
Pd-catalyzed intramolecular sequential Heck cyclization and oxidation reactions: a facile pathway for the synthesis of substituted cycloheptenone evaluated using computational studies†
Jayanta K.
Ray
*,
Sunanda
Paul
,
Paramita
Ray
,
Raju
Singha
,
Davuluri Yogeswara
Rao
,
Surajit
Nandi
 and
Anakuthil
Anoop
*
and
Anakuthil
Anoop
*
Department of Chemistry, Indian Institute of Technology Kharagpur, Kharagpur 721302, India. E-mail: jkray@chem.iitkgp.ernet.in; Tel: +91-3222 283326
First published on 10th November 2016
Abstract
A simple and convenient method for the construction of substituted cycloheptenones from 1-bromoocta-1,7-diene-3-ols has been developed. The reaction involves Pd(0)-catalyzed intramolecular 7-exo-trig cyclization followed by Pd(II)-catalyzed oxidation of cyclic alcohol. The course of the reaction pathway has been evaluated using DFT calculations.
Introduction
Seven-membered carbocyclic frameworks with ketone functionality appear as integral subunits in a variety of polycyclic natural products and many of them exhibit interesting biological activities. Examples of such natural products include Clavularin A and B,1 seven-membered vibsane-diterpenes,2 cartorimine,3 oxidative metabolites of sesquiterpene guaiol,4 ingenol diterpene5etc. Unlike other common ring sizes, such as five- and six-membered rings, the construction of a seven-membered ring is more challenging because of entropic reason, ring strain etc. Different methods and strategies aiming towards the synthesis of a cycloheptenone moiety have been reported. The most convergent approach for the synthesis of cycloheptenone points towards a variety of cycloaddition reactions6 leading to the synthesis of many natural products. There are also reports of acid-catalyzed ring expansion of bicyclo[3,2,0]heptanone, Rh(I)-catalyzed intramolecular hydroacylation of 4,6-dienals,7,8a a Pd-catalyzed Heck coupling reaction,8b and a microwave assisted tandem oxyanionic 5-exo dig cyclization-Claisen rearrangement9 to give cycloheptenone derivatives.The prospect of constructing carbocycles using an intramolecular Heck reaction makes it primarily versatile for organic synthesis.10a,b We demonstrated earlier that a substrate like 1 (n = 0 and 1) when subjected to Pd(0)-catalyzed intramolecular Heck cyclization afforded 2 (Scheme 1) as the cyclopentenone and cyclohexenone moieties starting from numerous β-bromo vinyl aldehydes.10c Therefore, following a similar methodology, we envisioned synthesizing a cycloheptane moiety with an α-β-unsaturated keto functionality.
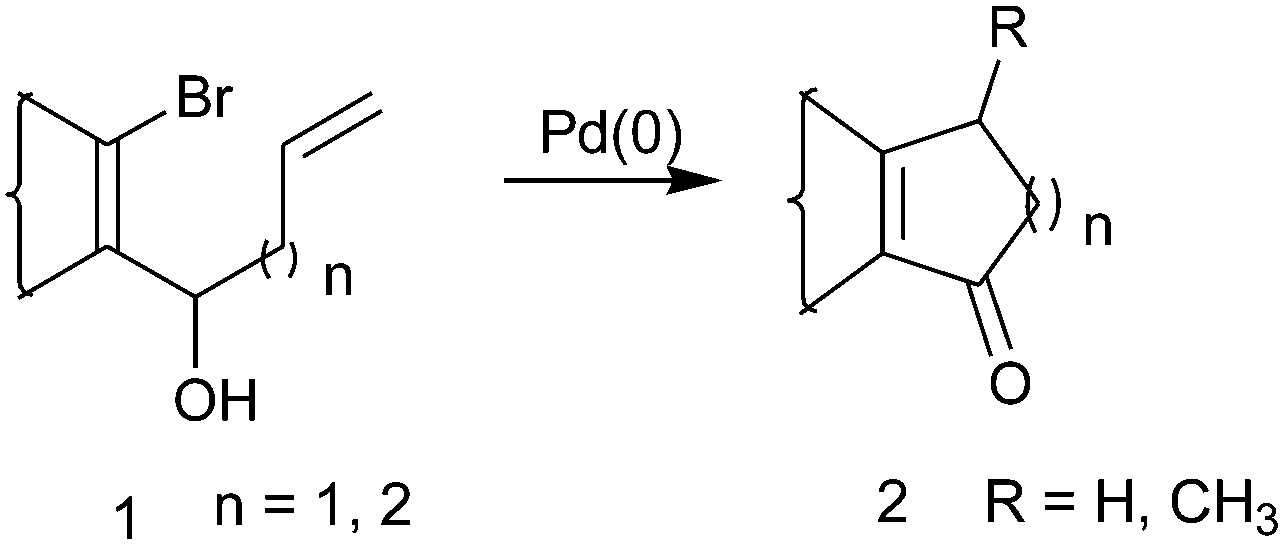 | ||
| Scheme 1 Intramolecular Heck cyclization. | ||
We have also studied the mechanism computationally, as such studies on intramolecular Heck cyclization are rare.
Results and discussion
The starting materials were synthesized by the treatment of β-bromo vinyl aldehydes with Grignard reagents derived from 1-bromo-pentene and Mg in dry diethyl ether at 0 °C for 1 h to afford bromo alcohols (Table 1, 4a–4h) in good yield.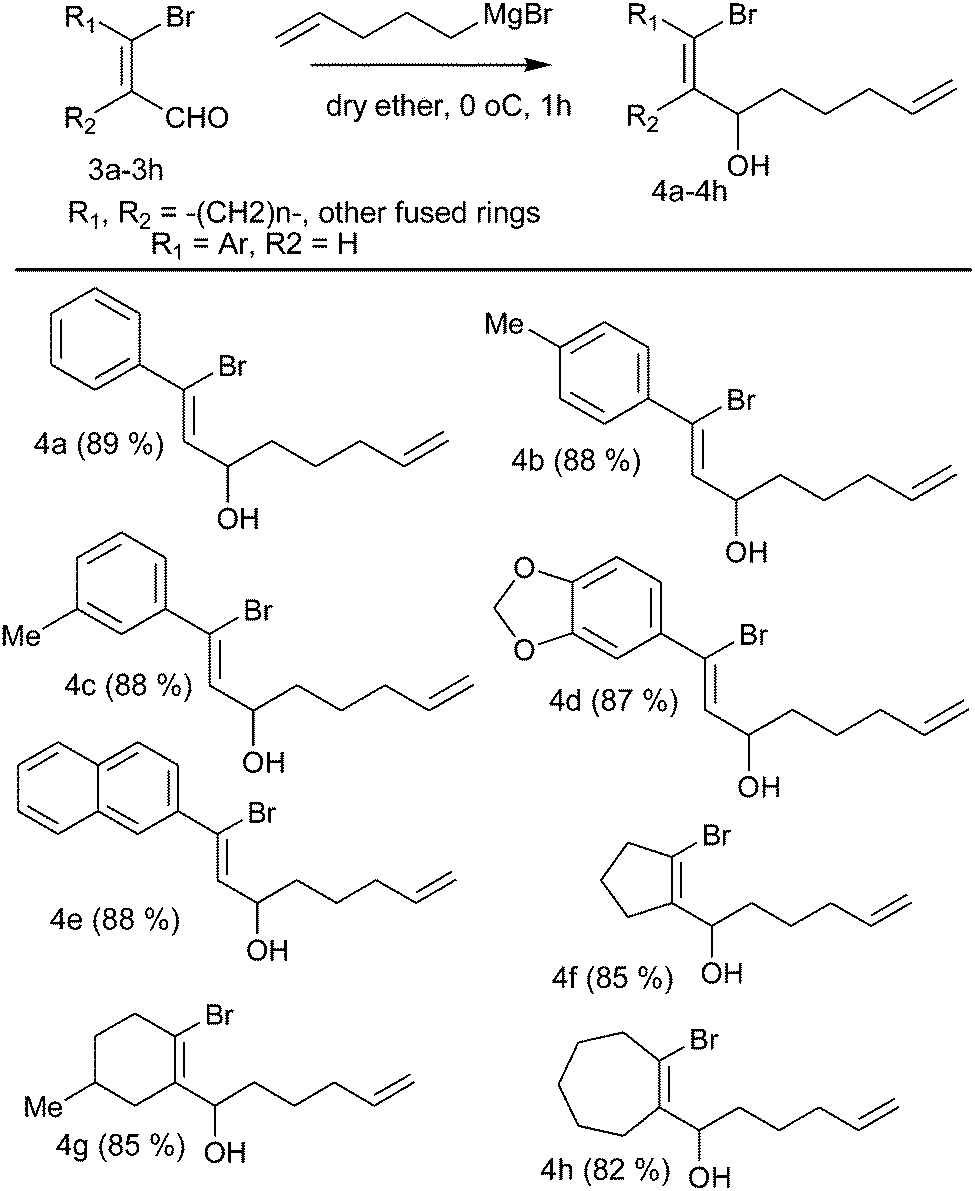
When intramolecular Heck cyclization was performed with the precursor 1-bromo-octa-1,7-diene-3-ol, which could cyclize via either 7-exo or 8-endo-trig pathways, it ultimately cyclizes exclusively in a 7-exo-trig way and unlike in the previous cases,10c 4-methylene-cyclohept-2-enol was obtained as the only isolable product (Scheme 2).
