
An organic polymer-grafted ionic liquid as a catalyst for the cycloaddition of CO2 to epoxides†
Dan
Xing
,
Bin
Lu
*,
Hongxia
Wang
,
Jingxiang
Zhao
and
Qinghai
Cai
*
Key Lab for Photonic & Electronic Bandgap Materials, Ministry of Education, School of Chemistry and Chemical Engineering, Harbin Normal University, No. 1 Shida Road Limin Development Zone, Harbin 150025, P. R. China. E-mail: caiqi1nghai@yahoo.com; lupin630411@163.com; Fax: +86-451-88060580
First published on 17th November 2016
Abstract
A facile method for synthesizing benzyl chloride polymer (BCP) has been developed and BCP immobilized N-methylimidazolium chloride (BCP-IMCl) was prepared by grafting N-methylimidazole ionic liquid on the BCP surface. The compositions and structures of BCP and BCP-IMCl were determined by gel permeation chromatography (GCP), FT-IR, NMR, and elemental analysis. BCP-IMBr, produced via the substitution of Cl− by Br−, was demonstrated to be an efficient catalyst for a solvent-free cycloaddition of CO2 to propylene epoxide (PO) to form propylene carbonate (PC), exhibiting a TON of 88.6 mol/molIL and a 98.7% selectivity towards PC formation. The catalyst can be easily recovered and effectively reused without a significant loss in its activity and selectivity, which would lead to its potential application foreground for the environmentally friendly synthesis of PC.
1. Introduction
Carbon dioxide is a kind of greenhouse gas and its large emissions can cause global warming, which has become a topic of concern in recent years. Despite being notorious for causing global warming, CO2 is an abundant, non-toxic, economical, and a regenerative sustainable carbon source.1–4 Chemical fixation of waste CO2 to produce valuable chemicals is attractive because such a strategy can be used to reduce the emission of CO2 into the atmosphere while tapping into an unlimited carbon source.5 One of the most efficient strategies is the cycloaddition of CO2 to epoxides to afford five-membered cyclic carbonates, which is a green and atom-economic reaction. Cyclic carbonates can serve as valuable organic intermediates, aprotic polar solvents, and monomers in polymerization reaction.6–9 Accordingly, various catalytic systems with different yields and turnover frequencies (TOFs) have been developed for the cycloaddition of CO2 to epoxides, including metal salts,10–13 transition metal complexes,14–16 zeolites,17 functional polymers,18 metal–organic frameworks,19–21 organocatalysts,22 graphene oxide23 and its functionalized materials,24 modified graphitic carbon nitride,25 N-heterocyclic carbenes,26–28etc.Recently, ionic liquids (IL) have received increasing interest in the area of green chemistry,29 and ionic liquids, such as bmimBF4,30 ZnCl2/BmimCl,31 and some functionalized (containing –OH, −NH2, etc.) ionic liquids have been reported as catalysts for the cycloaddition of CO2 to epoxides;32–34 moreover, they exhibit good catalytic performances. However, on the basis of economic criteria, the ease of separation and recyclability of the catalyst and the immobilization of the IL on the solid supports is highly desirable.35 Therefore, immobilized IL have attracted unprecedented attention because they present more advantages, such as high catalytic efficiency and easy recycling of the catalyst from the catalytic reaction as compared to the combination of inorganic and ionic liquid catalysts. To date, immobilized ionic liquids, including zeolite-, silica-, carbon nanotube-, polystyrene- and cross-linked polymer-supported IL materials,36–41 have been successfully introduced as catalysts for this cycloaddition reaction. However, these IL supports possess some disadvantages such as requiring a complicated preparation technology and a high production cost. Thus, the design and preparation of highly effective and cheap supports for the heterogeneous catalysts is still desirable. Although benzyl chloride polymers have been synthesized long before using aluminum chloride as a catalyst,42 homogeneous catalysis by aluminum chloride suffers from some disadvantages, such as the difficulty of separating and reusing the catalyst, corrosion of the reactor, as well as the low molecular mass of the polymer (about 1260–2250) obtained in this process. In this work, we polymerized benzyl chloride over α-Fe2O3 to prepare an organic polymer (BCP) with higher molecular mass via a simple method, followed by the covalent grafting of N-methylimidazole on BCP to form the polymer-supported N-methylimidazolium chloride or bromide materials. The catalytic performance of the BCP-supported IL materials for the cycloaddition of CO2 to epoxides was evaluated, and it was demonstrated that the catalyst was highly active, selective and stable, and could be easily separated and reused. To the best of our knowledge, this is the first demonstration of a facile synthesis for benzyl chloride polymers with an uncomplicated preparation technology and low production cost, followed by the grafting of IL on the polymer to prepare a heterogeneous catalyst.
