
Design, synthesis, anticancer activity and docking studies of theophylline containing 1,2,3-triazoles with variant amide derivatives†‡
Radhakrishnam Raju
Ruddarraju
a,
Adharvana Chari
Murugulla
*a,
Ravindar
Kotla
a,
Muni Chandra Babu
Tirumalasetty
b,
Rajendra
Wudayagiri
b,
Shobha
Donthabakthuni
a and
Ravichandar
Maroju
c
aDr.MACS Bio-Pharma Pvt. Ltd, Factory: Plot-79/B&C, Pasamylaram, Medak (Dist)-502307, Patancheru, Telangana, India. E-mail: drmac_s@yahoo.com
bBioinformatics Center, Division of Molecular Biology, Department of Zoology, Sri Venkateswara University, Tirupati-517 502, Andhra Pradesh, India
cMahatma Gandhi Institute of Technology, Gandipet, Hyderabad-500 075, Telangana, India
First published on 28th October 2016
Abstract
A new series of theophylline analogues containing 1,2,3-triazoles with different amide groups (22–41) has been designed and synthesized, and their biological activities have been evaluated as potential anticancer agents. The anticancer activities of the synthesized compounds were studied in four cancer cell lines viz. lung (A549), colon (HT-29), breast (MCF-7) and melanoma (A375). Furthermore, these compounds were screened for computational ADME and Lipinski's analysis followed by molecular docking and binding energy calculations against the various therapeutic targets involved in cell proliferation. The in vitro results demonstrate that compounds 22, 27, 36 and 40 have pivotal anticancer activity. Among these, compounds 22 and 27 have significant cytotoxic activity in all three cell lines; the in silico docking studies also reveal that compounds 22, 27 and 36 have good dock scores, binding affinities and binding energies towards human epidermal growth factor receptor 2. This is the first report to demonstrate theophylline hybrids containing 1,2,3-triazoles as potential anticancer agents.
Introduction
Cancer is considered as the second most lethal disease; it is responsible for 21% of annual deaths globally.1 Approximately 7.6 million people die of cancer every year worldwide, which is expected to reach 13 million in 2030. As per a report published in The Lancet,2 the total deaths due to cancer in India were 0.55 million in 2010. Lung, breast, colon and melanoma cancers are the most common cancers in the developing and underdeveloped countries. Lung and breast cancers among men and women are more prevalent in the developed countries. Overall, 16.8% of people in the United States diagnosed with lung cancer survive five years after the diagnosis.3 Whereas the breast cancer survival rates in the developed world are high;4 80% to 90% of patients in England and in the United States live for at least 5 years after diagnosis.5,6 However, globally, colon cancer is the third most common type of cancer, comprising about 10% of all cases.7 It is less common in women than in men and in developed countries. Melanoma is the most dangerous type of skin cancer. It is more common in men than in women.8 Globally, in 2012, it occurred in 2.32 lakhs people and resulted in 55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths. For this purpose, chemotherapy is the most commonly used treatment worldwide.9 However, it has several serious side effects and drawbacks. Thus, these limitations compelled us to search for novel anticancer agents with diverse chemical structures; herein, we report the synthesis and biological evolution of theophylline containing 1,2,3-triazoles with different amide derivatives as potential anticancer agents.
000 deaths. For this purpose, chemotherapy is the most commonly used treatment worldwide.9 However, it has several serious side effects and drawbacks. Thus, these limitations compelled us to search for novel anticancer agents with diverse chemical structures; herein, we report the synthesis and biological evolution of theophylline containing 1,2,3-triazoles with different amide derivatives as potential anticancer agents.
Analogues of naturally occurring purine bases are valuable tools in defining the chemical interactions involved in a specific biological response. Structural alterations of xanthine have resulted in several potent biological systems e.g. theophylline, also known as 1,3-dimethylxanthine, is a proven bronchodilator drug used in the therapy for respiratory diseases such as asthma or chronic pulmonary obstructive disease (COPD).10–12 Theophylline derivatives with alterations at their 7- and 8-positions have been investigated with respect to their bronchospasmolytic,13–16 anticancer17 and circulatory blood system activities.18 The aim of this study is to expand the pharmacological profile of theophylline to reduce its side effects and to identify possible candidates for further development.
Meanwhile, 1,2,3-triazoles have gained enormous attention in recent years owing to their broad spectrum of pharmaceutical activities and their highly diverse biological activities, such as anticancer,19–21 antifungal, antibacterial,22–24 antituberculosis25–27 and antivirus.28–30 Thus, click chemistry has attracted extensive interest in almost all aspects of drug discovery, such as lead discovery through combinatorial chemistry, target-templated in vitro chemistry, proteomics, and DNA research using bio-conjugation reactions. Bioactive theophylline and 1,2,3-triazole molecules are shown in Fig. 1.