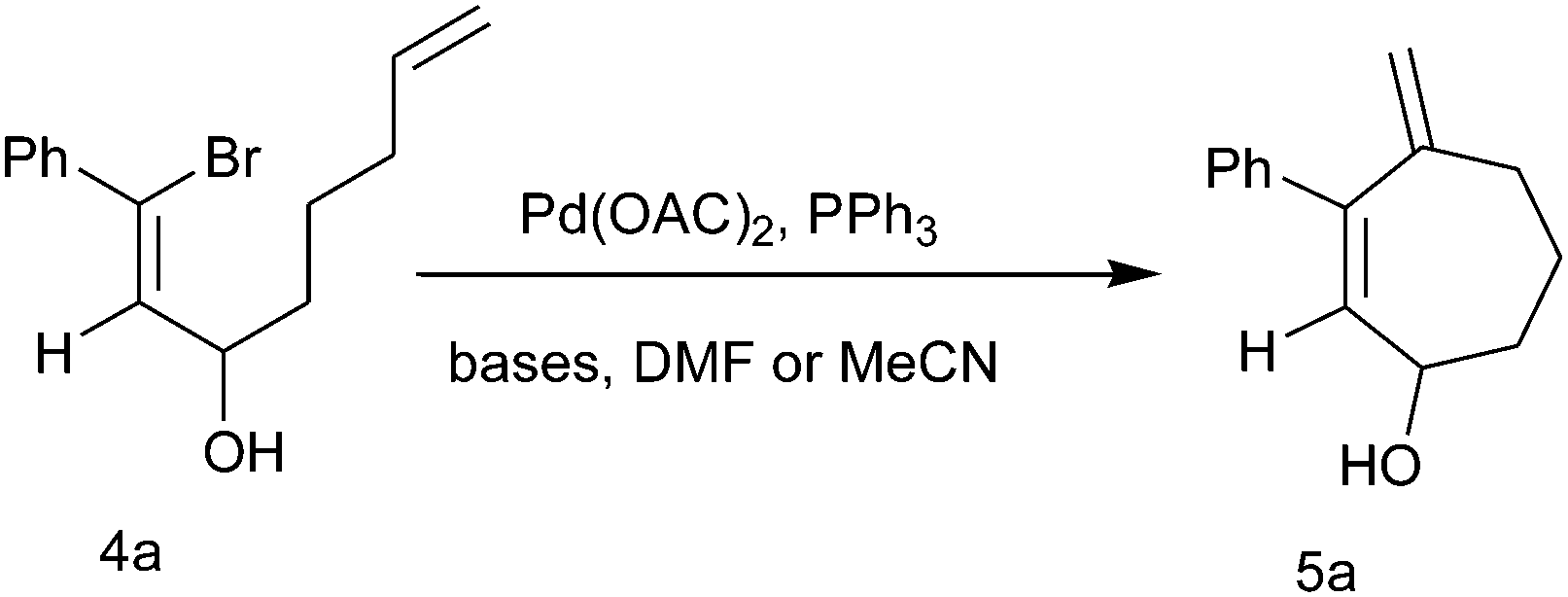 | ||
| Scheme 2 Intramolecular 7-exo-trig Heck cyclization. | ||
We have optimized the reaction conditions with substrate 4a by carrying out the cyclization reaction with several bases (Na2CO3, K2CO3, Cs2CO3, NaOAc, Et3N, HCOONa) in solvents such as DMF, acetonitrile using Pd(OAc)2 as the catalyst and PPh3 as the ligand (Table 2). We observed that carbonate bases are more effective than acetate or amine bases and among the carbonate bases, Na2CO3 gave the highest yield. Upon decreasing the amount of catalyst to 2 mol%, the reaction remained incomplete even after 24 h and we obtained a 76% yield. Thus among all these cases the highest yield of cyclic alcohol 5a (90%) was obtained when cyclization was carried out with 4a in the presence of 0.05 mmol Pd(OAc)2, 0.2 mmol PPh3 and 1 mmol Na2CO3 in DMF solvent at 80 °C for 2 h. Unlike in the previous publications where the methyl-substituted oxidized product through the 1,5-hydride shift of the five- and six-membered cyclic alcohol containing an exocyclic double bond formed by intramolecular Pd(0)-catalyzed cyclization of the allylated and homoallylated derivatives of vinyl bromoaldehyde were reported, here simple Pd(0)-catalyzed intramolecular 7-exo-trig cyclization occurs to afford a cycloheptenol moiety with an exocyclic double bond. Here with the seven-membered cyclic alcohol, a 1,5-hydride shift does not take place, not even upon prolonged heating.
| Entry | Catalyst | Solvent | Base | Yieldb (%) |
|---|---|---|---|---|
| a Reactions were carried out with 0.5 mmol of 4a in the presence of 0.05 mmol of Pd(OAc)2, 0.2 mmol of PPh3, in 3 mL of solvent at 80 °C for 2 h. b Isolated yield. c 2 mol% of the catalyst used, reaction time: 24 h. | ||||
| 1 | Pd(OAc)2 | DMF | Cs2CO3 | 76 |
| 2 | Pd(OAc)2 | DMF | K2CO3 | 83 |
| 3 | Pd(OAc)2 | DMF | Na2CO3 | 90 |
| 4 | Pd(OAc)2 | DMF | NaOAc | 65 |
| 5 | Pd(OAc)2 | DMF | Et3N | 58 |
| 6 | Pd(OAc)2 | MeCN | Na2CO3 | 81 |
| 7 | Pd(OAc)2 | DMA | Na2CO3 | 85 |
| 8 | PdCl2 | DMF | Na2CO3 | 79 |
| 9c | Pd(OAc)2 | DMF | Na2CO3 | 76 |
Therefore, after the completion of Heck cyclization, we attempted to oxidize the cyclic alcohol 5a into the corresponding keto derivative. Over the past decade, Pd(II)-catalyzed aerobic oxidation of alcohols has become well known.11 Here, we applied the Pd(OAc)2/pyridine/MS3A catalytic system under an O2 atmosphere for the oxidation of alcohol as reported by Uemura et al.12 After complete consumption of the starting material during the Heck cyclization of 4a, the reaction mixture was filtered and after usual workup, alcohol 5a was directly subjected to Pd(II)-catalyzed oxidation without further purification. When heated with 0.05 mmol Pd(OAc)2, 5 mmol pyridine and MS3A in toluene at 80 °C under an O2 atmosphere, alcohol 5a conveniently undergoes oxidation to give cyloheptenone 6a in a 74% yield (Scheme 3). Following the literature procedure, an excess of pyridine was also used here to oxidize this allylic alcohol, which resulted in better oxidation.