2. Experimental
2.1 Preparation of the catalytic materials
2.2 Characterization
The molecular mass of BCP was determined by gel permeation chromatography using an Agilent 1100 GPC with a PLgel 5 μm MIXED-C column of 300 × 7.5 mm at a column temperature of 30 °C and with tetrahydrofuran as the eluent. The FT-IR spectra of the samples were obtained using a Vertex 80 infrared spectrometer (Bruker Company). The 1H NMR spectra of BCP and BCP-IMCl were obtained using an AVANCE III 400 MHz NMR spectrophotometer (Bruker Company). Elemental analysis was performed using a Perkin-Elmer 2400 Analyzer. TGA was carried out using a Diamond 6300 thermal analyzer. The N2 adsorption–desorption technique was performed using a Quantachrome NOVA2000E instrument. The morphology of the catalysts was observed by scanning electron microscopy (SEM) using a Hitachi S-4800 instrument.2.3 Procedures for the catalytic reaction
The catalytic reaction was carried out in a 100 mL stainless steel autoclave equipped with a magnetic stirrer. Epoxy propane (10 mL, 143 mmol) and BCP-IL (0.3 g) were added to the reactor. After purging three times with CO2, the reactor was pressurized to an initial pressure of 2 MPa CO2 and heated to 140 °C in an oil bath. The reaction was carried out at this temperature for 5 h. When the reaction reached completion, the mixture solution was filtered to separate the catalyst and the liquid solution was analyzed by GC (Agilent 7820) using a HP-5 column (30 m × 0.32 mm × 0.25 μm) with an FID detector, and by GC-MS (GC7890-MS5975c, Agilent Company). The catalyst recovered after filtration was reused in the next run in the recovery and reuse experiments with BCP-IMBr.3. Results and discussion
3.1 Preparation and characterization
According to our previous work,43 α-Fe2O3 was so active that the benzylation of benzene with benzyl chloride reached completion within 1 min with 0.2 g of catalyst at 60 °C. The reaction rate was too high to be controlled, which is disadvantageous for the bulk preparation in the future industrial applications. The rapid benzylation was ascribed to the strong activation of α-Fe2O3 towards benzyl chloride. Therefore, benzyl chloride polymerization catalyzed by 0.2 g of α-Fe2O3 was much faster at 60 °C. To make this reaction milder, it was decided to perform the polymerization process at 55 °C using only 0.1 g of the catalyst. Under these reaction conditions the polymerization was completed within 20 min, which is acceptable for future industrial applications. The morphology of the BCP-supported ionic liquids was studied, and a basic uniform distribution of 500–800 nm particles was observed (Fig. S1, ESI†) with a specific surface area of 7.555 m2 g−1 for BCP-IMCl and 3.446 m2 g−1 for BCP-IMBr.In Fig. 1, the FT-IR spectra of BCP and BCP-IMCl are compared. As shown in Fig. 1a and b, the strong characteristic bands at 1583.5 and 1186.2 cm−1 were assigned to the in-plane C–H deformation vibration and the in-plane C–C and C–N stretching vibrations of the imidazole ring (Fig. 1b),46 respectively. Additional bands assignable to the CH3 bending vibrations were seen at 1493.2 and 1448.0 cm−1 (Fig. 1b), which were very similar to those for the bending vibration of C–H in Fig. 1a. Moreover, the characteristic band of the C–Cl bond at 787.5 cm−1 observed in Fig. 1a completely disappeared in the spectrum of BCP-IMCl. These findings proved the successful grafting of N-methylimidazole onto BCP, with the Cl atoms on the end groups of BCP completely replaced to form BCP-IMCl.
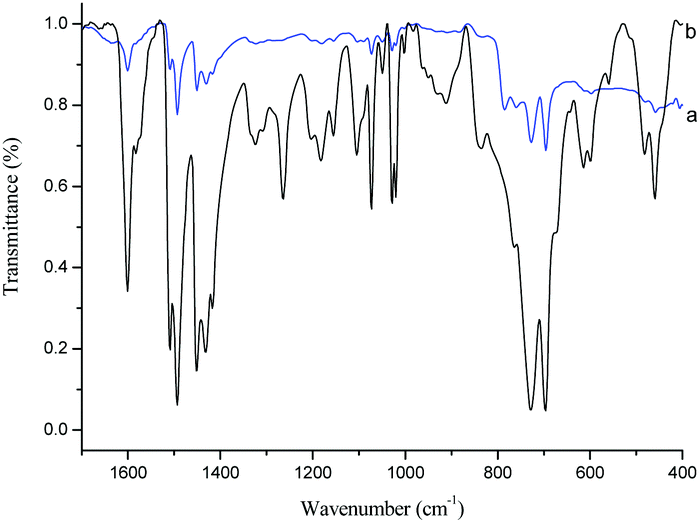 | ||
| Fig. 1 FT-IR spectra of BCP (a) and BCP-IMCl (b). | ||
The structure of the organic polymer and its N-methylimidazolium functionalized materials were confirmed by 1H-NMR spectra. As shown in Fig. 2b, for BCP-IMCl, the signals at 7.43–7.50 ppm corresponded to –C(2)H– (a in Scheme 1) in the imidazolium ring. The peaks at 5.58, 4.09 and 2.03 ppm were likely attributed to the chemical shifts of –C(4, 5)H– hydrogens in imidazolium, –CH2–N– and –CH3 groups, respectively (b, c and d in Scheme 1).45 On the other hand, the broad peaks at 6.99 and 3.74 ppm were assigned to H in the phenyl ring and the –CH2– linked to the phenyl ring, respectively, and were similar to those in the BCP spectrum (Fig. 2a). Thus, the presence of the imidazolium ring on BCP-IMCl was confirmed.
