
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal-mediated generation of triazapentadienate-terminated di- and trinuclear μ2-pyrazolate NiII species and control of their nuclearity†
Elena V.
Andrusenko
,
Evgeniy V.
Kabin
,
Alexander S.
Novikov
,
Nadezhda A.
Bokach
*,
Galina L.
Starova
and
Vadim Yu.
Kukushkin
*
Institute of Chemistry, Saint Petersburg State University, Universitetskaya Nab. 7/9, 199034, Saint Petersburg, Russian Federation. E-mail: n.bokach@spbu.ru; v.kukushkin@spbu.ru
First published on 17th November 2016
Abstract
1,3,5-Triazapentadienate-terminated di- and trinuclear nickel(II) complexes featuring bridging azolate ligands, [Ni2(μ2-azolate)2(TAP)2] (TAP = H![[N with combining low line]](https://www.rsc.org/images/entities/char_004e_0332.gif)
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(OMe)NC(OMe)H; azole = 3,5-Me2pyrazole 2, 3,5-Ph2pyrazole 3) and [Ni3(μ2-azolate)4(TAP)2] (azole = 3,5-Me2pyrazole 4, indazole 5), were obtained from systems Ni2+/NCNR2/azole systems in MeOH. The terminal TAP ligands in the [Ni2(μ2-azolate)2(TAP)2] and [Ni3(μ2-azolate)4(TAP)2] species originate from the previously unreported cascade NiII-mediated and chelation-driven reaction between cyanamides and methanol. The oligomeric species and also [Ni(TAP)2] (1) are subject to interconversions that depend on the reactants involved and the reaction conditions. The control of the nuclearity of the complexes can be achieved by changing the amount of azoles or by their protonation, alteration of the steric hindrance of the substituents in the heterocycles, and by changing the reaction temperature. Complexes 1–4 were characterized using elemental (C, H, N) analyses, 1H, 13C{1H} NMR, FTIR, HRESI-MS, TG-DTA, X-ray crystallography, and 5 was characterized using HRESI-MS and X-ray crystallography. Unconventional metallophilic contacts NiII⋯NiII were observed in dimer 3 in the solid state (the distance for Ni⋯Ni is 2.99 Å, whereas the double Bondi's vdW radius for Ni is 3.26 Å) and the reality of these interactions was confirmed theoretically by the topological analysis of the electron density distribution (AIM method). The estimated energy for these non-covalent Ni⋯Ni interactions (ca. 4 kcal mol−1) fills the gap in the reported energies of the metal⋯metal interactions in a series comprising of NiII⋯NiII (this work), PdII⋯PdII (4.3–6.0 kcal mol−1), and PtII⋯PtII (3.9–11.7 kcal mol−1).
C(OMe)NC(OMe)H; azole = 3,5-Me2pyrazole 2, 3,5-Ph2pyrazole 3) and [Ni3(μ2-azolate)4(TAP)2] (azole = 3,5-Me2pyrazole 4, indazole 5), were obtained from systems Ni2+/NCNR2/azole systems in MeOH. The terminal TAP ligands in the [Ni2(μ2-azolate)2(TAP)2] and [Ni3(μ2-azolate)4(TAP)2] species originate from the previously unreported cascade NiII-mediated and chelation-driven reaction between cyanamides and methanol. The oligomeric species and also [Ni(TAP)2] (1) are subject to interconversions that depend on the reactants involved and the reaction conditions. The control of the nuclearity of the complexes can be achieved by changing the amount of azoles or by their protonation, alteration of the steric hindrance of the substituents in the heterocycles, and by changing the reaction temperature. Complexes 1–4 were characterized using elemental (C, H, N) analyses, 1H, 13C{1H} NMR, FTIR, HRESI-MS, TG-DTA, X-ray crystallography, and 5 was characterized using HRESI-MS and X-ray crystallography. Unconventional metallophilic contacts NiII⋯NiII were observed in dimer 3 in the solid state (the distance for Ni⋯Ni is 2.99 Å, whereas the double Bondi's vdW radius for Ni is 3.26 Å) and the reality of these interactions was confirmed theoretically by the topological analysis of the electron density distribution (AIM method). The estimated energy for these non-covalent Ni⋯Ni interactions (ca. 4 kcal mol−1) fills the gap in the reported energies of the metal⋯metal interactions in a series comprising of NiII⋯NiII (this work), PdII⋯PdII (4.3–6.0 kcal mol−1), and PtII⋯PtII (3.9–11.7 kcal mol−1).
Introduction
The abundance of pyrazole(ate)-based ligands is due to the ability of these species to bind metals in a variety of coordination modes, in particular, as bridging ligands. In addition, pyrazole(ate) fragments can be integrated into metal species as they are or as a functionality of more complex polydentate ligands, e.g. polypyrazolylborates.1 Metal complexes bearing pyrazole(ates) of different types found applications as catalysts,2 magnetic,2–3 luminescent,2,4 gas storage or separation2,4a,d materials, useful compounds for medicine2 and biomimetic chemistry,5 and also as precursors for CVD.4b Several excellent reviews provide a general picture of the syntheses and application of pyrazole(ate) metal species.2,3,4a,b,5When pyrazolates act as bridging ligands, they are capable of forming metal complexes exhibiting different nuclearity.4a,6 In particular, nickel(II) centers react with unsubstituted pyrazole (HPz) giving the 1D-chain polymers, [Ni(Pz)2]n (Fig. 1, type A), as two thermochromic7 crystalline phases.7–8 Nickel(II) complexes featuring bis-pyrazolate ligands form porous MOFs possessing high thermal robustness and exhibiting adsorptive properties9 and, in particular, MOFs based upon Ni(bpb) (H2bpb = 1,4-(4-bispyrazolyl)benzene) act as a moisture resistant adsorbent toward thiophene.9a Nickel(II) and 3(5)-arylpyrazoles (HArPz) form binary cyclic trinuclear complexes [Ni3(ArPz)6] (type B).10
