DOI:
10.1039/C6NJ02246D
(Paper)
New J. Chem., 2017,
41, 271-277
Novel anti-algal nanocomposite hydrogels based on thiol/acetyl thioester groups chelating with silver nanoparticles†
Received
(in Montpellier, France)
19th July 2016
, Accepted 21st November 2016
First published on 22nd November 2016
Abstract
Novel nanocomposite (NC) hydrogels based on thiol/acetyl thioester groups chelating with silver nanoparticles (AgNPs) were designed and prepared. The AgNPs acted as both cross-linkers to produce stable NC hydrogels and antimicrobials to prevent the algal adhesion on the surface of hydrogels. The amount of AgNPs embedded in the NC hydrogels had a great influence on the mechanical properties of the NC hydrogels. When the feed molar ratio of the silver element to the sulfur element (Ag/S) approached 0.9, the hydrogels showed the highest mechanical properties. The dynamic nature of the interactions between the AgNPs and thiol/acetyl thioester groups endowed these NC hydrogels with interesting rapid mechanical recovery properties. The NC hydrogels containing thiol groups showed better self-recovery performance than the hydrogels with acetyl thioester groups. In addition, the NC hydrogels with AgNPs exhibited good anti-algal performance for Phaeodactylum tricornutum and Chlorella. When the Ag/S ratio exceeded 0.9, there were almost no algae on the surface of the NC hydrogels. Consequently, these hybrid hydrogels have promising applications in marine antifouling coating and the interfaces of biomaterials.
1. Introduction
Hydrogels with multiple functionalities and complexity have attracted considerable attention due to their potential applications in drug delivery, shape memory devices, tissue regeneration, sensors, wound healing, etc.1–4 The poly(N-isopropyl acrylamide) (PNIPAM) hydrogel is biocompatible and contains up to 90% (weight%) of water. Interestingly, the PNIPAM hydrogel is a typical thermo-responsive hydrogel. The hydrogels with PNIPAM as the polymer backbone can act as smart carriers for guests and have found applications in drug release,5 control switching6 and tissue engineering.7
Nanocomposite (NC) hydrogels were a combination of inorganic nanocomponents and soft, deformable, elastic three-dimensional hydrogel networks. Clearly, nanocomponents can endow the NC hydrogels with many entirely new properties, which are not accessible to conventional hydrogels.8–10 Nanocomponents, especially metallic nanoparticles (NPs), have already found widespread application in catalysis,11 electronics,12 photonics,13 biotechnology and medicine.14 Silver nanoparticles (AgNPs) have an extremely large surface and are being incorporated into products that range from photovoltaics to biological and chemical sensors.15–18 A large surface area permits the coordination of a vast number of ligands. The interactions between ligands and AgNPs can reach the strength of chemical bonds, while still being reversible. Their dynamic nature has proved to be a promising tool to build self-healing materials.19–21 Soft materials with self-healing ability are especially promising for a variety of medical applications.22–24 Moreover, another increasingly common application is the use of AgNPs for antimicrobial coatings.15,25 For example, Bajpai and coworkers reported the in situ formation of AgNPs in the PNIPAM hydrogel for antibacterial applications against Escherichia coli.15 The most common methods of fabricating such functional NC hydrogel systems were based on the physical incorporation of AgNPs into the polymer matrix.15,26,27 Namely, metal ionic precursors were firstly infiltrated into a preformed hydrogel, followed by chemical reduction or photoinduced nanoparticle growth. However, the nanoparticles in such prepared NC hydrogel systems often rapidly diffuse out of the hydrogel, partly because of the absence of a strong association between nanoparticles and polymer chains. Such uncontrolled loss of AgNPs resulted in unwanted agglomerations, toxicity problems as well as increasing material costs.28,29
To overcome the drawbacks of AgNP leakage, it is desirable for AgNPs to be immobilized and stabilized by a strong metal–ligand coordination with the polymeric chains. Various strategies have been used to develop chemically cross-linked NC hydrogels to prevent the leakage of AgNPs.30–34 For example, Gabilondo's group reported a bionanocomposite hydrogel with embedded AgNPs through the interaction between benzotriazole and AgNPs.33 Kim et al. prepared an AgNP-loaded hydrogel with minimal loss of nanoparticles, and this NC hydrogel successfully inhibited the bacterial growth.34
Gold(I)/silver(I)–thiolate (Au/Ag–S) compounds are currently of prime importance for the preparation of metallic nanoparticles.35,36 Recently, our group has developed a thermo-sensitive NC hydrogel based on the strong chelation between polythioether dendrons and Ag nanocrystals.37 The dynamic nature of the interaction between sulfurated polymers and AgNPs has the advantage of building self-healing materials. Odriozola developed a self-healing silicone elastomer with AgNPs as cross-linkers.20 However, the functional NC hydrogels based on the AgNPs–thiol/acetyl thioester group are rarely reported.
Herein, we designed a series of functional nanocomposite hydrogels based on thiol/acetyl thioester groups chelating with AgNPs (Scheme 1). The NC hydrogel systems exhibited interesting mechanical recovery properties and anti-algal behavior. The NC hydrogel design was made on the basis of the following considerations. Firstly, the strong coordination between the thiol/acetyl thioester groups and AgNPs can facilitate the NC hydrogel formation and effectively reduce the loss of silver elements. Secondly, the dynamic nature of the metal–ligand interaction may introduce self-healing properties to the hydrogel system. Thirdly, this NC hydrogel containing AgNPs could be used to prevent the bacterial or algal growth and adhesion on the material surface.