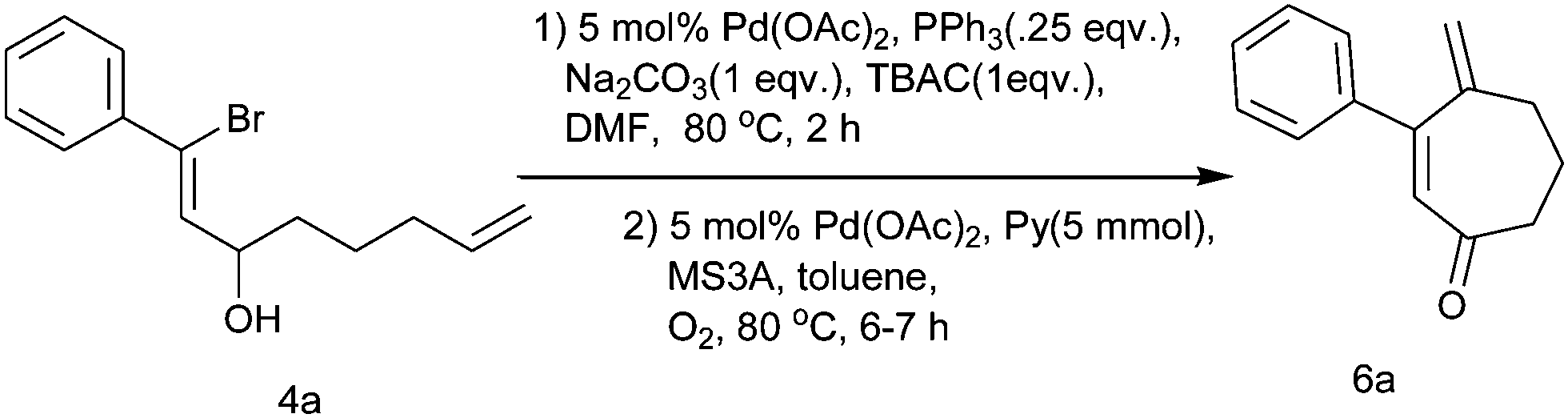 | ||
| Scheme 3 Synthesis of the cycloheptenone derivative. | ||
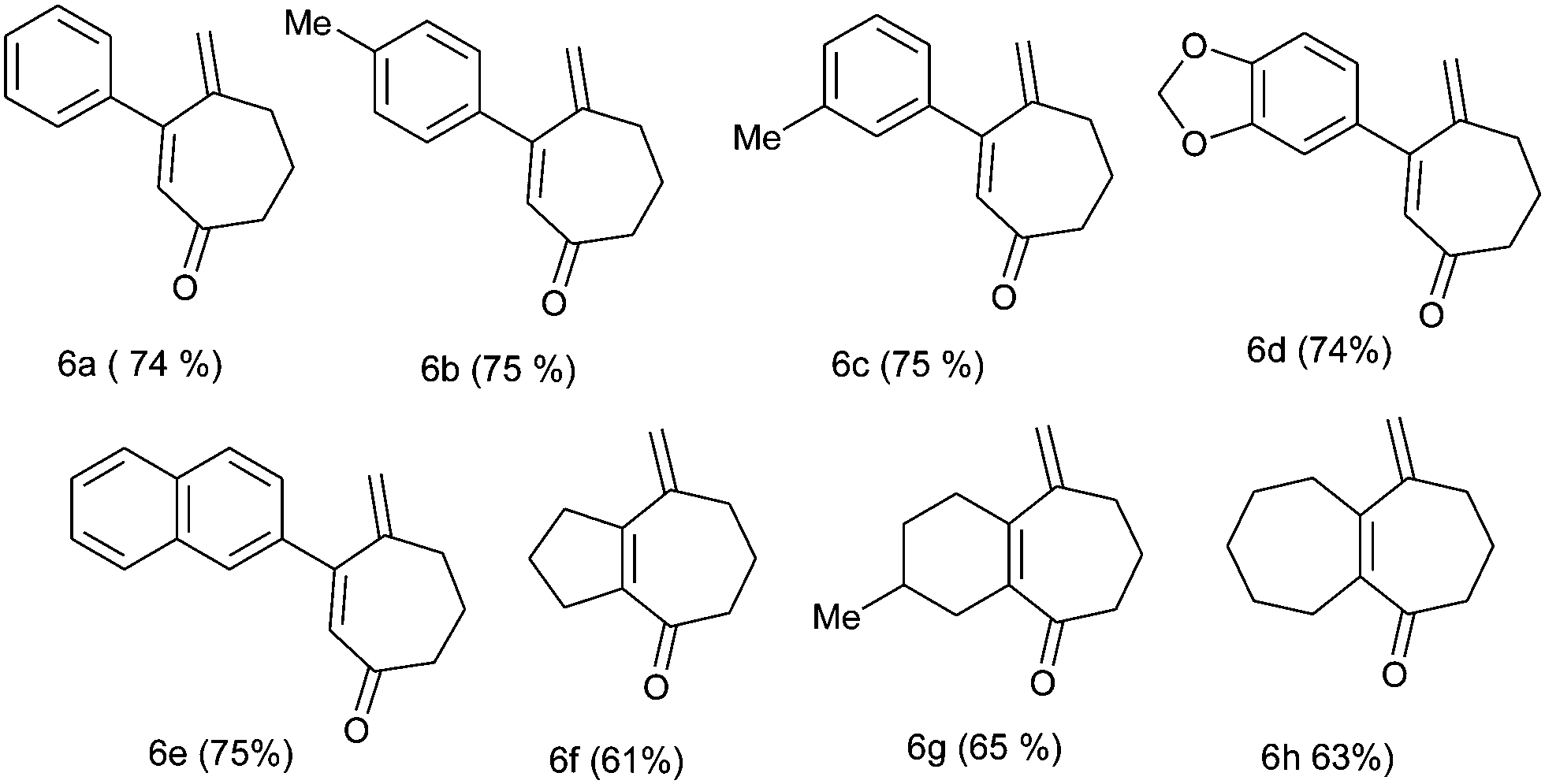
We further generalized the strategy using several other substrates (4b–4h) and obtained a number of substituted cycloheptenone derivatives in moderate to good yields (Table 3).
| a Yield refer to the isolated yield after purification. |
|---|
|
It should be noted that the formation of 3-aryl substituted cycloheptenone moieties (6a–6e) is associated with a somewhat higher yield compared to the cycloheptenone moiety containing 5,7; 6,7 and 7,7 fused bicyclic ring (6f–6h) systems. The structures of the products were supported by the analysis and spectral data.
We tried to understand the preference of 7-exo over the 8-endo product using Density Functional Theory based computations. Among the major steps in the catalytic cycle of the Heck reaction-catalyst coordination, oxidative addition, migratory insertion, reductive elimination etc. – cyclization occurs in the migratory insertion (MI) step, and hence, is the regeoselectivity deciding step of the reaction. Therefore, we focused our study on the MI step, considering 4g as the model precursor substrate. The substrate is bound to the Pd(II) centre through cyclopentadienyl carbon and the terminal olefin in the intermediate – named as int-OA-re/si – which undergo the MI step.
In the intermediate, Pd has other coordinating ligands in addition to the substrate. The identity of the other ligands depends on the active form of the catalyst. Although there have been many attempts reported in the literature, the nature of the active catalyst species is still elusive. One of the suggestions from previous experiments is Pd(OAc)L; where L is the triphenylphosphine ligand, as the active intermediate in the MI step (Scheme 4).13,14 The other possibilities suggested and used in theoretical studies include PdL2 or PdL as the active catalyst species.15,16 In this study, we have considered four such possibilities, viz, Pd(OAc)L, PdL2, PdL; and ClPdL (where L = PPh3) in the mechanistic study of the MI step.