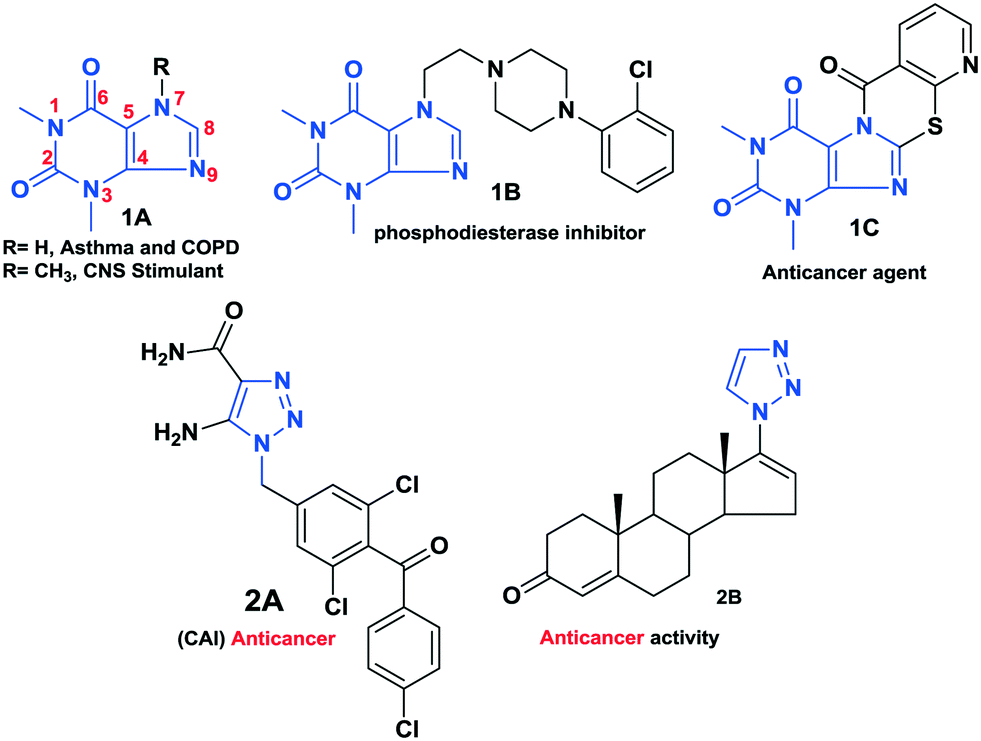 | ||
| Fig. 1 Bioactive theophylline and 1,2,3-triazole moieties. | ||
Based on their biological significance, the integration of theophylline and 1,2,3-triazole pharmacophore units with amide linkages in one molecular platform has been envisaged to generate new theophylline derivatives containing 1,2,3-triazoles with a variant amide analogue hybrid framework with anticancer activity. In addition, structure–activity relationship (SAR) studies, molecular docking, and toxicity screening were performed, and the ADME properties of the compounds were evaluated (Fig. 2).
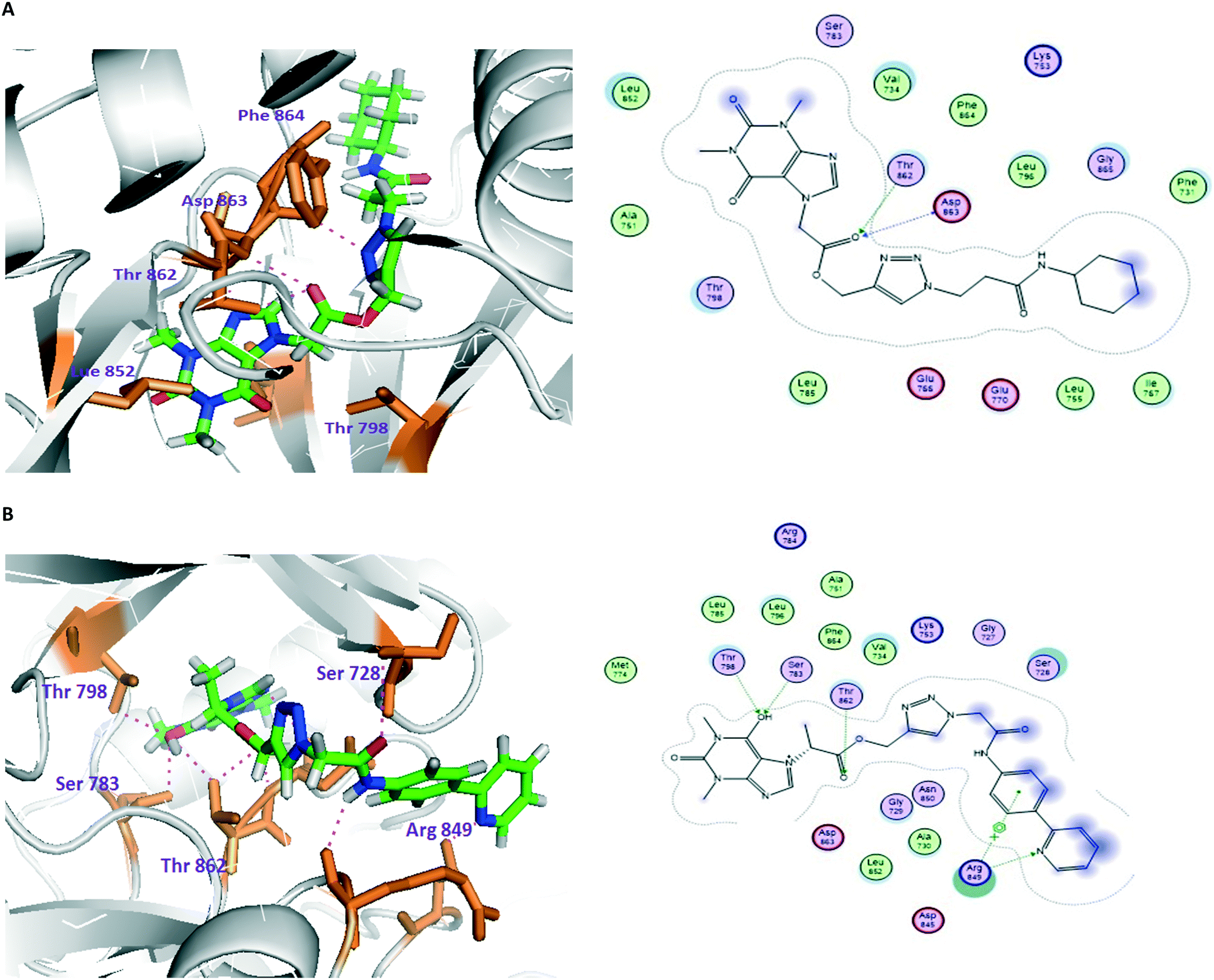 | ||
| Fig. 2 Molecular and LIGPLOT interactions of compounds 22 (A) and 41 (B) with the tyrosine kinase domain of human epidermal growth factor receptor 2 (HER2). | ||
Results and discussion
Chemistry
Different commercially available aryl halide compounds, 4, 6, 9, 11 and 13, were treated with boronic acids or boronate esters 3, 8 and 14 using potassium phosphate as a base and Pd(dppf)Cl2·CH2Cl2 as a catalyst in 1,4-dioxane and water at 100 °C for 10 to 16 hours; this gave biphenyl amine compounds 5, 7, 10, 12 and 15, as shown in Scheme 1.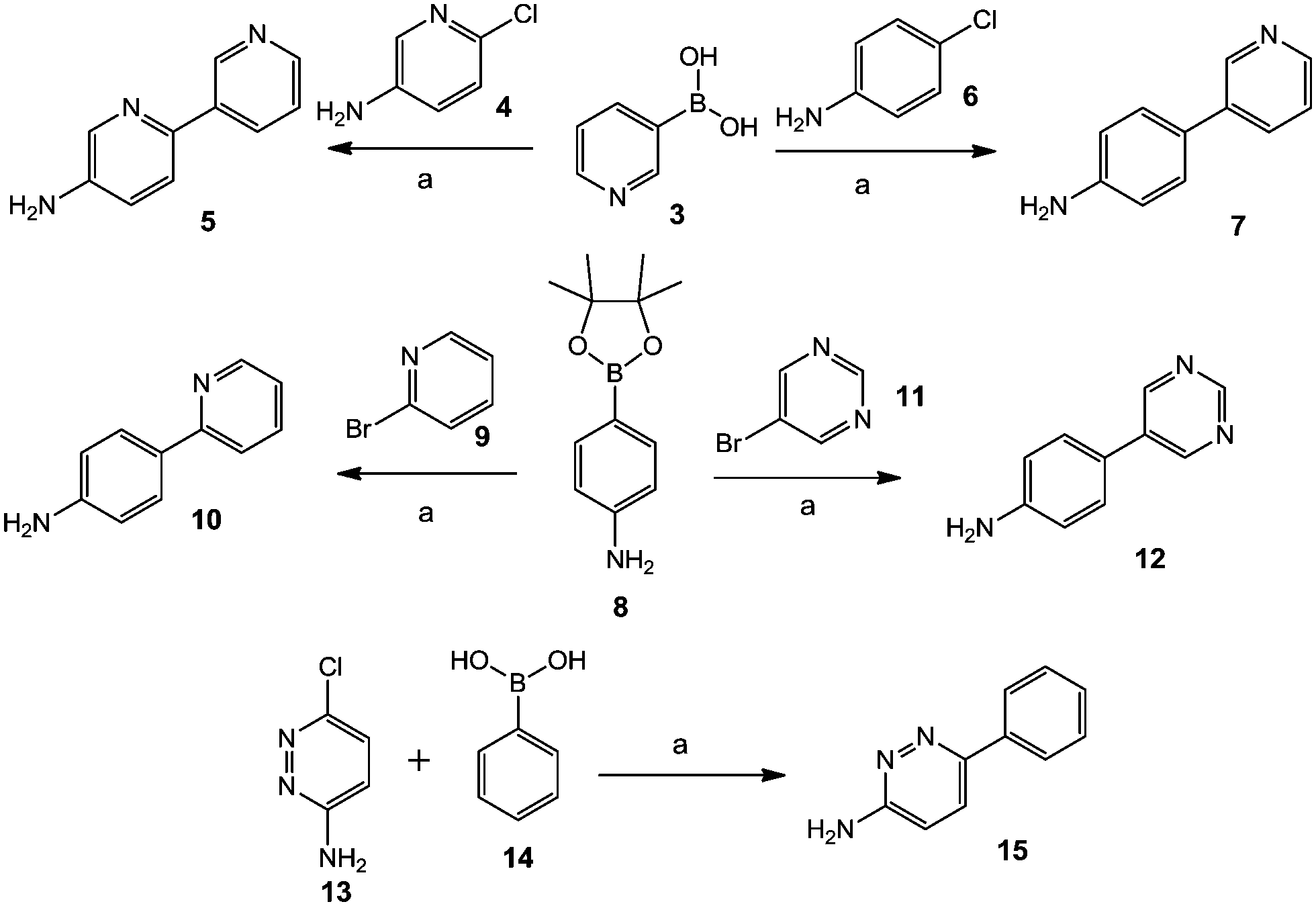 | ||