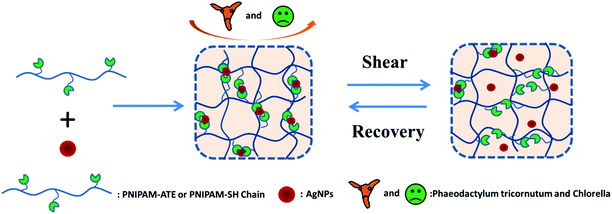 |
| | Scheme 1 Schematic illustration of the formation and self-recovery mechanism of NC hydrogels. | |
2. Experimental
2.1 Materials
Glycidyl methacrylate (GMA), bisacrylamide (BIS), N-isopropylacrylamide (NIPAM), thioacetic acid (TA) and the photo-initiator benzoin dimethyl ether (DMPA) were purchased from Aladdin (Shanghai, China). NIPAM was recrystallized in n-hexane to remove polymerization inhibitors. The reductant NaBH4 and trifluoromethanesulfonic acid silver salt were obtained from Sinopharm Chemical Reagent Co. Ltd (Shanghai, China). The other reagents were used as received. Artificial seawater is composed of NaCl (26.73 g L−1), MgCl2 (2.26 g L−1), MgSO4 (3.25 g L−1), CaCl2 (1.15 g L−1), NaHCO3 (0.20 g L−1), KCl (0.72 g L−1), NaBr (0.058 g L−1), H3BO3 (0.058 g L−1), Na2SiO3 (0.0024 g L−1), H3PO4 (0.0020 g L−1), Al2Cl6 (0.013 g L−1), NH3 (0.0020 g L−1) and LiNO3 (0.0013 g L−1).
2.2 Synthesis and characterization of polyPNIPAM-ATE and PNIPAM-SH
The synthetic procedures are shown in Scheme 2. 2-Methyl-acrylic acid 3-acetylsulfanyl-2-hydroxy-propyl ester (MAASE) was synthesized according to the literature.38 The sulfurated polymer poly(2-methyl-acrylic acid 3-acetylsulfanyl-2-hydroxy-propyl ester-co-N-isopropylacrylamide) (PNIPAM-ATE) was prepared by normal radical copolymerization of MAASE and NIPAM monomers. 10.00 g of NIPAM, 2.00 g of MAASE and 0.12 g of the photo-initiator DMPA were dissolved in 50.0 mL of tetrahydrofuran (THF). After purging with nitrogen gas for 10 min to eliminate oxygen, the mixture was irradiated in an ice-bath with a UV lamp (365 nm, 26 W) for 10 hours. The product PNIPAM-ATE was precipitated in anhydrous ether and dried in a vacuum oven. PNIPAM-SH was prepared from PNIPAM-ATE by a simple triethylamine catalyzed acetyl migration reaction. 9.00 g of PNIPAM-ATE was dissolved in 50.0 mL of chloroform (CH3Cl) and then 1.00 g of triethylamine was added dropwise to the reaction system. The nitrogen gas was purged for 10 min to eliminate oxygen. After stirring for 10 hours, the mixture was dropped in anhydrous ether to obtain the product PNIPAM-SH. The white precipitate was dried in a vacuum oven for 24 hours.
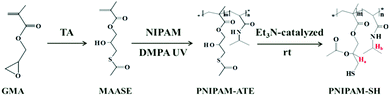 |
| | Scheme 2 Synthetic route of polymers PNIPAM-ATE and PNIPAM-SH. | |
1H Nuclear Magnetic Resonance Spectroscopy (1H NMR): compounds GMA, MASSE, PNIPAM-ATE and PNIPAM-SH were dissolved in D-choloform (CDCl3) or deuterium oxide (D2O). The solutions were measured on a Bruker 400 MHz AVANCE III spectrometer at 20 °C with tetramethylsilane (TMS) as an internal reference.
Gel Permeation Chromatography (GPC): the molecular weights of PNIPAM-ATE and PNIPAM-SH were determined by gel permeation chromatography (HLC-8320) with THF as an eluent.
2.3 Preparation of hydrogels
Silver nanoparticles (AgNPs) acted as both crosslinkers to prepare hydrogels by chelation with thiol/acetyl thioester groups and antiseptics to kill microorganisms. The experimental details are as follows: 1.50 g of sulfurated polymer PNIPAM-ATE (PNIPAM-SH) was dissolved into 6.0 mL of dimethylformamide (DMF). Different volumes (100 μL, 50 μL, 25 μL, 10 μL and 1 μL) of silver trifluoromethanesulfonate (1.95 mol L−1) in DMF solution were, respectively, added to 1.0 mL of the above prepared solution. A certain volume of DMF (0 μL, 50 μL, 75 μL, 90 μL and 99 μL) was, respectively, added to the mixed system. The mixed solutions were stirred and kept homogeneous. Then, sodium borohydride (NaBH4) in DMF solution (250 μL, 1.65 mol L−1) was added to the solution as described above. The obtained black mixtures were injected into molds and stable hydrogels were formed after 24 hours. These hydrogels were transferred to deionized water and immersed for one week to remove the DMF. A chemically cross-linked PNIPAM-ATE hydrogel with BIS as a cross-linker was prepared as a control sample.
2.4 Characterization of silver nanoparticles (AgNPs)
The morphology of AgNPs was investigated using Transmission Electron Microscopy (TEM, Tecnai F20). The hydrogel was dispersed in deionized water, and one drop of the dispersion was placed on a 3 mm carbon-coated Cu grid. The sample was dried at ambient temperature for 24 hours. Absorption spectra of the nanocomposite hydrogel were recorded in the wavelength range of 250 to 600 nm using a UV-Vis-NIR spectrophotometer (LABMDA 950).
