
Towards the rational design of novel drugs based on solubility, partitioning/distribution, biomimetic permeability and biological activity exemplified by 1,2,4-thiadiazole derivatives†‡
T. V.
Volkova
a,
I. V.
Terekhova
a,
O. I.
Silyukov
 a,
A. N.
Proshin
b,
A.
Bauer-Brandl
c and
G. L.
Perlovich
*a
a,
A. N.
Proshin
b,
A.
Bauer-Brandl
c and
G. L.
Perlovich
*a
aDepartment of Physical Chemistry of Drugs, Krestov's Institute of Solution Chemistry, Russian Academy of Sciences, 153045 Akademicheskaya str. 1, Ivanovo, Russia. E-mail: glp@isc-ras.ru
bInstitute of Physiologically Active Compounds, Russian Academy of Sciences, 142432, Chernogolovka, Russia
cDepartment of Physics, Chemistry and Pharmacy, University of Southern Denmark, 5230 Odense M, Denmark
First published on 28th October 2016
Abstract
Novel 1,2,4-thiadiazole derivatives as potent neuroprotectors were synthesized and identified. Their ability to inhibit the glutamate stimulated Ca2+ uptake was investigated. The solubility of thiadiazoles was measured in a buffer solution (pH 7.4) at 298 K. The distribution coefficients in 1-octanol/buffer (pH 7.4) and 1-hexane/buffer (pH 7.4) immiscible phases as model systems imitating the gastrointestinal tract epithelium and the blood–brain barrier were determined. Permeation experiments the new Permeapad™ barrier using Franz diffusion cells were conducted and the apparent permeability coefficients were obtained. The influence of the compound structure on the physicochemical properties determining the bioavailability of drug-like substances was revealed. Solubility–permeability interplay has been assessed to evaluate potential bioavailability of the compounds studied.
1. Introduction
In the opinion of leading experts, Alzheimer's disease is one of the most frequent and progressive conditions among elderly people, comparable by occurrence with cardiac and cerebral infarctions.1 As the etiology of Alzheimer's disease (Alzheimer type dementia) has not been found yet, there is no rational standard therapy of the disease. Currently, worldwide investigations in the sphere of neurodegenerative drug design are being performed mainly in the following directions: invention of neuroprotectors efficacious in terms of pathogenesis of the disease and capable of blocking a pathogenic mechanism, particularly, effective multitarget drugs;2 and searching for new targets participating in the course of the disease. Moreover, considering business purposes, repositioning the well-known formulations to novel applications is very attractive. During the creation of new neuroprotective agents, fast screening of a large number of structures which exhibit biological activity at the receptors is required. Investigation of the drug delivery processes (transport properties) is an important stage in the creation of bioavailable drugs. Therefore, obtaining the screening parameters characterizing solubility, partitioning, and membrane permeability is an essential part of drug design.The 1,2,4-thiadiazole derivatives are well known to have biologically active groups to specifically target different pathologies. Many of them reveal a wide spectrum of biological activity including antioxidant and neuroprotective ones.3–6 Analysis of the influence of 1,2,4-thiadiazole derivatives on the glutamate-induced calcium ion uptake into synaptosomes of the rat brain cortex is often used for primary evaluation of the biological activity of newly synthesized drug compounds.7 The glutamate receptors in the central nervous system are known to play an important role both in regular neurophysiological processes and pathogenesis of a series of neurodegenerative diseases.8 In this connection, the evaluation of a compound's ability to affect the calcium channels mediated by the glutamate receptors allows one to assess its common biological potential as a possible neuroprotector (in the case of inhibition) or activator of cognition functions (in the case of activation). Members of a newly discovered class of compounds, ampakines, boost the activity of glutamate and thus make it easier to form memories. The cognition enhancers work on the neural processes that underlie such mental activities as attention, perception, learning, memory, language, planning and decision-making, usually by altering the balance of the chemical neurotransmitters involved in these processes. At present, nootropics and some neuronotrophic agents represent the available approaches to symptomatic treatment of Alzheimer's disease.9 Attempts to improve cognitive function in patients with brain disorders have become the focus of intensive research efforts.10 The task of developing guidelines for creating effective modulators of brain cognitive functions based on the derivatives of 1,2,4-thiadiazole is relevant from the perspective of the creation of a new class of effective drugs for the treatment of Alzheimer's disease.
Lipophilicity is a property that has a major effect on in vivo processes of absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties as well as pharmacological activity.11 The partition coefficient is accepted by most scientists to be one of the most relevant lipophilicity descriptors applied in pharmaceutical research.12 It can be measured by a rather simple experiment or calculated. Lipophilicity has been correlated to many other properties, such as solubility, permeability, metabolism, toxicity, protein binding, and distribution. 1-Octanol is the preferred organic solvent for phase-distribution investigations or partition coefficient determination of drugs. The 1-octanol/water partition coefficient is reported in drug handbooks and is commonly used in various types of analysis of drug properties.13 The log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D7.4 (logDoct/buf in the present study) of a compound stands for its distribution coefficient at pH 7.4, and it is considered to be a property of utmost importance because of its high physiological relevance and its resemblance to real biological partitions in the body.14 As it has been emphasized in ref. 15, the implication of logD7.4 for drug development leads to a general rule for optimal intestinal absorption by passive diffusion permeability after oral dosing. Drug substances should have a moderate logD7.4. Namely, a good balance of permeability and solubility exists when logD7.4 = 0–3. At that, an optimum range for the CNS (central nervous system) narrows to logD7.4 = 1–3. Since all drug compounds for the prevention of Alzheimer's disease need to overcome the blood–brain barrier (BBB), the analysis of drug interactions with these barriers is an important objective in the design of effective drugs. As a rule, the water/alkane systems (usually 1-hexane/water distribution coefficients logDhex/w) are used for BBB simulation. Selecting the alkane phase is dictated by the fact that it is a good description of non-specific interactions occurring between the drug molecules and the rather tight and lipophilic BBB environment. A parameter describing the difference between the distribution coefficients in 1-octanol/water and 1-hexane/water phases is often used to account for specific interactions (donor–acceptor, hydrogen bonding) to drug transport properties.16
D7.4 (logDoct/buf in the present study) of a compound stands for its distribution coefficient at pH 7.4, and it is considered to be a property of utmost importance because of its high physiological relevance and its resemblance to real biological partitions in the body.14 As it has been emphasized in ref. 15, the implication of logD7.4 for drug development leads to a general rule for optimal intestinal absorption by passive diffusion permeability after oral dosing. Drug substances should have a moderate logD7.4. Namely, a good balance of permeability and solubility exists when logD7.4 = 0–3. At that, an optimum range for the CNS (central nervous system) narrows to logD7.4 = 1–3. Since all drug compounds for the prevention of Alzheimer's disease need to overcome the blood–brain barrier (BBB), the analysis of drug interactions with these barriers is an important objective in the design of effective drugs. As a rule, the water/alkane systems (usually 1-hexane/water distribution coefficients logDhex/w) are used for BBB simulation. Selecting the alkane phase is dictated by the fact that it is a good description of non-specific interactions occurring between the drug molecules and the rather tight and lipophilic BBB environment. A parameter describing the difference between the distribution coefficients in 1-octanol/water and 1-hexane/water phases is often used to account for specific interactions (donor–acceptor, hydrogen bonding) to drug transport properties.16
In the present study, we propose the synthesis of new 1,2,4-thiadiazole derivatives with different substitutions of the phenyl ring and replacement of the phenyl ring by cyclopropyl and isoxazole substituents. The aim focuses at investigating the biological activity of the synthesized 1,2,4-thiadiazoles and revealing correlations between the structures, solubilities, lipophilicities and membrane permeabilities of the respective substances. The present work is a continuation of our previous attempts to study the passive transport properties of 1,2,4-thiadiazoles17–23 and the relationship between the biological activity and transport characteristics of drugs and drug-like compounds.23,24
2. Materials and methods
2.1. Synthesis
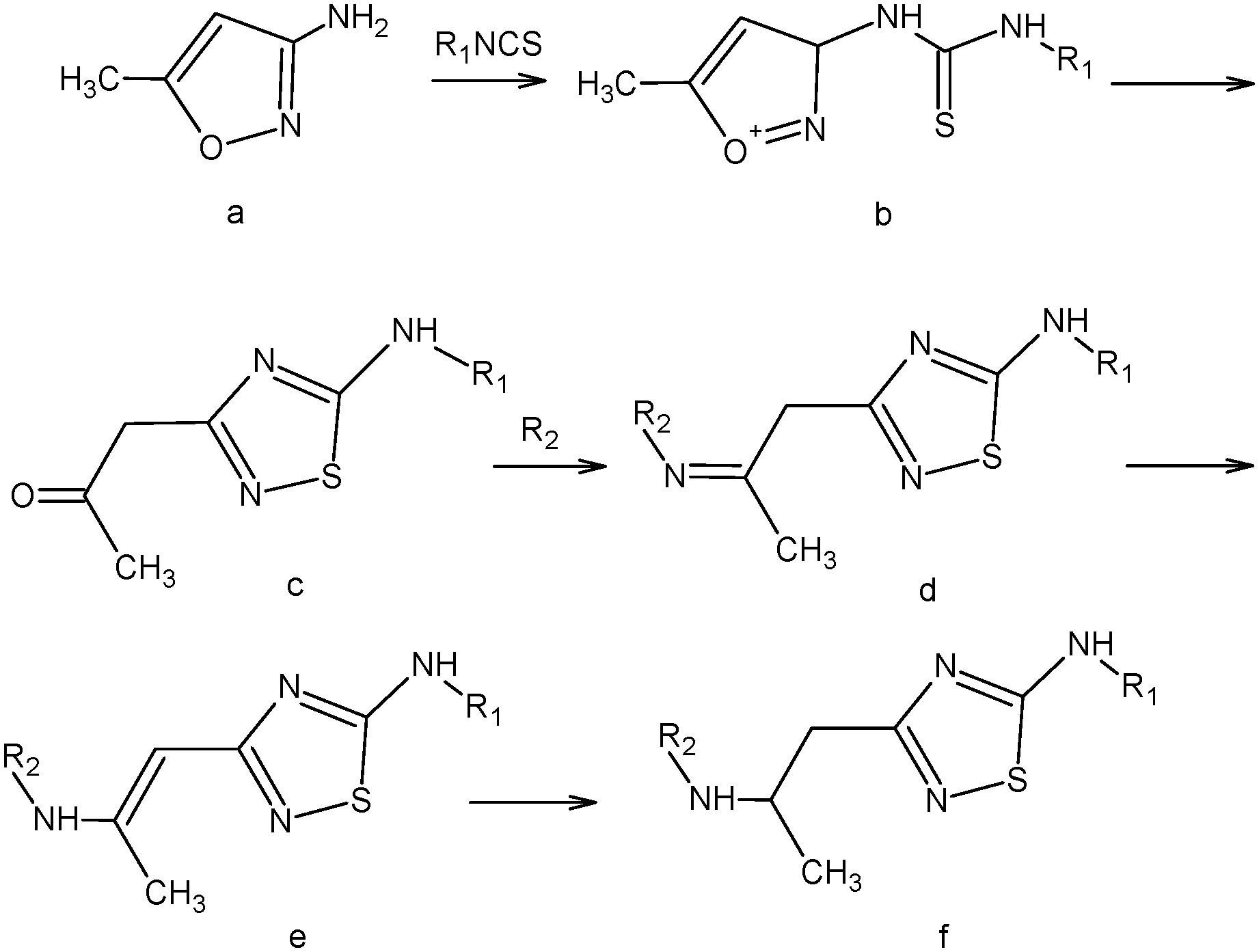 | ||
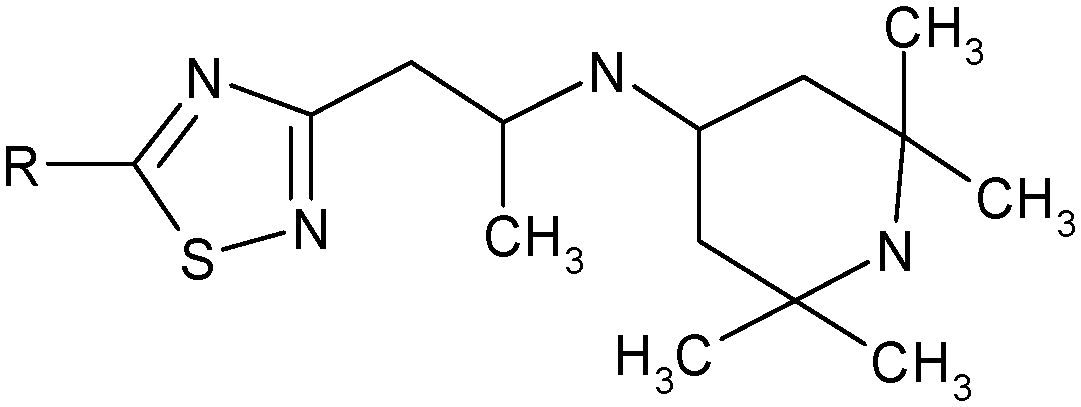
| Scheme 1 Synthesis of the compounds studied. | ||
5-N-Monoaminosubstituted 5-amino-3-(2-aminopropyl)-[1,2,4]thiadiazoles (f) were synthesized in the following manner:
A solution of 3-amino-5-methylisoxazole (a) (0.01 M) in 10 mL of acetonitrile was added dropwise to a solution of isothiocyanate (0.01 M) in 10 mL of acetonitrile under stirring. At that, the obtained isoxazole thiourea (b) in situ is transformed to a [1,2,4]thiadiazole ring according to Boulton–Katritzky.25 Upon completion of the dropping, the mixture was brought to boiling and left at room temperature until the precipitation of 5-amino-3-(2-oxopropyl)-[1,2,4]thiadiazole (c) occurs. The precipitate was filtered off. If the crystallization did not take place, the reaction mixture was evaporated and the resultant oil was triturated in diethyl ether.
The resulting 5-amino-3-(2-oxopropyl)-[1,2,4]thiadiazole (c) (0.01 M) and primary amine (0.01 M) were dissolved in 10 ml of methanol and left one day at room temperature. The precipitate was filtered off. According to NMR spectroscopy, the imine (d) forming during the reaction was rearranged to the corresponding vinylamine 5-amino-3-(2-amino-1-propenyl)-[1,2,4]thiadiazole (e) which is introduced into the reduction reaction.