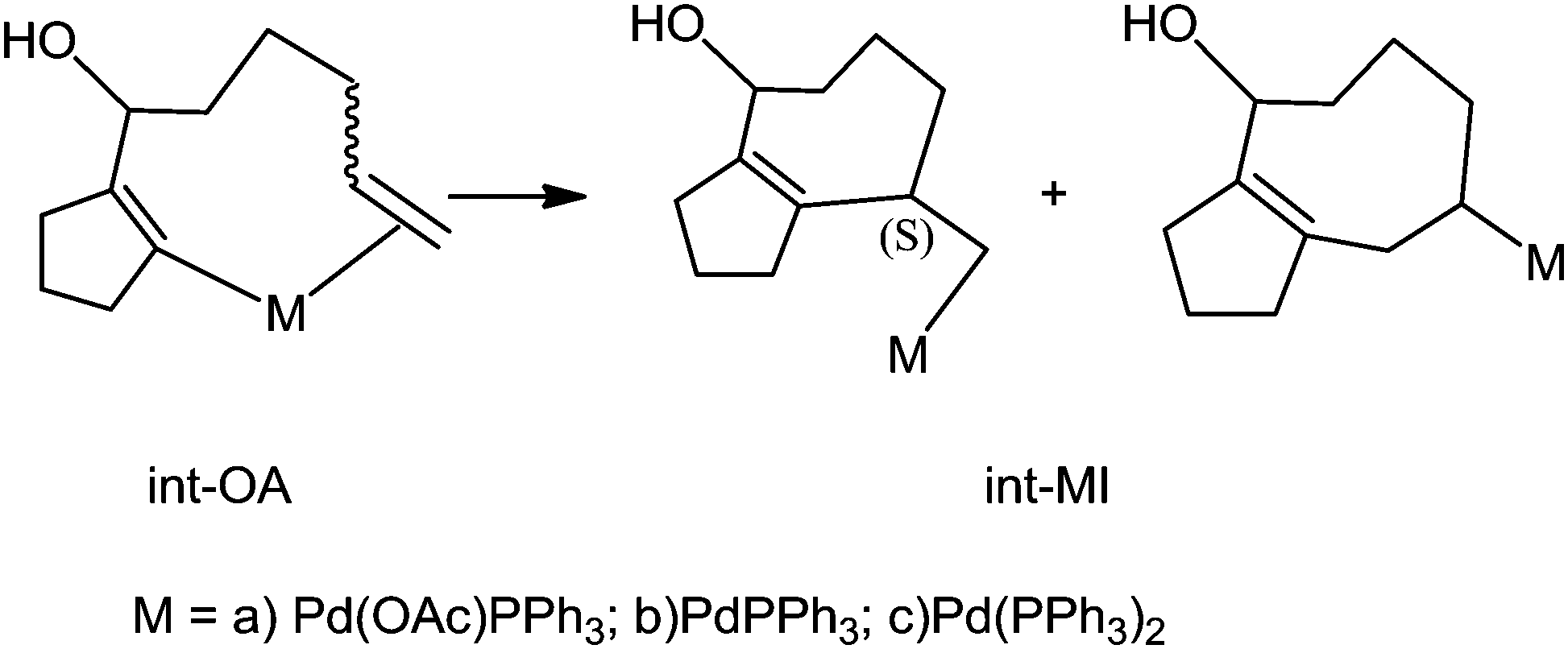 | ||
| Scheme 4 Migratory insertion. | ||
In all the pathways, there are some common geometrical features. In int-OA-re/si, Pd is coordinated with the substrate bound through cyclopentadienyl carbon and terminal olefin, in addition to the corresponding ligands. Depending on the face (re or si) through which the terminal olefinic double bond is coordinated with the Pd(II) centre, there are two possible isomers for this intermediate (Scheme 5). The names of the intermediates (int) and transition states (TS) are suffixed with re or si to indicate the originating intermediate. This naming convention is followed for all the catalyst pathways. From each of these isomeric intermediates, there are two possible pathways: coupling of C2 and C3 carbons leads to the 7-exo product, while coupling of C2 and C4 leads to the 8-endo product. The pathways with different active catalyst species are discussed below.
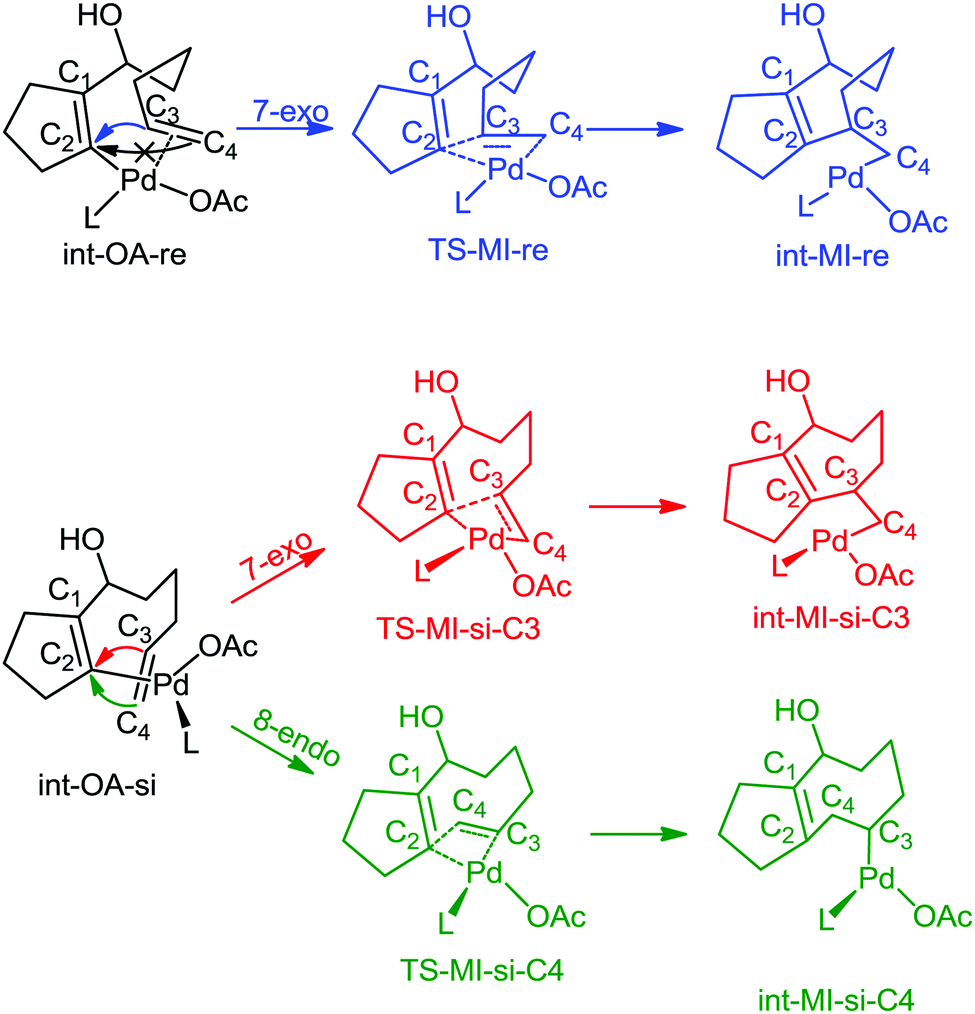 | ||
| Scheme 5 Mechanism of migratory insertion. Pd(OAc)L is shown as the active catalyst species in this scheme. The same mechanistic pathway was investigated for all the catalyst species. | ||
The Pd(OAc)L catalysed pathway
In this pathway, the coupling between C2 and C3 results in the intermediate int-MI-re. The activation free energy for this step is 14.19 kcal mol−1. The distance between C2 and C3 before coupling is 2.88 Å, which is reduced to 1.94 Å in the transition state, TS-MI-re. Optimized geometries are provided in the ESI.† The second pathway that leads to the formation of the 8-endo product from int-OA-re would require a major rotation of the olefin before the C4–C2 bond can be formed, because C2, C3 and C4 are in the same plane, and the C3–Pd bond hinders the direct coupling of the C4 atom with C2. Our attempts to find such a pathway were not successful. Thus the reaction of int-MI-re produces only the 7-exo product.On the other hand, the intermediate int-OA-si can lead to two different products via C3–C2 and C4–C2 coupling. The former leads to the intermediate int-MI-si-3, which eventually forms the 7-exo product and the latter forms int-MI-si-4, which leads to the 8-endo product. The C3–C2 and C4–C2 distances are 3.10 and 3.03 Å, respectively, in the OA intermediate. Here, we can anticipate a comparable barrier for these couplings. The activation free energy for the coupling between C3 and C2 is 32.18 kcal mol−1, whereas the coupling between C4 and C2 needs 34.93 kcal mol−1 of activation free energy. In TSs, the distance between C3 and C2 was reduced to 2.01 Å in TS-MI-si-C3 and the distance between C4 and C2 became 1.84 Å in TS-MI-si-C4. Both the activation barriers from int-OA-si are much higher than the activation barrier from int-OA-re.