2.5 Rheological measurements
Rheological and mechanical recoveries were characterized using a rheometer (RS6000, HAAKE) equipped with a steel parallel-plate geometry. The samples were cut into discs of similar size (∅ = 15 mm and 0.15–0.30 mm gap). Firstly, strain amplitude sweeps (γ = 0.1–100%) were performed at a constant angular frequency (ω = 1 rad s−1) to confirm the linear viscoelastic region (LVR). Frequency sweep experiments were performed in the LVR (γ = 1%) from 0.1 to 100 Hz. The cycle of deformation and the recovery of the hydrogels include two steps: (1) deformation: a constant oscillatory strain (200%) that is enough to destroy the gel is applied to the fresh gel in the sample holder for 100 seconds. The frequency of the measurement is 6.28 rad s−1. (2) Modulus recovery in a time sweep: the recovery of the storage modulus is monitored at a constant frequency (6.28 rad s−1) under a low strain (1%) within 50 seconds. The storage modulus G′ and the loss modulus G′′ are recorded as a function of time in both processes.
2.6 Algae static settlement assay
Phaeodactylum tricornutum and Chlorella were chosen to evaluate the algal settlements on the surface of hydrogels. The experimental details were as follows: these nanocomposite hydrogels were immersed in seawater for 24 hours in order to keep their shape stable and then were cut into disks and fixed at the bottom of a 24-well plate. 1.0 mL of the microorganism suspension was added to each well and the plate was incubated in a biochemical incubator for 24 hours. The initial concentrations of Phaeodactylum tricornutum and Chlorella were (2.7 ± 0.3) × 108 and (1.7 ± 0.3) × 108 cells per mL, respectively. After incubation for 24 hours, these hydrogels were washed three times with artificial seawater to remove the cells that did not adhere or adhered loosely. The cells on the surface of the samples were fixed by a 2.5 vol% glutaraldehyde solution for 15 min. Phaeodactylum tricornutum and Chlorella on the surface of hydrogels were observed using a Laser Scanning Confocal Microscope (LSCM, Dimension 3100v) analysis system.
3. Results and discussion
3.1 Synthesis and characterization of the sulfurated polymer
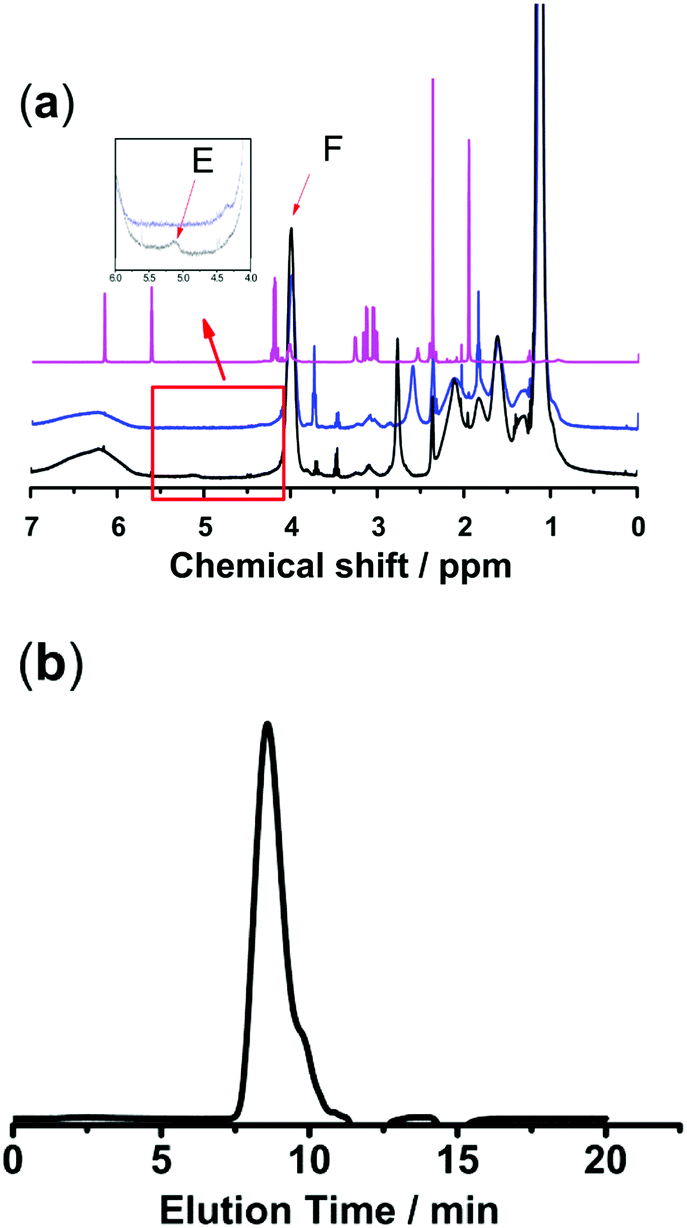
Polymer networks with AgNPs as cross-linking sites to build self-healing elastomers have already been reported.16,17 Inspired by this method of developing self-healing materials, we designed a new class of nanocomposite hydrogels based on thiol/acetyl thioester groups chelating with AgNPs. Two kinds of sulfurated polymers PNIPAM-ATE and PNIPAM-SH were synthesized successfully. Fig. 1a shows the 1H NMR spectra of MAASE, PNIPAM-ATE and PNIPAM-SH. The peak E at 5.15 ppm of compound 3 corresponded to the Ha proton in the tertiary carbon of the adjacent acetyl group. The peak F at 4.05 ppm was attributed to the Hb proton of the PNIPAM segment in PNIPAM-SH. The molar ratio of NIPAM to the MAASE segment in the polymer PNIPAM-SH calculated using the integral area ratio of peaks F and E is approximately 15. The molecular weight and distribution of PNIPAM-ATE are shown in Fig. 1b. Combined with the analysis results from the above 1H NMR spectra, it can be calculated that there is on average 3 chain units of MAASE and 48 chain units of NIPAM in a single PNIPAM-SH chain.