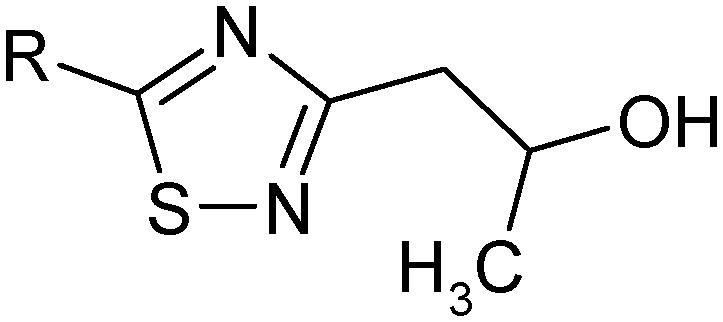
The resulting 5-amino-3-(2-amino-1-propenyl)-[1,2,4]thiadiazole (e) (0.01 M) was suspended in 30 mL of methanol, heated to 50 °C. Then sodium borohydride (0.01 M) was added portionwise under vigorous stirring. During the addition of sodium borohydride the precipitate was dissolved. Upon the completion of the reaction, methanol was evaporated, 50 mL of methylene chloride was added and washed with water (2 × 50 mL), and then the organic layer was separated, and dried over sodium sulfate. Sodium sulfate was filtered off, and the filtrate was evaporated to obtain 5-amino-3-(2-hydroxypropyl)-[1,2,4]thiadiazole (f).
| Group I | Group II | ||||||
|---|---|---|---|---|---|---|---|
|
|
|
|
|
||||
| No | R- | No | R- | No | R- | No | R- |
| 1 |
|
7 |
|
13 |
|
18 |
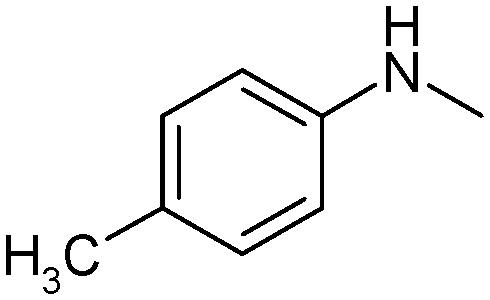
|
| 2 |
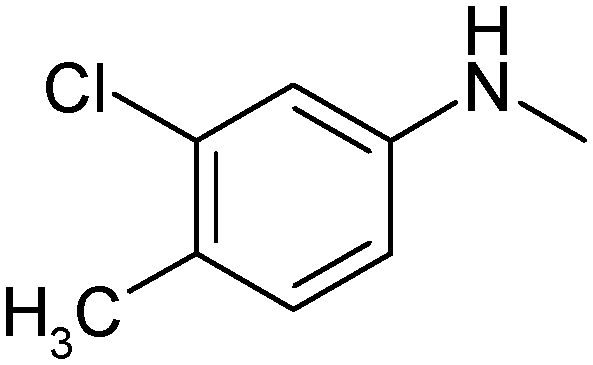
|
8 |
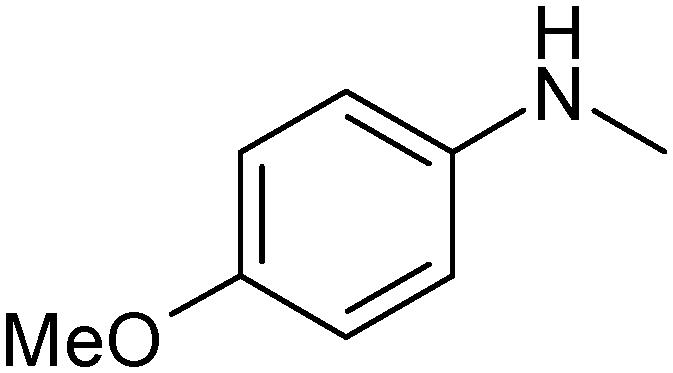
|
14 |
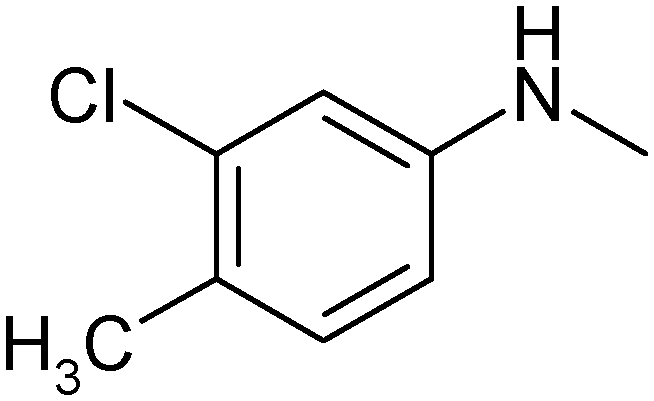
|
19 |
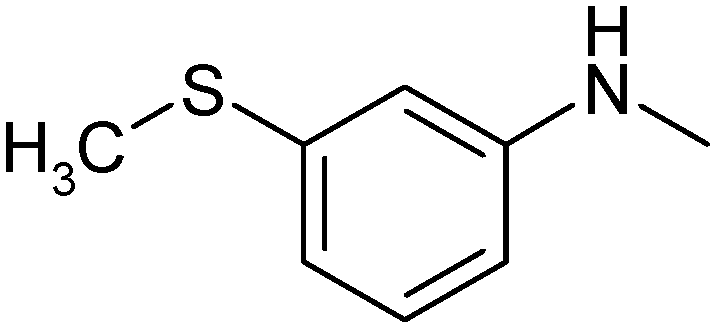
|
| 3 |
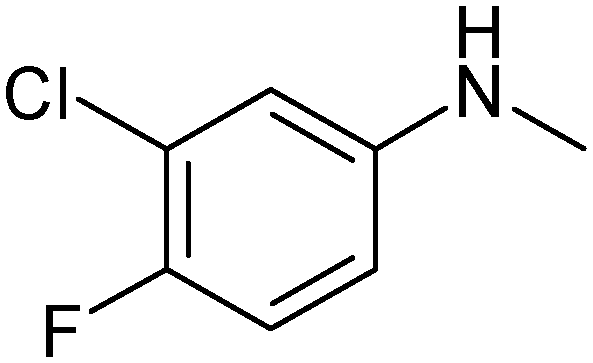
|
9 |
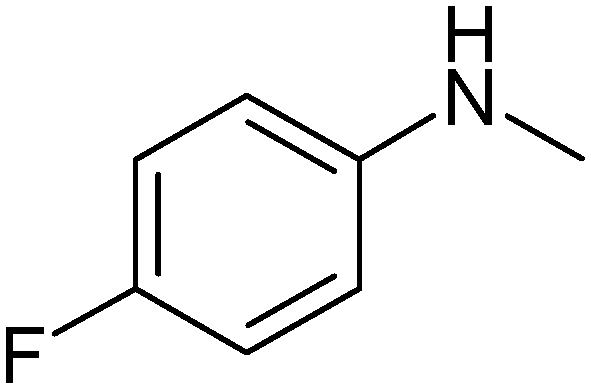
|
15 |
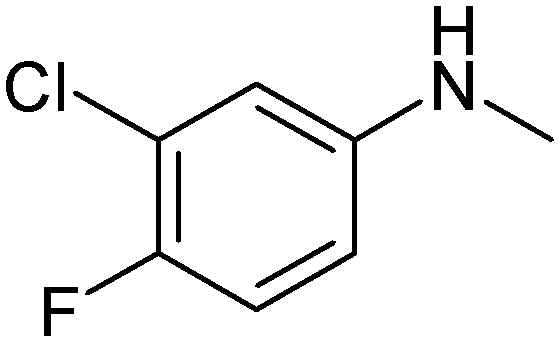
|
20 |
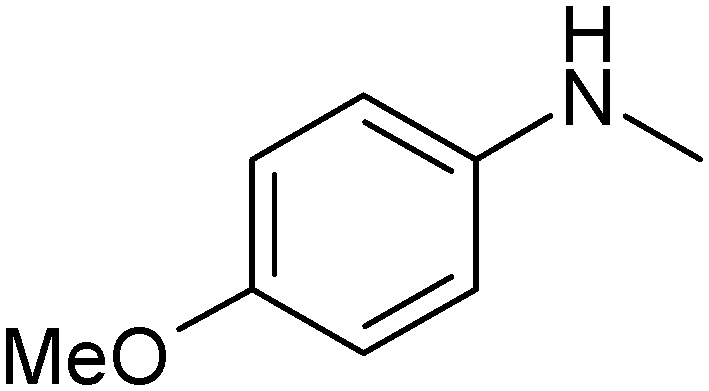
|
| 4 |
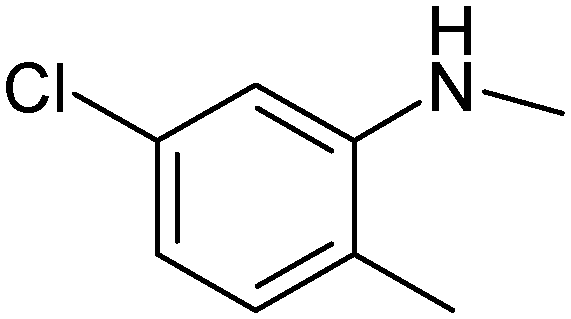
|
10 |
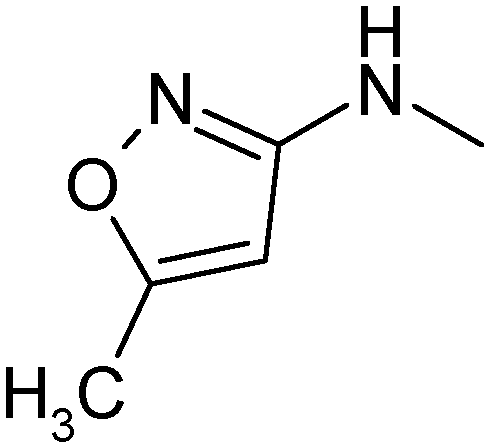
|
16 |
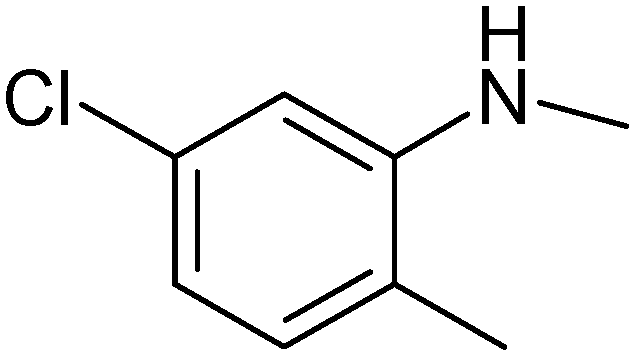
|
21 |
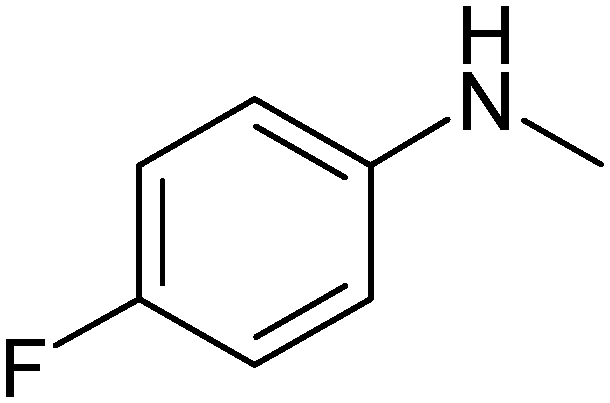
|
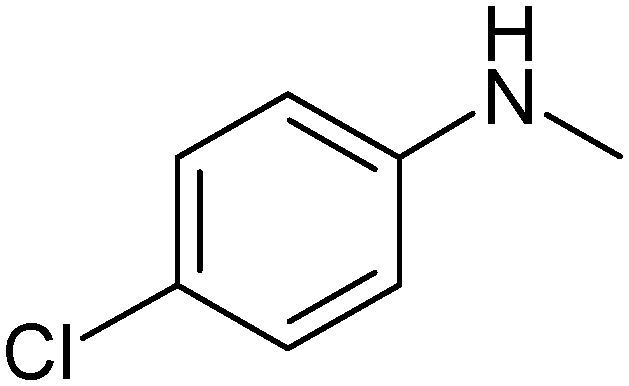
| 5 |
|
11 |
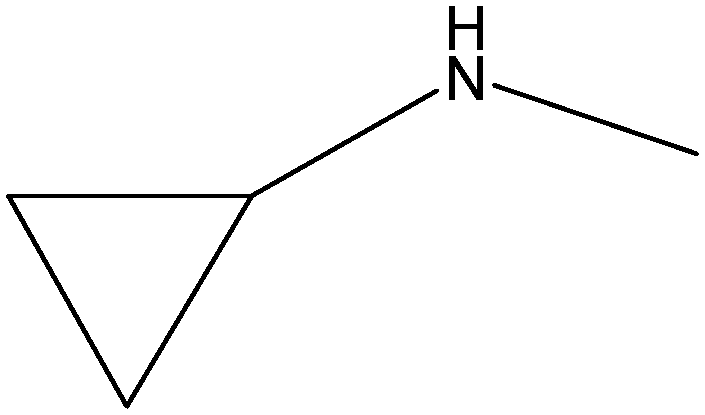
|
17 |
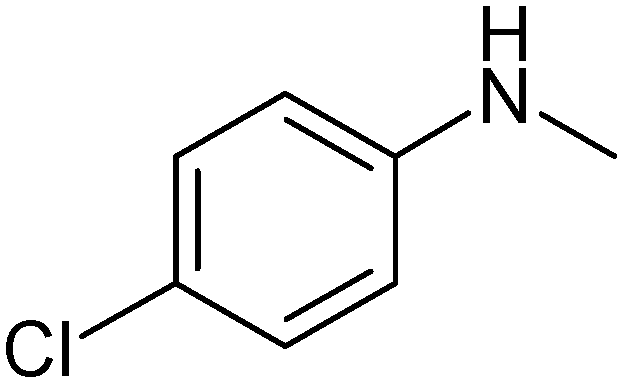
|
22 |
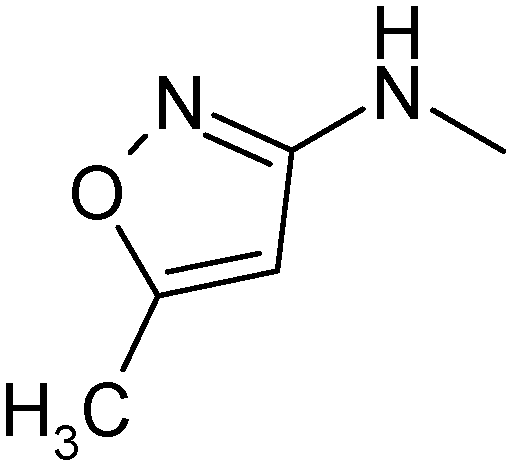
|
| 6 |
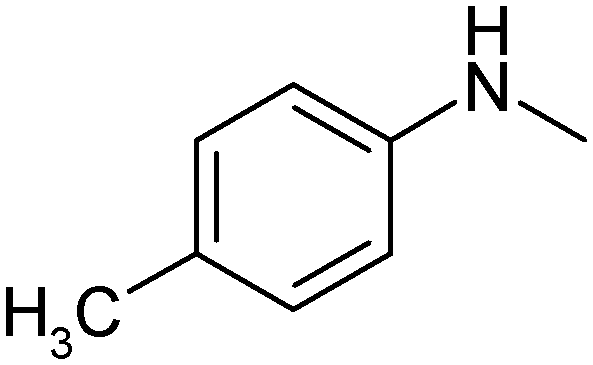
|
12 |
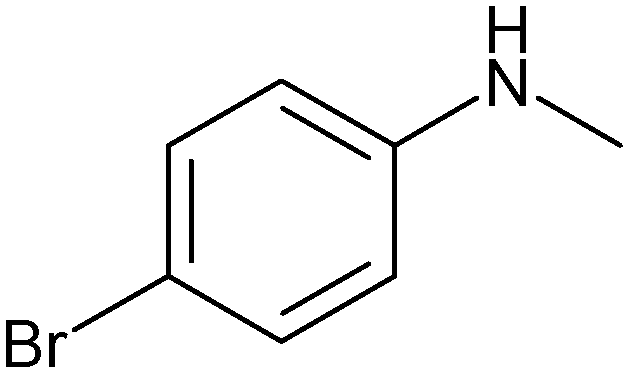
|
||||
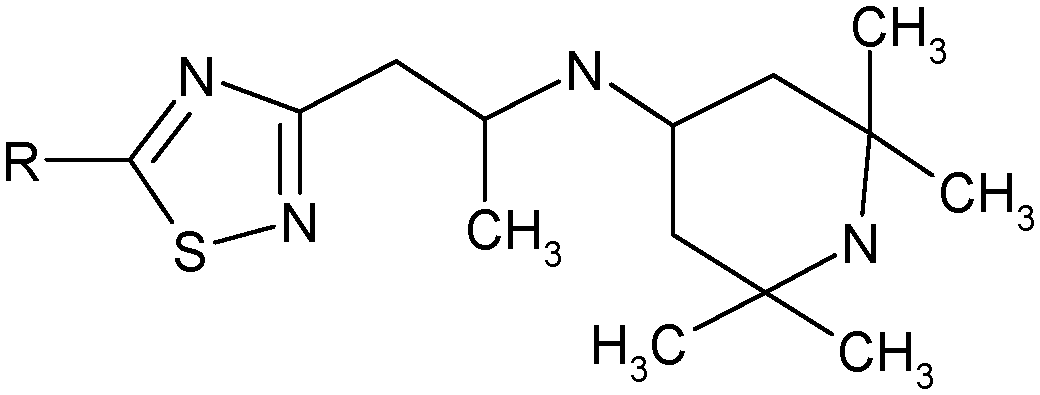
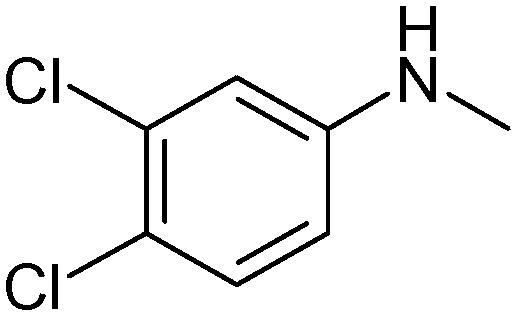
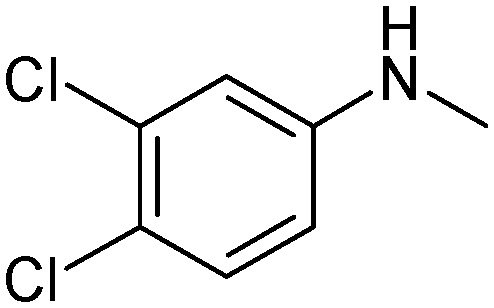
N-{1-[5-(3,4-Dichlorophenylamino)-[1,2,4]thiadiazol-3-yl]-propan-2-yl}-(2,2,6,6-tetramethylpiperidine-4-yl)amine (1). White powder. Yield 67%. Calcd., %: C 54.29, H 6.61, N 15.83 C20H29Cl2N5S. Found, %: C 54.67, H 6.66, N 15.48.