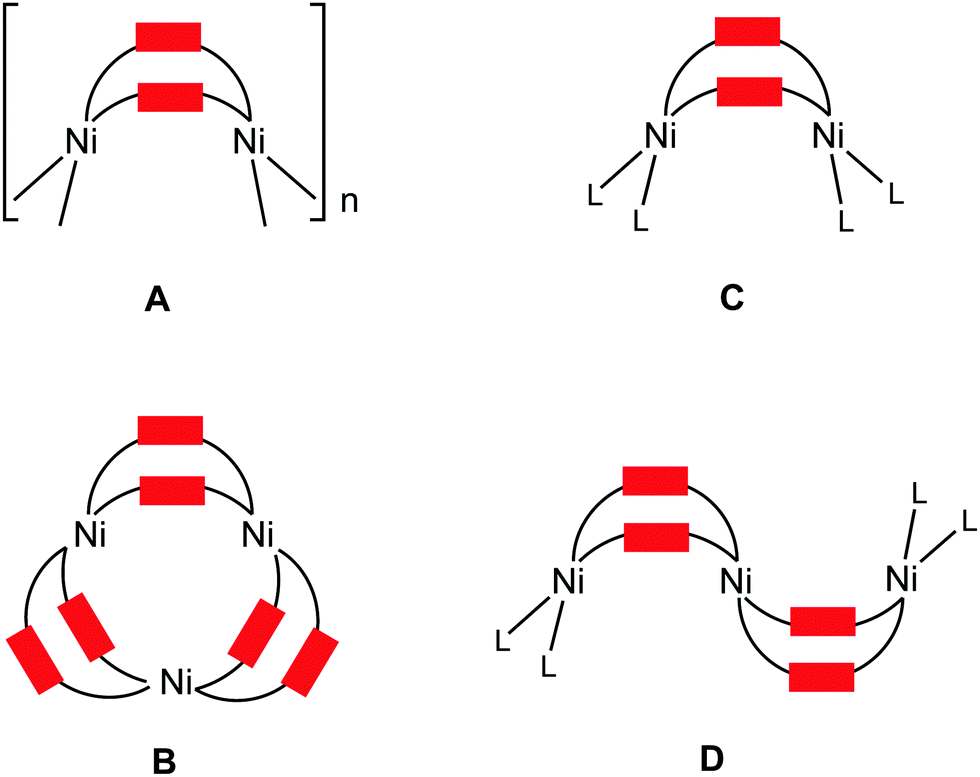 | ||
| Fig. 1 Selected types of nickel(II) complexes featuring the {Ni(μ-RPz)2Ni} moiety. | ||
Examples of oligomeric species incorporating the {Ni(μ-RPz)2Ni} fragment supported by other ligands include the dinuclear [Ni2Cp2(RRR′Pz)2] (L = 1/2Cp, type C) and the trinuclear [Ni3Cp2(RRR′Pz)4] (L = 1/2Cp, type D) species that were obtained from the 3,4,5-substituted pyrazoles HRRR′Pz and Cp2Ni.11 The nuclearity of these complexes depends on the bulkiness of the 3- and 5-substituents in the pyrazolate moiety11a and also on the reaction temperature.11b It is reported that the sterically hindered substituents and low synthesis temperature favor dimetallic rather than trimetallic species. It is noteworthy that before our current work, interconversion of di- and trinuclear species (see later) had never been studied.11
The homodinuclear complexes of the type [Ni2(RRR′Pz)2L4] (RRR′ = Me3, L = 1/2 bis(imidazolyl)(pyrazolyl)borate;12 R = Me, R′ = H, L = 1/2 3,5-dimethylpyrazole-1-carbodithioate;13 R = H, R′ = Me, or R = R′ = H, L = C6F5;14RRR′PzL2 = 3,5-bis((1-(2,6-diisopropylphenyl)-1H-imidazol-3-ium-3-yl)methyl)pyrazol-1-ate;15RRR′PzL2 = 3,5-bis((1-methyl-1H-imidazol-3-ium-3-yl)methyl)pyrazol-1-ide;16 type C) were also reported. Two dinuclear NHC/pyrazolate complexes [Ni2L](PF6)2 (type C) bearing the macrocyclic ligand were generated from [H6L](PF6)4 ([H6L]4+ = methylene- and ethylene-bridged calix[4]imidazolium[2]-pyrazole cyclophane),17 and the latter complex is able to selectively recognize halides and self-assemble with guest anions (Cl− and Br−).17b The {Ni(μ-PzR)2Ni} motif was identified in the highly porous and robust [Ni8(OH)4(OH2)2(4,40-(buta-1,3-diyne-1,4-diyl)bispyrazolate)6]n MOF,18 in the octanuclear [Ni8(μ4-O)6(Pz)12]2− complex possessing a cube structure,3e and in a Ni9 cluster-bearing bridged μ2-pyrazolate, μ6-CO32−, and μ3-OH− ligands which display cluster glass-like magnetic behavior.19 The indazole (IndH) analog featuring the {Ni(μ-Ind)2Ni} group was only observed for the [Ni2(μ-Ind)2(C6F5)4]2− complex.14
In view of our general interest in ligand reactivity and, in particular, in metal-mediated and metal-catalyzed transformations of conventional nitriles and cyanamides20 with pyrazolate-based systems,21 we extended our project to reactions of (azole)NiII (azole = pyrazole, indazole) species with NCNR2. Taking into account that (azolate)2NiII complexes may have some applied significance, for instance in anion recognition chemistry (see, e.g., ref. 17b), we planned to understand how to control the nuclearity of nickel(II)–azole systems in order to obtain a rational approach for the generation of well-defined (azolate)2NiII species. Our goals were to develop a rational route to oligomeric NiII complexes featuring the {Ni(μ2-azolate)2}n fragment and to study how the nuclearity and structure of these NiII-based pyrazolate oligomers depend on reaction conditions and the nature of the reactants.
Results and discussion
To study the NiII-involving reactions between cyanamides and azole-type heterocycles we chose nickel salts NiX2·nH2O (X = Cl, OTf, n = 0; Br, n = 3; I, n = 6), N,N-disubstituted cyanamides NCNR2 (R2 = Me2, Et2, C5H10, C2H4OC2H4), and also pyrazoles and indazole as depicted in Fig. 2.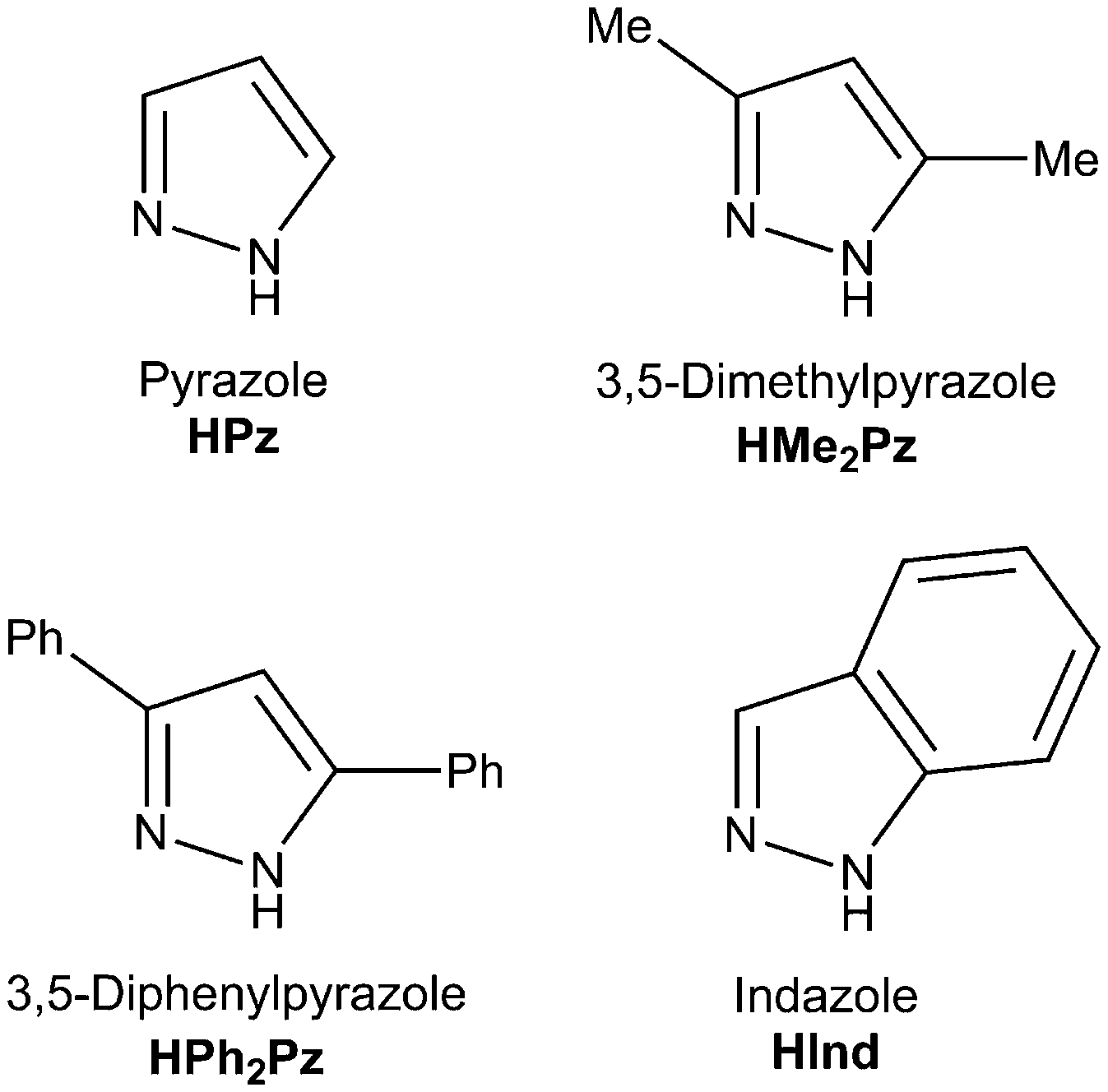 | ||
| Fig. 2 The azoles applied in this study, their IUPAC names, and abbreviations. | ||
We studied the reaction of dialkylcyanamides with the azoles in the presence of nickel(II) salts in methanol, where the nickel salts are well soluble. In these reactions, di- and trinuclear nickel(II) complexes bearing bridging μ2-azolate and terminal 1,3,5-triazapentadienate (abbreviated as TAP) ligands were obtained as major products. We observed (see later) that the TAP ligand originates from an unusual cascade of NiII-mediated and chelation-driven cyanamide–methanol integration, and that TAP generation is not affected by the presence of any one of the azoles. Methanol in the reaction serves as both a reagent and a solvent and in view of that we decided to study the NiII-mediated reaction of dialkylcyanamides with different alcohols, whereupon we included the azoles in the tested systems. Our results are sequentially disclosed in the sections that follow.
Nickel(II)-mediated and chelation-driven cascade reaction of NCNR2 and MeOH
The NiII-mediated integration between cyanamides and alcohols was conducted in alcohols R′OH (R′ = Me, Et, iPr). The reactions of NiX2·nH2O and NCNR2 (Scheme 1) were attempted at molar ratios in the range between 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 and 1:10 and all tests were conducted either at RT or at 50 °C (for a detailed discussion of the reaction conditions see the ESI†).
:2 and 1:10 and all tests were conducted either at RT or at 50 °C (for a detailed discussion of the reaction conditions see the ESI†).