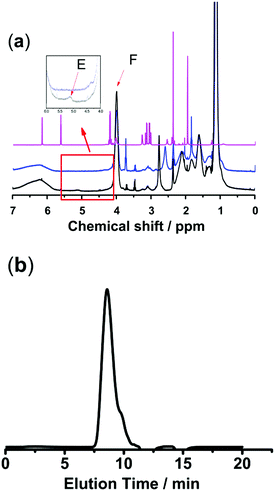 |
| | Fig. 1 (a) 1H NMR spectra of compounds (1) MAASE, (2) PNIPAM-ATE and (3) PNIPAM-SH. (b) THF-GPC trace of PNIPAM-ATE showed a monomodal signal: Mn = 6104, Mw = 44![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 262, and Mw/Mn = 7.251. 262, and Mw/Mn = 7.251. | |
3.2 Gelation performance and analysis
The molar ratio of the silver element to sulfurated groups (thiol or acetyl thioester) greatly influenced the macrostate of the polymeric system. Table 1 displays PNIPAM-ATE and PNIPAM-SH hydrogels with different ratios of Ag and sulfurated groups (thiol or acetyl thioester). When the ratio of Ag to sulfurated groups was too low, the materials presented a viscous fluid-like state and poor mechanical properties. When the Ag/S ratio reached 0.018, a black and stable NC hydrogel was formed. However, if the ratio exceeded 5, a solid grain-like state instead of the hydrogel was found. The reasonable explanation was that if the molar ratio was too small, the lack of AgNPs led to a reduction of the crosslinker and finally resulted in a viscous liquid. Upon increasing the ratio, the crosslinker sites between the polymer chains and AgNPs increased and finally produced a stable hydrogel. In contrast, when the ratio was too large, the aggregation of AgNPs resulted in low chain density and low crosslinking. Therefore, the molar ratio of the silver element to sulfurated groups was a key factor in the formation of hydrogels.
Table 1 Optimal design of AgNP-cross-linked materials by tuning Ag/S
| Ag/S |
12.7 |
5.6 |
1.8 |
0.9 |
0.46 |
0.18 |
0.018 |
| PNIPAM-ATE hydrogel |
A |
B |
C |
D |
E |
F |
G |
| PNIPAM-SH hydrogel |
A1 |
B1 |
C1 |
D1 |
E1 |
F1 |
G1 |
| State |
Solid grain |
Hydrogel |
Viscous fluid-like |
| Optical photograph |

|

|
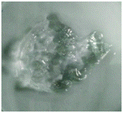
|
| Schematic illustration |
|
|
|
In addition, the Ag/S ratio also has a great influence on the solid contents and the equilibrium swelling ratio of the hydrogels. Fig. S1a (ESI†) clearly shows that the solid contents of the hydrogels increased upon increasing the Ag/S ratio, and the PNIPAM-SH hydrogel showed higher solid contents than the PNIPAM-ATE hydrogel at the same Ag/S ratio. The equilibrium swelling ratio of the hydrogels decreased upon increasing the Ag/S ratio, and the PNIPAM-SH hydrogel showed higher solid contents than the PNIPAM-ATE hydrogel at the same Ag/S ratio (Fig. S1b, ESI†).
3.3 The characterisation of AgNPs
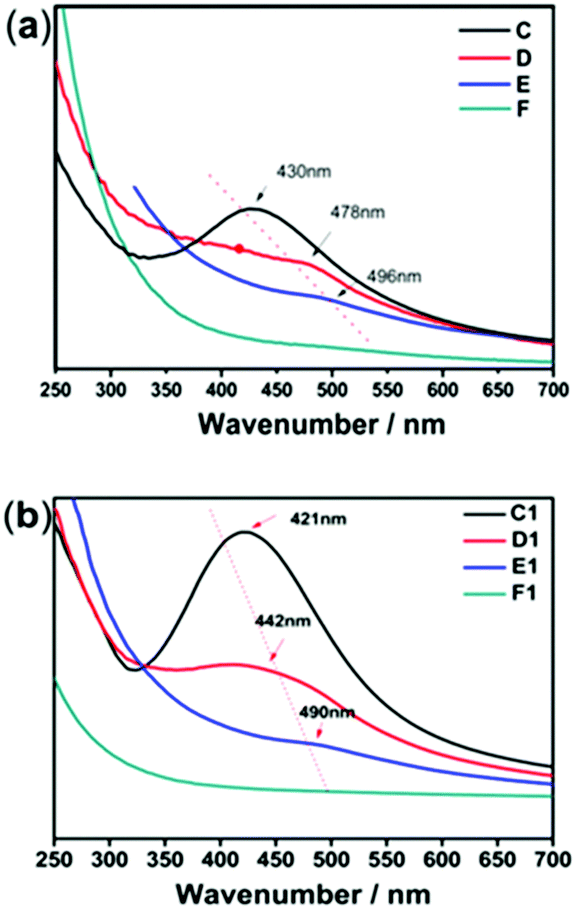
The size, morphology and content of AgNPs embedded in the hydrogel were characterized by UV-Vis absorption spectra, TEM and TGA. Fig. 2 shows the UV-Vis spectra of the hydrogel samples containing AgNPs. The characteristic absorption bands of PNIPAM-ATE hydrogels C, D, and E were at 430, 478, and 496 nm, respectively, which indicated the formation of AgNPs. It is reported that the absorption band shifting to longer wavelengths indicates that the metal particle size increases.39 Therefore, the size of AgNPs decreased upon increasing the content of AgNPs. The size of the nanoparticles in the PNIPAM-SH system showed a similar trend. Compared with PNIPAM-ATE hydrogels, the characteristic absorption bands of the PNIPAM-SH hydrogel shifted to lower wavelengths. We speculated that the stronger chelating ability of the thiol groups and AgNPs could disperse silver ions well in the hydrogel system and finally result in the formation of small-sized AgNPs. This hypothesis was further confirmed by TEM images. Fig. 3 displays TEM images of AgNPs in PNIPAM-ATE and PNIPAM-SH hydrogels. The sizes of Ag particles in all hydrogels presented are less than 100 nm. In detail, the size of AgNPs in PNIPAM-SH hydrogels is slightly smaller than that in the PNIPAM-ATE hydrogel (Fig. S2, ESI†). From Fig. S3 (ESI†), it can be observed that the hydrogels containing acetyl thioester groups showed a lower cumulative released amount of silver elements than the pristine PNIPAM hydrogel.