| Scheme 1 Synthesis of biphenyl amine compounds 5, 7, 12 and 15. Reagents and conditions: (a) K3PO4, Pd(dppf)Cl2·CH2Cl2, 1,4-dioxane:H2O, 100 °C, 10–16 h. | ||
The synthesis of theophylline containing 1,2,3-triazoles with different aliphatic cyclic amides and biphenyl amide analogues (compounds 22 to 41) is outlined in Scheme 2. Initially, the commercially available starting material theophylline 16 was alkylated with ethyl 2-bromoacetate and K2CO3 in N,N-dimethylformamide (DMF) at 85 °C for 12 hours, which gave 17a in 84% yield; in contrast, 17b was prepared by a Mitsunobu reaction. Theophylline 16 was reacted with ethyl 2-hydroxypropanoate using triphenylphosphine (TPP) and diisopropyl azodicarboxylate (DIAD) at room temperature, affording 17b in 50% yield. The ester compounds 17a and 17b were hydrolyzed with LiOH·H2O in THF and H2O at room temperature for 2 hours to afford 18a and 18b in 91% and 98% yield, respectively. The propynyl compounds were prepared by coupling acid compounds 18a and 18b with propargyl alcohol using N,N′-dicyclohexylcarbodiimide (DCC) and 4-dimethylaminopyridine (DMAP) at room temperature for 16 hours; this gave 19a and 19b in 78% and 83% yield, respectively. Lastly, the 1,2,3-triazole ring was