The intermediate int-OA-re was found to be 5.68 kcal mol−1 higher in energy than the intermediate int-OA-si. However, the reactivity of the intermediate rather than the stability is important in a multistep reaction with short-lived intermediates. The reactivity depends on the activation barrier for the migratory insertion. The reaction profile diagram for the MI step from the OA intermediates is given in Fig. 1. The lowest barrier is found for the formation of the 7-exo product from the re- intermediate. The corresponding TS, TS-MI-re, is the most stable among the three TSs. Thus, the preference for the 7-exo product over the 8-endo product is in line with the experimental observation.
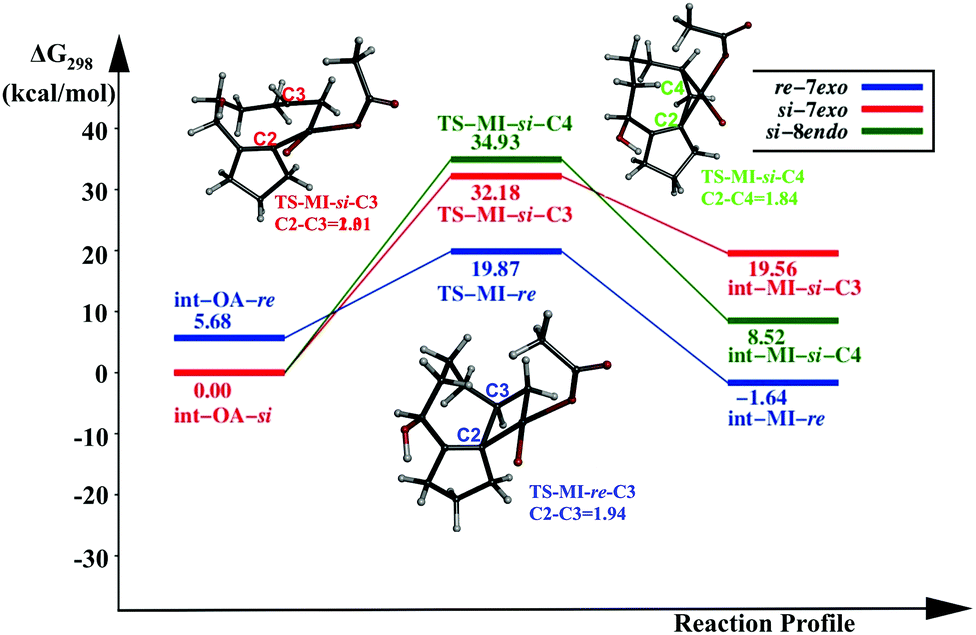 | ||
| Fig. 1 Reaction profile of the migration insertion (MI) step with [AcOPdL] as the catalyst. | ||
The PdL2 catalysed pathway
The catalytic cycle involving a PdL2 catalyst is another possible reaction pathway. We discuss here only the migratory insertion step for the reaction involving the PdL2 catalyst, following the same naming convention as in the previous section. In int-MI-re/si, the Pd(II) centre is coordinated with two triphenyl phosphine ligands, and the substrate is coordinated through cyclopentenyl carbon and the terminal olefin. The reaction profile diagram is given in Fig. 2.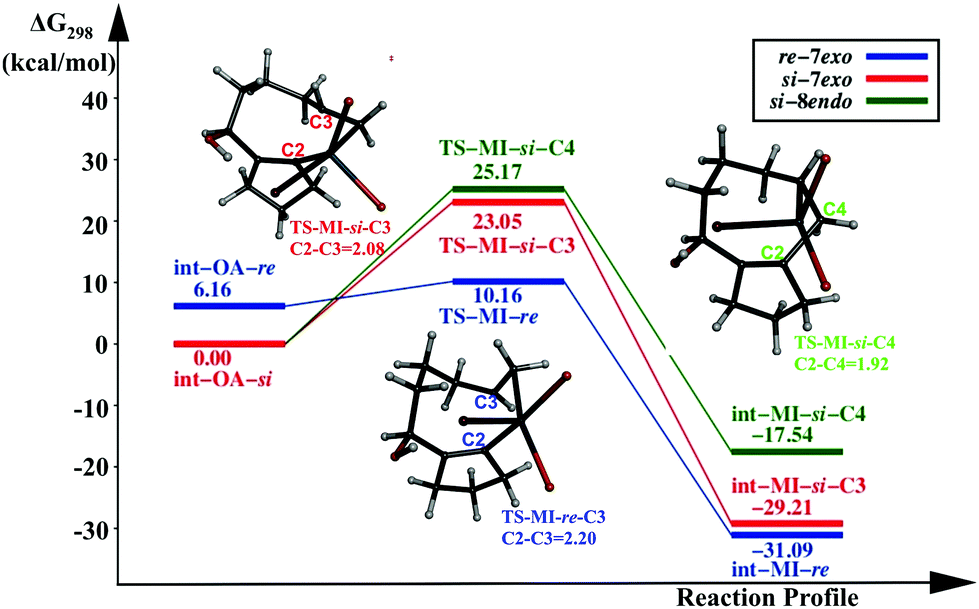 | ||
| Fig. 2 Reaction profile of the migration insertion (MI) step with [PdL2] as the catalyst. | ||
Two intermediates, similar to the Pd(OAc)L pathway, were considered for the MI step, viz, int-OA-re and int-OA-si. The C2–C3 coupling that leads to the formation of the intermediate int-MI-re, and eventually leads to the formation of the 7-exo product, requires an activation free energy of 4.0 kcal mol−1. Similar to the Pd(OAc)L pathway, int-OA-re does not lead to the 8-endo product, as the C4 atom is hindered from coupling with the C2 atom. The other intermediate, int-OA-si, is capable of undergoing two coupling reactions; C2 can couple with either C3 or C4 giving rise to 7-exo or 8-endo products, respectively. The corresponding activation energies are 23.05 and 25.17 kcal mol−1. Overall, comparing the three pathways, the formation of 7-exo is favored by the lowest activation barrier and the lowest energy TS. Thus, the PdL2 pathway also supports the experimental finding.
The PdL catalysed pathway
Another less explored pathway is the catalysis by PdL. The Pd centre is coordinated with one triphenylphosphine, and Br, in addition to the substrate. Three possible pathways originating from int-OA are investigated, similar to the previous cases. The reaction profile diagram is given in Fig. 3.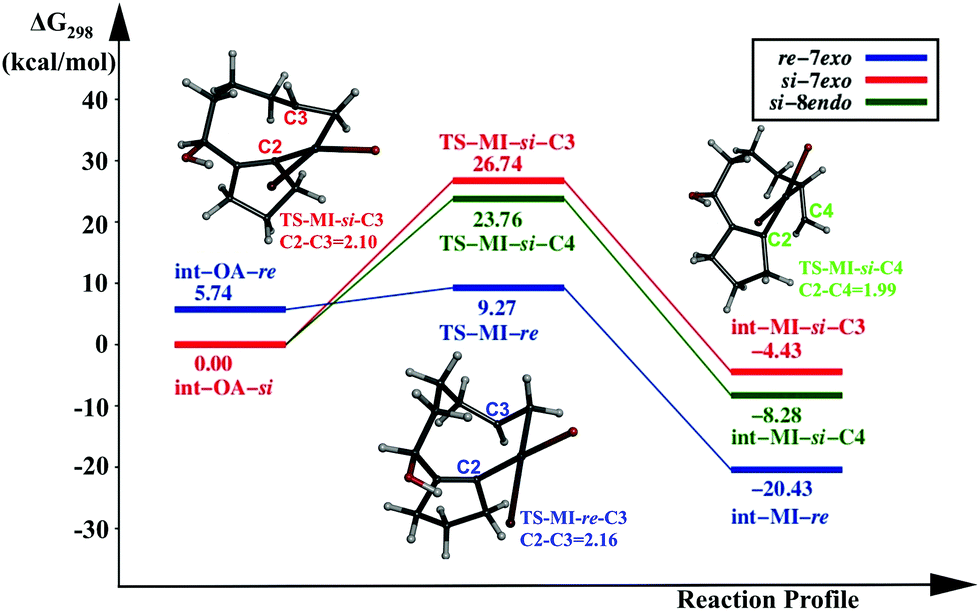 | ||
| Fig. 3 Reaction profile of the migration insertion (MI) step with [PdL] as the catalyst. | ||
The activation energy for the first pathway, i.e., C2–C3 coupling from int-OA-re., is 3.53 kcal mol−1, the lowest among all the pathways. The other two pathways from int-OA-si: (i) the coupling between C2 and C3, which leads to the 7-exo product, and (ii) the coupling between C2 and C4, which leads to the 8-endo product. The activation free energies for these two paths are 26.74 and 23.76 kcal mol−1, respectively. Here, the 8-endo pathway is lower in energy than the 7-exo pathway, unlike in the two previous catalyst systems. Although the -si reaction path indicates the preference for the 8-endo product over the 7-exo product, the -re path prefers the 7-exo product formation. The pathway from int-MI-re is the preferred one among the three pathways, with PdL as the active catalyst.