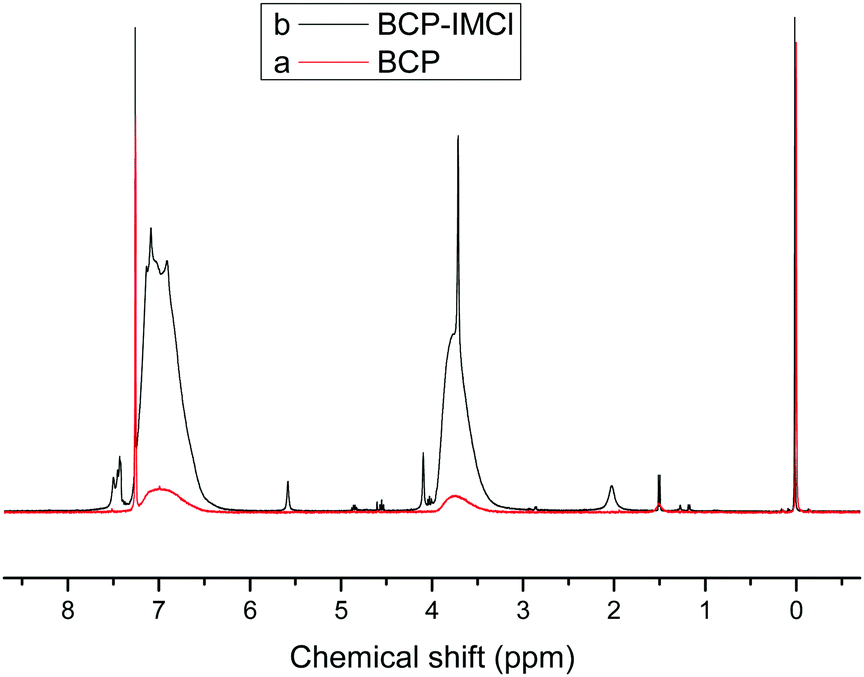 | ||
| Fig. 2 1H-NMR of BCP (a) and BCP-IL (b). | ||
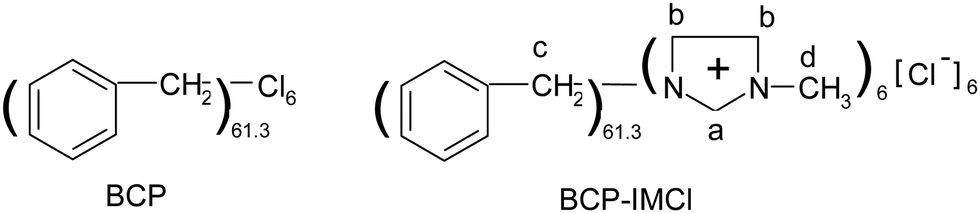 | ||
| Scheme 1 Formula of BCP and BCP-IMCl. | ||
The molecular mass of BCP was measured by GPC (Fig. S2, ESI†). According to the result, Mn (number-average molecular mass) and Mw (weight-average molecular mass) of BCP were estimated to be 5732.3 and 36334.0 g mol−1, respectively. It can be seen in the chromatogram that the molar mass distribution in the polymer was wide, in the range from 103 to 105 g mol−1. PDI was calculated to be 6.34, which also confirmed the wide molar mass distribution of BCP. Elemental analysis results showed that the content of carbon in the polymer was 90.38%, and the content of hydrogen was 7.52%. Similarly, the composition of BCP-IMCl was also determined; the C and N content were 89.83 and 2.80%, respectively. As estimated from the N content, the percentage of grafted methylimidazolium chloride approached to 11.75%, which equals to a -IMCl loading of 1.0 mmol g−1. According to these results, the formulas of BCP and BCP-IMCl were estimated as shown in Scheme 1. It was estimated that the average polymerization degree of BCP was about 61 benzyl groups with six chloride atoms, and for BCP-IMCl, six methylimidazolium chloride moieties were linked to the BCP surface, which was concluded from the disappearance of the C–Cl bond in the IR spectrum of BCP-IMCl. This result implied that the branched-chain reactions in the polymerization of benzyl chloride were taking place via benzylation over the α-Fe2O3 catalyst.42 It was reported that α-Fe2O3 was a highly efficient catalyst for the benzylation of aromatics with benzyl chloride.43 Based on the formula, the content of carbon in BCP was calculated to be 89.87%, which agrees well with the measured value (90.38%). In BCP-IMCl, the carbon and nitrogen content were 87.27% and 2.70%, respectively, which is quite close to the measured values (89.83 and 2.80%, respectively) obtained by the elemental analysis. Also, a –IMCl loading of 0.96 mmol g−1 was obtained with the theoretical calculation. This indicated that the abovementioned estimation is in agreement with the experimental results.
The TGA curves of BCP and BCP-IMCl are shown in Fig. 3. Apparently, BCP exhibited an excellent stability below 410 °C. The weight loss was observed between 410 °C and 540 °C, which was attributed to the thermal decomposition of BCP (Fig. 3a). By contrast, for BCP-supported –IMCl, weight loss was observed in the temperature range from 180 to 410 °C, as seen in the thermal evolution shown in Fig. 3b. The percentage of weight loss was estimated to be 11.03%, which approaches the loading percentage (11.75%) of –IMCl on the BCP surface. This finding also proved the reliability of the elemental analysis results. The TGA curve of BCP-IMBr was similar to that of BCP-IMCl. A weight loss of 15.54% was observed in the TGA curve, which confirmed the formation of BCP-IMBr (Fig. S3, ESI†).
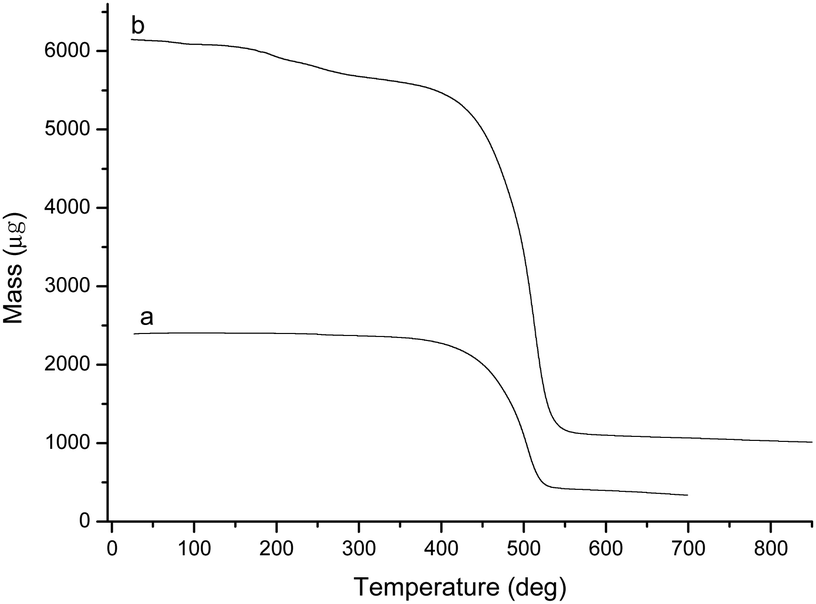 | ||
| Fig. 3 TG curves of BCP (a) and BCP-IMCl (b). | ||
3.2 Catalytic activity of various catalysts
The catalytic activities of various grafted ionic liquids were evaluated for the cycloaddition of CO2 to propylene oxide (PO) to yield PC, and 0.1–0.5 g of the specified catalysts were used. The results, summarized in Table 1, showed that BCP was completely inactive (entry 1), whereas BCP-IMCl was highly active, yielding a 51.8% conversion and a 96.9% selectivity towards PC formation (entry 2), thus confirming the activating role of the imidazolium chloride ionic liquid for the reaction between CO2 and PO. However, materials with a metal-chloride coordinated to BCP-IMCl, such as BCP-IMCl-FeCl3 and BCP-IMCl–ZnCl2, exhibited almost the same activity as BCP-IMCl (entries 3 and 4). The relatively high activity of BCP-IMCl–ZnCl2 as compared to that of BCP-IMCl–FeCl3 was likely attributed to its higher Lewis acidity.41,44 As expected, the metathesis of Cl− by Br− to form BCP-IMBr resulted in a surprisingly high activity that afforded a 71.4% conversion for PO and a TON of 37.0 mol/molIL(entry 5). This dramatic effect may be explained by the nature of the attack of the halide ion on the carbon atom of the epoxide, which is facilitated with the more nucleophilic bromide ions.47,48 On the other hand, BCP-IMOH and BCP-IMBF4 showed a much lower activity than BCP-IMCl (entries 6 and 7), revealing that the relevant properties of ionic liquids for solute interaction and their effect on the catalytic reaction are significantly determined by the nature of the anion rather than the cation.49,50 Metal chloride coordinated BCP-IMBr ionic liquids, such as BCP-IMBr–ZnCl2, BCP-IMBr–FeCl3 and BCP-IMBr–CuCl2, also exhibited a slightly higher activity as compared with BCP-IMBr, and the activity order was correlated with their Lewis acidity order: BCP-IMBr–ZnCl2 > BCP-IMBr–FeCl3 > BCP-IMBr–CuCl2 (entries 8–10).| Entry | Catal. (g) | t (h) | P (MPa) | T (deg) | Con. (%) | Sel. (%) | TON |
|---|---|---|---|---|---|---|---|
| t – Reaction time; P – pressure; T – reaction temperature; Con. – conversion; Sel. – selectivity; TON – converted moles of PO per moles of IMX (IMCl, IMOH, IMBF4 or IMBr). | |||||||
| 1 | BCP | 5 | 2 | 140 | — | — | — |
| 2 | BCP-IMCl | 5 | 2 | 140 | 51.8 | 96.9 | 25.7 |
| 3 | BCP-IMCl–ZnCl2 | 5 | 2 | 140 | 57.9 | 96.7 | 27.0 |
| 4 | BCP-IMCl–FeCl3 | 5 | 2 | 140 | 51.3 | 95.8 | 24.0 |
| 5 | BCP-IMBr | 5 | 2 | 140 | 71.4 | 96.8 | 37.0 |
| 6 | BCP-IMOH | 5 | 2 | 140 | 38.9 | 97.3 | 18.9 |
| 7 | BCP-IMBF4 | 5 | 2 | 140 | 25.8 | 93.4 | 11.5 |
| 8 | BCP-IMBr–ZnCl2 | 5 | 2 | 140 | 79.9 | 96.8 | 46.4 |
| 9 | BCP-IMBr–FeCl3 | 5 | 2 | 140 | 77.9 | 96.5 | 46.2 |
| 10 | BCP-IMBr–CuCl2 | 5 | 2 | 140 | 72.5 | 96.4 | 42.1 |
| 11 | BCP-IMBr | 5 | 2 | 130 | 62.9 | 97.9 | 32.6 |
| 12 | BCP-IMBr | 5 | 2 | 150 | 70.1 | 96.9 | 36.3 |
| 13 | BCP-IMBr | 4 | 2 | 140 | 66.9 | 97.4 | 34.7 |
| 14 | BCP-IMBr | 6 | 2 | 140 | 70.1 | 96.4 | 36.3 |
| 15 | BCP-IMBr(0.1) | 5 | 2 | 140 | 57.0 | 98.6 | 88.6 |
| 16 | BCP-IMBr(0.2) | 5 | 2 | 140 | 63.5 | 97.8 | 49.4 |
| 17 | BCP-IMBr(0.5) | 5 | 2 | 140 | 69.7 | 96.2 | 21.7 |
| 18 | BCP-IMBr | 5 | 1.5 | 140 | 67.7 | 97.0 | 35.1 |
| 19 | BCP-IMBr | 5 | 2.5 | 140 | 70.0 | 96.5 | 36.3 |
| 20 | SBA-15-IMCl | 5 | 2 | 140 | 49.9 | 96.3 | 7.5 |
| 21 | SBA-15-IMBr | 5 | 2 | 140 | 58.8 | 96.7 | 9.6 |
It has been widely known that reaction temperature has a significant impact on the reaction rate and product selectivity. It could be seen that the catalytic activity greatly increased from 62.9% at 130 °C to 71.4% at 140 °C, exhibiting a TON of 32.6 and 37.0 mol/molIL, respectively (entries 5 and 11). Further increasing the reaction temperature from 140 to 150 °C almost had no effect on the catalytic performance (entry 12), showing that the appropriate reaction temperature had a positive impact on the cycloaddition. Moreover, the conversion and selectivity also depend on the reaction time; the conversion was increased from 66.9% to 71.4% when the reaction time was increased from 4 to 5 h (entries 13 and 5, respectively). Thereafter, a thermodynamic equilibrium was achieved upon further prolonging the reaction time (entry 14), achieving a 70.1% conversion and a 36.1 mol/molIL TON after 6 h.
The increase in the catalyst dose used in this system would lead to an increase in the amount of active species on the catalyst surface. There was an increase in the conversion of PO as the catalyst amount increased from 0.1 to 0.3 g (entries 15, 16 and 5). Similar conversions were achieved when increasing the catalyst dose from 0.3 to 0.5 g, with a 71.4% and 70.1% conversion, respectively, although the TON remarkably decreased from 37.0 to 21.7 mol/molIL. Note that, the highest TON (88.6 mol/molIL) with the highest selectivity was obtained when using 0.1 g of BCP-IMBr as the catalyst. Besides, the impact of the CO2 pressure on the reactivity was similar to the abovementioned effects of the reaction time and catalyst dose. Higher pressures of CO2 (more than 2 MPa) had almost no effect on the activity of the catalyst (entries 18, 19 and 5). In addition, SBA-15-supported ionic liquids SBA-15-IMCl and SBA-15-IMBr also yielded higher conversions, 49.9% and 58.8%, respectively (entries 20 and 21), whereas their TONs (7.5 and 9.6 mol/molIL, respectively) were much lower as compared to those of BCP-IMCl and BCP-IMBr.
Note that the selectivities to PC formation using BCP-IMBr under various conditions were all higher than 96.0%. The only by-product identified in this process was propanediol. As compared with previous reports on CO2 cycloaddition, the high selectivity to PC is the superiority in this case. Furthermore, the BCP-IMBr system possesses some advantages such as low cost of raw materials, a simple and highly efficient preparation technology with a high TON.51,52
3.3 Cycloaddition of other epoxides
To broaden the scope of the cycloaddition reaction, various mono- and di-substituted epoxides, such as epoxy ethane, epoxy chloropropane, styrene oxide and cyclohexene oxide, were also used in the cycloaddition. As shown in Table 2, the monosubstituted terminal epoxides were readily converted to the corresponding cyclic carbonates, but the reactivity of the epoxides was affected by the substituent at the carbon atom adjacent to the epoxide. Monosubstituted terminal epoxides with less bulky substituents exhibited a higher reactivity (entries 1–3), whereas styrene oxide, with a bulky substituent, was found to have a relatively poor reactivity (entry 4). Furthermore, disubstituted epoxide (cyclohexene oxide) showed little reactivity, and a long reaction time was needed (entry 5), which may have resulted from steric hindrance and electronic effects.| Entry | Epoxides | Products | t (h) | Con. (%) | Sel. (%) |
|---|---|---|---|---|---|
| Reaction conditions: epoxides (143 mmol), amount of catalyst (0.3 g), temperature of 140 °C, initial pressure of 2.0 MPa CO2. | |||||
| 1 |
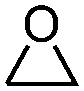
|
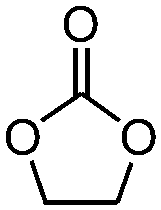
|
3 | 82.1 | 98.1 |
| 2 |
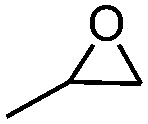
|
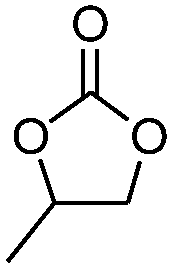
|
5 | 71.4 | 96.8 |
| 3 |
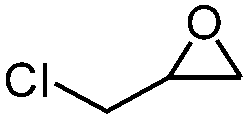
|
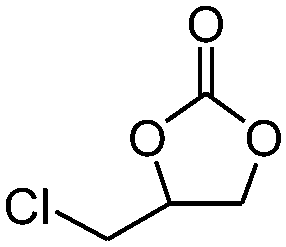
|
4 | 78.7 | 97.3 |
| 4 |
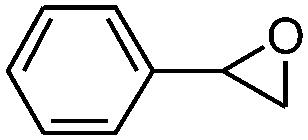
|
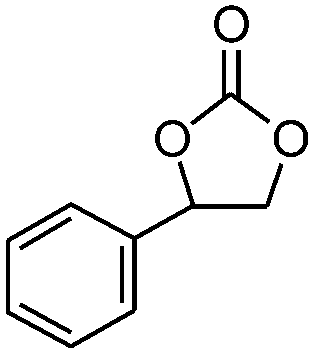
|
5 | 67.8 | 95.6 |
| 5 |
|
|
12 | 36.3 | 83.2 |
3.4 The reusability of the catalyst
To further evaluate the performance of the catalyst, recovery and reuse of the BCP-IMBr catalyst were studied. The catalyst was recovered by filtration after each reaction, and then it was reused in the next run under the same conditions. It was clear that the activity of the catalyst was unaffected even at the fifth run (Fig. 4). This showed that the prepared BCP-IMBr catalyst was highly stable at least for up to five repeated runs. The slightly lower activity in the fifth run was likely ascribed to a reduction in the recovered amount of catalyst.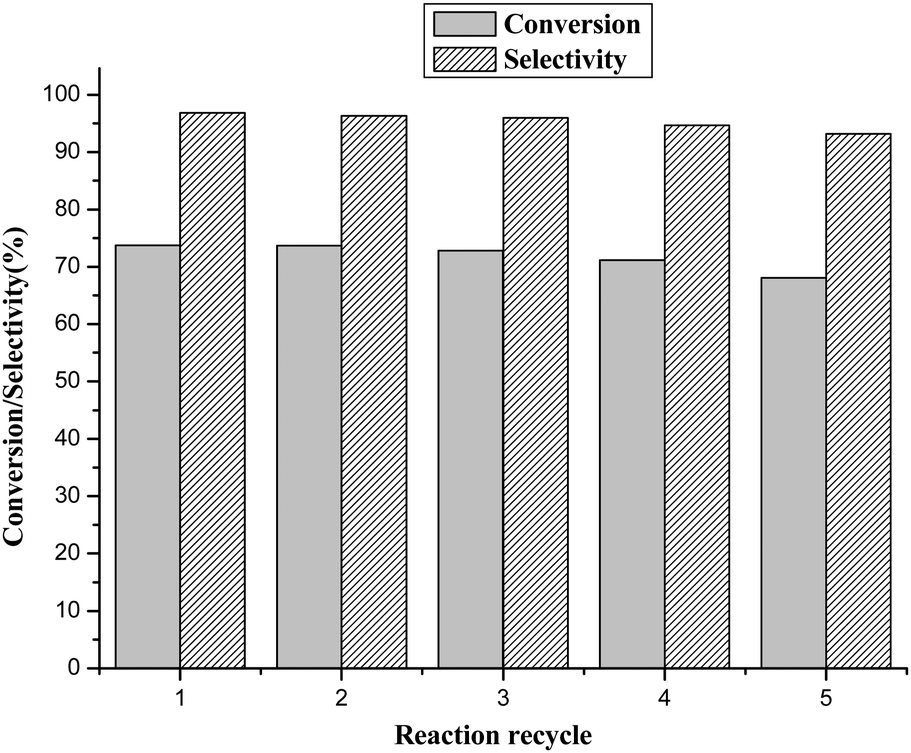 | ||
| Fig. 4 Reusability of the catalyst. | ||
4. Conclusion
An organic polymer, benzyl chloride polymer (BCP), was developed by a simple method and the BCP-immobilized ionic liquid N-methylimidazolium chloride (BCP-IMCl) was prepared by grafting N-methylimidazole on the BCP surface. The composition, structure and thermal stability of BCP and BCP-IMCl were determined. The BCP-IMBr catalyst, obtained by Br− substituting Cl−, exhibited an excellent catalytic performance for the solvent-free cycloaddition of CO2 to epoxides. Especially, high selectivity to propylene carbonate was achieved when using BCP-IMBr. In addition, the catalyst was prepared via an uncomplicated technology and was easily reused, which showed its high potential application foreground in industry.Acknowledgements
We highly acknowledge the National Natural Science Foundation of China (No. 21671050), the Discipline Leader Foundation of Harbin (No. 2013RFXXJ009) and the Natural Science Foundation of the Heilongjiang Province (No. B201119) for the financial support.References
- M. R. Kember, A. Buchard and C. K. Williams, Chem. Commun., 2011, 47, 212–214 RSC.
- C. Maeda, T. Taniguchi, K. Ogawa and T. Ema, Angew. Chem., 2015, 127, 136–140 CrossRef.
- R. J. Wei, X. H. Zhang, B. Y. Du, Z. Q. Fan and G. R. Qi, J. Mol. Catal. A: Chem., 2013, 379, 38–45 CrossRef CAS.
- X. Zhou, Y. Zhang, X. G. Yang, J. Yao and G. Y. Wang, Chin. J. Catal., 2010, 31, 765–768 CrossRef CAS.
- M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514–1539 RSC.
- T. Sakakura and K. Kohno, Chem. Commun., 2009, 1312–1330 RSC.
- K. A. Salmeia, S. Vagin, C. E. Anderson and B. Rieger, Macromolecules, 2012, 45, 8604–8613 CrossRef CAS.
- P. P. Pescarmona and M. Taherimehr, Catal. Sci. Technol., 2012, 2, 2169–2187 CAS.
- B. Wang, X. Feng, L. Zhang, S. Yang, X. Jiang, J. Zhou and G. Gao, J. CO2 Util., 2013, 1, 88–91 CrossRef CAS.
- K. Yamaguchi, K. Ebitani, T. Yoshida, H. Yoshida and K. Kaneda, J. Am. Chem. Soc., 1999, 121, 4526–4527 CrossRef CAS.
- J. Song, B. Zhang, P. Zhang, J. Ma, J. Liu, H. Fan, T. Jiang and B. Han, Catal. Today, 2012, 183, 130–135 CrossRef CAS.
- J. Tharun, G. Mathai, A. C. Kathalikkattil, R. Roshan, J.-Y. Kwak and D.-W. Park, Green Chem., 2013, 15, 1673–1679 RSC.
- Q. Song, B. Yu, X. Li, R. Ma, Z. Diao, R. Li, W. Li and L. He, Green Chem., 2014, 16, 1633–1638 RSC.
- J. Melendez, M. North and R. Pasquale, Eur. J. Inorg. Chem., 2007, 3323–3325 CrossRef CAS.
- M. North and R. Pasquale, Angew. Chem., 2009, 121, 2990–2992 CrossRef.
- L. Jin, T. Chang and H. Jing, Chin. J. Catal., 2007, 28, 287–289 CrossRef CAS.
- Y. Du, F. Cai, D. L. Kong and L. N. He, Green Chem., 2005, 7, 518–522 RSC.
- Y. Zhao, H. S. Tian, X. H. Qi, Z. N. Han and Y. Y. Zhuang, J. Mol. Catal. A: Chem., 2007, 271, 284–289 CrossRef CAS.
- J. Song, Z. Zhang, S. Hu, T. Wu, T. Jiang and B. Han, Green Chem., 2009, 11, 1031–1036 RSC.
- Y. Ren, Y. Shi, J. Chen, S. Yang, C. Qi and H. Jiang, RSC Adv., 2013, 3, 2167–2173 RSC.
- A. C. Kathalikkattil, R. Roshan, J. Tharun, H.-G. Soek, H.-S. Ryu and D.-W. Park, ChemCatChem, 2014, 6, 284–292 CrossRef CAS.