constructed based on Cu(I)-catalyzed 1,3-dipolar cycloaddition between the appropriate alkyne and azide as the key step. The 1,2,3-triazoles were synthesized by reacting propynyl compounds 19a and 19b with 3-azidopropanoic acid in the presence of CuSO4·5H2O and sodium ascorbate in t-BuOH and H2O for 16 hours; this gave 20a and 20b in 85% and 78% yield, respectively, whereas 2-azido acetic acid in the presence of CuSO4·5H2O and sodium ascorbate in t-BuOH and H2O for 16 hours gave 21a and 21b in 82% and 77% yield, respectively.
 | ||
| Scheme 2 Synthesis of aliphatic cyclic amide and biphenyl amide compounds 22–31 and 32–41. Reagents and conditions: (a) (i) K2CO3, DMF, 85 °C, 12 h; (ii) TPP, DIAD, THF, rt, 5 h; (b) LiOH·H2O, THF:H2O, rt, 2 h; (c) DCC, DMAP, DCM, rt, 16 h; (d and e) CuSO4·5H2O, sodium ascorbate, t-BuOH:H2O, rt, 16 h; (f, g and h) HATU, DIPEA, DCM, rt, 16 h. | ||
Finally, the 1,2,3-triazole acid compounds 20a and 20b or 21a and 21b were coupled with different aliphatic cyclic amines or biphenyl amines using 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium-3-oxide hexafluorophosphate (HATU) and N,N-diisopropylethylamine (DIPEA) as a base in dichloromethane to afford the final compounds 22–41 in 44% to 86% yield.