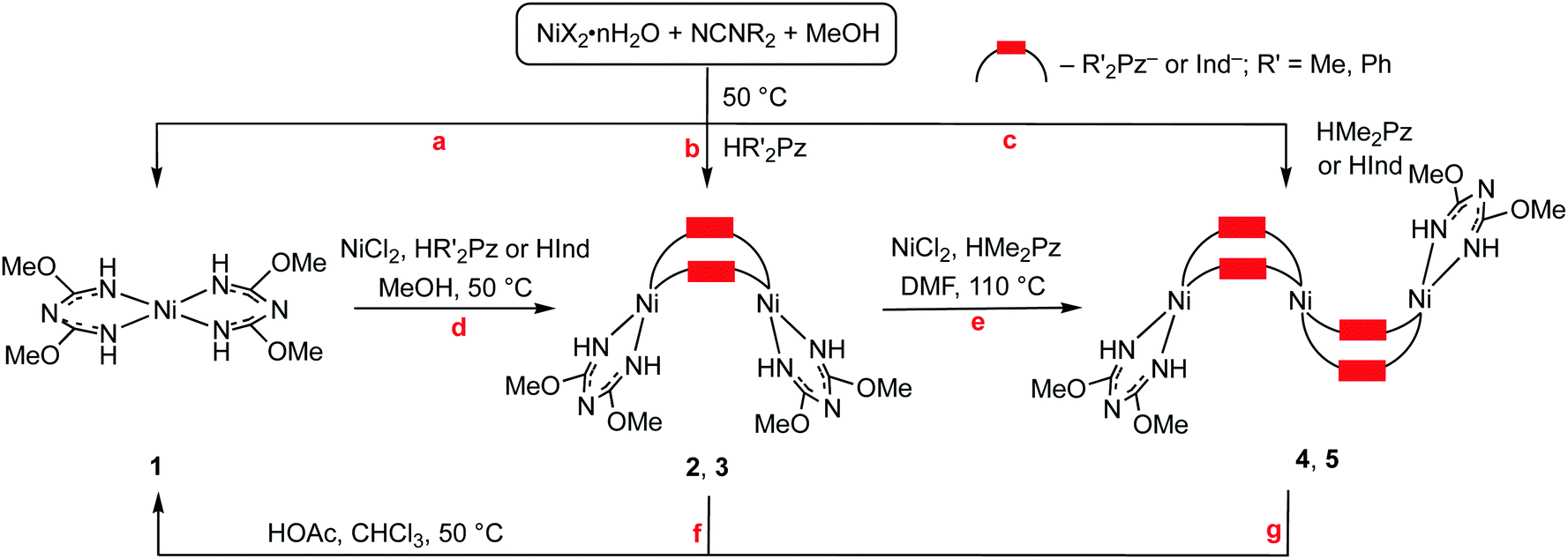 | ||
| Scheme 1 Reaction between the azoles, cyanamides, and nickel salts in methanol. | ||
We observed that the cascade generation of (TAP)NiII from NCNR2 proceeds in MeOH after maintaining the reaction mixture for two weeks at RT or for 3 d at 50 °C and this reaction gives the complex [Ni(TAP)2] (1; TAP = HC(OMe)NC(OMe)H; Scheme 1, a). Compound 1 was isolated as orange crystals after complete evaporation of the solvent and separation of the residue via column chromatography (yields up to 40%; for detailed characterization of 1 see the ESI†).
Bis-chelated complex 1 can be formed in a NiII-mediated and chelation-driven cascade reaction of dimethylcyanamide with MeOH. Moreover, MeOH plays the role of an alkylation reagent supporting NR2/OMe replacement in the formed species, and other products derived from the methylation of the NR2 group, viz. [Me2NR2]X were identified using HRESI+-MS (for a detailed discussion of the postulated reaction mechanism see the ESI†).
Complexes of the type M{HC(OR)NC(OR)H} have been reported for late transition metals, e.g. CoIII,22 NiII,22b CuII,22,23 ZnII,22b,23a,b,24 and PdII.25 These chelates were obtained either through a chelation-driven reaction of the dicyanimides, (NC)2NNa or (NC)2NH, with alcohols in the presence of a transition metal salt (CuII,14 ZnII10,12) or via the transmetalation of [M{HC(OR)NC(OR)H}2] (M = CuII or ZnII) with other metal centers such as, for instance, CoIII.22a,23a,b
Nickel(II)-mediated reaction of NCNR2 with the azoles in alcohols
Mixing NiX2·nH2O (X = Cl, OTf, n = 0; Br, n = 3; I, n = 6) with NCNR2 (R = Me2, Et2, C5H10, C2H4OC2H4) and the azoles in methanol at RT or 50 °C (see the more detailed discussion later) results in the generation of the di- and trimeric complexes [Ni2(μ2-R′2Pz)2(TAP)2] (R′ = Me 2, Ph 3) and [Ni3(μ2-Me2Pz)4(TAP)2] (4) (Scheme 1, b and c). The reactions were performed in all possible combinations of the reactants and molar ratios between NiX2·nH2O and NCNR2 that range from 1:2 to 1:6 and between NCNR2 and the azoles that are in the interval from 1:0.5 to 1:4 and two temperatures, i.e. RT and 50 °C.
Although all four cyanamides underwent the reaction, two of them (R2 = Me2, C2H4OC2H4) demonstrated greater reactivity and gave higher yields of 2–4; these two cyanamides were also the most efficient in the generation of the TAP ligand (see previous section). The di- and trinuclear complexes were obtained for the substituted pyrazoles, i.e. HMe2Pz and HPh2Pz. Among the tested nickel salts, the highest yields of 2–4 were obtained with NiX2 (X = Cl, OTf). Concurrently, the reaction with the unsubstituted pyrazole (R′ = H) after 10 min (MeOH, either RT or 50 °C) gave the previously reported crystalline polymeric α-[Ni(Pz)2]n (for its identification by powder XRD see Fig. S25, ESI†),8 which is poorly soluble in all commonly used solvents.
For the systems NiX2/NCNR2/azole (X = Cl, OTf; R2 = Me2), the composition of the product depends on the temperature, anion X, and substituent R′ in the heterocycle. For HMe2Pz and NiCl2 at 50 °C, only trimer 4 was isolated (43%), while at RT both 2 and 4 species were obtained. Pure dimer 2 was obtained when excess of NCNR2 was applied in the reaction with NiCl2 (NiCl2:NCNR2 ratio was 1:4 or 1:6) at RT and when Ni(OTf)2 reacted with HMe2Pz and NCNR2 at either RT, or at 50 °C (yield up to 25%). The HPh2Pz forms dimer 3 in all tested combinations of NCNR2 and NiX2 and the highest yield of 3 (34%) was obtained for the system NiCl2/NCNMe2/HPh2Pz (molar ratio 1:4:2). In other cases, the obtained optimal molar ratios between NCNR2 and the azoles were 4:2 (2) and 1:4 (4) at a 0.1 M concentration of NiX2 (1 equiv.).
The reaction that involves the unsymmetrical indazole and NCNR2 should potentially give a mixture of regiosiomeric species. Indeed, we observed using both NMR and HRESI-MS the generation of a broad spectrum of products that we failed to separate using column chromatography due to similar retention or to isolate the products in the pure form via fractional crystallization. A few single-crystals of [Ni3(μ2-Ind)4(TAP)2] (5) were mechanically picked out from the NiCl2/NCNMe2/HInd (1:1:4, 5 d, 50 °C) reaction mixture and were characterized by XRD (see later). The solid residue obtained after evaporation of the solvent contains a mixture of 5 with other yet unidentified products. The HRESI+-MS of this residue displays several groups of peaks corresponding to [HNC(OMe or NMe2)NHC(NMe2)NH2]+, [Ni(C2H2N3)2(OMe)x(NMe2)4−x + H]+ (x = 1–4), [Ni2(μ2-Ind)2(C2H2N3)2(OMe)x(NMe2)4−x + H]+ (x = 1–3), and [Ni3(μ2-Ind)4(C2H2N3)2(OMe)x(NMe2)4−x + H]+ (x = 1–3) (ESI,† Fig. S8).