The PdLCl catalysed pathway
In addition to the three pathways mentioned above, we also investigated the reaction by taking LPdCl as the active catalyst, considering the possibility of the involvement of chloride from TBAC. In this case (Fig. 4), the activation energy for the C2–C3 coupling from int-OA-re is 9.86 kcal mol−1. The activation energies for the C2–C3 and C2–C4 coupling from int-OA-si are 24.83 and 25.28 kcal mol−1, respectively. So, if the chloride ion takes part in the reaction, it will facilitate it compared to the acetate ion.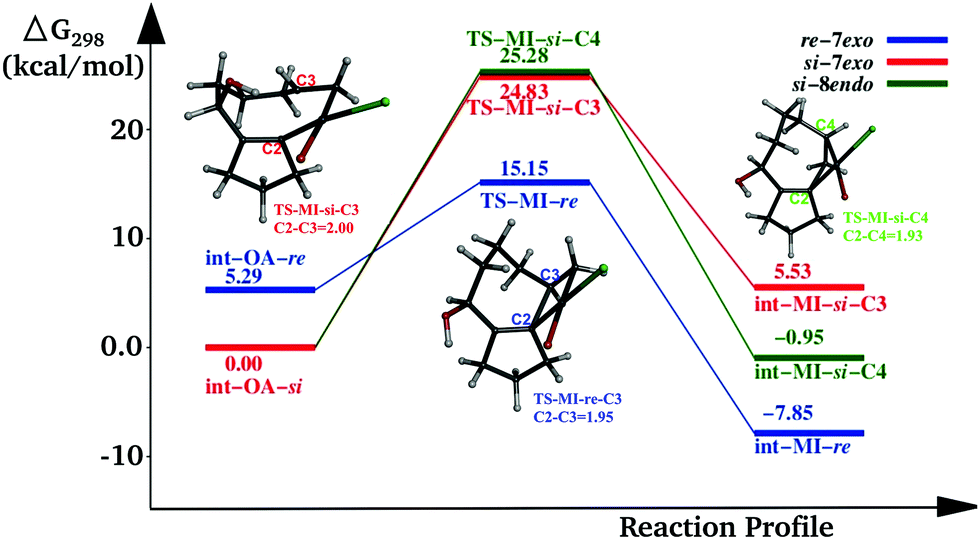 | ||
| Fig. 4 Reaction profile of the migration insertion (MI) step with [Cl-Pd-L] as the catalyst. | ||
Conclusions
In conclusion we have shown that 1-bromo-octa-1,7-diene-3-ol systems, readily prepared from the corresponding β-bromo vinyl aldehydes, serve as useful precursors to a number of novel cycloheptenone derivatives via Pd(0)-catalyzed 7-exo-trig cyclization and subsequent Pd(II)-catalyzed oxidation of cyclized alcohol using the Pd(OAc)2/Py/MS3A catalytic system under an O2 atmosphere. This methodology can be applied for the synthesis of various types of cycloheptenone moieties containing natural products in due time.From this study it can be concluded that the selectivity prefers to form the 7-exo intermediate rather than the 8-endo intermediate irrespective of the mode of the initial substrate coordination.
Experimental section
General procedure for the synthesis of 1-bromo-octa-1,7-diene-3-ol derivatives
5-Bromo-1-propene (2 mmol) was added to magnesium turnings (2.2 mmol) in diethyl ether for the formation of Grignard reagents. The reaction mixture was refluxed for 1 h and then stirred for 2 h at r.t. The Grignard reagent was added to a solution of bromoaldehyde (1 mmol) in diethyl ether at 0 °C. The resulting solution was allowed to warm at 25 °C and quenched with saturated ammonium chloride solution. The aqueous layer was extracted with diethyl ether. The combined ethereal extracts were washed with saturated sodium chloride solution, dried over (Na2SO4), and evaporated to give the crude alcohol which was purified using column chromatography.1-Bromo-1-phenyl-octa-1,7-diene-3-ol (4a)
Colorless liquid; yield = 89% (249 mg, 0.89 mmol); 1H NMR (CDCl3, 200 MHz) δ: 1.45–1.49 (2H, m), 1.66–1.73 (2H, m), 2.05–2.10 (2H, m), 4.64–4.74 (1H, m), 4.93–5.06 (2H, m), 5.70–5.92 (1H, m), 6.25 (1H, d, J = 7.6), 7.30–7.38 (3H, m), 7.52–7.57 (2H, m). 13C NMR (CDCl3, 50 MHz) δ: 24.5, 33.6, 35.7, 72.3, 114.8, 125.9, 127.6, 128.3, 128.9, 133.8, 138.5, 139.1. Anal. calcd for C14H17BrO: C, 59.80; H, 6.09. Found: C, 59.70; H, 6.00.1-Bromo-1-p-tolyl-octa-1,7-diene-3-ol (4b)
Colorless liquid; yield = 88% (219 mg, 0.88 mmol); 1H NMR (CDCl3, 200 MHz) δ: 1.52–1.78 (4H, m), 2.09–2.19 (2H, m), 2.37 (3H, s), 4.65–4.75 (1H, m), 4.93–5.09 (2H, m), 5.74–5.94 (1H, m), 6.22 (1H, d, J = 7.6), 7.15 (2H, d, J = 8), 7.45 (2H, d, J = 8.2). 13C NMR (CDCl3, 50 MHz) δ: 21.1, 24.5, 33.6, 35.8, 72.3, 114.8, 126.2, 127.5 (2 × C), 129.0 (2 × C), 132.9, 136.3, 138.6, 139.0. Anal. calcd for C15H19BrO: C, 61.03; H, 6.49. Found: C, 61.20; H, 6.33.1-Bromo-1-m-tolyl-octa-1,7-diene-3-ol (4c)
Colorless liquid; yield = 88% (259 mg, 0.88 mmol); 1H NMR (CDCl3, 200 MHz) δ: 1.52–1.78 (4H, m), 2.12–2.19 (2H, m), 2.37 (3H, s), 4.65–4.72 (1H, m), 4.95–5.10 (2H, m), 5.77–5.90 (1H, m), 6.24 (1H, d, J = 8), 7.12–7.16 (1H, m), 7.21–7.27 (1H, m), 7.33–7.36 (2H, m). 13C NMR (CDCl3, 50 MHz) δ: 21.4, 24.5, 33.6, 35.8, 72.3, 114.8, 124.8, 126.3, 128.2, 128.3, 129.7, 133.6, 138.0, 138.5, 139.1.1-Benzo[1,3]-dioxol-5-yl-1-bromo-octa-1,7-diene-3-ol (4d)
Colorless liquid; yield = 87% (282 mg, 0.87 mmol); 1H NMR (CDCl3, 200 MHz) δ: 1.50–1.78 (4H, m), 2.06–2.19 (2H, m), 4.63–4.73 (1H, m), 4.96–5.11 (2H, m), 5.75–5.92 (1H, m), 6.00 (3H, s), 6.16 (1H, d, J = 8), 6.75–6.81 (1H, m), 7.05–7.09 (2H, m). 13C NMR (CDCl3, 50 MHz) δ: 24.5, 33.6, 35.8, 72.3, 101.5, 107.9, 108.2, 114.9, 121.9, 125.5, 132.8, 133.5, 138.6, 147.6, 148.2.1-Bromo-1-naphthalene-2-yl-octa-1,7-diene-3-ol (4e)
Colorless liquid; yield = 88% (290 mg, 0.88 mmol); 1H NMR (CDCl3, 200 MHz) δ: 1.56–1.79 (4H, m), 2.11–2.21 (2H, m), 4.72–4.82 (1H, m), 4.96–5.12 (2H, m), 5.75–5.95 (1H, m), 6.41 (1H, d, J = 7.6), 7.46–7.55 (2H, m), 7.65 (1H, dd, J = 1.8, 8.6), 7.81–7.88 (3H, m), 8.03 (1H, d, J = 1.6). 13C NMR (CDCl3, 50 MHz) δ: 24.5, 33.6, 35.8, 72.4, 114.9, 124.8, 126.3, 126.7, 126.8, 127.5, 127.6, 128.0, 128.5, 132.9, 133.4, 134.2, 136.3, 138.5.1-(1-Bromo-cyclopent-1-enyl)-hex-5-en-1-ol (4f)
Colorless liquid; yield = 85% (207 mg, 0.85 mmol); 1H NMR (CDCl3, 200 MHz) δ: 1.34–1.65 (4H, m), 1.88–1.99 (2H, m), 2.01–2.11 (2H, m), 2.26–2.29 (2H, m), 2.57–2.65 (2H, m), 4.54 (1H, bs), 4.91–5.04 (2H, m), 5.68–5.88 (1H, m). 13C NMR (CDCl3, 50 MHz) δ: 21.6, 24.7, 29.4, 33.5, 34.3, 40.2, 69.3, 114.7, 117.3, 138.5, 142.3.1-(1-Bromo-5-methyl-cyclohex-1-enyl)-hex-5-en-1-ol (4g)
Colorless liquid; yield = 85% (231 mg, 0.85 mmol); 1H NMR (CDCl3, 200 MHz) δ: 0.97 (3H, d, J = 6), 1.25–1.74 (7H, m), 1.99–2.21 (4H, m), 2.51–2.53 (2H, m), 4.77 (1H, bs), 4.94–5.07 (2H, m), 5.71–5.91 (1H, m). 13C NMR (CDCl3, 50 MHz) δ: 21.2, 24.9, 28.2, 32.7, 33.3, 33.6, 33.9, 36.8, 74.1, 114.7, 119.4, 136.9, 138.7.1-(2-Bromo-cyclohept-1-enyl)-hex-5-en-1-ol (4h)