Biological activity
| Compound
|
||||||
|---|---|---|---|---|---|---|
| R1 | R2 | No. | A549 (lung) | HT-29 (colon) | MCF-7 (breast) | A375 (melanoma) |
| All assays were performed in triplicate; IC50 values are reported as mean ± SD. | ||||||
| H | Cyclohexyl | 22 | 1.40 ± 0.09 | 52.3 ± 1.20 | 2.20 ± 0.32 | 2.51 ± 0.58 |
| H | Cyclopentyl | 23 | 4.70 ± 0.14 | 15.4 ± 1.29 | 12.8 ± 0.33 | 60.8 ± 4.61 |
| H | Cyclobutyl | 24 | 5.24 ± 0.49 | 19.2 ± 2.81 | 9.18 ± 0.99 | 61.2 ± 3.51 |
| H | Morpholine | 25 | 7.24 ± 1.02 | 38.2 ± 3.22 | 18.2 ± 1.81 | 58.2 ± 1.90 |
| CH3 | Cyclohexyl | 26 | 34.6 ± 2.49 | 21.4 ± 0.60 | 7.54 ± 1.36 | 3.92 ± 0.59 |
| CH3 | Cyclopentyl | 27 | 1.21 ± 0.16 | 2.30 ± 0.16 | 2.31 ± 0.41 | 63.2 ± 2.30 |
| CH3 | Cyclobutyl | 28 | 3.54 ± 0.44 | 55.2 ± 2.76 | 6.33 ± 1.06 | 8.52 ± 0.34 |
| CH3 | Cyclopropyl | 29 | 10.2 ± 0.68 | 14.8 ± 0.34 | 16.8 ± 0.85 | 68.2 ± 2.71 |
| CH3 | Morpholine | 30 | 5.82 ± 0.39 | 6.2 ± 0.59 | 5.92 ± 0.30 | 24.8 ± 1.83 |
| CH3 | N-Ethylpiperazine | 31 | 6.82 ± 0.41 | 24.2 ± 1.94 | 38.2 ± 2.62 | 62.3 ± 4.30 |
| Combretastatin-A4 | 0.11 ± 0.19 | 0.93 ± 0.82 | 0.18 ± 0.5 | 0.21 ± 0.62 | ||

| Compound
|
||||||
|---|---|---|---|---|---|---|
| R1 | R4 | No. | A549 (lung) | HT-29 (colon) | MCF-7 (breast) | A375 (melanoma) |
| All assays were performed in triplicate; IC50 values are reported as mean ± SD. | ||||||
| H |

|
32 | 2.12 ± 0.26 | 54.3 ± 2.10 | 53.8 ± 3.5 | 3.80 ± 0.31 |
| H |

|
33 | 4.70 ± 0.35 | 56.8 ± 2.36 | 8.41 ± 0.63 | 9.34 ± 0.55 |
| H |

|
34 | 53.4 ± 2.02 | 17.3 ± 0.43 | 2.40 ± 0.17 | 3.62 ± 0.34 |
| CH3 |

|
35 | 6.33 ± 0.18 | 50.2 ± 0.90 | 62.8 ± 3.27 | 2.80 ± 0.29 |
| CH3 |

|
36 | 4.90 ± 0.09 | 2.90 ± 0.73 | 2.71 ± 0.44 | 58.2 ± 1.58 |
| CH3 |
|
37 | 48.2 ± 0.26 | 27.5 ± 2.06 | 5.91 ± 0.28 | 10.31 ± 0.25 |
| CH3 |

|
38 | 6.82 ± 0.12 | 5.82 ± 0.46 | 18.4 ± 0.51 | 50.4 ± 3.41 |

|
|
||||||
|---|---|---|---|---|---|---|
| R1 | R3 | No. | ||||
| H |
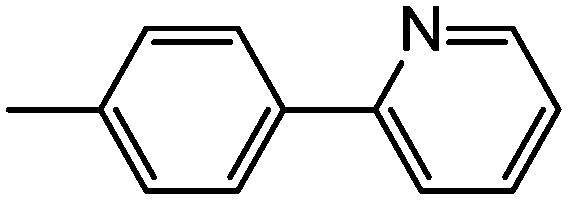
|
39 | 4.20 ± 0.18 | 2.96 ± 0.10 | 5.72 ± 0.54 | 61.4 ± 2.56 |
| H |
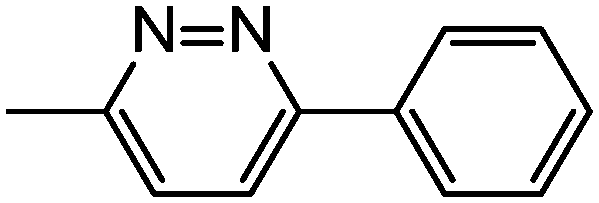
|
40 | 1.34 ± 0.14 | 11.7 ± 0.19 | 13.5 ± 0.49 | 48.2 ± 2.86 |
| CH3 |

|
41 | 8.24 ± 0.05 | 7.23 ± 0.5 | 10.2 ± 0.31 | 63.4 ± 2.8 |
| Combretastatin-A4 | 0.11 ± 0.19 | 0.93 ± 0.82 | 0.18 ± 0.5 | 0.21 ± 0.62 | ||

With regard to the amide analogues (22–31), the series of aliphatic cyclic amides showed massive differences in potency and selectivity with decreasing cyclic ring size. Here, a slight difference in two series of compounds, 22–25 and 26–30, which contain a methyl group on the α-carbon at position 7 of the theophylline ring, resulted in a dramatic change in activity. Among these, compounds 22 and 27 showed the highest anticancer activity against three cancer cell lines, i.e. lung, breast, melanoma for 22 (IC50 = 1.40 ± 0.09, 2.20 ± 0.32, and 2.51 ± 0.58 μM, respectively) and lung, colon, breast for 27 (IC50 = 1.21 ± 0.16, 2.30 ± 0.16, and 2.31 ± 0.41 μM, respectively) compared to the other compounds. In comparison, compound 22, in which the cyclohexyl ring was replaced with cyclopentyl and cyclobutyl rings to form compounds 23 and 24, respectively, displayed less activity against the four cancer cell lines. Similarly, compounds 26, 28 and 29, which contain cyclohexyl, cyclobutyl and cyclopropyl rings, displayed less activity against the four cancer cell lines than compound 27. Here, the anticancer activity of the cyclic amides 22–24 and 27–29 clearly showed a decreasing trend with decreasing ring size, as displayed by their losses of activity. Meanwhile, the other cyclic amides, such as the morpholine analogues in 25 and 30 and the ethyl piperazine derivative in 31, showed no improvement in potency or selectivity.