NMR 1H [200 MHz] δ: 0.79 (2H, dt, J = 12.0, 21.2 Hz, C(3)![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) H, C(5)H), 1.08 and 1.12 (both s, 6 H, 2 CH3), 1.15 (3H, d, J = 6.6 Hz, CH2CHC3), 1.84 (2H, dd, J = 3.3, 12.6 Hz, C(3)H, C(5)H), 2.84 (1H, dd, J = 6.0, 14.2 Hz, CHCHMe), 2.92 (1H, dd, J = 4.0, 14.2 Hz, CHCHMe), 3.00 (1H, m, C(4)H), 3.42 (1H, m, CH2CMe), 7.17 (1H, dd, J = 2.2, 8.6 Hz, Harom), 7.45 (1H, d, J = 8.6 Hz, Harom), 7.51 (1H, d, J = 2.2 Hz, Harom).
H, C(5)H), 1.08 and 1.12 (both s, 6 H, 2 CH3), 1.15 (3H, d, J = 6.6 Hz, CH2CHC3), 1.84 (2H, dd, J = 3.3, 12.6 Hz, C(3)H, C(5)H), 2.84 (1H, dd, J = 6.0, 14.2 Hz, CHCHMe), 2.92 (1H, dd, J = 4.0, 14.2 Hz, CHCHMe), 3.00 (1H, m, C(4)H), 3.42 (1H, m, CH2CMe), 7.17 (1H, dd, J = 2.2, 8.6 Hz, Harom), 7.45 (1H, d, J = 8.6 Hz, Harom), 7.51 (1H, d, J = 2.2 Hz, Harom).
{2-[5-(4-Methyl-3-chlorophenylamino)-[1,2,4]thiadiazol-3-yl]-1-methylethyl}-(2,2,6,6-tetramethylpiperidine-4-yl)-amine (2). White powder. Yield 77%. Calcd., %: C 59.77, H 7.64, N 16.59 C21H32ClN5S. Found, %: 59.87, H 7.34, N 16.44.
NMR 1H [200 MHz] δ: 0.65 (1H, t, J = 12.0 Hz, CHH), 0.75 (1H, t, J = 12.0 Hz, CHH), 1.00 (6H, s, (CH3)2), 1.06 (3H, d, J = 6.3 Hz, CCH3), 1.10 (6H, s, (CH3)2), 1.71 (2H, dd, J = 3.5, 12.3 Hz, 2CH), 2.31 (3H, s, Ph-CH3), 2.71 (2H, dd, J = 6.3, 14.2 Hz, CH2), 2.89 (1H, tt, J = 3.5, 11.6 Hz, NCH), 3.32 (1H, m, NCH), 7.17 (1H, d, J = 8.4 Hz, Harom), 7.29 (1H, dd, J = 2.2, 8.4 Hz, Harom), 7.71 (1H, d, J = 2.2 Hz, Harom).
{2-[5-(4-Fluoro-3-chlorophenylamino)-[1,2,4]thiadiazol-3-yl]-1-methylethyl}-(2,2,6,6-tetramethylpiperidine-4-yl)-amine (3). White powder. Yield 67%. Calcd., %: C 56.39, H 6.86, N 16.44 C20H29ClFN5S. Found, %: C 56.26, H 6.81, N 16.55.
NMR 1H [200 MHz,] δ: 0.77 (2H, dt, J = 12.1, 21.0 Hz, C(3)H, C(5)H), 1.06 and 1.14 (12H, c and c, 4 × CH3), 1.15 (3H, d, J = 6.6 Hz, CH2CHC3), 1.81 (2H, dd, J = 3.4, 12.6 Hz, C(3)H, C(5)H), 2.83 (1H, dd, J = 6.0, 14.0 Hz, CHCHMe), 2.97 (1H, dd, J = 4.1, 14.0 Hz, CHCHMe), 3.00 (1H, m, C(4)H), 3.40 (1H, m, CH2CMe), 7.16 (2H, m, Harom), 7.45 (1H, d, J = 8.0 Hz, Harom).
{2-[5-(2-Methyl-5-chlorophenylamino)-[1,2,4]thiadiazol-3-yl]-1-methylethyl}-(2,2,6,6-tetramethylpiperidine-4-yl)-amine (4). White powder. Yield 72%. Calcd., %: C 59.77, H 7.64, N 16.59 C21H32ClN5S. Found, %: C 59.80, H 7.30, N 16.49. C21H32ClN5S. Found, %: 59.87, H 7.34, N 16.44.
NMR 1H [200 MHz] δ: 0.71 (1H, t, J = 12.0 Hz, CHH), 0.85 (1H, t, J = 12.0 Hz, CHH), 1.12 (6H, s, (CH3)2), 1.14 (3H, d, J = 6.2 Hz, CCH3), 1.19 (6H, s, (CH3)2), 1.85 (2H, dd, J = 3.5, 12.3 Hz, 2CH), 2.32 (3H, s, Ph-CH3), 2.831 (2H, m, CH2), 2.99 (1H, tt, J = 3.5, 11.6 Hz, NCH), 3.41 (1H, m, NCH), 7.17 (2H, m, Harom), 7.52 (1H, s, Harom).
{2-[5-(4-Chlorophenylamino)-[1,2,4]thiadiazol-3-yl]-1-methylethyl}-(2,2,6,6-tetramethylpiperidine-4-yl)-amine (5). White powder. Yield 70%. Calcd., %: C 58.88, H 7.41, N 17.16 C20H30ClN5S. Found, %: C 58.80, H 7.40, N 17.34.
NMR 1H [200 MHz] δ: 0.86 (2H, m, C(3)H, C(5)H), 1.14 and 1.20 (12H, s and s, 4 × CH3), 1.27 (3H, d, J = 6.65 Hz, CH2CHC3), 1.87 (2H, d, J = 12.5 Hz, C(3)H, C(5)H), 2.84 (2H, d, J = 6.5 Hz, C2CHMe), 3.03 (1H, m, C(4)H), 3.45 (1H, m, CH2CMe), 7.40 and 7.50 (4H, d and d, J = 8.4 Hz, Harom).
[1-Methyl-2-(5-p-tolylamino-[1,2,4]thiadiazol-3-yl)-ethyl]-(2,2,6,6-tetramethylpiperidine-4-yl)-amine (6). Light crystals. Yield 69%. Calcd., %: C 65.08, H 8.58, N 18.07. C21H33N5S. Found, %: C 65.01, H 8.52, N 18.09.
NMR 1H [200 MHz] δ: 0.71 (2H, m, C(3)H, C(5)H), 1.02, 1.05 (6H, s + s, 2 × CH3), 1.07 (3H, d, J = 6.5 Hz, CH2CHC3), 1.12, 1.14 (6H, s + s, 2 × CH3), 1.75 (2H, dd, J = 3.1, 12.5 Hz, C(3)H, C(5)H), 2.73 (2H, dd, J = 2.3, 6.5 Hz, C2CHMe), 2.91 (1H, m, C(4)H), 3.32 (1H, m, CH2CMe), 7.09 and 7.38 (4H, d + d, J = 8.6 Hz, Harom), 10.56 (1H, br. s, ArNH).
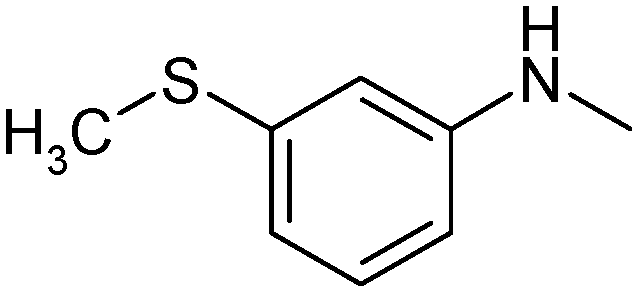
{2-[5-(3-Methylthiophenylamino-[1,2,4]thiadiazol-3-yl)-ethyl]-1-methylethyl}-(2,2,6,6-tetramethylpiperidine-4-yl)-amine (7). White powder. Yield 70%. Calcd., %: C 60.10, H 7.93, N 16.69 C21H33N5S2. Found, %: C 60.28, H 7.99, N 16.64.
NMR 1H [200 MHz] δ: 0.81 (2H, m, C(3)H, C(5)H), 1.13 and 1.20 (12H, s and s, 2 × CH3), 1.21 (3H, d, J = 6.5 Hz, CH2CHC3), 1.86 (2H, d, J = 12.4 Hz, C(3)H, C(5)H), 2.54 (3H, s, Ph-SCH3), 2.88 (2H, m, C2CHMe), 3.01 (1H, m, C(4)H), 3.47 (1H, m, CH2CMe), 7.01 (2H, m, Harom), 7.34 (2H, m, Harom).
{2-[5-(3-Methoxyphenylamino)-[1,2,4]thiadiazol-3-yl]-1-methylethyl}-(2,2,6,6-tetramethylpiperidine-4-yl)-amine (8). White powder. Yield 70%. Calcd., %: C 62.50, H 8.24, N 17.35 C21H33N5OS. Found, %: C 62.66, H 8.14, N 17.34.
NMR 1H [200 MHz] δ: 0.81 (2H, m, C(3)H, C(5)H), 1.09 and 1.17 (12H, c and c, 4 × CH3), 1.20 (3H, d, J = 6.2 Hz, CH2CHC3), 1.83 (2H, d, J = 12.2 Hz, C(3)H, C(5)H), 2.83 (2H, m, C2CHMe), 2.97 (1H, m, C(4)H), 3.39 (1H, m, CH2CMe), 3.81 (3H, c, Ph-OCH3), 6.73 (2H, m, Harom), 7.29 (2H, m, Harom).
N-{1-[5-(4-Fluorophenylamino)-[1,2,4]thiadiazol-3-yl]-propane-2-yl}-(2,2,6,6-tetramethylpiperidine-4-yl)-amine (9). Light crystals. Yield 69%. Calcd., %: C 61.35, H 7.72, N 17.89 C20H30FN5S. Found, %: C 61.23, H 7.87, N 17.92.
NMR 1H [200 MHz] δ: 0.71 (2H, m, C(3)H, C(5)H), 1.02, 1.04 (12H, s, 4 × CH3), 1.07 (3H, d, J = 6.6 Hz, CH2CHC3), 1.72 (2H, dd, J = 3.0, 12.6 Hz, C(3)H, C(5)H), 2.72 (2H, dd, J = 2.3, 6.6 Hz, C2CHMe), 2.91 (1H, m, C(4)H), 3.34 (1H, m, CH2CMe), 7.04 (2H, t, J = 8.6 Hz, Harom), 7.58 (2H, dd, J = 4.7, 9.1 Hz, Harom).
{2-[5-(5-Methylisoxazol-3-ylamino)-[1,2,4]thiadiazol-3-yl]-1-methylethyl}-(2,2,6,6-tetramethylpiperidine-4-yl)-amine (10). White powder. Yield 56%. Calcd., %: C 57.11, H 7.99, N 22.20, C18H30N6OS. Found, %: C 56.91, H 8.10, N 22.44.
NMR 1H [200 MHz] δ: 0.74 (2H, m, C(3)H, C(5)H), 1.00 and 1.10 (12H, c and c, 4 × CH3), 1.12 (3H, d, J = 6.2 Hz, CH2CHC3), 1.76 (2H, d, J = 12.2 Hz, C(3)H, C(5)H), 2.33 (1H, s, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CCH3), 2.83 (2H, m, C2CHMe), 3.00 (1H, m, C(4)H), 3.42 (1H, m, CH2CMe), 4.12 (2H, br. s, 2NH), 5.87 (1H, s, CCH).
CCH3), 2.83 (2H, m, C2CHMe), 3.00 (1H, m, C(4)H), 3.42 (1H, m, CH2CMe), 4.12 (2H, br. s, 2NH), 5.87 (1H, s, CCH).
{2-[5-Cyclopropylamino-[1,2,4]thiadiazol-3-yl]-1-methylethyl}-(2,2,6,6-tetramethylpiperidine-4-yl)-amine (11). White powder. Yield 63%. Calcd., %: C 60.49, H 9.26, N 20.75 C17H31N5S. Found, %: C 60.66, H 9.11, N 20.74.