X-ray crystallographic studies for 2–5
Single-crystal XRD studies were performed for 2–5 (Fig. 3–6). The structure of 3 is built up of two crystallographically independent molecular units, whereas all other structures consist of one molecular unit. In the structures of the dimers and trimers, the nickel atoms exhibit square-planar environments formed with four N atoms from the TAP ligand or a substituted azolate (see more information regarding the X-ray crystallographic studies in the ESI†).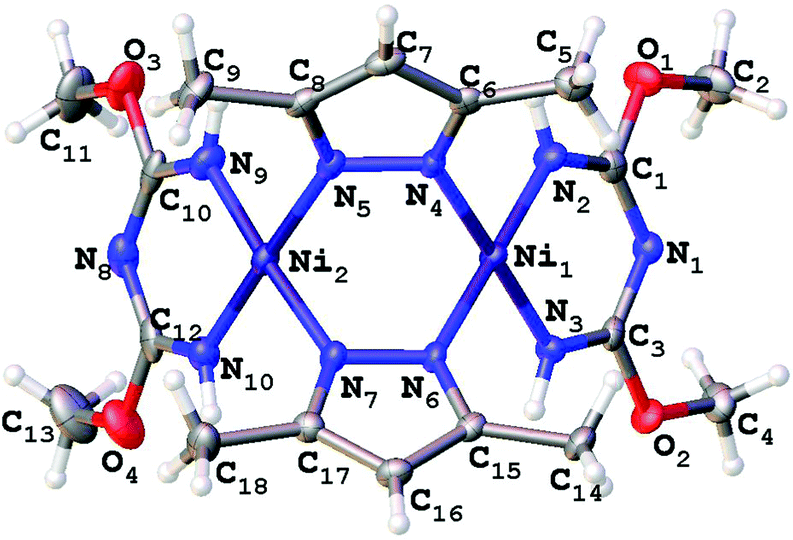 | ||
| Fig. 3 Molecular structure of 2 with the atomic numbering scheme. Thermal ellipsoids are given at the 50% probability level. | ||
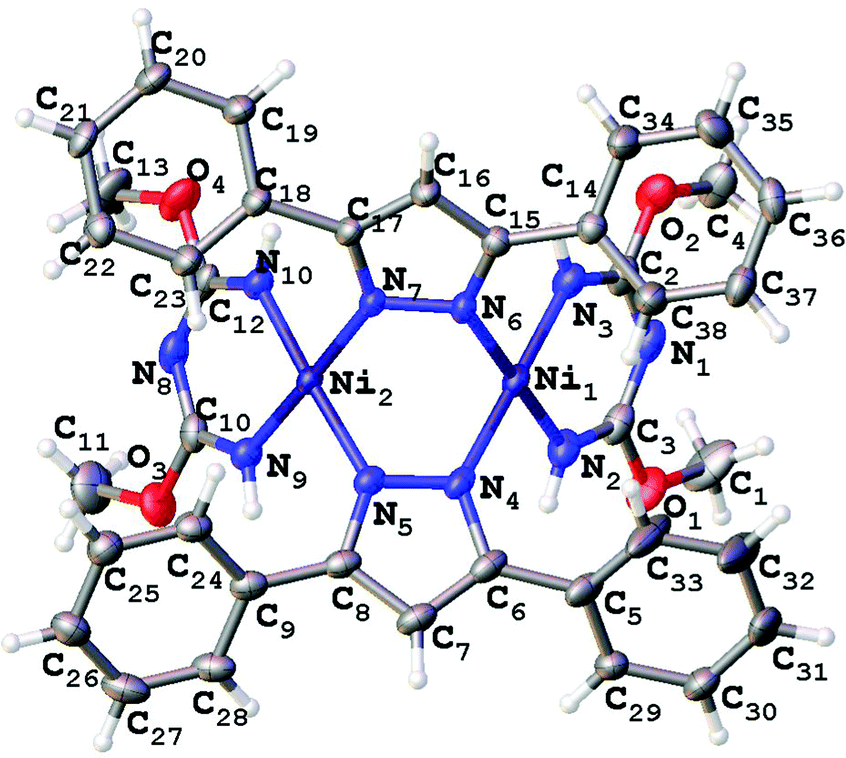 | ||
| Fig. 4 Molecular structure of 3 with the atomic numbering scheme. Thermal ellipsoids are given at the 50% probability level. | ||
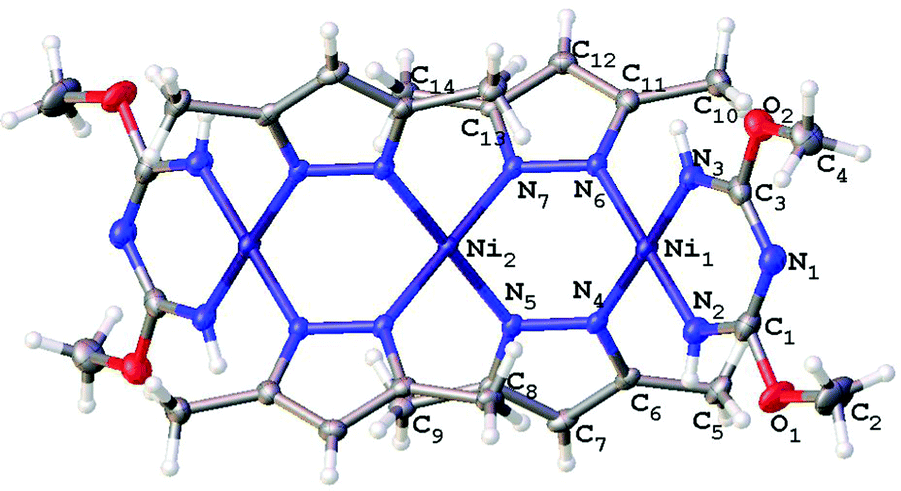 | ||
| Fig. 5 Molecular structure of 4 with the atomic numbering scheme. Thermal ellipsoids are given at the 50% probability level. | ||
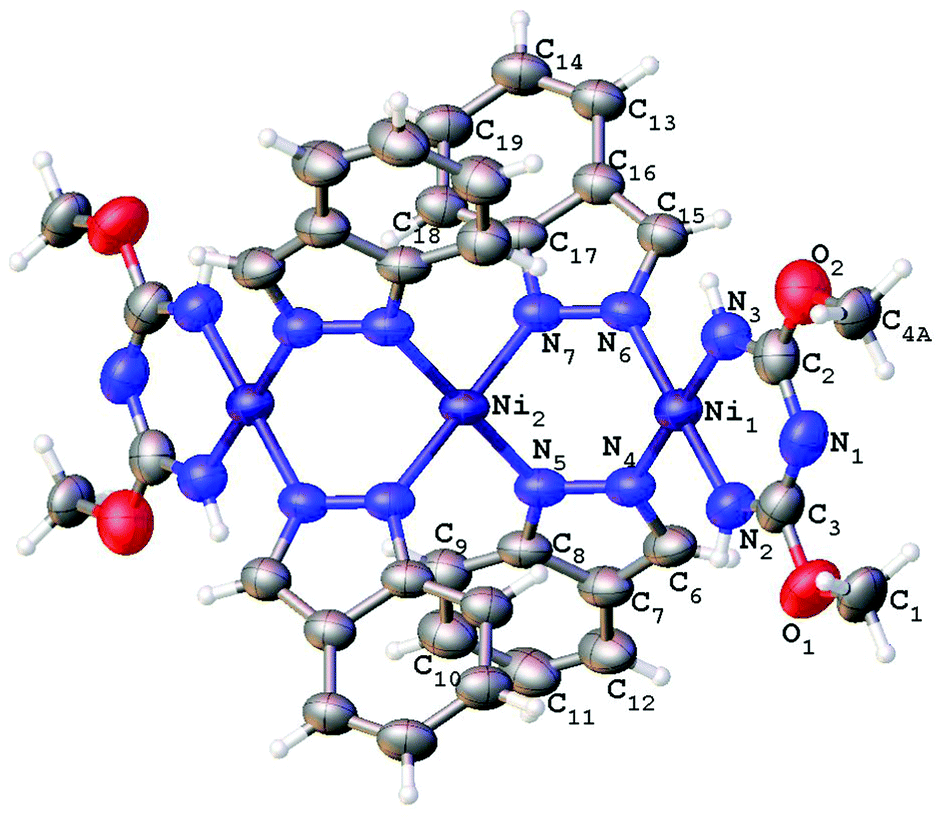 | ||
| Fig. 6 Molecular structure of 5 with the atomic numbering scheme. Thermal ellipsoids are given at the 50% probability level. | ||
Consideration of the crystallographic data suggests the presence of Ni⋯Ni metallophilic interactions in the di- and trinuclear structures. Indeed, inspection of the XRD data reveals that the interatomic distances between the Ni atoms in 2 (3.10 Å), 3 (2.99 Å), and 4 (3.25 Å) are less than the sum of their Bondi's (the shortest) van der Waals radii26 (3.26 Å for Ni + Ni atoms). In order to confirm or deny the assumption on the presence of Ni⋯Ni metallophilic interactions in 2–4 and to quantitatively estimate their energies we carried out theoretical DFT calculations and performed topological analysis of the electron density distribution within the formalism of Bader's theory (AIM method; for more detailed information see the ESI†).27 This approach has already been successfully used by us to study the non-covalent interactions and properties of the coordination bonds in transition metal complexes.28
Theoretical consideration of Ni⋯Ni metallophilic interactions in 3
Metallophilic interactions impart important properties to functional materials such as luminescence,29 polychromism,30 magnetism,31 or one-dimensional electrical conductivity.32 This phenomenon is generally considered as weak attractive interactions between adjacent metal centers due to overlapping of their dz2 and pz orbitals33 and it has been intensively studied in recent years. Most reports in this area are related to the solid state, where structural parameters obtained by XRD provide precise information on metal⋯metal separations.The strength of such M⋯M interactions is comparable to weak or medium hydrogen bonding, e.g., for Au⋯Au aurophilic interactions (the strongest one) the appropriate bond energy is ca. 7–12 kcal mol−1,34 whereas other metallophilic interactions are usually weaker because the influence of relativistic effects in such cases is lower, and the ability of atoms to be involved in this type of bonding increases down through the group. The MI⋯MI (M = Cu, Ag, Au) metallophilic interactions are certainly most studied.35 For group 10 elements, metallophilic interactions of heavy elements (i.e. PtII⋯PtII (ref. 36) and PdII⋯PdII(ref. 37)) are quite common, whereas NiII⋯NiII short contacts were detected for a significantly lower number of systems.12,38 Their metallophilic nature was never confirmed using theoretical studies and, consequently, their energies were not estimated.