In the next series of compounds, substituting the aliphatic cyclic amide moiety with biphenyl amide structures, such as those found in 32–38, typically resulted in less anticancer activity compared with the cyclic amide analogues. Meanwhile, some of the biphenyl series (32–38) still displayed good inhibition activities and showed values comparable to some of the better amide analogues; the reason for this is unclear. Compounds 32, 34, 35 and 36 showed higher anticancer activities than compounds 33, 37 and 38. A vital comparison between the two series of compounds, 32–34 and 35–37, with α-carbons at position 7 of the theophylline ring, displayed a change in activity. Comparing compounds 32 and 35, both of which contain 2,3-bipyridine, 32 showed activity in the lung cell line (IC50 = 2.12 ± 0.26 μM) and 35 showed anti-melanoma activity (IC50 = 2.80 ± 0.29 μM); both compounds were inactive against the remaining three cell lines. Similarly, among compounds 33 and 36, containing 2-phenylpyridine, and 34 and 37, containing 3-phenylpyridine, compounds 33 and 37 showed no activity in all four cancer cell lines, whereas compounds 34 and 36, interestingly, showed activity in the breast and colon cancer cell lines; 34 was active against breast cancer (IC50 = 2.40 ± 0.17 μM), while 36 was active against colon and breast cancer (IC50 = 2.90 ± 0.73 and 2.71 ± 0.44 μM, respectively).
Other synthesized biphenyl amide analogues (i.e., 39–41) were based on the aliphatic cyclic amide scaffold, which has been shown to display promising anticancer activity and selectivity. Compound 40 was shown to have slightly better anticancer activity towards lung cancer (IC50 = 1.34 ± 0.14 μM) but less activity towards other cell lines, i.e., colon, breast and melanoma, compared to the aliphatic cyclic amide analogues 22 and 27. Comparing compounds 39 and 41, compound 39, containing 2-phenylpyridine, showed activity against colon cancer cells (IC50 = 2.96 ± 0.10 μM), whereas compound 41 showed no activity in all four cancer cell lines compared to compounds 39 and 40. According to this data, the compounds which contain aliphatic cyclic amide analogues showed higher anticancer activity in all three cancer cell lines than those containing biphenyl amide analogues.
In conclusion, theophylline compounds containing 1,2,3-triazoles with different amide analogues have been designed, synthesized and evaluated; these contain systematic variations, such as electron donating groups, e.g. a methyl group, on the α-carbon atom at position 7 of the theophylline ring, and substitutions such as cyclic rings and biphenyls. Structure activity relationships were also established for the compounds. Although the role of the methyl group is still not clear, substitution of cyclic rings and biphenyl rings seems to be important for their activities against cancer cell lines.
From a medicinal chemistry perspective, each functional group provides specific properties and behaviours that allow drug molecules to exert their desired pharmacodynamic and pharmacokinetic effects. Any given functional group may play a significant role in overall water/lipid solubility, electronic effects, steric effects and ability to interact with specific biological targets.
Molecular docking studies
In order to better understand their structure–activity relationships, molecular docking studies of compounds 22–41 with various therapeutic targets of cancer were observed; this demonstrated that human epidermal growth factor receptor 2 shows significant dock scores, binding energies and binding affinities with all the compounds. Meanwhile, the majority of the compounds show moderate scores with other targets, i.e., EGFR, VEGFR2 and aromatase.Regarding the docking results and the binding affinity and binding energy calculations against HER2, 22, 27 and 36 showed the highest dock scores. Compound 22 shows two H-bonds with amino acids in the active site of HER2; its dock score, binding energy and binding affinity are −15.42, −25.43 and 10.29, respectively. The docking results revealed that amino acids Thr862 and Asp863, located in the binding pocket of the protein, played important roles. The oxygen atom of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group acts as an H-bond acceptor; its oxygen atom formed two H-bonds, one with the OH of Thr862 (bond length 2.49 Å), and another with the N of Asp863 (bond length 2.82 Å). Similarly, compound 27 showed two H-bonds, with a dock score, binding energy and binding affinity of −15.23, −9.673 and 10.95, respectively. The hydrogen atom of the amine group formed one H-bond with the oxygen atom of Asp863 by donating an electron pair; the bond length is 1.55 Å. The nitrogen atom of the theophylline ring formed one H-bond with the OH of Ser783 by accepting an electron pair; the bond length is 2.71 Å.