NMR 1H [200 MHz] δ: 0.76 (6H, m, C(3)H, C(5)H, CH2CH2), 1.07 and 1.13 (12H, c and c, 4 × CH3), 1.13 (3H, d, J = 6.35 Hz, CH2CHC3), 1.79 (2H, m, C(3)H, C(5)H), 2.61 (1H, m, NCH), 2.75 (2H, m, C2CHMe), 2.91 (1H, m, C(4)H), 3.33 (1H, m, CH2CMe), 6.87 (1H, br. s, NH).
N-{1-[5-(4-Bromophenylamino)-[1,2,4]thiadiazol-3-yl]-propane-2-yl}-(2,2,6,6-tetramethylpiperidine-4-yl)-amine (12). Yellow powder. Yield 72%. Calcd., %: C 53.09, H 6.68, N 15.48 C20H30BrN5S. Found, %: C 53.28, H 6.56, N 15.64.
NMR 1H [200 MHz] δ: 0.73 (2H, m, C(3)H, C(5)H), 1.02 and 1.05 (12H, c and c, 4 × CH3), 1.08 (3H, d, J = 6.5 Hz, CH2CHC3), 1.72 (2H, dd, J = 3.1, 12.5 Hz, C(3)H, C(5)H), 2.75 (2H, dd, J = 2.3, 6.5 Hz, C2CHMe), 2.91 (1H, m, C(4)H), 3.33 (1H, m, CH2CMe), 7.40 and 7.52 (4H, d and d, J = 8.8 Hz, Harom).
2.2. Materials and solvents
1-Octanol (, CH3(CH2)7OH, MW 130.2, lot 11K3688) ARG was from Sigma Chemical Co. (USA). 1-Hexane (C6H14, MW 86.18, lot 07059903 C) ARG was from SDS (Peypin, France).Bidistilled water (with an electrical conductivity of 2.1 μS cm−1) was used for preparation of buffer solutions.
Phosphate buffer pH 7.4 (I = 0.15 mol L−1) for distribution and solubility experiments was prepared by combining a potassium dihydrogen phosphate solution (KH2PO4) (9.1 g in 1 L) and a disodium hydrogen phosphate dodecahydrate solution (Na2HPO4·12H2O) (23.6 g in 1 L).26 The pH values were measured by using a pH meter FG2-Kit (Mettler Toledo, Switzerland) standardized with pH 1.68, 6.86 and 9.22 solutions.
Phosphate buffer saline for permeation experiments was prepared as described in ref. 27 by mixing a 2.5% (weight/volume) sodium dihydrogen phosphate dehydrate solution with a 1.8% (weight/volume) disodium hydrogen phosphate dodecahydrate solution in a ratio of 1 to 4 in order to obtain a 74 mM phosphate solution.
The pH of the solution was measured using a pH meter FG2-Kit (Mettler Toledo, Switzerland) and adjusted to physiologic conditions (pH 7.3–7.4) by the addition of sodium hydroxide. The osmolality of the buffer solution was measured by a Semi-Micro Osmometer K-7400 (Herbert Knauer GmbH, Berlin, Germany) and adjusted to 280–290 mOsm by the addition of NaCl.
Permeapad™ barrier was prepared according to the description of di Cagno and Bauer-Brandl.27 In brief, a thin layer of phosphatidylcholine (S-100) was applied on a support sheet (Pütz GmbH, Taunusstein, Germany). The final barrier was composed of a support layer and a dried layer of lipid. The final barrier appears mechanically flexible and resistant, and can be cut to size by scissors or a punching tool.
2.3. Solubility experiments
All the experiments were carried out by the shake flask method28 at 298 K (±0.1 K). The essence of the above mentioned method includes determination of the compound concentration in the saturated solution. Glass ampoules containing the test substance and the solvent were placed into an air thermostat supplied by a stirring device. To ensure the equilibration conditions, the dissolution profiles of the compounds were investigated. Thermodynamic equilibrium was determined from preliminary experiments including measuring the kinetic dependences of the concentration of the saturated solutions for the investigated substances. The time needed to reach the plateau of drug concentration against time was considered a suitable equilibration time. The incubation time of 24 h was estimated to be high enough for the equilibrium to be reached and all the solubility experiments lasted 24 h before the solubility measurements were performed. An aliquot of the saturated solution was taken and centrifuged under temperature control (Biofuge stratus, Germany) for 5 minutes at 298 K temperature. The solid phase was removed through isothermal filtration by the filter MILLEX® HA 0.45 μm (Ireland). The saturated solution was diluted with the corresponding solvent to the required concentration. The molar solubility of the drugs was determined by means of a Cary-50 (USA) spectrophotometer in the UV spectral region (λ = 200–400 nm) with an accuracy of 2–4%. The experimental results are reported as an average value of at least three replicated experiments.2.4. Permeation studies
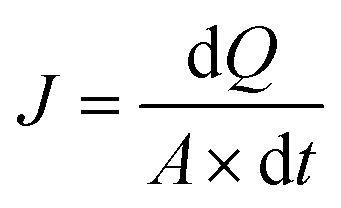 | (1) |
Secondly, the obtained flux was applied to calculate the apparent permeability coefficient (Papp) using the following equation:
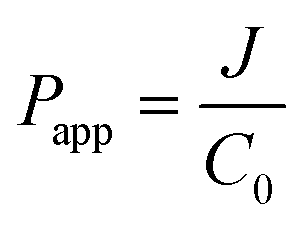 | (2) |
Lastly, based on the permeability coefficients measured through the Permeapad™ barrier (Papp) and the support layer (Psup), the permeability coefficient through the lipid layer (Plip) was determined by the following equation:29,30
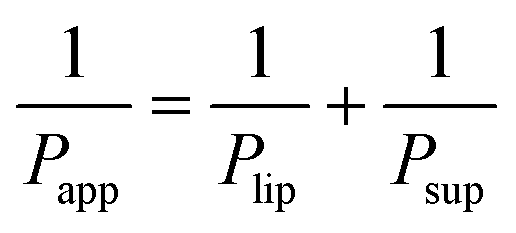 | (3) |
Standard deviation calculations and student's t-test were evaluated; P ≤ 0.05 was considered as significantly different. The Thomson Tau test was applied in order to identify possible outlier values.
2.5. Distribution coefficient determination
The shake flask method was used to determine the distribution coefficients (Doct/buf) in the 1-octanol/buffer (pH 7.4) system.31–34 The procedure was performed as follows. Buffer (pH 7.4) and 1-octanol were mixed vigorously for 24 h at 298 K (±0.1 K) to promote solvent saturation in both phases. The solvents were left standing long enough to allow the phases to separate, and the compound was dissolved in the 1-octanol phase to obtain the stock solution. The buffer saturated 1-octanol phase with the dissolved substance and the 1-octanol saturated buffer phase were placed in glass vials and mixed for 24 hours at 298 K. The substance concentrations were determined spectrophotometrically using calibration curves. The distribution coefficient was calculated by the following equation:| Doct/buf = Coct/buf/Cbuf/oct | (4) |
| mi = moct/buf + mbuf/oct | (5) |
2.6. Calcium-blocking property experiments
Synaptosomes of the rat brain cortex were isolated according to a standard procedure from the brain of newborn rats (9–10 days old). For accumulation of the radioactive label, the P2-fraction of synaptosomes was suspended in the incubation buffer A: 132 mM NaCl, 5 mM KCl, 5 mM HEPES, 10 mM glucose, pH 7.4 (protein concentration – 1.5–2 mg mL−1). The calcium concentration in the final volume was 1.25 mM. A glutamate concentration of 200 μM was applied for stimulation of 45Ca2+ uptake. After 5 min incubation at 335 K, the process was terminated by filtration on GF/B-filters (Whatman, UK) and washing three times with cold buffer B (145 mM HEPES, 10 mM Tris, 5.4 mM Trilon B, pH 7.4). All the experiments were performed in triplicate. The samples were measured by means of a fluid scintillator beta-analyzer SL-4000 (TriCarb 2800, Perkin Elmer).In preliminary experiments, all the compounds were tested for their ability to inhibit glutamate stimulated Ca uptake at the concentration of 100 μM. If the inhibition of Glu-Ca-uptake was 50% and more, then further studies were carried out to determine the concentration dependence of inhibition. The amount of the 45Ca2+ uptake in the synaptosomes was determined by the difference of the label content with and without uptake stimulation and expressed in % control (control – 100%). The corresponding value of K43/21 was calculated according to the following equation:
| K43/21 = [(Ca4 − Ca3)/(Ca2 − Ca1)] × 100% | (6) |
2.7. Calculation procedure
All the descriptors were calculated by the program package HYBOT-PLUS (version 2003) in Windows.35Equilibrium constants for the compounds synthesized were calculated by the ACD/ChemSketch program.36 Analysis of the equilibrium of the charged state of the molecules was conducted by using the program package presented in ref. 37.
2.8. DSC experiments
The fusion temperatures of the compounds under investigation have been determined using a Perkin-Elmer Pyris 1 DSC differential scanning calorimeter (Perkin-Elmer Analytical Instruments, Norwalk, Connecticut, USA) with Pyris software for Windows NT. DSC experiments were conducted in an atmosphere of flowing (20 cm3 min−1) dry helium gas of high purity 0.99996 (mass fraction) using standard aluminum sample pans and a heating rate of 2 K min−1. The accuracy of weight measurements was 0.005 mg. The DSC was calibrated with an indium sample from Perkin-Elmer (P/N 0319-0033). The value determined for the fusion enthalpy corresponded to 28.48 J g−1 (the reference value was 28.45 J g−1). The fusion temperature was 429.5 ± 0.1 K (determined from ten measurements).3. Results and discussion
3.1. Distribution experiments
The processes of distribution of compounds in biological media play an important role in drug delivery. These processes contribute significantly to membrane permeability determining the concentrations of drug compounds in the membrane/lipophilic compartments (biological medium phases). In addition, the distribution coefficient of the compounds in 1-octanol/buffer (Doct/buf) and 1-hexane/buffer (Dhex/buf) systems are important characteristics commonly used as a measure to describe many biological, chemical and physical processes. For example, Doct/buf is often used to estimate the processes of intestinal tract membrane permeability, whereas Dhex/buf for the processes occurring in the BBB. Moreover, the parameter characterizing the difference between the 1-hexane phase and the 1-octanol one (logDoct/hex) is often used to distinguish the contributions of non-specific and specific interactions (donor–acceptor, hydrogen bonds) of the drug molecules with biological environments. Based on this, we tried to describe the distribution of the selected class of compounds in the assigned model systems of immiscible solvents. The experimental data for Doct/buf and Dhex/buf are presented in Table 2. It should be noted that for almost all the compounds (except 14), the Dhex/buf-values are less than 1. This indicates that these substances predominantly dissolve in the aqueous phase. In contrast, in the 1-octanol/buffer system, the Doct/buf-values exceed 1 (except 10), therefore, the substances prefer the 1-octanol phase.
| S 2982/mol l−1 | X 2982/mol fract | D oct/buf | K oct/buf0 | D hex/buf | K hex/buf0 | K 43/21/% | T m/K | α | ∑(Ca) | ∑(Ca)/α | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Relative standard uncertainty for solubility values ur(S2982) = 0.03; ur(X2982) = 0.03. b nd – not determined. c Ref. 23. d Ref. 20. e Ref. 19. f Ref. 17. g Ref. 18. | |||||||||||
| 1 | 7.45 × 10−3 | 1.35 × 10−4 | 12.60 ± 0.02 | 8.28 × 104 | 0.011 ± 0.003 | 72.3 | 305 ± 18 | 347.2 ± 0.5 | 47.113 | 7.68 | 0.163 |
| 2 | 5.57 × 10−3 | 1.00 × 10−4 | 11.01 ± 0.03 | 7.31 × 104 | 0.0031 ± 0.0002 | 20.6 | 214 ± 12 | 319.3 ± 0.5 | 47.02 | 7.75 | 0.165 |
| 3 | 9.24 × 10−3 | 1.67 × 10−4 | 5.0 ± 0.4 | 3.25 × 104 | 0.0064 ± 0.0005 | 42.0 | 189 ± 14 | 333.5 ± 0.5 | 45.094 | 7.76 | 0.172 |
| 4 | 4.86 × 10−3 | 8.77 × 10−5 | 6.9 ± 0.2 | 4.56 × 104 | 0.017 ± 0.004 | 112.8 | 177 ± 9 | 334.2 ± 0.5 | 47.02 | 7.83 | 0.166 |
| 5 | 3.72 × 10−3 | 6.71 × 10−5 | 1.83 ± 0.04 | 1.22 × 104 | 0.011 ± 0.005 | 73.0 | 172 ± 3 | 323.2 ± 0.5 | 45.185 | 7.69 | 0.17 |
| 6 | 8.37 × 10−3 | 1.51 × 10−4 | 6.6 ± 0.4 | 4.44 × 104 | 0.013 ± 0.001 | 89.3 | 128.0 ± 0.1 | 320.2 ± 0.5 | 45.092 | 7.73 | 0.171 |
| 7 | 1.20 × 10−2 | 2.17 × 10−4 | 4.4 ± 0.2 | 2.92 × 104 | 0.014 ± 0.002 | 92.9 | 117 ± 2 | 309.1 ± 0.5 | 47.792 | 8.16 | 0.171 |
| 8 | 2.13 × 10−2 | 3.87 × 10−4 | 3.5 ± 0.3 | 2.34 × 104 | 0.013 ± 0.003 | 86.3 | 112 ± 25 | 314.7 ± 0.5 | 45.366 | 8.33 | 0.184 |
| 9 | 1.82 × 10−2 | 3.30 × 10−4 | 7.6 ± 0.3 | 5.06 × 104 | 0.024 ± 0.003 | 158.0 | 112 ± 12 | 317.6 ± 0.5 | 43.166 | 7.66 | 0.177 |
| 10 | 1.41 × 10−2 | 2.55 × 10−4 | 0.375 ± 0.005 | 2.41 × 103 | 0.041 ± 0.002 | 258.6 | 108 ± 6 | 342.6 ± 0.5 | 41.113 | 8.48 | 0.206 |
| 11 | 3.10 × 10−1 | 6.19 × 10−3 | 1.21 ± 0.02 | 3.19 × 103 | 0.097 ± 0.004 | 655.5 | 86 ± 14 | 341.4 ± 0.5 | 38.328 | 7.32 | 0.191 |
| 12 | 3.49 × 10−3 | 6.29 × 10−5 | 2.041 ± 0.004 | 1.36 × 104 | 0.013 ± 0.001 | 85.0 | 82 ± 5 | 318.5 ± 0.5 | 45.883 | 7.69 | 0.168 |
| 13 | ndb | ndb | 610 ± 30c | 6.10 × 102 | 0.17 ± 0.01c | 0.17 | 51 ± 9c | nd | 29.307 | 5.21 | 0.178 |
| 14 | 4.83 × 10−4d | 8.70 × 10−6d | 1038 ± 10c | 1.038 × 103 | 1.68 ± 0.07c | 1.68 | 87 ± 2c | 402.3 ± 0.2e | 29.214 | 5.28 | 0.181 |
| 15 | nd | nd | 226 ± 5c | 2.26 × 102 | 0.16 ± 0.04c | 0.16 | 75 ± 6c | nd | 27.288 | 5.29 | 0.194 |
| 16 | 4.45 × 10−4d | 8.00 × 10−6d | 1049 ± 50c | 1.049 × 103 | 0.56 ± 0.01c | 0.56 | 88 ± 16c | 374.5 ± 0.2e | 29.214 | 5.35 | 0.183 |
| 17 | 4.836 × 10−4d | 8.30 × 10−6d | 380 ± 5c | 3.80 × 102 | 0.46 ± 0.05c | 0.46 | 89 ± 13c | 408.4 ± 0.2e | 27.379 | 5.22 | 0.191 |
| 18 | 2.19 × 10−3f | 3.95 × 10−5f | 445 ± 28c | 4.45 × 102 | 0.31 ± 0.02c | 0.31 | 85 ± 4c | 390.1 ± 0.2f | 27.286 | 5.26 | 0.193 |
| 19 | nd | nd | 94 ± 13c | 9.4 × 10 | 0.48 ± 0.02c | 0.48 | 86.7 ± 0.5c | nd | 29.986 | 5.68 | 0.19 |
| 20 | 3.04 × 10−3d | 5.48 × 10−5d | 205 ± 5c | 2.05 × 102 | 0.58 ± 0.03c | 0.58 | 108 ± 11c | 363.4 ± 0.2e | 27.56 | 5.93 | 0.215 |
| 21 | 3.40 × 10−3g | 6.13 × 10−4g | 316 ± 3c | 3.16 × 102 | 0.32 ± 0.02c | 0.32 | 88 ± 16c | 376.3 ± 0.2g | 25.36 | 5.18 | 0.204 |
| 22 | nd | nd | 45 ± 1c | 4.5 × 10 | 0.18 ± 0.01c | 0.18 | 109 ± 20c | nd | 23.307 | 6.01 | 0.258 |
We studied the trends between Doct/buf and selected physicochemical descriptors. The main purpose of such a search was connected with an idea to develop an algorithm for prediction of the experimental data on the basis of just molecular structure knowledge. The HYBOT35 descriptors were selected as independent parameters, because they describe the interaction of drug molecules with biological media. It should be noted that the access to obtain the correlation equation was complemented by Doct/buf-values for the related compounds which were prepared in the present work (group I) and those which have been published by us previously (group II)23 (Table 1). However, the compounds of group I are in the ionized state in buffer (pH 7.4), whereas drugs of group II are in the form of neutral entities. The appropriate calculation of the equilibrium forms of the compounds by using the approach described in ref. 37 and taking into account the calculated pKa (pKa1 = 14.01–14.04; pKa2 = 1.29–4.67)36 confirms the neutral forms of the compounds of group II. For this group of compounds, the equality is realized: Koct/buf0 = Doct/buf and Khex/buf0 = Dhex/buf.