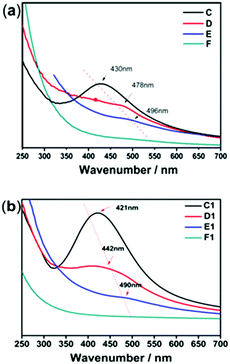 |
| | Fig. 2 UV-Vis spectra of (a) PNIPAM-ATE hydrogels and (b) PNIPAM-SH hydrogels. | |
 |
| | Fig. 3 TEM images of AgNPs in PNIPAM-ATE and PNIPAM-SH hydrogels. (a) and (c) represent PNIPAM-ATE hydrogels C1 and E1, respectively, (b) and (d) represent PNIPAM-SH hydrogels C and E, respectively. | |
3.4 Rheological studies of AgNP hybrid gels
The mechanical and self-recovery properties of these hydrogels were investigated by rheological measurements. Dynamic frequency sweep data (Fig. 4) showed that the storage modulus G′ is greater than the loss modulus G′′ in all tested hydrogel samples, indicating dominant elastic characteristics of the NC hydrogels. As shown in Fig. 4a, the storage modulus G′ increased firstly and then decreased upon increasing the content of AgNPs. The C1 sample showed the highest storage modulus at low frequency in all hydrogel samples, indicating the best elastic character. One reasonable explanation is that too high or low Ag/S ratio endows the system with low cross-linking density and results in poor mechanical properties. Besides, the storage modulus G′ of the PNIPAM-SH hydrogels had a similar trend (Fig. 4b). As shown in Fig. 4c, the storage modulus G′ of PNIPAM-SH hydrogels is greater than that of PNIPAM-ATE hydrogels with the same content of AgNPs. It is speculated that the chelating ability of thiol groups to the AgNP surface is stronger than that of the acetyl thioester groups, thus resulting in improved mechanical properties.
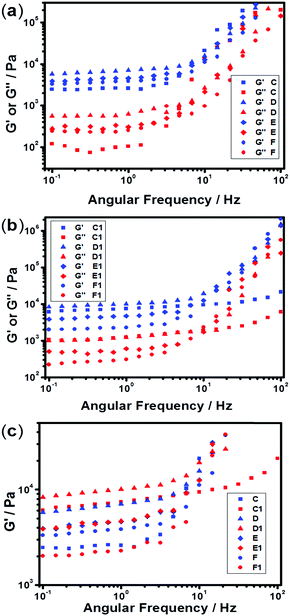 |
| | Fig. 4 Frequency dependence of storage modulus G′ and loss modulus G′′ for NC hydrogels at 25 °C with a frequency from 0.01 to 100 rad s−1 and strain γ = 1%. (a) PNIPAM-ATE and (b) PNIPAM-SH hydrogels with different contents of AgNPs. (c) The comparison of G′ in PNIPAM-ATE and PNIPAM-SH hydrogels. | |
3.5 Self-recovery of the NC hydrogels under continuous step strain
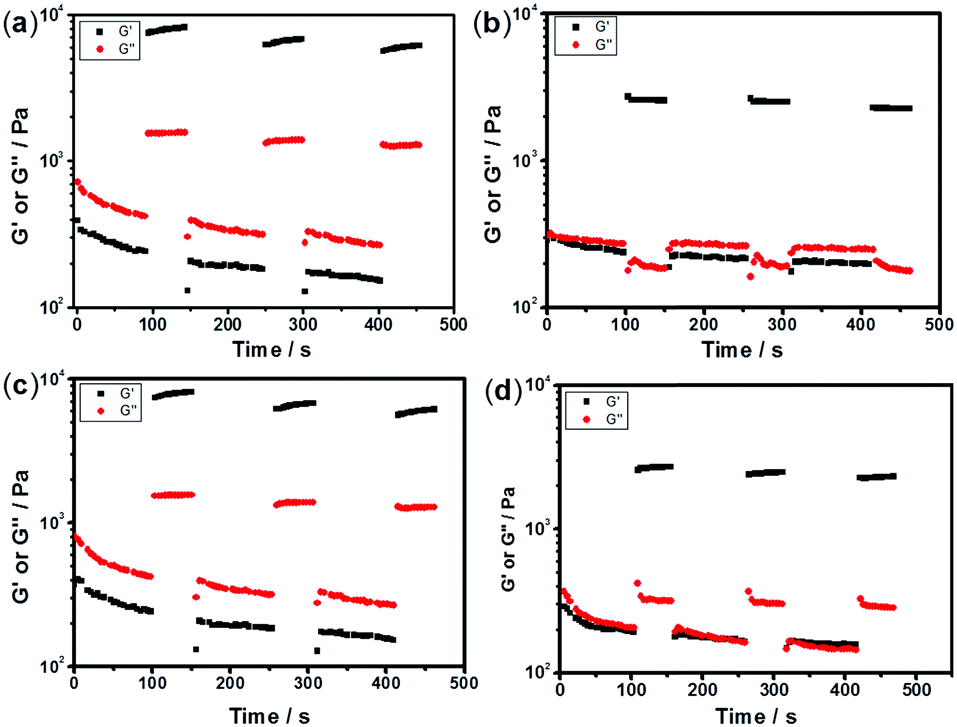
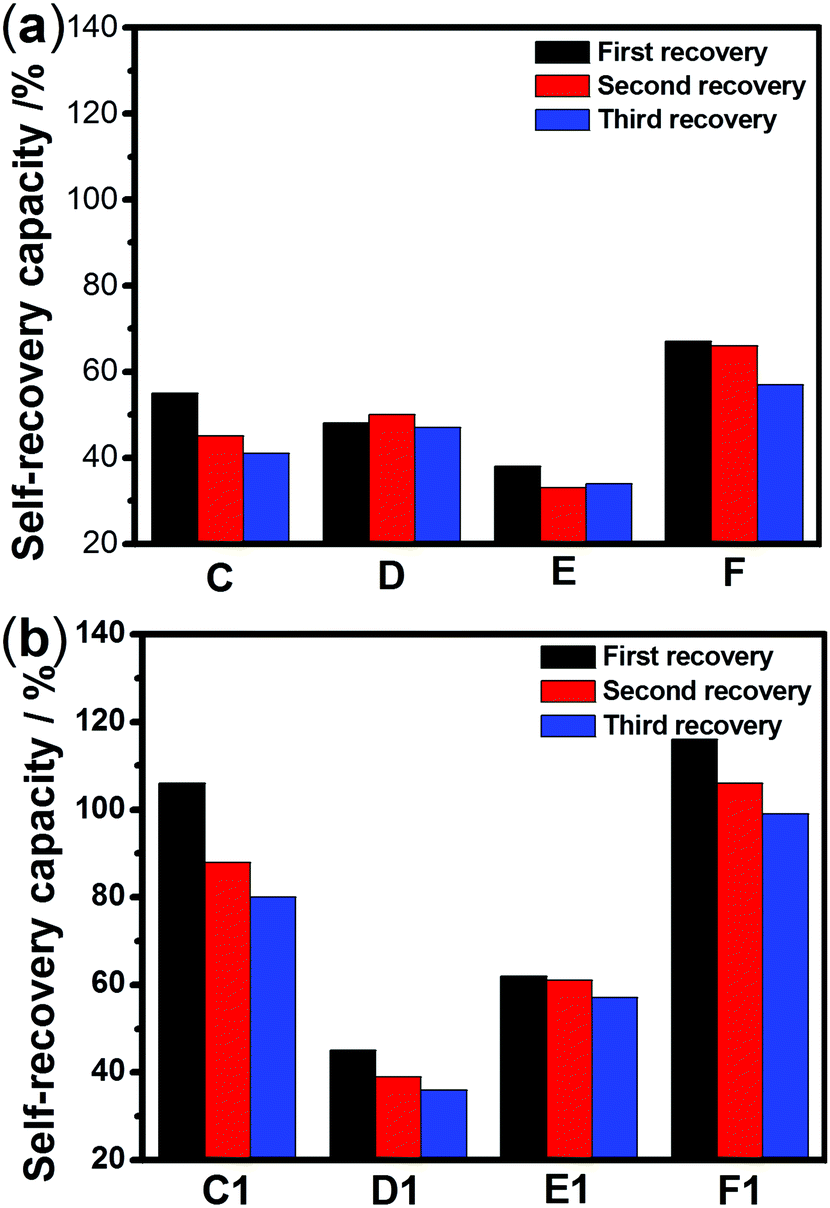
The sulfurated polymer hybrid hydrogel systems with AgNPs as crosslinkers are scarcely reported. The NC hydrogels showed rapid recovery of their mechanical properties after being broken under the application of a large-amplitude oscillatory (Fig. 5). Under the application of a large-amplitude oscillatory force (γ = 200%), the storage modulus and loss modulus simultaneously decreased and G′′ is greater than G′, and the materials showed a quasi-liquid state. When the amplitude oscillatory force decreased to 1%, the storage and loss modulus recovered and G′ is larger than the G′′, indicating that the quasi-liquid state transformed into a quasi-solid state. In order to compare the self-recovery capacity of these hydrogels, we here defined the self-recovery capacity as the ratio of recovered G′ to initial G0′, where G0′ was obtained from Fig. 4 (the G′ at an angular frequency of 1.0 Hz). Fig. 6 shows the calculated self-healing capacity of all PNIPAM-ATE and PNIPAM-SH hydrogel samples. Upon increasing the cycle of measurements, the self-recovery ability of the hydrogel samples decreased. It can be explained that the microstructure of the hydrogel was slightly damaged irreversibly when applied with a large-amplitude oscillatory force. The stronger interaction of silver nanoparticles and thiol groups of PNIPAM-SH hydrogels resulted in better self-recovery performances than those of PNIPAM-ATE gels. As shown in Fig. 6, the self-recovery capacity of PNIPAM-SH or PNIPAM-ATE hydrogels decreased at first and then increased upon increasing the Ag/S ratio. More free AgNPs and free polymer chains in the hydrogels are beneficial to the recovery of the damaged hydrogels.
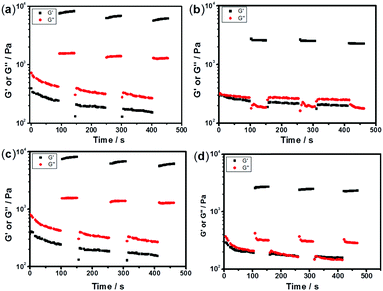 |
| | Fig. 5 The time dependence of G′ and G′′ in alternate strain measurements; the gel was first given a strong strain of 200% for 100 s and then subjected to a low strain of 1% for 50 s, following a small strain of 1% for its recovery. This experiment was repeated twice to detect the reversibility, with the frequency being maintained at 6.28 rad s−1. (a), (b), (c) and (d) represent the self-recovery performance of hydrogels C, F, C1, and F1 respectively. | |
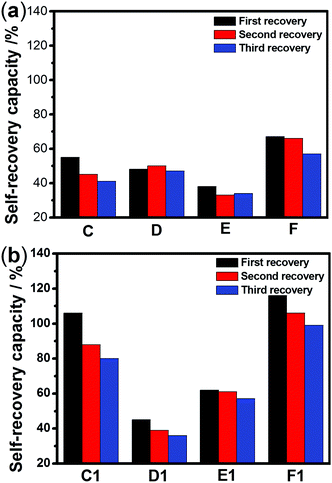 |
| | Fig. 6 The self-recovery capacity of (a) PNIPAM-ATE hydrogels and (b) PNIPAM-SH hydrogels. | |
3.6 Resistance to macrobiofouling: Phaeodactylum tricornutum and Chlorella
Silver nanoparticles (AgNPs) are unique and very effective broad-spectrum anti-algal agents.15,25 The settlement of Chlorella and Phaeodactylum tricornutum on these AgNP hybrid hydrogel surfaces was further studied (Fig. 7). Herein, we also prepared a control sample in which BIS instead of AgNPs was used to as a chemical crosslinker to produce a hydrogel system. Fig. 7 clearly shows that the control sample was highly susceptible to adhesion and colonization of Chlorella and Phaeodactylum tricornutum. Compared with the control sample, the coverage areas of Chlorella and Phaeodactylum tricornutum on the surfaces of the NC hydrogel decreased sharply, indicating a better anti-algal performance. The amount of AgNPs embedded in the hydrogel is a key factor affecting the anti-algal performance. The settlements of Chlorella and Phaeodactylum tricornutum on these NC hydrogels decreased upon increasing the AgNPs. When the Ag/S ratio exceeded 0.9, there were almost no algae on the surface of the PNIPAM-ATE or PNIPAM-SH hydrogel, indicating its excellent anti-algal performance. These hybrid hydrogels have promising applications in marine antifouling coating and interfaces of biomaterials.
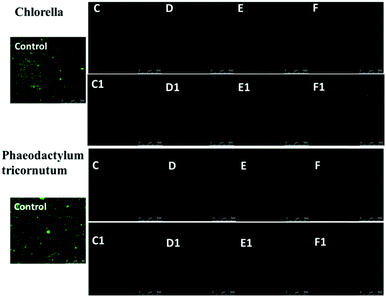 |
| | Fig. 7 Confocal images of Chlorella and Phaeodactylum tricornutum adhering to the surfaces of PNIPAM-ATE and PNIPAM-SH hydrogels after one day. A chemically cross-linked PNIPAM-ATE hydrogel was used as a control sample. | |
4. Conclusions
A series of functional nanocomposite hydrogels based on thiol/acetyl thioester groups chelating with silver nanoparticles were designed and prepared. The silver nanoparticles (AgNPs) acted as both crosslinkers to produce a sell-recovery nanocomposite hydrogel and antimicrobials to prevent the algal adhesion on the surface. The amount of AgNPs embedded in the hydrogel had a great influence on the macroscopic state and the mechanical properties of the hydrogel. When the feed molar ratio of the silver element to the sulfur element (Ag/S) approached 0.9, the hydrogel showed the best mechanical properties. The mechanical properties of these nanocomposite hydrogels increased firstly and then decreased upon increasing the AgNPs. The hydrogels containing thiol groups showed better self-recovery performance than the corresponding hydrogels with acetyl thioester groups. In addition, these nanocomposite hydrogels with AgNPs exhibited good anti-algal performance for Phaeodactylum tricornutum and Chlorella. When the Ag/S ratio exceeded 0.9, there were almost no algae on the surface of these NC hydrogels. Therefore, these AgNP hybrid hydrogels have promising applications in marine antifouling coating and interfaces of biomaterials.
Acknowledgements
The authors gratefully acknowledge financial support from the National Key Basic Research Program of China (2014CB643305), the National Natural Science Foundation of China (Grant No. 51603217), the Ningbo Natural Science Foundation (2016A610258), and the China Postdoctoral Science Foundation funded project (2016M592023).
Notes and references
- D. S. W. Benoit, M. P. Schwartz, A. R. Durney and K. S. Anseth, Nat. Mater., 2008, 7, 816–823 CrossRef CAS PubMed.
- I. Strehin, Z. Nahas, K. Arora, T. Nguyen and J. Elisseeff, Biomaterials, 2010, 31, 2788–2797 CrossRef CAS PubMed.
- N. A. Peppas, K. M. Wood and J. O. Blanchette, Expert Opin. Biol. Ther., 2004, 4, 881–887 CrossRef CAS PubMed.
- R. J. Mart, R. D. Osborne, M. M. Stevens and R. V. Ulijn, Soft Matter, 2006, 2, 822–835 RSC.
- S. Ashraf, H. K. Park, H. Park and S. H. Lee, Macromol. Res., 2016, 24, 297–304 CrossRef CAS.
- R. E. Rivero, M. A. Molina, C. R. Rivarola and C. A. Barbero, Sens. Actuators, B, 2014, 190, 270–278 CrossRef CAS.
- L. Han, Y. N. Zhang, X. Lu, K. F. Wang, Z. M. Wang and H. P. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 29088–29100 CAS.
- R. Barbucci, D. Pasqui, G. Giani, M. D. Cagna, M. Fini, R. Giardino and A. Atrei, Soft Matter, 2011, 7, 5558–5565 RSC.