O group acts as an H-bond acceptor; its oxygen atom formed two H-bonds, one with the OH of Thr862 (bond length 2.49 Å), and another with the N of Asp863 (bond length 2.82 Å). Similarly, compound 27 showed two H-bonds, with a dock score, binding energy and binding affinity of −15.23, −9.673 and 10.95, respectively. The hydrogen atom of the amine group formed one H-bond with the oxygen atom of Asp863 by donating an electron pair; the bond length is 1.55 Å. The nitrogen atom of the theophylline ring formed one H-bond with the OH of Ser783 by accepting an electron pair; the bond length is 2.71 Å.
Compound 36 formed four H-bonds with active site amino acid residues, with a dock score, binding energy and binding affinity of −16.71, −25.52 and 14.10, respectively. The 6th position-OH of theophylline acts as an H-bond acceptor; also, its oxygen atom formed two H-bonds, one with the OH of Ser783 (bond length: 2.66 Å) and another with the OH of Thr798 (bond length: 2.76 Å). The oxygen atom of the CO group formed one H-bond with the OH of Thr862 by accepting an electron pair; the bond length is 2.86 Å. In the biphenyl ring, the nitrogen atom formed one H-bond with the NE of Arg849 by accepting an electron pair; the bond length is 2.89 Å. Based on this characterization of protein–ligand interactions, it is evident that theophylline, 1,2,3-triazole and amide analogues played key roles in forming H-bond interactions in both in vitro and in silico studies. The docking calculations of all the compounds (22–41) are also depicted in Table S5 in the ESI.‡
Toxicity risk assessment screening
The amide analogues (22–41) were screened for their toxic properties, such as irritant, mutagenic, tumorigenic and reproductive effects, using Molinspiration and the Osiris server. The server contains a built-in list of about 5300 distinct substructure fragments created from 15000 commercially available fragments with reported drug scores and drug likenesses. A drug score associated with drug likeness, clogP, molecular weight and toxicity risks may be used as a total value to judge the overall potential of a compound for use as a drug. The toxicity screening of all the compounds (22–41) showed no risk of mutagenic, tumorigenic, irritant or reproductive toxicity (Table S3 in the ESI‡).
Theoretical evolution of ADME properties
The bioavailability of amide analogues (22–41) was determined by ADME (Adsorption, Distribution, Metabolism and Excretion) using the MOE QSAR (Molecular Operating Environment Quantitative structure–activity relationship) model descriptor module. In order to explore the drug-like properties of compounds (22–41), the lipophilicity, expressed as the octanol/water partition coefficient and referred to here as logP(o/w), as well as other theoretical calculations, such as molecular size, TPSA, number of hydrogen bond acceptors and donors, and number of rotatable bonds, were calculated for each compound (Table S4 in ESI‡).
Lipinski and colleagues defined the properties and molecular features that are associated with orally active drugs in humans and summarized then in the rule-of-five.31 Veber and co-workers later added additional related criteria.32 These parameters have been widely used as a filter for new potential drugs. Therefore, a computational study was performed to predict the physico-chemical parameters of the synthesized compounds to verify if the substances could full fill Lipinski's rule-of-five.
The physico-chemical properties of the studied amide analogues (22–41) were summarized (Table S4‡). Compounds (22–41) showed logP(o/w) values below 5 which is favorable features for their oral bioavailability. TPSA represents the hydrogen bonding capacity of molecules with less than 140 Å are recognized to have good intestinal absorption. It is being expressed here as topological surface area (TPSA) which is sum of the surface areas occupied by oxygen and nitrogen atoms and the hydrogen's attached to them.
On the other hand hydrogen bonding capacity of drug candidate is also described by the number of hydrogen donors and acceptors. The compounds in this series showed H-donors >5 and H-acceptors >10. The number of rotatable bonds is also important factor for the coefficient binding to receptors and channels as well as for the oral bioavailability.32 Molecules with more than 10 rotatable bonds tend to show poor oral bioavailability. All studied compounds showed low conformational flexibility with less than 10 rotatable bonds.
The violation of more than one of these rules may indicate problems in the bioavailability of the potential drugs. The results showed that all compounds complied with Lipinski’s rule (Table S4‡), with exception slightly higher values were observed in TPSA, even in high molecular size more than 500. Finally summarizing the physico-chemical properties of the amide analogues (22–41), we could conclude that almost they obey the rule-of-five and meet all criteria for good solubility and permeability.