Colorless liquid; yield = 82% (223 mg, 0.82 mmol); 1H NMR (CDCl3, 200 MHz) δ: 1.26–1.50 (8H, m), 1.58–1.79 (2H, m), 2.04–2.11 (2H, m), 2.21–2.35 (2H, m), 2.74–2.79 (2H, m), 4.72–4.78 (1H, m), 4.93–5.06 (2H, m), 5.71–5.91 (1H, m). 13C NMR (CDCl3, 50 MHz) δ: 24.8, 25.3, 26.5, 27.3, 31.7, 33.5, 33.7, 41.6, 75.4, 114.6, 123.2, 138.6, 142.8.General experimental procedure for the Heck cyclization and Pd(II)-catalyzed oxidation of cyclic alcohol
The appropriate bis-homoallylated β-bromovinyl aldehyde (1 mmol), Pd(OAc)2 (∼11 mg,0.05 mmol), PPh3 (∼65 mg, 0.25 mmol), Na2CO3 (106 mg, 1 mmol) and TBAC (278 mg, 1 mmol) in acetonitrile were placed in a two necked flask. After degassing with N2 it was heated at 80 °C for 2 h. After complete consumption of the starting material (observed using TLC), the reaction mixture was cooled, filtered on a celite bed and washed with diethyl ether. The filtrate was washed with water and after drying (Na2SO4) the solvent was evaporated under reduced pressure to obtain the cyclized alcohol. Then, for the oxidation of cycloheptenol, pyridine (5 mmol) and MS3A (500 mg) were added to a mixture of Pd(OAc)2 (∼11 mg,0.05 mmol) and toluene (6 mL) in a two necked flask. O2 gas was introduced into the flask from an O2 balloon under atmospheric pressure. Then the mixture was heated to 80 °C for 10 min, cycloheptenol (1 mmol) in toluene (4 mL) was added in a dropwise manner and the mixture was heated for 6–7 h at the same temperature. After cooling, the reaction mixture was filtered through a pad of florisil. The solvent was evaporated under reduced pressure and the residue was then purified using column chromatography (silica gel, 5% ethyl acetate/petroleum ether) to afford the desired product.4-Methylene-3-phenyl-cyclohept-2-enone (6a)
Yellow liquid; yield = 74% (146 mg, 0.74 mmol); 1H NMR (CDCl3, 200 MHz) δ: 1.86–2.01 (2H, m), 2.55–2.67 (4H, m), 5.00 (1H, d, J = 1.4), 5.44 (1H, s), 6.19 (1H, s), 7.23–7.42 (5H, m). 13C NMR (CDCl3, 100 MHz) δ: 22.7, 36.7, 41.6, 122.3, 128.3 (2 × C), 128.7, 128.8 (2 × C), 129.3, 141.0, 147.0, 157.9, 203.0. Anal. calcd for C14H14O: C, 84.81; H, 7.12. Found: C, 84.69; H, 7.00.4-Methylene-3-p-tolyl-cyclohept-2-enone (6b)
Yellow liquid; yield = 75% (159 mg, 0.75 mmol); 1H NMR (CDCl3, 400 MHz) δ: 1.91–1.98 (2H, m), 2.37 (3H, s), 2.61–2.66 (4H, m), 5.04 (1H, s), 5.46 (1H, s), 6.22 (1H, s), 7.16 (2H, d, J = 8), 7.33 (2H, d, J = 8). 13C NMR (CDCl3, 100 MHz) δ: 22.1, 22.7, 36.7, 41.5, 122.0, 127.9, 128.8 (2 × C), 129.1 (2 × C), 138.0, 139.6, 147.1, 158.0, 203.0. Anal. calcd for C15H16O: C, 84.87; H, 7.60. Found: C, 84.79; H, 7.50. HRMS (ESI, 70 eV): m/z = 213.1275 [M+ + H] (calculated mass for C15H17O+: 213.1279 [M+ + H]).4-Methylene-3-m-tolyl-cyclohept-2-enone (6c)
Yellow liquid; yield = 75% (159 mg, 0.75 mmol); 1H NMR (CDCl3, 200 MHz) δ: 1.92–2.02 (2H, m), 2.36 (3H, s), 2.59–2.69 (4H, m), 5.03 (1H, d, J = 1.6), 5.46 (1H, s), 6.21 (1H, s), 7.16–7.25 (4H, m). 13C NMR (CDCl3, 50 MHz) δ: 21.4, 22.7, 36.7, 41.6, 122.2, 126.0, 128.2, 128.6, 129.5, 130.1, 137.9, 141.0, 147.1, 158.0, 203.1.Anal. calcd for C15H16O: C, 84.87; H, 7.60. Found: C, 84.90; H, 7.59.3-Benzo[1,3]-dioxol-5-yl-4-methylene-cyclohept-2-enone (6d)
Yellow semisolid; yield = 74 (178 mg, 0.74 mmol)%; 1H NMR (CDCl3, 200 MHz) δ: 1.87–2.00 (2H, m), 2.58–2.65 (4H, m), 5.08 (1H, d, J = 1.4), 5.47 (1H, s), 6.00 (2H, s), 6.18 (1H, s), 6.79 (1H, d, J = 8), 6.91–6.99 (2H, m). 13C NMR (CDCl3, 50 MHz) δ: 22.7, 36.6, 41.4, 101.5, 108.1, 108.9, 122.1, 123.3, 127.5, 134.9, 147.0, 147.7, 148.8, 157.6, 202.9. Anal. calcd for C15H14O3: C, 74.36; H, 5.82. Found: C, 74.50; H, 5.70. HRMS (ESI, 70 eV): m/z = 243.1030 [M+ + H] (calculated mass for C15H15O3+: 243.1021 [M+ + H]).4-Methylene-3-naphthalene-2-yl- cyclohept-2-enone (6e)
Yellow semisolid; yield = 75% (186 mg, 0.75 mmol); 1H NMR (CDCl3, 200 MHz) δ: 1.93–2.07 (2H, m), 2.65–2.77 (4H, m), 5.08 (1H, d, J = 1.4), 5.53 (1H, d, J = 1), 6.37 (1H, s), 7.43–7.55 (3H, m), 7.78–7.87 (3H, m), 7.93 (1H, d, J = 1.6). 13C NMR (CDCl3, 50 MHz) δ: 22.8, 36.8, 41.7, 122.4, 126.2, 126.5, 126.9, 127.6, 127.9, 128.5, 128.7, 129.1, 133.0, 133.7, 138.3, 147.0, 157.9, 202.9. HRMS (ESI, 70 eV): m/z = 249.1272 [M+ + H] (calculated mass for C18H16O: 249.1279 [M+ + H]).8-Methylene-2,3,5,6,7,8-hexhydro-1H-azulene-4-one (6f)
Yellow liquid; yield = 61% (98 mg, 0.61 mmol); 1H NMR (CDCl3, 200 MHz) δ: 1.77–1.91 (4H, m), 2.54–2.83 (8H, m), 5.19 (2H, s). 13C NMR (CDCl3, 50 MHz) δ: 20.9, 22.0, 35.2, 36.5, 37.7, 44.2, 118.4, 139.9, 144.6, 151.2, 202.0. Anal. calcd for C11H14O: C, 81.44; H, 8.70. Found: C, 81.49; H, 8.89.3-Methyl-9-methylene-1,2,3,4,6,7,8,9-octahydro-benzocyclohepten-5-one (6g)
Yellow liquid; yield = 65% (119 mg, 0.63 mmol); 1H NMR (CDCl3, 200 MHz) δ: 0.99 (3H, d, J = 6.2), 1.51–1.83 (5H, m), 2.19–2.67 (8H, m), 5.13 (1H, s), 5.18 (1H, d, J = 1.2). 13C NMR (CDCl3, 50 MHz) δ: 21.4, 22.3, 27.9, 30.8, 31.6, 33.3, 36.2, 41.5, 115.0, 135.9, 148.6, 151.3, 204.0. Anal. calcd for C13H18O: C, 82.06; H, 9.53. Found: C, 82.11; H, 9.69. HRMS (ESI, 70 eV): m/z = 205.1589 [M+ + H] (calculated mass for C18H16O: 205.1592 [M+ + H]).5-Methylene-3,4,5,6,7,8,9,10-octahydro-2H-heptalen-1-one (6h)