In order to recalculate the distribution coefficients, the ionization state of the molecules was determined taking into account that the presently investigated thiadiazoles (group I) are bases and have several ionizable groups. The macroscopic dissociation constants were calculated according to ref. 36.
Based on the calculated pKa-values, we determined the content of a number of the forms with different degrees of protonation in aqueous solutions depending on the pH of the media by using ref. 37, taking into account the following ionization processes:
| HL+ = H+ + L (pK1) | (7) |
| H2L2+ = HL+ + H+ (pK2) | (8) |
| H3L3+ = H2L+ + H+ (pK3) | (9) |
The pKa1 = 10.95 corresponds to the protonation process of the nitrogen atom of the NH-group of the bipyridine ring, pKa2 = 7.31–7.36 to the protonation of the bridge NH-group and pKa3 = 0.45–3.83 to the protonation process of a nitrogen atom of the thiadiazole ring. K1, K2 and K3 are equilibrium constants of the mentioned equations. The ionized/unionized particle distributions (%) as a function of pH for compound 2 (as an example) is presented in Fig. 1SI (ESI‡). It is found that all the compounds have similar dissociation constant values and, respectively, the protonation degree. As follows from Fig. 1SI,‡ all the compounds are completely ionized at pH 7.4 and their molecules exist as approximately equal shares in the form of mono- and di-cations.
For ionizable compounds, the distribution coefficient describing the overall lipophilicity of drugs is the ratio of the concentrations of all molecular forms of compounds existing in equilibrium in the aqueous phase and uncharged particles in 1-octanol. For the investigated thiadiazoles, the equilibrium distribution can be represented by an equation by which the apparent distribution coefficient (Doct/buf) can be experimentally determined:
| Doct/buf = [L]o/([L]w + [HL+]w + [HL2+]w + [HL3+]w) | (10) |
| Doct/buf = [L]o/[L]w + [L][H+]/K1 + [L][H+]2/K2 + [L][H+]3/K3 | (11) |
Further transformation leads to the following equation, which relates the intrinsic distribution coefficients to the experimentally determined apparent distribution coefficients, hydrogen ion concentration (H+) and the dissociation constants, K1, K2 and K3:
| Koct/buf0 = Doct/buf(1 + [H+]/K1 + [H+]2/K2 + [H+]3/K3) | (12) |
To make the conditions of the distribution processes comparable, we recalculated the values of Doct/buf and Dhex/buf for the neutral forms of the compounds of group I by eqn (12) (in Table 2, these coefficients are designated as Koct/buf0 and Koct/buf0).
The data in Table 2 show that the calculated intrinsic distribution coefficients (normalized to neutral species) of the studied thiadiazoles significantly exceed (more than 4 times) the experimental apparent ones (belonging to the ionized forms of the molecules). The estimated ratios indicate greater lipophilicity of the neutral particles compared with the charged ones as expected.
The Koct/buf0-values have been analyzed concerning 32 descriptors. A suitable correlation was obtained for the independent variables which include polarizability (α) and the acceptor molecular ability to create hydrogen bonds (∑(Ca)). The results of the correlation analysis can be represented by the equation:
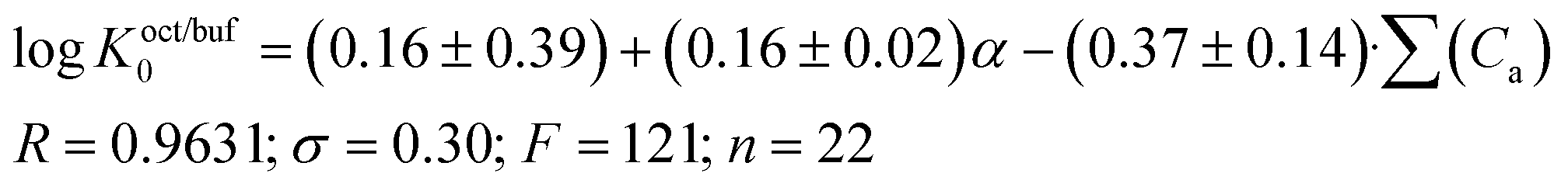 | (13) |
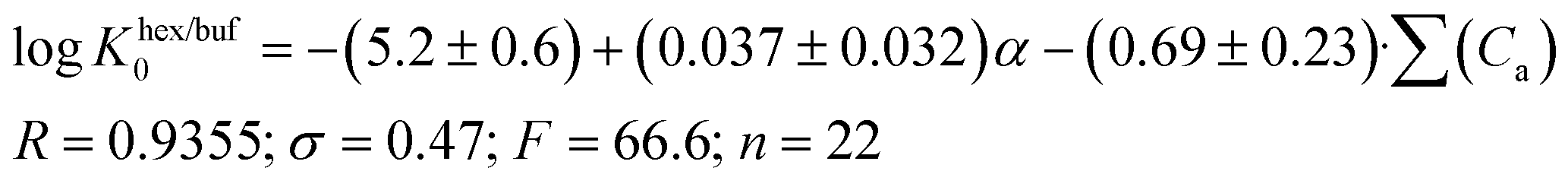 | (14) |
A comparison of eqn (13) and (14) shows that Khex/buf0 is approximately two times more sensitive to the variation of the ∑(Ca)-descriptor than Koct/buf0. Most probably, this phenomenon can be explained by the specific interactions of the dissolved molecule with the solvent both in 1-octanol and buffer phases in the 1-octanol/buffer system. Therefore, the contribution of the descriptor is partially equalized. As opposed to this, in the 1-hexane/buffer system the dissolved molecules interact specifically only with one phase (buffer) which leads to a higher regression coefficient value. Thus, the correlation of eqn (13) and (14) allow predicting the distribution coefficients for the studied class of compounds both in 1-octanol/water and 1-hexane/water systems. It is interesting to compare the distribution coefficients for the molecules of group I compounds (normalized to the neutral species) with those of group II which differ from each other by only one fragment (a tetramethylpiperidine fragment (group I) instead of a hydroxyl group (group II)) both in 1-octanol/water and 1-hexane/water systems. The results of the comparative analysis are given in Fig. 1. In the 1-octanol/water system, the difference in the distribution coefficients between group I and group II compounds is positive with an average ΔlogKoct/buf0-value of 2.58 ± 0.20 (excluding the pair of compounds 10/22). A similar tendency for the 1-hexane/water system with an average ΔlogKhex/buf0-value of 2.47 ± 0.23 (excluding the pair of compounds 2/14) is observed. The derived values of ΔlogK0 can be determined by: firstly, conformational differences of the molecules of groups I and II in water and 1-octanol and also water and 1-hexane phases; secondly, variation of the energy of the hydrogen bond forming by a hydroxyl-group in the considered phases.
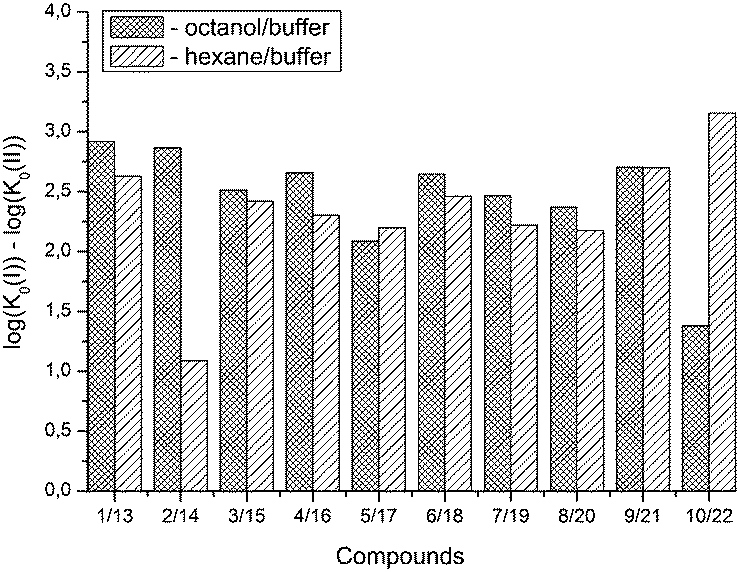 | ||
| Fig. 1 Comparative analysis of distribution coefficients for the molecules of group I compounds (normalized to the neutral particles) with those of group II, which differ from each other by only one fragment (a tetramethylbiperidine fragment (group I) instead of a hydroxyl group (group II)) both in 1-octanol/buffer and 1-hexane/buffer systems. | ||
Analysis of Doct/hex-values showed the existence of strong specific interactions of the drug molecules with the 1-octanol (1.5 < logDoct/hex < 3.6). On this ground, it may be assumed that the redistribution of molecules from blood plasma through the blood–brain barrier will be aggravated.
3.2. Solubility in buffer and melting temperatures
The results of solubility experiments in buffer (pH 7.4) at 298 K and melting points of the compounds studied are presented in Table 2.It is well known that the nature and position of the substituent may influence essentially the solubility of the compounds in a structure analogue series. In this connection, the comparative solubility analysis for the synthesized thiadiazoles was performed. It was found that the solubility among the investigated compounds with the tetramethylpiperidine fragment differs within two orders of magnitude. Compound (11) with a cyclopropyl substituent has the maximal solubility, and compounds (12) and (5) with 4-bromophenyl and 4-chlorophenyl substituents, respectively, have the minimal ones. The replacement of a chlorine atom in the para-position of the phenyl ring by a methyl group (compound 6) increases the solubility twice, and by a methoxy group (compound 8) or a fluorine atom (compound 9) – about 6 times. Changing a methylphenyl- to a methylisoxazole substituent slightly enhances the solubility (about 1.5 times). Interestingly, the introduction of the second chlorine atom in the meta-position of the phenyl ring of compound 5 (3,4-dichloro-derivative (1)) leads to double the solubility. At that, the solubility values of the structural isomers 3-chloro-4-methyl- (2) and 3-chloro-6-methyl- (4) are approximately equal. According to the solubility increase, the investigated thiadiazoles are arranged in the following order: 12 (4-bromophenyl-) ≤ 5 (4-chlorophenyl-) < 4 (3-chloro-6-methylphenyl-) ≤ 2 (3-chloro-4-methylphenyl-) ≤ 1 (3,4-dichlorophenyl-) ≤ 6 (4-methylphenyl-) ≤ 3 (3-chloro-4-fluorophenyl-) < 7 (3-methylthiaphenyl-) ≤ 10 (methylisoxazol-) < 9 (4-fluorophenyl-) ≤ 8 (4-methoxyphenyl-) < 11 (cyclopropyl-).
The solubility change regularities described above do not have a forecasting power. Therefore, it was important to obtain correlation equations between the solubility characteristics and any kinds of descriptors. HYBOT descriptors were used as physicochemical descriptors.25
Crystal lattice thermodynamic characteristics are important parameters in solid substance dissolution. To take this factor into account, Yalkowsky et al.38 introduced crystal melting temperature as a descriptor into the correlation equation (general solubility equation). Using this descriptor to analyze the solubility thermodynamics is justified by the fact that in many cases there is a correlation between the molecular crystal sublimation Gibbs energies and melting temperatures.39 Besides, Yalkowsky et al.38 described water solubility of the compounds using 1-octanol/water partitioning coefficients as descriptors (for imitation of the hydration term). The descriptor series suggested by Yalkowsky was used to describe the solubility of selected compounds. As a result, the correlation equation looks as follows:
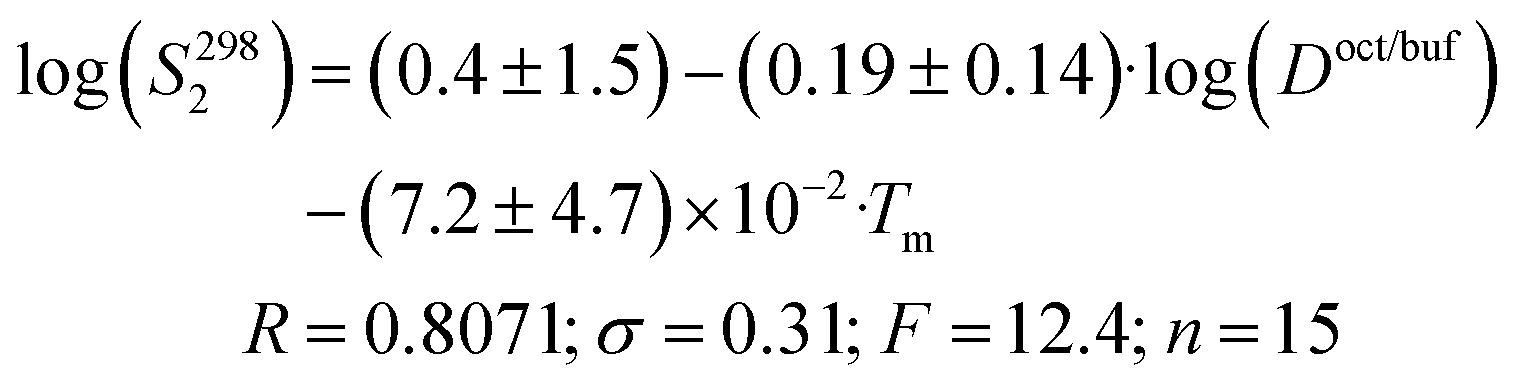 | (15) |
Thus, the solubility of this class of compounds decreases with an increase in the lipophilicity (due to reduction of the hydration term) and the melting temperature growth (crystal lattice energy strengthening).
We also tried to find a correlation equation using HYBOT descriptors. Analysis of all the available descriptors found that the best correlations were obtained with the polarizability (α) and the acceptor molecular ability to create hydrogen bonds (∑(Ca)):
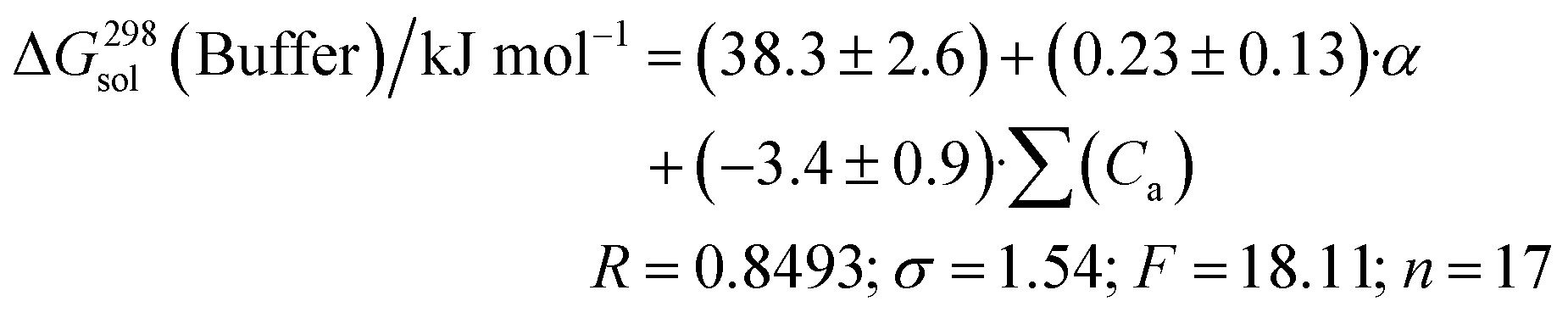 | (16) |
Thus, the solubility-values of this class of compounds in a buffer solution can be estimated based on the knowledge of the structural formula only.
3.3. Permeability properties
Recently, much attention has been paid on the development of better bioavailable drugs, which significantly reduces both dosage and side effects. Water solubility and permeability are considered to be the main parameters determining the bioavailability of drugs. Therefore, in our work we have tried to analyze these specified parameters. The Permeapad™ barrier developed and described in ref. 27 was chosen as a biomimetic artificial barrier to study the permeability. The advantages of Permeapad™ (compared to other artificial systems such as PAMPA and PVPA) are: a unique design, usability, functional stability in a wide range of pH values, compatibility with biomimetic media, adaptability to a Franz diffusion cell and shelf life (more than 1 year). The apparent permeability coefficients through the Permeapad™ barrier (Papp), support layer (Psup) and lipid layer (Plip) of the selected compounds, measured in a buffer solution (pH 7.4) at T = 298 K are summarized in Table 3.| Compound | Apparent permeability coefficients | ||
|---|---|---|---|
| Permeapad™ barrier | Support layer | Lipid layer | |
| P app × 106/cm s−1 | P sup × 106/cm s−1 | P lip × 106/cm s−1 | |
| 1 | 9.8 ± 0.8 | 15.7 ± 1.2 | 26.1 ± 4.0 |
| 2 | 13.5 ± 0.4 | 20.6 ± 0.1 | 39.5 ± 3.1 |
| 3 | 13.9 ± 2.3 | 25.4 ± 1.5 | 30.9 ± 7.3 |
| 4 | 14.4 ± 0.1 | 30.8 ± 3.0 | 26.9 ± 4.8 |
| 5 | 8.6 ± 0.9 | 21.9 ± 1.4 | 14.1 ± 0.2 |
| 6 | 12.2 ± 2.7 | 26.3 ± 1.9 | 22.8 ± 7.7 |
| 7 | 10.1 ± 0.5 | 22.8 ± 1.4 | 18.2 ± 1.9 |
| 8 | 9.4 ± 1.1 | 16.5 ± 1.3 | 21.7 ± 0.7 |
| 9 | 8.7 ± 1.1 | 16.3 ± 1.1 | 18.6 ± 2.0 |
| 10 | 3.4 ± 0.3 | 31.7 ± 4.4 | 3.7 ± 0.4 |
| 11 | 5.4 ± 0.4 | 27.6 ± 1.3 | 6.8 ± 0.6 |
| 12 | 6.5 ± 0.7 | 25.8 ± 2.2 | 9.5 ± 0.6 |
The results showed in ref. 27 indicate that the permeability coefficients through the sheer support layer (Psup) were in all cases higher than the respective Papp measured for the entire Permeapad™ barrier. Our data in Table 3 confirmed these conclusions. This fact is a clear indication of a significant retention effect induced by the lipids present in the artificial barrier and that the lipid layer is the dominant impediment to drug permeation. From this it can be concluded that for all compounds investigated, the rate limiting step in permeability was represented by the lipid layer of the artificial barrier. Due to this fact it is proposed that for screening purposes, it is reasonable to go without measuring the Psup-value.
The values of the permeation coefficients vary depending on the nature and number of substituents in the molecule. The maximum permeability coefficient (Plip) is observed for the 3-chloro-4-methylphenylderivative (compound 2). Replacement of the methyl group in this compound by a chlorine atom (dichlorophenyl-substituted compound (1)) leads to a small reduction of permeability. Thus, the permeability coefficient of the 3-chloro-4-methylphenylsubstituted compound is about 1.5 times higher than that of its 3-chloro-6-methylisomer (4), indicating the effect of not only the nature but also the position of the substituent on the permeability of thiadiazoles. Significant reduction in permeability (minimum values among all compounds) occurs in substances with isoxazole (10) and cyclopropyl (11) substituents. According to the permeability values the investigated substances can be arranged in the following order: 3-chloro-4-methyl- (2) > 3-chloro-4-fluoro- (3) > 3-chloro-6-methyl- (4) ≥ 3,4-dichloro- (1) > 4-methyl- (6) ≥ 3-methyloxy- (8) > 4-fluoro- (9) ≥ 3-methylthia- (7) > 4-chloro- (5) > 4-bromo- (12).
Due to the fact that the distribution and permeability processes for the selected compounds were studied in the same buffer solution (pH 7.4) (i.e. ionization degree of the particles and their ratio remained the same for the two designated processes), we tried Doct/buf and Dhex/buf as descriptors for the permeability. In turn, logPapp, logPsup and logPlip were selected as the analyzed functions. It should be noted that there are no correlations between the selected functions and Dhex/buf (Fig. 2SI‡). In contrast, good correlations between logPapp and logDoct/buf, as well as between logPlip and logDoct/buf (Fig. 2), exist, which can be described by the following equations:
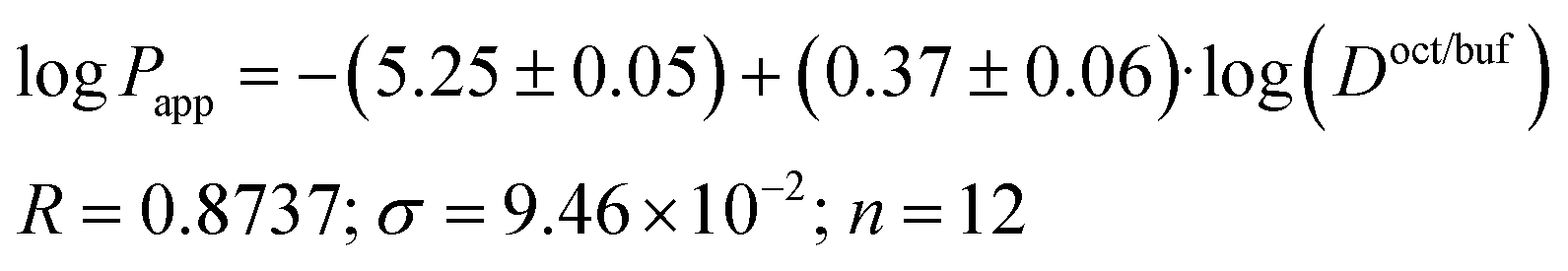 | (17) |
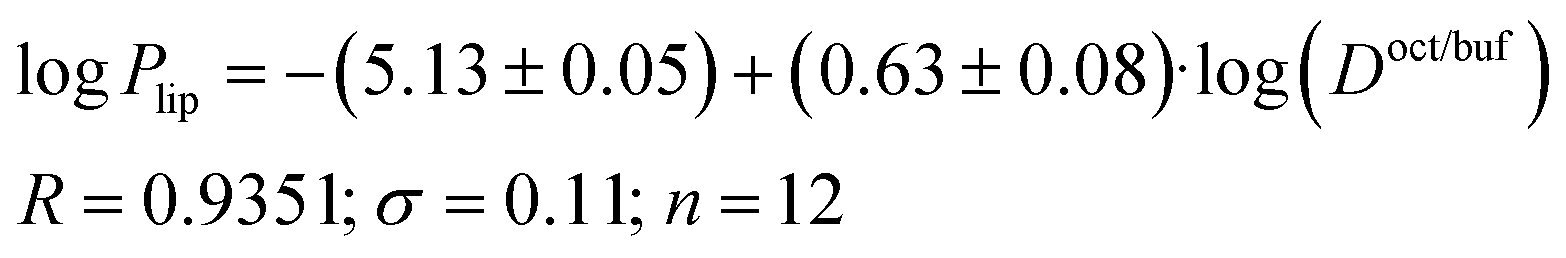 | (18) |
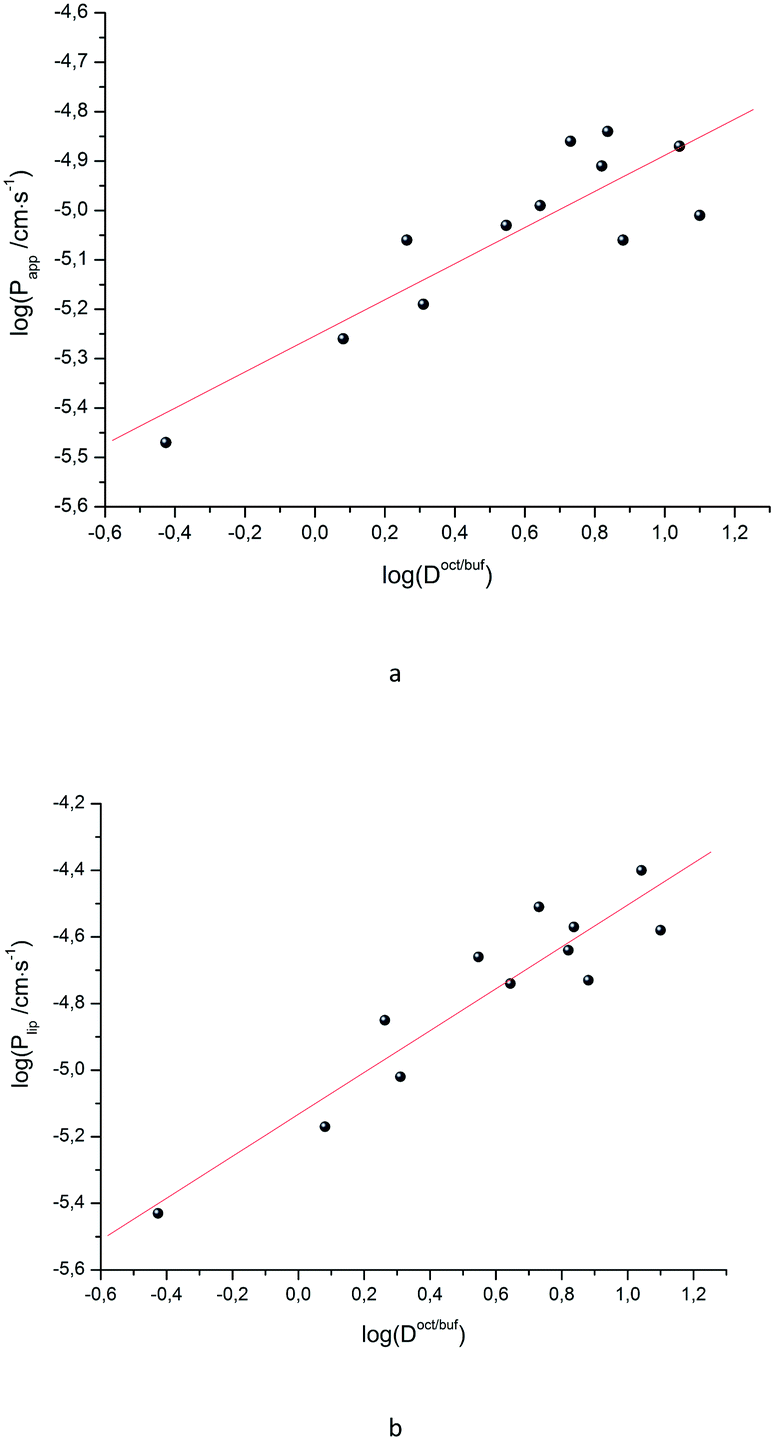 | ||
| Fig. 2 LogPappvs. logDoct/buf (a) and logPlipvs. logDoct/buf (b) dependences. | ||
It is easy to see that logPlip is 1.7 times more sensitive to log(Doct/buf) changes than logPapp. Thus, the supporting layer contributes to the sensitivity of this coefficient, but, the correlation parameters are not much deteriorated. Consequently, logPapp can be used to analyze the lipid layer permeability. If we take into account that the logPlip-value is burdened with two experimental errors (sequential execution of two experiments: permeability of the Permeapad™ barrier and support layer), then the Papp value is more accurate (and the total time of the experiment is halved).