Conclusion
In the present work, an efficient, clean and convenient synthesis of novel theophylline analogues containing 1,2,3-triazoles with different amide groups has been designed. The procedure offers several advantages, including simple reaction conditions as well as simple experimental and product isolation procedures. Here, a series of twenty novel compounds were synthesized in good yield and characterized. This work focused on the anticancer activities of chemically diverse theophylline analogues containing 1,2,3-triazoles; the synthesized compounds 22, 27, 36 and 40 showed significant inhibitory activity against one or more tested cancer cell lines viz., lung (A549), colon (HT-29), breast (MCF-7) and melanoma (A375). Also, the docking results correlated with the IC50 data and enabled analysis of the importance of specific interactions for describing the binding energies and binding affinities of different amide derivatives. The obtained results also showed that structural modification of theophylline can yield products with different pharmacological activities than those commonly associated with theophylline analogues, such as asthma, COPD and adenosine receptor antagonism. Therefore, this class of compounds could be a good starting point to develop new leads in the treatment of cancer. To demonstrate the utility of this chemistry, several biologically important molecules were synthesized; further studies are required to design more specific and efficient molecules.Acknowledgements
The authors are thankful to Dr. V. Koteswara Rao, Postdoctoral Research Associate, University of Kansas, USA, for providing MOE software.Notes and references
- World Health Statistics 2012, World Health Organization, Geneva, 2012 Search PubMed.
- R. Dikshit, P. C. Gupta, C. Ramasundarahettige, V. Gajalakshmi, L. Aleksandrowicz, R. Badwe, R. Kumar, S. Roy, W. Suraweera, F. Bray, M. Mallath, P. K. Singh, D. N. Sinha, A. S. Shet, H. Gelband and P. Jha, Lancet, 2012, 379, 1807–1816 CrossRef.
- National Cancer Institute, Retrieved, 15 July 2014.
- World Cancer Report, International Agency for Research on Cancer, 2008, Retrieved, 26 February 2011 Search PubMed.
- Cancer Survival in England: Patients Diagnosed (2007–2011) and Followed up to 2012, Office for National Statistics, 29 October 2013, Retrieved, 29 June 2014 Search PubMed.
- SEER Stat Fact Sheets: Breast Cancer, NCI, Retrieved 18 June (2014) Search PubMed.
- World Cancer Report (2014), World Health Organization, (2014), ch. 1.1 Search PubMed.
- S. C. Azoury and J. R. Lange, Surg. Clin. North Am., 2014, 94, 945–962 CrossRef PubMed.
- S. J. Isakoff, Cancer J., 2010, 16, 53–61 CrossRef CAS PubMed.
- K. Ohta, Y. Fukuchi, L. Grouse, R. Mizutani, K. F. Rabe, S. I. Rennard and N. S. Zhong, Respir. Med., 2004, 98, 1016–1024 CrossRef PubMed.
- T. T. Hansel, R. C. Tennant, A. J. Tan, L. A. Higgins, H. Neighbour, E. M. Erin and P. J. Barnes, Drugs Today, 2004, 40, 55–69 CrossRef CAS PubMed.
- Y. Nuhoglu and C. Nuhoglu, Expert Rev. Respir. Med., 2008, 2, 305–313 CrossRef CAS PubMed.
- S. Corsano, R. Scapiochi and G. Strappaghetii, Arch. Pharm., 1994, 327, 411 CrossRef.
- G. Baziard-Mouysset, A. Rached, S. Younes, C. Tournaire, J. L. Stigliani, M. Payard, J. C. Yavo and C. Advenier, Eur. J. Med. Chem., 1995, 30, 253–260 CrossRef CAS.
- A. G. Sandoz, Israeli IL, 1992, 83, 659 ( Chem. Abstr. , 1992 , 117 , 7957k ) Search PubMed.
- L. Gajewczyk and A. Zejc, Acta Pol. Pharm., 1992, 49, 61–63 CAS.
- R. Radhakrishnam Raju, M. Adharvana Chari, K. Ravindar, T. Muni Chandra Babu, W. Rajendra, D. Shobha, M. Ravichandar, K. Baburao and P. Lakshmana Swamy, Eur. J. Med. Chem., 2016, 123, 379–396 CrossRef PubMed.
- P. R. Bovy, J. T. Collins, T. S. Chamberlain and B. K. Gheng, PCT Int ppl WO9207852, 1992, Chem. Abstr., 1992, 117, 131218j Search PubMed.
- M. J. Fray, D. J. Bull, C. L. Carr, E. C. Gautier, C. E. Mowbray and A. Stobie, J. Med. Chem., 2001, 44, 1951–1962 CrossRef CAS PubMed.
- G. Smith, M. Glaser, M. Perumal, Q. D. Nguyen, B. Shan, E. Arstad and E. O. Aboagye, J. Med. Chem., 2008, 51, 8057–8067 CrossRef CAS PubMed.
- C. B. Yim, I. Dijkgraaf, R. Merkx, C. Versluis, A. Eek, G. E. Mulder, D. T. Rijkers, O. C. Boerman and R. M. Liskamp, J. Med. Chem., 2010, 53, 3944–3953 CrossRef CAS PubMed.
- N. S. Vatmurge, B. G. Hazra, V. S. Pore, F. Shirazi, P. S. Chavan and M. V. