Comb. Sci., 2016, 18, 1–14 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR, HRMS, and HPLC purity checks. See DOI: 10.1039/c6nj02862d |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2017 |