- C. J. Whiteoak, A. H. Henseler, C. Ayats, A. W. Kleij and M. A. Pericàs, Green Chem., 2014, 16, 1552–1559 RSC.
- D. H. Lan, F. M. Yang, S. L. Luo, C. T. Au and S. F. Yin, Carbon, 2014, 73, 351–360 CrossRef CAS.
- D. H. Lan, L. Chen, C. T. Au and S. F. Yin, Carbon, 2015, 93, 22–31 CrossRef CAS.
- D. H. Lan, H. T. Wang, L. Chen, C. T. Au and S. F. Yin, Carbon, 2016, 100, 81–89 CrossRef CAS.
- H. Zhou, W.-Z. Zhang, C.-H. Liu, J.-P. Qu and X.-B. Lu, J. Org. Chem., 2008, 73, 8039–8044 CrossRef CAS PubMed.
- M. J. Ajitha and C. H. Suresh, Tetrahedron Lett., 2011, 52, 5403–5406 CrossRef CAS.
- R. Lo and B. Ganguly, New J. Chem., 2012, 36, 2549–2560 RSC.
- T. Welton, Chem. Rev., 1999, 99, 2071–2083 CrossRef CAS PubMed.
- J. Peng and Y. Deng, New J. Chem., 2001, 25, 639–641 RSC.
- F. Wang, C. Xu, Z. Li, C. Xia and J. Chen, J. Mol. Catal. A: Chem., 2014, 385, 133–140 CrossRef CAS.
- J. Sun, S. Zhang, W. Cheng and J. Ren, Tetrahedron Lett., 2008, 49, 3588–3591 CrossRef CAS.
- J. Wang, W. Cheng, J. Sun, T. Shi, X. Zhang and S. Zhang, RSC Adv., 2014, 4, 2360–2367 RSC.
- L. Xiao, D. Lv, D. Su, W. Wu and H. Li, J. Cleaner Prod., 2014, 67, 285–290 CrossRef CAS.
- C. P. Mehnert, R. A. Cook, N. C. Dispenziere and M. Afeworki, J. Am. Chem. Soc., 2002, 124, 12932–12933 CrossRef CAS PubMed.
- S. Udayakumar, M.-K. Lee, H.-L. Shim and D.-W. Park, Appl. Catal., A, 2009, 365, 88–95 CrossRef CAS.
- H.-J. Choi, M. Selvaraj and D.-W. Park, Chem. Eng. Sci., 2013, 100, 242–248 CrossRef CAS.
- L. Han, H. Li, S.-J. Choi, M.-S. Park, S.-M. Lee, Y.-J. Kim and D.-W. Park, Appl. Catal., A, 2012, 429–430, 67–72 CrossRef CAS.
- J. Q. Wang, X. D. Yue, F. Cai and L. N. He, Catal. Commun., 2007, 8, 167–172 CrossRef CAS.
- L. Han, S. W. Park and D. W. Park, Energy Environ. Sci., 2009, 2, 1286–1292 CAS.
- Y. Xie, Z. Zhang, T. Jiang, J. He, B. Han, T. Wu and K. Ding, Angew. Chem., Int. Ed., 2007, 46, 7255–7258 CrossRef CAS PubMed.
- R. A. Jacobson, J. Am. Chem. Soc., 1932, 54, 1513–1518 CrossRef CAS.
- X. Li, X. You, B. Lu, X. Wu, J. Zhao and Q. Cai, Ind. Eng. Chem. Res., 2014, 53, 20085–20091 CrossRef CAS.
- B. Lu, N. Cai, J. Sun, X. Wang, X. Li, J. Zhao and Q. Cai, Chem. Eng. J., 2013, 225, 266–270 CrossRef CAS.
- R. Cang, B. Lu, X. Li, R. Niu, J. Zhao and Q. Cai, Chem. Eng. Sci., 2015, 137, 268–275 CrossRef CAS.
- N. Nanbu, Y. Sasaki and F. Kitamura, Electrochem. Commun., 2003, 5, 383–387 CrossRef CAS.
- L. Han, S. W. Park and D. W. Park, Energy Environ. Sci., 2009, 2, 1286–1292 CAS.
- H. S. Kim, J. J. Kim, H. Kim and H. G. Jang, J. Catal., 2003, 220, 44–46 CrossRef CAS.
- C. Chiappe and D. J. Pieraccini, Phys. Org. Chem., 2005, 18, 275–297 CrossRef CAS.
- R. Lungwitz and S. Spange, New J. Chem., 2008, 32, 392–394 RSC.
- W. L. Dai, B. Jin, S. Luo, S. Yin, X. Luo and C. T. Au, J. CO2 Util., 2013, 3–4, 7–13 CrossRef CAS.
- W. L. Dai, L. Chen, S. Yin, W. Li, Y. Zhang, S. Luo and C. T. Au, Catal. Lett., 2010, 137, 74–80 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6nj02691e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2017 |