Yellow liquid; yield = 63% (120 mg, 0.63 mmol); 1H NMR (CDCl3, 200 MHz) δ: 1.42–1.83 (8H, m), 2.33 (2H, t), 2.49–2.64 (6H, m), 5.16 (1H, s), 5.19 (1H, s). 13C NMR (CDCl3, 50 MHz) δ: 22.6, 26.1, 26.2, 28.2, 32.2, 36.6, 37.1, 41.6, 115.6, 141.6, 150.4, 157.6, 204.3. Anal. calcd for C13H18O: C, 82.06; H, 9.53. Found: C, 82.09; H, 9.37. HRMS (ESI, 70 eV): m/z = 191.1440 [M+ + H] (calculated mass for C13H19O+: 191.1436 [M+ + H]).Computational methods
All calculations were performed using Density Functional Theory (DFT) and the TURBOMOLE V6.517,18 and ChemShell-3.5.019 program packages. All the optimizations were carried out at the BP8620,21/def2-SVP22 level of theory along with the resolution of identity (RI) approximation.23 Dispersion correction24 with BJ25 damping was used. Effective core potential (ECP) was used for Palladium. Analytical vibrational frequency calculations were performed at the same level to confirm the stationary points as minima (no imaginary frequency) or transition states (one imaginary frequency that corresponds to the reaction mode). The dynamic reaction coordinate (DRC)26 calculations were performed to further confirm that the transition state connects the correct reactants and intermediates in each elementary steps of the reaction profile. The Climbing Image Nudged Elastic Band (CI-NEB) method27 and the dimer method28 were used to obtain the guess transition state geometries as implemented in the ChemShell program package. Frequency and DRC calculations were carried out using TURBOMOLE. Furthermore, single point energy calculations were carried out at the B3LYP29/def2-SVP level of theory for all the geometries.Acknowledgements
We thank the Department of Science & Technology, Government of India, New Delhi for HPC facilities and financial support. S. P. thanks the Council of Scientific & Industrial Research, New Delhi, and DYR thanks the University Grants Commission (UGC) for the fellowship.References
- M. Endo, M. Nakagawa, Y. Hamamoto and Y. Nakanishi, J. Chem. Soc., Chem. Commun., 1983, 322 RSC.
- Y. Fukuyama, H. Minami, A. Matsuo, K. Kitamura, M. Akizuki, M. Kubo and M. Kodama, Chem. Pharm. Bull., 2002, 50, 368 CrossRef CAS PubMed.
- H. B. Yin, Z. S. He and Y. Ye, J. Nat. Prod., 2000, 63, 1164 CrossRef CAS.
- M. I. Choudhary, I. Batool, M. Atif, S. Hussain and A. U. Rahman, J. Nat. Prod., 2007, 70, 849 CrossRef CAS PubMed.
- X. L. Li, Y. Li, S. F. Yang, Y. L. Zhao, K. C. Liu, X. M. Wang and Y. P. Yang, J. Nat. Prod., 2009, 72, 1001 CrossRef CAS PubMed.
- (a) A. P. Wender, J. A. Dyckman, O. C. Husfeld and J. C. M. Scanio, Org. Lett., 2000, 2, 1609 CrossRef PubMed; (b) K. Tanino, F. Kondo, T. Shimizu and M. Miyashita, Org. Lett., 2002, 4, 2217 CrossRef CAS PubMed; (c) M. A. Battiste, P. A. Pelphrey; and D. L. Wright, Chem. – Eur. J., 2006, 12, 3438 CrossRef CAS PubMed.
- K. E. Hine and R. F. Childs, Can. J. Chem, 1976, 54, 12 CrossRef CAS.
- (a) Y. Oonishi, M. Mori and Y. Sato, Synthesis, 2007, 2323 CAS; (b) B. M. Trost and T. A. Runge, J. Am. Chem. Soc., 1981, 103, 7559 CrossRef CAS.
- X. Li, R. E. Kyne and T. V. Ovaska, J. Org. Chem., 2007, 72, 6624 CrossRef CAS PubMed.
- (a) S. K. Mal, D. Ray and J. K. Ray, Tetrahedron Lett., 2004, 45, 277 CrossRef CAS; (b) D. Ray, S. K. Mal and J. K. Ray, Synlett, 2005, 2135 CrossRef CAS; (c) D. Ray, S. Paul, S. Brahma and J. K. Ray, Tetrahedron Lett., 2007, 48, 8005 CrossRef CAS.
- (a) S. S. Stahl, Angew. Chem., Int. Ed., 2004, 43, 3400 CrossRef CAS PubMed; (b) B. Karimi and A. Zamani, J. Iran. Chem. Soc., 2008, 5, 1 CrossRef.
- T. Nishimura, T. Onoue, K. Ohe and S. Uemura, J. Org. Chem., 1999, 64, 6750 CrossRef CAS PubMed.
- C. Amatore, E. Carré, A. Jutand, M. A. M'Barki and G. Meyer, Organometallics, 1995, 14, 5605 CrossRef CAS.
- C. Amatore and A. Jutand, Acc. Chem. Res., 2000, 33, 314 CrossRef CAS PubMed.
- P. Vidossich, G. Ujaque and A. Lledós, Chem. Commun., 2014, 50, 661 RSC.
- R. Grüber and P. Fleurat-Lessard, Organometallics, 2014, 33, 1996 CrossRef.
- TURBOMOLE V6.5, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 19892007, TURBOMOLE GmbH since 2007.
- R. Ahlrichs, M. Br, M. Hser, H. Horn and C. Khmel, Chem. Phys. Lett., 1989, 162, 165 CrossRef CAS.
- ChemShell, a Computational Chemistry Shell, see http://www.chemshell.org.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822 CrossRef.
- A. Schaefer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571 CrossRef CAS.
- K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chem. Acc., 1997, 97, 119 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 1541 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 CrossRef CAS PubMed.
- K. M. Dieter and J. J. P. Stewart, J. Mol. Struct., 1988, 163, 143 CrossRef.
- G. Henkelman, B. P. Uberuaga and H. Jnsson, J. Chem. Phys., 2000, 113, 9901 CrossRef CAS.
- A. Heyden, A. T. Bell and F. J. Keil, J. Chem. Phys., 2005, 123, 224101 CrossRef PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra and DFT data. See DOI: 10.1039/c6nj02694j |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2017 |