As the next step, it was interesting to analyze the relationships of the HYBOT physicochemical descriptors with the obtained permeability characteristics. Testing all the descriptors revealed that it is only for logPlip and logPapp (as in the previous cases for logDoct/buf) in which the correlation with the acceptor molecular ability to create hydrogen bonds normalized to molecular polarizability (∑(Ca)/α) was observed. The results of the analysis for logPlip and logPapp are presented in Fig. 3, whereas the relationship between logPsup and ∑(Ca)/α is shown in Fig. 3SI.‡ The received correlation can be described by the following equations:
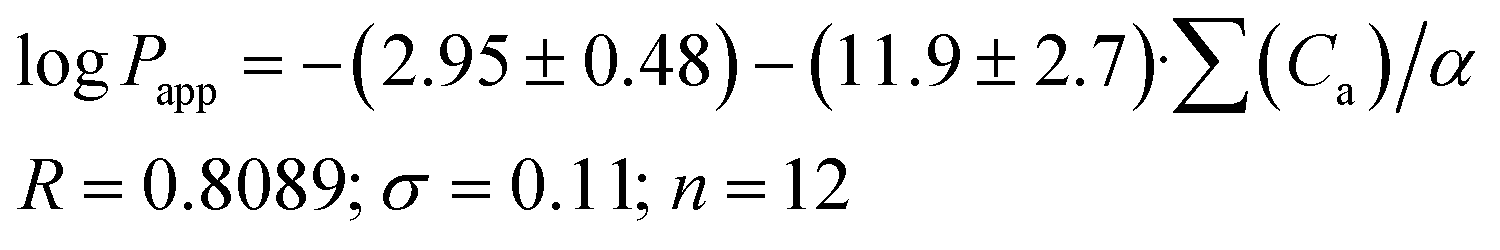 | (19) |
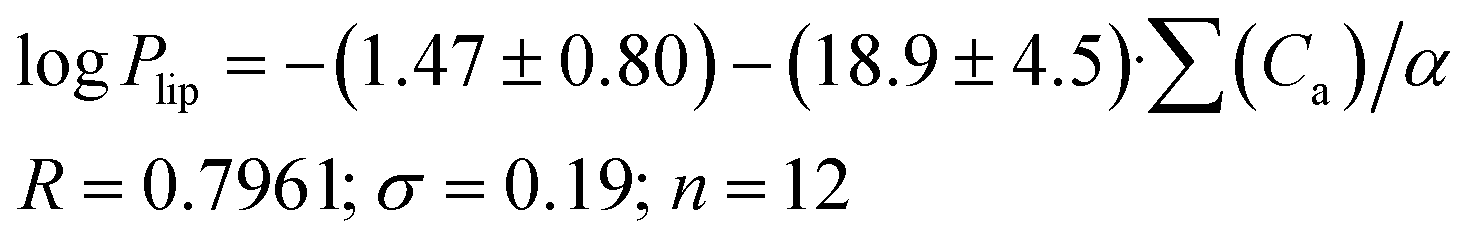 | (20) |
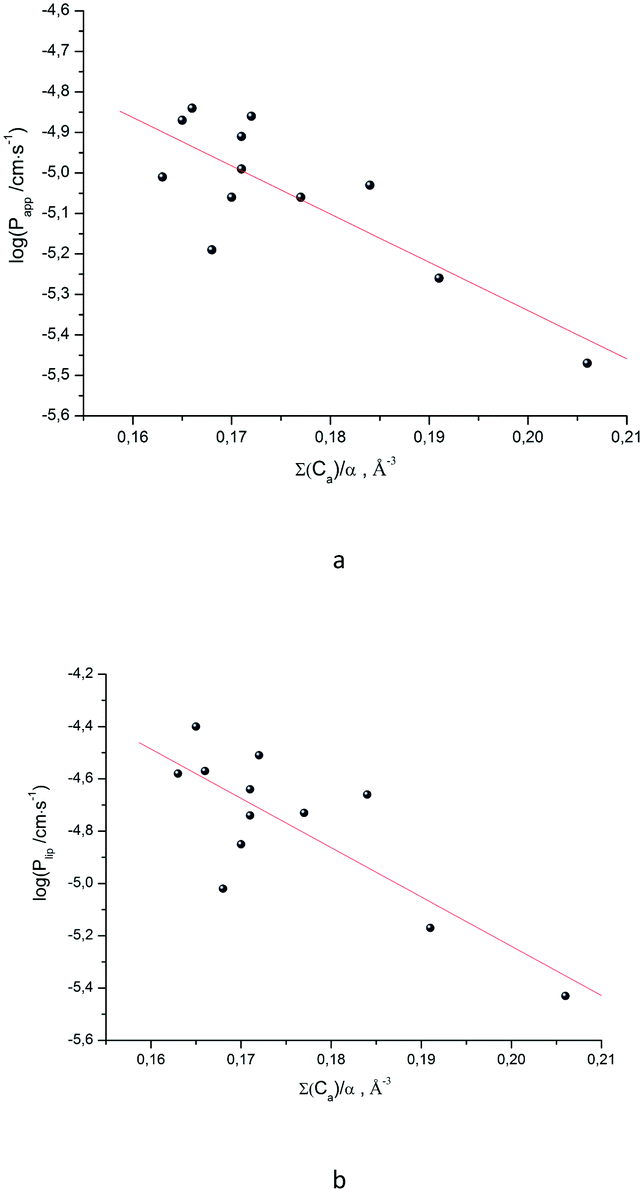 | ||
| Fig. 3 LogPappvs. ∑(Ca)/α (a) and logPlipvs. ∑(Ca)/α (b) dependences. | ||
Thus, the permeability coefficients of this class of compounds may be predicted based solely on their structural formulas. Due to the small number of data points, the paired correlation coefficients are not rather high. However, improvement should be expected if the number of data points would increase. Just as in the case of the distribution coefficients, the correlation equations for logPapp and logPlip retain the same trends on the selected descriptor.
3.4. Integral solubility – permeability properties
The available data on the solubility of the test compounds and the apparent permeability coefficients make it possible to evaluate the potential bioavailability of the substances, similarly to Sanphui et al.40 The approach consists of the estimation of diffusion fluxes through the lipid barrier (Jlip) and Permeapad (Japp) (area of 1 cm2 and nominal thickness of 1 cm) on the assumption that at one end of the barrier the concentration is kept equal to the thermodynamic solubility value, while at the other end, the substance is effectively removed and its concentration is practically equal to zero (sink conditions). In other words, the value of the product of the thermodynamic solubility (S2982 in mol l−1), lipid permeability coefficient (Plip in cm s−1) and Permeapad permeability coefficient (Papp in cm s−1) is analyzed. The analysis in the logarithmic scale yields the following equations:| logJlip = log(S2982·Plip) = logS2982 + logPlip | (21a) |
| logJapp = log(S2982·Papp) = logS2982 + logPapp | (21b) |
Fig. 4 depicts the analyzed values. It is easy to see that according to the degree of decreasing diffusion flux value (a measure of absorption), the compounds can be placed in the following order: for the lipid layer – 11 > 8 > 9 > 3 > 2 > 7 > 1 ≈ 6 > 4 > 5 ≈ 10 > 12; for Permeapad – 11 > 8 > 9 > 3 > 7 ≈ 6 > 2 ≈ 1 ≈ 4 > 10 > 5 > 12.
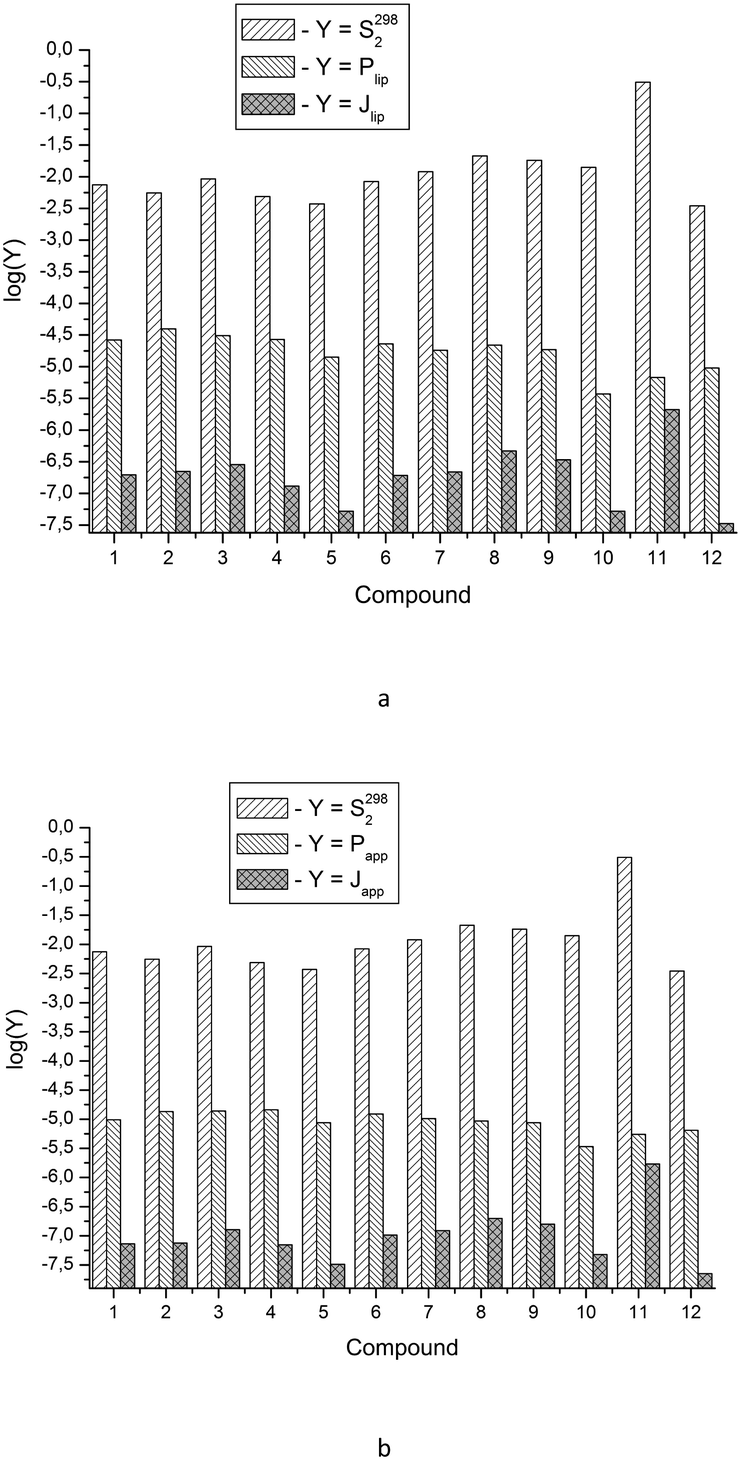 | ||
| Fig. 4 Experimental solubility data (S2982), lipid layer permeability coefficients (Plip) and diffusion fluxes through the lipid layer (Jlip) for the compounds studied (a); experimental solubility data (S2982), Permeapad layer permeability coefficients (Papp) and diffusion fluxes through Permeapad (Japp) for the compounds studied (b). | ||
With this range of changing the Plip and Papp-values, it turns out that the solubility values have the main contribution to the diffusion fluxes (Fig. 5).
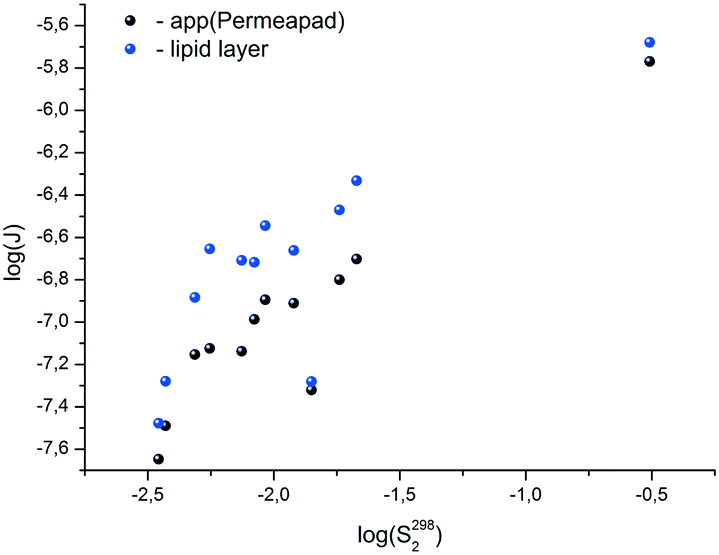 | ||
| Fig. 5 LogJlipvs. logS2982 and logJappvs. logS2982 dependences. | ||
3.5. Calcium-blocking properties
The substances synthesized in the present study belong to an essentially new group of compounds in a series of 1,2,4-thiadiazole derivatives which, being bioisosteric analogues of a model “ampakine” cyclothiazide, as proposed, can be the positive modulators of AMPA-receptors. First of all, the ability of the synthesized substances to modulate glutamate-dependent Ca-uptake in synaptosomes of the rat brain cortex was studied. The biological activity results (K43/21-values) are presented in Table 2.The obtained results clearly indicate that the variation of the structure of the substituent in the phenyl ring as well as the replacement of the phenyl ring by the cyclopropyl- and isoxazole-one influences the ability of thiadiazole derivatives to inhibit calcium ion uptake. The biological activity (K43/21-values) of the investigated substances is presented in Fig. 6 as a histogram to compare the compounds of groups I and II with the same substituents.
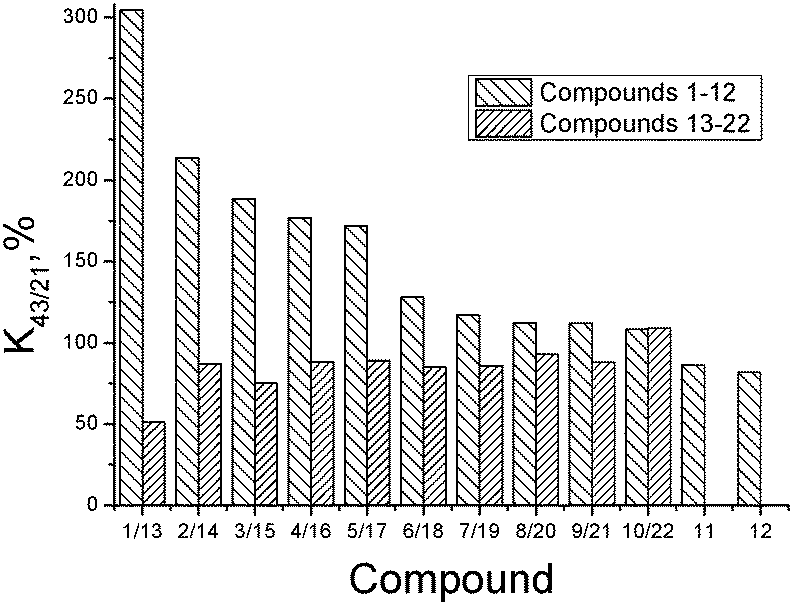 | ||
| Fig. 6 Log(1/K43/21) vs. logDoct/buf dependence. | ||
The comparative analysis of the activity of thiadiazole derivatives newly synthesized and investigated by us before indicated that, on the whole, the presence of the tetramethylpiperidine ring and an additional NH-group instead of a hydroxyl group facilitates a considerable growth of K43/21-values (up to 6 times for the compounds with the dichlorophenyl substituent (compounds 1 and 13)). However, it is interesting to note that if the replacement of the phenyl ring by isoxazole substituent takes place, the activities of the respective structural analogues of groups I and II practically coincide. The analysis of the structure–activity relationship was conducted for thiadiazoles synthesized in the present study (group I – compounds 1–12). Compound 12 with a bromine atom as a substituent in the para-position of the phenyl ring, revealing maximal inhibiting activity (minimal K43/21), was chosen as a reference. As seen from the histogram data, replacing a bromophenyl substituent by a cyclopropyl- and methylisoxazole-one leads to a slight consistent increase in K43/21 (compounds 11 and 10). Further reduction of the inhibitory activity is observed for compounds with 4-fluorophenyl-, 3-methoxyphenyl- and 3-methylthiaphenyl-substituents, which have similar activity values (9 (112%) = 8 (112%) ≤ 7 (117%)). Introduction of the methyl group (compound 6) in the para-position of the phenyl ring contributes to a further increase of K43/21. At that, the inhibitory activity of the compound with a methylisoxazole fragment (10) is higher than that of the methylphenyl-substituted one (6).