The AIM analysis (ESI,† Fig. S7) did not reveal Ni⋯Ni interactions in 2 and 4. However, in 3, exhibiting the shortest Ni⋯Ni distance, we succeeded to find an appropriate bond critical point (3, −1) (BCP) (Fig. 7). The low magnitude of the electron density (0.016–0.017 Hartree), positive values of the Laplacian (0.046–0.052 Hartree), and close to zero energy density (−0.001) in this BCP are typical for non-covalent interactions. We have defined energies (3.5–4.4 kcal mol−1) for this contact according to the procedures proposed by Espinosa et al.39 and Vener et al.40 The covalent contribution in the Ni⋯Ni metallophilic interactions in 3 is negligible (for details see the ESI†). It is noteworthy that the estimate of the properties of electron density in BCP and energies for the metallophilic interactions Ni⋯Ni in 3 weakly depends on the basis set used (relativistic or non-relativistic approaches).
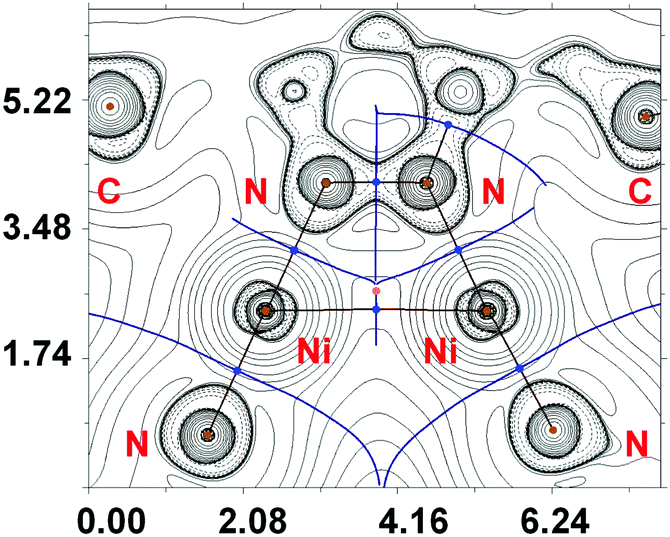 | ||
| Fig. 7 Contour line diagrams of the Laplacian distribution ∇2ρ(r), bond paths and selected zero-flux surfaces for 3. Bond critical points (3, −1) are shown in blue, nuclear critical points (3, −3) in pale brown and ring critical points (3, +1) in orange; the length unit is Å. | ||
Although the analysis of the XRD data for 2–4 indicates that in all three cases Ni⋯Ni separations are less that the double Bondi's van der Waals radius for Ni, the conducted DFT calculations allowed the reliable identification of these metallophilic contacts only in one case (complex 3 with the shortest Ni⋯Ni distance). Even for 3, the theoretically determined energies of the nickel(II)⋯nickel(II) interactions are rather low and certainly lower than those found for the PdII⋯PdII (4.3–6.0 kcal mol−1, ref. 37) and PtII⋯PtII (3.9–11.7 kcal mol−1, ref. 36a–g) systems. Taking all these into account, we believe that the emergence of nickel(II)⋯nickel(II) interaction in 3 is, at least partially, determined by crystal packing effects.
Interconversions of 1–4
We found that di- and trinuclear complexes 2–4 could be converted to monomeric 1. In contrast, 1 could give 2–4 when reacting with the appropriate azole and NiCl2. Thus, complexes 2 and 4 in a chloroform solution transform to 1 by acidification with 1 equiv. HOAc and by keeping the mixture at 50 °C for 2 d (yields of 1 are 19 and 22%, respectively; Scheme 1, f and g); formation of 1 was confirmed using HRESI-MS and single-crystal XRD. Acidification of a solution of 4 gives solely 1 with no traces of 2.Heating of a mixture of 1 and NiCl2 (a molar ratio of 1:1; MeOH solution) with HR′2Pz (R′ = Me, Ph; 8 equiv.) for 1 d at 50 °C leads to the gradual release of yellow crystals of 2 or 3 (yields 19 and 14%, respectively; Scheme 1, d). The formation of 2 and 3 was confirmed using HRESI-MS and by both powder and single-crystal XRD. HRESI-MS analysis of the mother liquor demonstrates the presence of unreacted 1 and some intermediate products, e.g. the cationic [Ni(Ph2Pz)2(TAP)]+ species. The HRESI+-MS spectra of the reaction mixture comprising of 1, NiCl2, and IndH (a molar ratio of 1:1:8; 7 d, 50 °C, MeOH) exhibit several groups of peaks corresponding to the [TAP + 2H]+, [Ni(TAP)2 + H]+, [Ni2(μ2-Ind)2(TAP)2 + H]+, and [Ni3(μ2-Ind)4(TAP)2 + H]+ ions (Fig. S16, ESI†). Therefore, although the formation of indazolate-based oligomers takes place under the reaction conditions, all our attempts to isolate individual products were, unfortunately, unsuccessful.
Dimer 2 was converted to trimer 4 when a mixture of 2 and NiCl2 (in a molar ratio of 1:1) was heated with 3 equiv. NCNMe2 and excess HMe2Pz at 110 °C in DMF for 1 d (yield 52%; Scheme 1, e). Formation of 4 was confirmed using HRESI+-MS and powder XRD, while HRESI+-MS analysis of the mother liquor demonstrates the presence of unreacted 2.
These interconversions of 1–4 explain the relatively low yields of 2–4 and the strong dependence of the outcome of the reaction on the molar ratio of the reactants. Analysis of the reaction mixtures using HRESI-MS indicates the co-existence of several mono-, di-, and trimeric (azolate)2NiII-based species, while their corresponding tetramers and nickel-containing species of higher nuclearities were not detected even upon prolonged heating of the resection mixtures at 50 °C in DMF or at 140–160 °C in HOCH2CH2OH.
Conclusions
The results of this work can be considered from the following main perspectives. We synthesized two triazapentadienate-terminated di- and two trinuclear NiII complexes featuring the bridging azolate ligands, [Ni2(μ2-azolate)2(TAP)2] and [Ni3(μ2-azolate)4(TAP)2] (TAP = HC(OMe)NC(OMe)H). We observed that these dimers and trimers and also [Ni(TAP)2] could undergo interconversions depending on the reactants involved and the reaction conditions (Scheme 1).
The nuclearity of the complexes depends on the amount of azoles (or, in contrast, acid) in the system, steric hindrance of the substituents in the ring systems, and the temperature of the reaction. A higher amount of azoles leads to higher nuclearity systems, whereas the addition of an acid promotes conversion of trimers to dimers and then to [Ni(TAP)2]. As it is more sterically demanding, due to the restricted rotational freedom in the complexes, H(3,5-Ph2)Pz forms only dimer 3, whereas H(3,5-Me2)Pz gives dimeric 2 and trimeric 4 species. Higher temperature favors the generation of trimeric 4 rather than dimeric 2.
All these observations open up a route to control both the nuclearity of nickel(II)–azole complexes and allows the synthesis of well-defined (azolate)2NiII species. Partially our observations are in agreement with those reported previously for azolate species bearing the terminal CpNi fragment.11 Although in these two studies the dependence of nuclearity on the amount of azole/acid was not established, the authors observed the dependence of the nuclearity of (azole)NiII species on both the steric hindrance of azoles and temperature. We expect that our data might be useful in the generation of nickel(II) systems of higher nuclearity in, e.g. solvothermal syntheses.41
Using XRD we detected unconventional nickel(II)⋯nickel(II) metallophilic interactions in 3, and provided the first case where the availability of these metallophilic interactions was confirmed theoretically using a DFT method. Furthermore, we succeeded in getting an estimate of the energy for these non-covalent contacts (3.5–4.4 kcal mol−1)—that is lower than those for PdII⋯PdII (4.3–6.0 kcal mol−1, ref. 37) and PtII⋯PtII (3.9–11.7 kcal mol−1, ref. 36a–g) systems—and it is believed that the existence of these NiII⋯NiII interactions is mainly determined by the packing effects. It has now been proven that the strength of the metallophilic interactions for compounds of group 10 elements increases down through the group according to their ability to overlap dz2 and pz orbitals of the neighboring metal centers.
Experimental
Materials and instrumentation
Solvents, dialkylcyanamides, and nickel(II) salts NiBr2·3H2O, NiI2·6H2O, and Ni(OTf)2 were obtained from commercial sources and used as received. Anhydrous nickel chloride was prepared according to a previously reported procedure.42 FTIR spectra (4000–400 cm−1) were recorded on a Shimadzu IR Prestige-21 instrument in KBr pellets (Fig. S9–S12 of the ESI†). Microanalyses (C, H, N) were carried out on a Euro EA3028-HT instrument. A standard EDTA method was utilized for Ni analysis of complex 1.43 Electrospray ionization mass-spectra were obtained on a Bruker micrOTOF spectrometer equipped with an electrospray ionization (ESI) source. The instrument was operated both in positive and negative ion modes in the m/z range 50–3000. For ESI, the complexes were dissolved in MeOH. In the isotopic pattern, the most intensive peak is reported (Fig. S8 and S13–S17, ESI†). GC-MS identification of the by-products from the synthesis of 1 was performed on a Shimadzu GCMS-QP2010 Ultra instrument equipped with a Stabilwax 30 m × 0.32 × 0.50 μm column. The temperature program was set at 70–230 °C with a linear rate of 10 °C min−1 and the column was held at 230 °C for 75 min. 1H and 13C{1H} NMR (400.13 and 100.613 MHz, respectively) spectra were measured on a Bruker Avance 400 spectrometer in Me2SO-d6 (for 1) and CDCl3 (for 2–4) at ambient temperature; residual solvent signals were used as the internal standard (Fig. S17–S24, ESI†). The TG-DTA study (Fig. S28–S31, ESI†) was performed using a NETZSCH TG 209 F1 Libra thermoanalyzer and MnO2 powder was used as a standard. The initial weights of the samples were in the range of 0.9–1.4 mg. The experiments were run in an open aluminum crucible in a stream of argon at a heating rate of 10 K min−1. The final temperature was 610 °C. Processing of the thermal data was performed using Proteus analysis software.44 TLC was performed on Merck Silica gel 60G F254 plates.X-ray crystal structure determination
For single-crystal XRD experiments all crystals were fixed on a micro mount and placed on an Agilent Technologies SuperNova diffractometer using CuKα monochromated radiation (1, 3, 5) and on a Xcalibur, Eos diffractometer using MoKα monochromated radiation (2, 4); the crystals were measured at a temperature of 100 K. The structures were solved using the direct methods on a ShelXS45 structure solution program (2, 3) and Superflip46 structure solution program (1 and 4). All structures were refined by means of the SHELX program45 incorporated into the OLEX2 program package.47 The crystallographic data and some refinement parameters are given in Table S3 (ESI†). The carbon-bound and nitrogen-bound H atoms were placed in the calculated positions and were included in the refinement in the ‘riding’ model approximation, with Uiso(H) set to 1.5Ueq(C) and C–H 0.96 Å for the CH3 groups, with Uiso(H) set to 1.2Ueq(C) and C–H 0.93 Å for the CH groups, and with Uiso(H) set to 1.2Ueq(N) and N–H 0.86 Å for the NH groups. Empirical absorption correction was applied in the CrysAlisPro program complex21b using spherical harmonics, implemented in the SCALE3 ABSPACK scaling algorithm. The unit cell of 3 also contains disordered molecules of the solvent that have been treated as a diffuse contribution to the overall scattering without specific atom positions using SQUEEZE/PLATON.48 Crystallographic data for this paper were deposited at the Cambridge Crystallographic Data Centre and the CCDC numbers are 1440113, 1440114, 1440621, 1442880, and 1453875. Polycrystalline samples were studied in the 2θ range of 5–45° with a Bruker “D2 Phaser” powder diffractomer equipped with CuKα source monochromated radiation.Computational details
The single point calculations for 2–4 have been carried out at the DFT level of theory using the M06 functional (this functional describes reasonably weak dispersion forces and non-covalent interactions)49 with the help of the Gaussian-09 program package.50 The experimental X-ray geometries were taken as starting points. Two approaches were used in the present work, viz. (i) the Douglas–Kroll–Hess 2nd order scalar relativistic calculations51 requested relativistic core Hamiltonian were carried out using the DZP-DKH basis sets52 for all atoms; (ii) the non-relativistic calculations were carried out using the standard 6-311+G* basis sets for all atoms. The topological analysis of the electron density distribution with the help of the atoms in molecules (AIM) method developed by Bader has been performed by using the Multiwfn program (version 3.3.4).53 The Cartesian atomic coordinates for 2–4 are given in Table S2 (ESI†).Syntheses and characterization
:hexane, 10:1, v/v). The red fraction was collected and evaporated to dryness at RT in air giving a crystalline product, which was dried over CaCl2 in a desiccator at 20–25 °C.
:hexane, 10:1, v/v). The complex sublimes at 175 °C upon heating. Calcd. for C8H16N6NiO4: C, 30.13; N, 26.35; H, 5.06; Ni, 18.40; Found: C, 30.19; N, 26.29; H, 5.04; Ni, 17.98. HRESI+-MS, m/z: 319.0670 ([M + H]+, requires 319.0670). νmax(KBr)/cm−1: 3364 m, 3344 m ν(N–H); 2994 s, 2950 s ν(C–H), 1614 vs. ν(CN), 1394 s ν(C–O from OCH3), 1214 s ν(C–N). 1H NMR (DMSO-d6): 3.58 (s, 3H, OMe), 4.66 (br s, 1H, NH). 13C{1H} NMR (DMSO-d6): 54.0 (OCH3), 163.1 (CN). Crystals of 1 suitable for XRD were obtained by the slow evaporation of the reaction mixture at RT.
NiII-mediated reaction of cyanamides with the azoles in methanol
Complexes 3 and 4 were prepared similarly to 2. NiCl2 (64 mg, 0.5 mmol), MeOH (3 mL), NCNR2 (R2 = Me2 or C2H4OC2H4; 2 mmol), and HPh2Pz (220 mg, 1 mmol; for generation of 3) or HMe2Pz (192 mg, 2 mmol; for generation of 4) were used. The reaction occurred for 4 (3) and 5 (4) days.
N); 1526 s ν(C–N)Pz; 1398 s ν(C–O from OMe), 1220 m ν(C–N). 1H NMR (CDCl3, δ): 5.38 (s, 1H, CH), 3.78 (s, 6H, OCH3), 2.46 (s, 6H, C–CH3). 13C{1H} NMR (CDCl3, δ): 163.8 (CN), 148.0 (CH(Pz)), 105.4 (C(Pz)), 54.3 (CH3O), 13.5 (CH3).
N); 1522 s ν(C–N)Pz; 1400 s ν(C–O from OMe), 1232 m ν(C–N). 1H NMR (CDCl3, δ): 8.73 (d, J = 7.3 Hz, 4H, CHAr), 7.60 (t, J = 7.0 Hz, 4H, CHAr), 7.46 (t, J = 6.6 Hz, 2H, CHAr), 6.37 (s, 1H, CH), 3.58 (s, 6H, OCH3). 13C{1H} NMR (CDCl3, δ): 163.4 (CN), 153.4 (CH(Pz)), 132.7 (C(Ph)), 128.5, 127.8 (CH(Ph)), 105.5 (C(Pz)), 54.9 (CH3O).
N); 1524 s ν(C–N)Pz; 1398 s ν(C–O from OMe), 1222 m ν(C–N). 1H NMR (CDCl3, δ): 5.38 (s, 1H, CH), 3.82 (s, 3H, OCH3), 2.56, 2.22 (two s, 3H, C–CH3). 13C{1H} NMR (CDCl3, δ): 163.9, 161.5 and 161.1 (CN) and (CO)Me2NCONH2, 150.1, 149.9, 149.8, 149.8, 147.3, 147.3, 147.2 (CH(Pz)), 106.4, 106.2, 106.2, 106.2, 106.0 (C(Pz)), 55.0, 54.4, 53.8 (CH3O), 15.3, 14.3, 14.2, 14.0, 13.9, 13.8 (CH3).
Acknowledgements
The synthetic part of this combined project was supported by the Russian Science Foundation (grant 14-13-00060), whereas the theoretical part was conducted under the Russian Foundation for Basic Research project (grant 16-33-60063). The authors thank the Center for X-ray Diffraction Studies, the Center for Magnetic Resonance, the Center for Thermogravimetric and Calorimetric Research, the Center for Chemical Analysis and Materials Research, the Chemistry Educational Center, and “Geomodel” (which all belong to Saint Petersburg State University) for the physicochemical measurements.References
- (a) S. Trofimenko, Scorpionates: The Coordination Chemistry of Polypyrazolylborate Ligands, Imperial College Press, London, 1999 Search PubMed; (b) S. Trofimenko, J. Chem. Educ., 2005, 82, 1715–1720 CrossRef CAS.
- C. Pettinari, A. Tabacaru and S. Galli, Coord. Chem. Rev., 2016, 307, 1–31 CrossRef CAS.
- (a) E. D. Doidge, J. W. Roebuck, M. R. Healy and P. A. Tasker, Coord. Chem. Rev., 2015, 288, 98–117 CrossRef CAS; (b) M. Viciano-Chumillas, S. Tanase, L. J. de Jongh and J. Reedijk, Eur. J. Inorg. Chem., 2010, 3403–3418, DOI:10.1002/ejic.201000412; (c) I. Castro, W. P. Barros, M. L. Calatayud, F. Lloret, N. Marino, G. De Munno, H. O. Stumpf, R. Ruiz-García and M. Julve, Coord. Chem. Rev., 2016, 315, 135–152 CrossRef CAS; (d) W. P. Barros, M. Luisa Calatayud, F. Lloret, M. Julve, N. Marino, G. De Munno, H. O. Stumpf, R. Ruiz-Garcia and I. Castro, CrystEngComm, 2016, 18, 437–449 RSC; (e) J.-Y. Xu, X. Qiao, H.-B. Song, S.-P. Yan, D.-Z. Liao, S. Gao, Y. Journaux and J. Cano, Chem. Commun., 2008, 6414–6416, 10.1039/B813705F.
- (a) M. A. Halcrow, Dalton Trans., 2009, 2059–2073, 10.1039/b815577a; (b) J. Perez and L. Riera, Eur. J. Inorg. Chem., 2009, 4913–4925, DOI:10.1002/ejic.200900694; (c) L. Hou, W.-J. Shi, Y.-Y. Wang, H.-H. Wang, L. Cui, P.-X. Chen and Q.-Z. Shi, Inorg. Chem., 2011, 50, 261–270 CrossRef CAS PubMed; (d) L.-Y. Du, W.-J. Shi, L. Hou, Y.-Y. Wang, Q.-Z. Shi and Z. Zhu, Inorg. Chem., 2013, 52, 14018–14027 CrossRef CAS PubMed.
- K. E. Dalle and F. Meyer, Eur. J. Inorg. Chem., 2015, 3391–3405, DOI:10.1002/ejic.201500185.
- (a) J. Tong, S.-Y. Yu and H. Li, Chem. Commun., 2012, 48, 5343–5345 RSC; (b) G.-H. Ning, L.-Y. Yao, L.-X. Liu, T.-Z. Xie, Y.-Z. Li, Y. Qin, Y.-J. Pan and S.-Y. Yu, Inorg. Chem., 2010, 49, 7783–7792 CrossRef CAS PubMed.
- N. Masciocchi, G. A. Ardizzoia, S. Brenna, G. LaMonica, A. Maspero, S. Galli and A. Sironi, Inorg. Chem., 2002, 41, 6080–6089 CrossRef CAS PubMed.
- C. J. Adams, M. A. Kurawa and A. G. Orpen, Dalton Trans., 2010, 39, 6974–6984 RSC.
- (a) S. Galli, N. Masciocchi, V. Colombo, A. Maspero, G. Palmisano, F. J. Lopez-Garzon, M. Domingo-Garcia, I. Fernandez-Morales, E. Barea and J. A. R. Navarro, Chem. Mater., 2010, 22, 1664–1672 CrossRef CAS; (b) E. Albanese, B. Civalleri, M. Ferrabone, F. Bonino, S. Galli, A. Maspero and C. Pettinari, J. Mater. Chem., 2012, 22, 22592–22602 RSC.
- K. S. M. Salih, S. Bergner, H. Kelm, Y. Sun, A. Grün, Y. Schmitt, R. Schoch, M. Busch, N. Deibel, S. Bräse, B. Sarkar, M. Bauer, M. Gerhards and W. R. Thiel, Eur. J. Inorg. Chem., 2013, 6049–6059 CrossRef CAS.
- (a) W. Tan, Z. Yu, B. Liu, K. Wu, Z. Liu and J. Chen, J. Organomet. Chem., 2009, 694, 199–206 CrossRef CAS; (b) S. J. Rettig, A. Storr, D. A. Summers, R. C. Thompson and J. Trotter, Can. J. Chem., 1997, 75, 949–958 CrossRef CAS.
- K. Fujita, S. Hikichi, M. Akita and Y. Moro-oka, J. Chem. Soc., Dalton Trans., 2000, 117–119, 10.1039/A908181J.
- S. Mukhopadhyay, U. Mukhopadhyay, T. C. W. Mak and D. Ray, Inorg. Chem., 2001, 40, 1057–1059 CrossRef CAS.
- P. Espinet, A. M. Gallego, J. M. Martínez-Ilarduya and E. Pastor, Inorg. Chem., 2000, 39, 975–979 CrossRef CAS PubMed.
- S.-J. Jeon and R. M. Waymouth, Dalton Trans., 2008, 437–439, 10.1039/B715212D.
- Y. Zhou, Z. Xi, W. Chen and D. Wang, Organometallics, 2008, 27, 5911–5920 CrossRef CAS.
- (a) P. J. Altmann, C. Jandl and A. Pothig, Dalton Trans., 2015, 44, 11278–11281 RSC; (b) P. J. Altmann and A. Pöthig, Chem. Commun., 2016, 52, 9089–9092, 10.1039/C6CC00507A.
- E. Quartapelle Procopio, S. Rojas, N. M. Padial, S. Galli, N. Masciocchi, F. Linares, D. Miguel, J. E. Oltra, J. A. R. Navarro and E. Barea, Chem. Commun., 2011, 47, 11751–11753 RSC.
- J.-Y. Xu, H.-B. Song, G.-F. Xu, X. Qiao, S.-P. Yan, D.-Z. Liao, Y. Journaux and J. Cano, Chem. Commun., 2012, 48, 1015–1017 RSC.
- (a) N. A. Bokach, Russ. Chem. Rev., 2010, 79, 89–100 CrossRef CAS; (b) N. A. Bokach, M. L. Kuznetsov and V. Y. Kukushkin, Coord. Chem. Rev., 2011, 255, 2946–2967 CrossRef CAS; (c) N. A. Bokach and V. Y. Kukushkin, Russ. Chem. Rev., 2005, 74, 153–170 CrossRef CAS; (d) N. A. Bokach and V. Y. Kukushkin, Coord. Chem. Rev., 2013, 257, 2293–2316 CrossRef CAS; (e) V. Y. Kukushkin and A. J. L. Pombeiro, Chem. Rev., 2002, 102, 1771–1802 CrossRef CAS PubMed; (f) V. Y. Kukushkin and A. J. L. Pombeiro, Inorg. Chim. Acta, 2005, 358, 1–21 CrossRef CAS; (g) A. J. L. Pombeiro and V. Y. Kukushkin, Compr. Coord. Chem. II, 2004, 1, 639–660 CAS.
- (a) A. V. Khripun, V. Y. Kukushkin, S. I. Selivanov, M. Haukka and A. J. L. Pombeiro, Inorg. Chem., 2006, 45, 5073–5083 CrossRef CAS PubMed; (b) M. A. Kinzhalov, K. V. Luzyanin, V. P. Boyarskiy, M. Haukka and V. Y. Kukushkin, Russ. Chem. Bull., 2013, 62, 758–766 CrossRef CAS.
- (a) R. Boca, M. Hvastijova and J. Kozisek, J. Chem. Soc., Dalton Trans., 1995, 1921–1923 RSC; (b) G. Goetz and H. U. Kibbel, Z. Chem., 1975, 15, 278–280 CrossRef CAS.
- (a) I. M. Atkinson, M. M. Bishop, L. F. Lindoy, S. Mahadev and P. Turner, Chem. Commun., 2002, 2818–2819, 10.1039/b206898b; (b) M. M. Bishop, L. F. Lindoy, S. Mahadev and P. Turner, J. Chem. Soc., Dalton Trans., 2000, 233–234 RSC; (c) M. N. Kopylovich, Y. Y. Karabach, M. F. C. Guedes da Silva, P. J. Figiel, J. Lasri and A. J. L. Pombeiro, Chem. – Eur. J., 2012, 18, 899–914 CrossRef CAS PubMed; (d) L.-L. Zheng, J.-D. Leng, W.-T. Liu, W.-X. Zhang, J.-X. Lu and M.-L. Tong, Eur. J. Inorg. Chem., 2008, 4616–4624, DOI:10.1002/ejic.200800486; (e) M.-L. Tong, Y.-M. Wu, Y.-X. Tong, X.-M. Chen, H.-C. Chang and S. Kitagawa, Eur. J. Inorg. Chem., 2003, 2385–2388 CrossRef CAS.