- C. A. Demarchi, A. Debrassi, F. de Campos Buzzi, R. Corrêa, V. C. Filho, C. A. Rodrigues, N. Nedelko, P. Demchenko, A. Slawska-Waniewska, P. Dłuzewski and J. M. Greneche, Soft Matter, 2014, 10, 3441–3450 RSC.
- A. K. Gaharwar, N. A. Peppas and A. Khademhosseini, Biotechnol. Bioeng., 2014, 111, 441 CrossRef CAS PubMed.
- S. Butun and N. Sahiner, Polymer, 2011, 52, 4834–4840 CrossRef CAS.
- A. Kiesow, J. E. Morris, C. Radehaus and A. Heilmann, J. Appl. Phys., 2003, 94, 6988–6990 CrossRef CAS.
- S. Xu, J. Zhang, C. Paquet, Y. Lin and E. Kumacheva, Adv. Funct. Mater., 2003, 13, 468–472 CrossRef CAS.
- J. Ramos, T. Potta, O. Scheideler and K. Rege, ACS Appl. Mater. Interfaces, 2014, 6, 14861–14873 CAS.
- V. Thomas, M. M. Yallapu, B. Sreedhar and S. K. Bajpai, J. Colloid Interface Sci., 2007, 315, 389–395 CrossRef CAS PubMed.
- Y. Xiang and D. Chen, Eur. Polym. J., 2007, 43, 4178–4187 CrossRef CAS.
- S. J. Devaki, R. K. Narayanan and S. Sarojam, Mater. Lett., 2014, 116, 135–138 CrossRef CAS.
- Y. Zheng and A. Wang, J. Mater. Chem., 2012, 22, 16552–16559 RSC.
- S. Bokern, Z. Y. Fan, C. Mattheis, A. Greiner and S. Agarwal, Macromolecules, 2011, 44, 5036–5042 CrossRef CAS.
- R. Martin, A. Rekondo, J. Echeberria, G. Cabanero, H. J. Grande and I. Odriozola, Chem. Commun., 2012, 48, 8255–8257 RSC.
- A. Skardal, J. X. Zhang, L. McCoard, S. Oottamasathien and G. D. Prestwich, Adv. Mater., 2010, 22, 4736–4740 CrossRef CAS PubMed.
- Y. Zhang, B. Yang, X. Zhang, L. Xu, L. Tao, S. Li and Y. Wei, Chem. Commun., 2012, 48, 9305–9307 RSC.
- N. Holten-Andersen, M. J. Harrington, H. Birkedal, B. P. Lee, P. B. Messersmith, K. Y. C. Lee and J. H. Waite, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2651–2655 CrossRef CAS PubMed.
- I. Irwansyah, Y. Q. Li, W. Shi, D. Qi, W. R. Leow, M. B. Y. Tang, S. Li and X. Chen, Adv. Mater., 2015, 27, 648–654 CrossRef CAS PubMed.
- M. Pollini, F. Paladini, M. Catalano, A. Taurino, A. Licciulli and A. Maffezzoli, J. Mater. Sci.: Mater. Med., 2011, 22, 2005–2012 CrossRef CAS PubMed.
- E. M. Organomet, J. Inorg. Organomet. Polym. Mater., 2011, 21, 297–305 CrossRef.
- B. Hu, S. B. Wang, K. Wang, M. Zhang and S. H. Yu, J. Phys. Chem. C, 2008, 112, 11169–11174 CAS.
- M. Mahmoudi, A. Simchi, M. Imani and U. O. Häfeli, J. Phys. Chem. C, 2009, 113, 8124–8131 CAS.
- P. V. AshaRani, G. Low Kah Mun, M. P. Hande and S. Valiyaveettil, ACS Nano, 2009, 3, 279–290 CrossRef CAS PubMed.
- D. Pasqui, A. Atrei, G. Giani, M. De Cagna and R. Barbucci, Mater. Lett., 2011, 65, 392–395 CrossRef CAS.
- R. Messing, N. Frickel, L. Belkoura, R. Strey, H. Rahn, S. Odenbach and A. M. Schmidt, Macromolecules, 2011, 44, 2990–2999 CrossRef CAS.
- Y. Zhou, N. Sharma, P. Deshmukh, R. K. Lakhman, M. Jain and R. M. Kasi, J. Am. Chem. Soc., 2011, 134, 1630–1641 CrossRef PubMed.
- C. García-Astrain, C. Chen, M. Burón, T. Palomares, A. Eceiza, L. Fruk, M. Ángeles Corcuera and N. Gabilondo, Biomacromolecules, 2015, 16, 1301–1310 CrossRef PubMed.
- K. Baek, J. Liang, W. T. Lim, H. Zhao, D. H. Kim and H. Kong, ACS Appl. Mater. Interfaces, 2015, 7, 15359–15367 CAS.
- C. F. Shaw, Chem. Rev., 1999, 99, 2589–2600 CrossRef CAS.
- R. Sardar, A. M. Funston, P. Mulvaney and R. W. Murray, Langmuir, 2009, 25, 13840–13851 CrossRef CAS PubMed.
- D. Y. Jiang, G. Wang, F. Zheng, J. Han and X. D. Wu, Polym. Chem., 2015, 6, 625–632 RSC.
- G. Wang, L. Peng, Y. C. Zheng, Y. Q. Gao, X. D. Wu, T. H. Ren, C. Gao and J. Han, RSC Adv., 2015, 5, 5674–5679 RSC.
- B. Manjula, K. Varaprasad, R. Sadiku, K. Ramam, G. Reddy and K. M. Raju, J. Biomed. Mater. Res., Part A, 2014, 102, 928–934 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6nj02246d |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2017 |
Click here to see how this site uses Cookies. View our privacy policy here.