The results presented in the histogram allowed us to evaluate the total biological potential of the compounds, not only as possible neuroprotectors, but also as cognitive stimulants (nootrops). Thus, for five of the compounds (1–5), the values of K43/21 > 150 suggest that they be subjected for further testing as stimulants of cognitive functions using a recognition test. It is interesting to note that these compounds have chlorine atoms as substituents in the phenyl ring. At the same time, di-substituted substances with a chlorine atom in the meta-position reveal the maximum K43/21-values and according to the reduction of the activation function of glutamate-dependent uptake of calcium ions, the thiadiazoles can be arranged in the following order: 1 (3-chloro-4-chloro) > 2 (3-chloro-4-methyl) > 3 (3-chloro-4-fluoro) > 4 (3-chloro-6-methyl) > 5 (4-chloro).
3.6. Activity correlations
During scientific prognostication of physiological activity in QSAR,41 the influence of the following phenomena is usually assessed: electronic effects (influencing the ionization or polarity of the substance), steric characteristics of the structure (playing an important role in the evaluation of the binding strength of the investigated compound with the biological target), and lipophilicity (ability to dissolve in fats, characterizing the properties of the substance to penetrate through the cell membranes). The distribution coefficient (and its logarithm) in the 1-octanol/buffer system is well known to be the most often used descriptor during the estimation of the quantitative structure–activity correlations.In order to describe graphically the activity–lipophilicity dependence, we applied an approach introduced in ref. 42 for compounds of a specific type of activity and a close chemical structure according to the equation:
| log(1/Kba) = a0 + a1·logDoct/buf − a2·(logDoct/buf)2 + a3·σ + a4·Es | (22) |
Fig. 7 depicts the dependence of the biological activity of the investigated thiadiazoles on the lipophilicity of the molecules characterized by the distribution coefficient in the 1-octanol/buffer system.
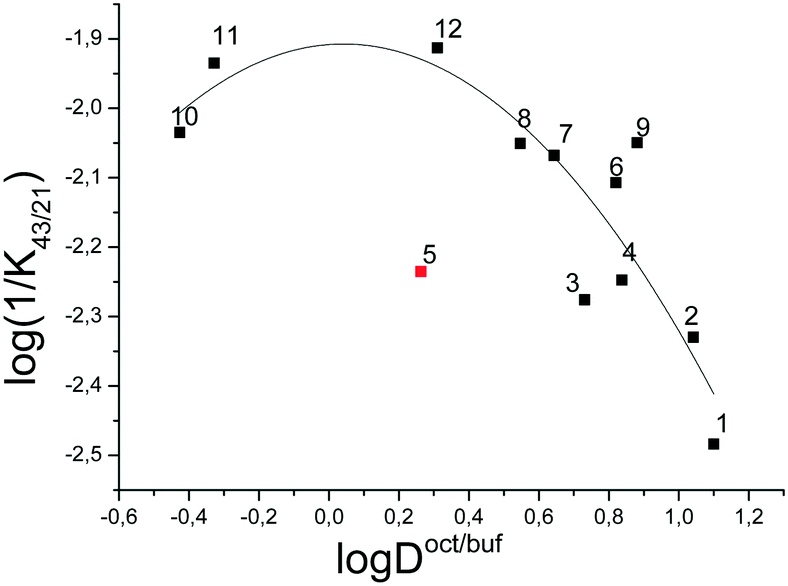 | ||
| Fig. 7 Log(1/K43/21) vs. logDoct/buf dependence. | ||
The correlation equation, in which the biological activity acts as a dependent variable and the distribution coefficient in the 1-octanol/buffer system acts as an independent one, is as follows (compound 5 is excluded from the correlation):
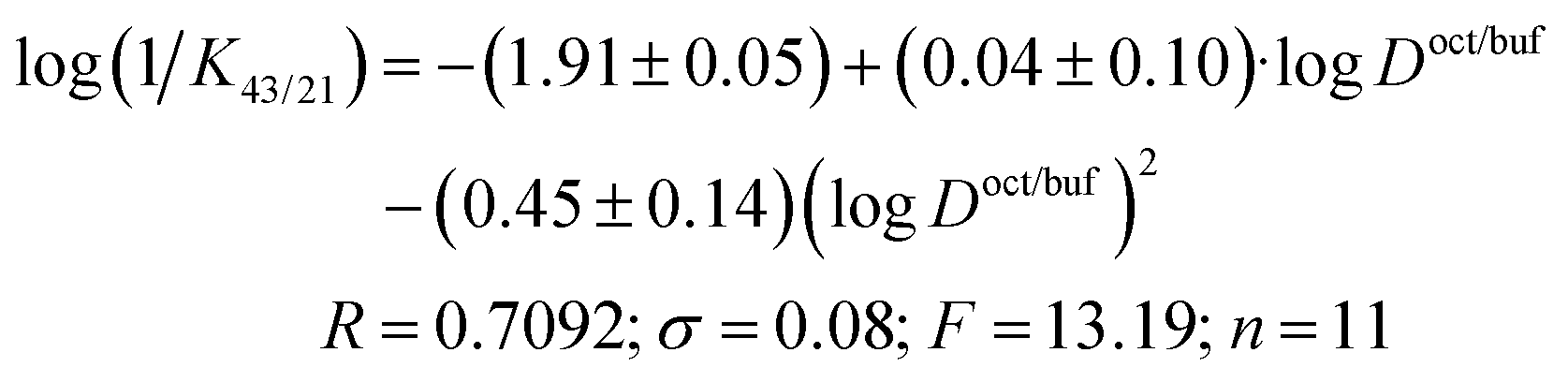 | (23) |
It should be noted that the obtained correlation is not high; this fact may be explained by the rather small number of the experimental points. Enhancement of the correlation can be expected at the extension of the number of the compounds investigated. However, the obtained regularities support the parabolic model to be an appropriate manner for describing the dependence of the specific activity of thiadiazoles synthesized in the present study on the lipophilicity expressed by the distribution coefficient in the 1-octanol/water system.
4. Conclusions
Novel 1,2,4-thiadiazole derivatives as potent neuroprotectors were synthesized and identified. Their ability to inhibit the glutamate stimulated Ca2+ uptake was investigated in vitro. The structure of thiadiazole molecules was shown to influence the inhibition properties. Activity–lipophilicity correlation was estimated based on the parabolic model. The solubility of 1,2,4-thiadiazoles was measured in a buffer solution pH 7.4 at 298 K. The distribution coefficients in 1-octanol/buffer (pH 7.4) and 1-hexane/buffer (pH 7.4) were determined. Apparent permeability coefficients were obtained in a Franz diffusion cell using the new Permeapad™ barrier. A search for the correlations between the distribution coefficients in 1-octanol/buffer and 1-hexane/buffer systems and physicochemical descriptors was performed. A suitable correlation was obtained for the independent variables which include polarizability (α) and the acceptor molecular ability to create hydrogen bonds (∑(Ca)). The appropriate correlation equations were derived and the predictive force of the models was assessed. Analysis of the relationships of physicochemical descriptors with the permeability characteristics was fulfilled. Testing all the descriptors revealed the correlation with the (∑(Ca)/α)-parameter. The obtained correlation equations confirmed the possibility of permeability coefficients of this class of compounds to be predictable based only on their structural formulas. Potential bioavailability of the substances was determined by the solubility–permeability combination through the estimation of the diffusion flux across the synthetic lipid membrane. The solubility values were revealed to have the main contribution to the diffusion fluxes of the investigated compounds. The results obtained in the present study will be useful when fast bioavailability screening of newly synthesized substances is required.Acknowledgements
This work was supported by the Russian Science Foundation (No 15-13-10017).References
- R. A. Jellinger, G. Ladurner and M. Windisch, New Trends in the Diagnosis and Therapy of Alzheimer's Disease, Springer-Verlag, Wien-New-York, 1994 Search PubMed.
- N. Guzior, A. Wieckowska, D. Panek and B. Malawska, Curr. Med. Chem., 2015, 22(3), 373 CrossRef CAS PubMed.
- A. Castro, T. Castano, A. Encinas, W. Porcal and C. Gil, Bioorg. Med. Chem., 2006, 14, 1644 CrossRef CAS PubMed.
- A. Martinez, M. Alonso, A. Castro, C. Perez and F. J. Moren, J. Med. Chem., 2002, 45, 1292 CrossRef CAS PubMed.
- A. M. MacLeod, R. Baker, S. B. Freedman, S. Patel, K. J. Merchant, M. Roe and J. Saunders, J. Med. Chem., 1990, 33, 2052 CrossRef CAS PubMed.
- Y. Iizawa, K. Okonogi, R. Hayashi, T. Iwahi, T. Yamazaki and A. Imada, Antimicrob. Agents Chemother., 1993, 37, 100 CrossRef CAS PubMed.
- L. N. Petrova and S. O. Bachurin, Bull. Exp. Biol. Med., 2006, 142, 51 CrossRef.
- J. N. Kew and J. A. Kemp, Psychopharmacology, 2005, 179(1), 4 CrossRef CAS PubMed.
- L. Parnetti, U. Senin and P. Mecocci, Drugs, 1997, 53(5), 752 CrossRef CAS PubMed.
- M. Husain and M. A. Mehta, Trends Cognit. Sci., 2011, 15(1), 28 CrossRef CAS PubMed.
- E. Kerns and L. Di, Druglike Properties: Concepts, Structure Design and Methods, Academic Press, 2008 Search PubMed.
- A. Avdeef, Absorption and Drug Development: Solubility, Permeability, and Charge State, Wiley, Hoboken, 2003 Search PubMed.
- M. Masson, B. V. Sigurdadottir, K. Matthiasson and T. Loftsson, Chem. Pharm. Bull., 2005, 53, 958 CrossRef CAS PubMed.
- A. Andrés, M. Rosés, C. Ràfols, E. Bosch, S. Espinosa, V. Segarra and J. M. Huerta, Eur. J. Pharm. Sci., 2015, 76, 181 CrossRef PubMed.
-
J. E. Comer, High-throughput measurement of logD and pKa, ed. H. van de Waterbeemd, H. Lennernäs and P. Artursson, In: Drug Bioavailability: Estimation of Solubility, Permeability, Absorption, and Bioavailability, Wiley-VCH, Weinheim, 2007 Search PubMed.
- P. Seiler, Eur. J. Med. Chem., 1974, 9(5), 473 CAS.
- G. L. Perlovich, T. V. Volkova, A. N. Proshin, D. Yu. Sergeev, B. T. Cong, L. N. Petrova and S. O. Bachurin, J. Pharm. Sci., 2010, 99(9), 3754 CrossRef CAS PubMed.
- G. L. Perlovich, A. N. Proshin, T. V. Volkova, B. T. Cong and S. O. Bachurin, Mol. Pharmaceutics, 2011, 8, 1807 CrossRef CAS PubMed.
- A. O. Surov, B. T. Cong, A. N. Proshin, P. Roussel, A. Idrissi and G. L. Perlovich, J. Phys. Chem. B, 2013, 117, 10414 CrossRef CAS PubMed.
- A. O. Surov, B. T. Cong, T. V. Volkova, A. N. Proshin and G. L. Perlovich, Phys. Chem. Chem. Phys., 2015, 17, 20889 RSC.
- A. O. Surov, B. T. Cong, T. V. Volkova, A. N. Proshin and G. L. Perlovich, J. Chem. Thermodyn., 2016, 96, 57 CrossRef CAS.
- G. L. Perlovich, A. N. Proshin, T. V. Volkova, S. V. Kurkov, V. V. Grigoriev, L. N. Petrova and S. O. Bachurin, J. Med. Chem., 2009, 52, 1845 CrossRef CAS PubMed.
- G. L. Perlovich, A. N. Proshin, T. V. Volkova, L. N. Petrova and S. O. Bachurin, Mol. Pharmaceutics, 2012, 9(8), 2156 CrossRef CAS PubMed.
- S. V. Blokhina, M. V. Ol'khovich, A. V. Sharapova, A. N. Proshin and G. L. Perlovich, J. Chem. Eng. Data, 2012, 57, 1996 CrossRef CAS.
- A. J. Boulton, A. R. Katritzky and A. M. Hamid, J. Chem. Soc. C, 1967, 2005 RSC.
- In: Chemical equilibrium. Properties of solutions, ed. C. A. Simanova, Manual chemist and technologist. S.-Pt: Professional, 2004, p. 194 Search PubMed.
- M. di Cagno, H. A. Bibi and A. Bauer-Brandl, Eur. J. Pharm. Sci., 2015, 73, 29 CrossRef CAS PubMed.
- T. Higuchi and K. A. Connors, Adv. Anal. Chem. Instrum., 1965, 4, 117 CAS.
- M. Brandl, G. E. Flaten and A. Bauer-Brandl, Passive diffusion across membrane, Wiley Encyclopedia Chem Biol., 2009, vol. 3, pp. 541–500.
- E. B. Ghartey-Tagoe, J. S. Morgan, A. S. Neish and M. R. Prausnitz, J. Controlled Release, 2005, 103, 177 CrossRef CAS PubMed.
- A. J. Leo, J. Pharm. Sci., 1987, 76, 166 CrossRef CAS PubMed.
- A. J. Leo, C. Hansch and D. Elkins, Chem. Rev., 1972, 71, 525 CrossRef.
- A. Andrés, M. Roses, C. Rafols, E. Bosch, S. Espinosa, V. Segarra and J. M. Huerta, Eur. J. Pharm. Sci., 2015, 76, 181 CrossRef PubMed.
- OECD guideline for the testing of chemicals adopted by the Council on 27th July 1995 Partition Coefficient (n-octanol/water): Shake Flask Method. Available from: http://www.oecd.org/chemicalsafety/risk-assessment/1948169.pdf.
- O. A. Raevsky, V. J. Grigor'ev and S. V. Trepalin, HYBOT (Hydrogen Bond Thermodynamics) program package, Registration by Russian State Patent Agency N 990090 of 26.02.99 Search PubMed.
- ACD/ChemSketch program. Available from: http://eduinf.waw.pl/che/inne/prgchem/pages/chsk_eng.pdf.
- V. A. Borodin, E. V. Kozlovsky and V. P. Vasil'ev, Russ. J. Inorg. Chem., 1982, 27, 2169 CAS.
- S. H. Yalkowsky and S. C. Valvani, J. Pharm. Sci., 1980, 69, 912 CrossRef CAS PubMed.
- G. L. Perlovich, A. M. Ryzhakov, V. V. Tkachev, L. K. Hansen and O. A. Raevsky, Cryst. Growth Des., 2013, 13(9), 4002 Search PubMed.
- P. Sanphui, V. K. Devi, D. Clara, N. Malviya, G. S. Somnath and G. R. Desiraju, Mol. Pharmaceutics, 2015, 12(5), 1615 CrossRef CAS PubMed.
- C. Hansch and T. Fujita, J. Am. Chem. Soc., 1964, 86, 1616 CrossRef CAS.
- C. Hansch, In: Quantitative structure–activity relationship in drug design, ed. E. J. Ariens, Academic Press, New York, 1971, pp. 271–342 Search PubMed.
Footnotes |
| † The authors declare no competing interests. |
| ‡ Electronic supplementary information (ESI) available: Dependence of the content of the ionized forms of the molecules on the pH of a buffer solution of compound 2 (Fig. 1SI); relationships of the permeability coefficients with the distribution coefficients (Fig. 2SI); relationship of the permeability coefficient with physicochemical descriptor HYBOT (Fig. 3SI). See DOI: 10.1039/c6md00545d |
| This journal is © The Royal Society of Chemistry 2017 |