- G. Rembarz, E. Fischer, K. C. Roeber, R. Ohff and H. Crahmer, J. Prakt. Chem., 1969, 311, 889–892 CrossRef CAS.
- M. N. Kopylovich, J. Lasri, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Eur. J. Inorg. Chem., 2011, 377–383, DOI:10.1002/ejic.201000898.
- S. Alvarez, Dalton Trans., 2013, 42, 8617–8636 RSC.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, Oxford, 1990 Search PubMed.
- (a) X. Ding, M. J. Tuikka, P. Hirva, V. Y. Kukushkin, A. S. Novikov and M. Haukka, CrystEngComm, 2016, 18, 1987–1995 RSC; (b) A. S. Novikov, M. L. Kuznetsov and A. J. L. Pombeiro, Chem. – Eur. J., 2013, 19, 2874–2888 CrossRef CAS PubMed; (c) D. M. Ivanov, A. S. Novikov, I. V. Ananyev, Y. V. Kirina and V. Y. Kukushkin, Chem. Commun., 2016, 52, 5565–5568 RSC; (d) T. V. Serebryanskaya, A. S. Novikov, P. V. Gushchin, M. Haukka, R. E. Asfin, P. Tolstoy and V. Y. Kukushkin, Phys. Chem. Chem. Phys., 2016, 18, 14104–14112 RSC; (e) V. N. Mikhaylov, V. N. Sorokoumov, K. A. Korvinson, A. S. Novikov and I. A. Balova, Organometallics, 2016, 35, 1684–1697 CrossRef CAS; (f) D. M. Ivanov, A. S. Novikov, G. L. Starova, M. Haukka and V. Y. Kukushkin, CrystEngComm, 2016, 18, 5278–5286 RSC.
- (a) V. W. Yam and K. K. Lo, Chem. Soc. Rev., 1999, 28, 323–334 RSC; (b) N. L. Coker, J. A. Krause Bauer and R. C. Elder, J. Am. Chem. Soc., 2004, 12–13 CrossRef CAS PubMed; (c) B. Liu, W. Chen and S. Jin, Organometallics, 2007, 26, 3660–3667 CrossRef CAS.
- V. W.-W. Yam, K. M.-C. Wong and N. Zhu, J. Am. Chem. Soc., 2002, 124, 6506–6507 CrossRef CAS PubMed.
- D. B. Leznoff, B.-Y. Xue, C. L. Stevens, A. Storr, R. C. Thompson and B. O. Patrick, Polyhedron, 2001, 20, 1247–1254 CrossRef CAS.
- (a) S. Myllynen and M. Wasberg, Electrochem. Commun., 2009, 11, 1453–1456 CrossRef CAS; (b) M. Mitsumi, H. Ueda, K. Furukawa, Y. Ozawa, K. Toriumi and M. Kurmoo, J. Am. Chem. Soc., 2008, 130, 14102–14104 CrossRef CAS PubMed; (c) M. Mitsumi, H. Goto, S. Umebayashi, Y. Ozawa, M. Kobayashi, T. Yokoyama, H. Tanaka, T. Kuroda and K. Toriumi, Angew. Chem., Int. Ed., 2005, 44, 4164–4168 CrossRef CAS PubMed.
- W. B. Connick, R. E. Marsh, W. P. Schaefer and H. B. Gray, Inorg. Chem., 1997, 36, 913–922 CrossRef CAS.
- A. Orthaber, S. Borucki, W. Shen, R. Réau, C. Lescop and R. Pietschnig, Eur. J. Inorg. Chem., 2014, 1751–1759 CrossRef CAS.
- S. Sculfort and P. Braunstein, Chem. Soc. Rev., 2011, 40, 2741–2760 RSC.
- (a) M. R. R. Prabhath, J. Romanova, R. J. Curry, S. R. P. Silva and P. D. Jarowski, Angew. Chem., Int. Ed., 2015, 54, 7949–7953 CrossRef CAS PubMed; (b) A. Aliprandi, D. Genovese, M. Mauro and L. De Cola, Chem. Lett., 2015, 44, 1152–1169 CrossRef CAS; (c) V. V. Sivchik, E. V. Grachova, A. S. Melnikov, S. N. Smirnov, A. Y. Ivanov, P. Hirva, S. P. Tunik and I. O. Koshevoy, Inorg. Chem., 2016, 55, 3351–3363 CrossRef CAS PubMed; (d) B.-H. Xia, C.-M. Che, D. L. Phillips, K.-H. Leung and K.-K. Cheung, Inorg. Chem., 2002, 41, 3866–3875 CrossRef CAS PubMed; (e) B.-H. Xia, C.-M. Che and Z.-Y. Zhou, Chem. – Eur. J., 2003, 9, 3055–3064 CrossRef CAS; (f) H.-K. Yip, T.-F. Lai and C.-M. Che, J. Chem. Soc., Dalton Trans., 1991, 1639–1641 RSC; (g) I. M. Sluch, A. J. Miranda, O. Elbjeirami, M. A. Omary and L. M. Slaughter, Inorg. Chem., 2012, 51, 10728–10746 CrossRef CAS PubMed.
- (a) M. Weber, J. E. M. N. Klein, B. Miehlich, W. Frey and R. Peters, Organometallics, 2013, 32, 5810–5817 CrossRef CAS; (b) D. L. J. Broere, S. Demeshko, B. de Bruin, E. A. Pidko, J. N. H. Reek, M. A. Siegler, M. Lutz and J. I. van der Vlugt, Chem. – Eur. J., 2015, 21, 5879–5886 CrossRef CAS PubMed; (c) A. Gouranourimi, M. Ghassemzadeh and S. Bahemmat, Monatsh. Chem., 2015, 146, 57–67 CrossRef CAS.
- (a) M. Himmelsbach, R. L. Lintvedt, J. K. Zehetmair, M. Nanny and M. J. Heeg, J. Am. Chem. Soc., 1987, 109, 8003–8011 CrossRef CAS; (b) S. J. R. Kenneth, S. Chong, A. Storr and J. Trotter, Can. J. Chem., 1981, 59, 996–1006 CrossRef; (c) B. Ding, L. Yi, W.-Z. Shen, P. Cheng, D.-Z. Liao, S.-P. Yan and Z.-H. Jiang, J. Mol. Struct., 2006, 138–143, DOI:10.1016/j.molstruc.2005.08.025; (d) M. Maekawa, M. Munakata, T. Kuroda and Y. Nozaka, Inorg. Chim. Acta, 1993, 208, 243–244 CrossRef CAS.
- E. Espinosa, E. Molins and C. Lecomte, Chem. Phys. Lett., 1998, 285, 170–173 CrossRef CAS.
- M. V. Vener, A. N. Egorova, A. V. Churakov and V. G. Tsirelson, J. Comput. Chem., 2012, 33, 2303–2309 CrossRef CAS PubMed.
- (a) J. Lai, W. Niu, R. Luque and G. Xu, Nano Today, 2015, 10, 240–267 CrossRef CAS; (b) Y. Zhao, K. Li and J. Li, Z. Naturforsch., 2010, 976–998 Search PubMed.
- A. R. Pray, R. F. Heitmiller, S. Strycker, V. D. Aftandilian, T. Muniyappan, D. Choudhury and M. Tamres, Inorg. Synth., 2007, 28, 321 CrossRef.
- D. Harvey, Modern analytical chemistry, McGraw-Hill Companies Inc., International ed, 2000 Search PubMed.
- NETZSCH Proteus Software v.6.1., Netzsch-Gerätebau, Bayern, Germany, 2013 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., 2008, A64, 112 CrossRef PubMed.
- (a) L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS; (b) L. Palatinus and A. van der Lee, J. Appl. Crystallogr., 2008, 41, 975–984 CrossRef CAS; (c) L. Palatinus, S. J. Prathapa and S. van Smaalen, J. Appl. Crystallogr., 2012, 45, 575–580 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- A. L. Spek, PLATON, A Multipurpose Crystallographic Tool, Utrecht University, Utrecht, The Netherlands, 2005 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- M. Reiher, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 139–149 CrossRef CAS.
- (a) F. E. Jorge, A. Canal Neto, i. G. G. Camilett and S. F. Machado, J. Chem. Phys., 2009, 130, 064108 CrossRef CAS PubMed; (b) A. Canal Neto and F. E. Jorge, Chem. Phys. Lett., 2013, 582, 158–162 CrossRef CAS; (c) R. C. de Berredo and F. E. Jorge, THEOCHEM, 2010, 961, 107–112 CrossRef CAS; (d) C. L. Barros, P. J. P. de Oliveira, F. E. Jorge, A. C. Neto and M. Campos, Mol. Phys., 2010, 108, 1965–1972 CrossRef CAS.
- T. Lu and F. W. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC 1440113, 1440114, 1440621, 1442880, and 1453872. For ESI and crystallographic data in CIF or other electronic formats see DOI: 10.1039/c6nj02962k |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2017 |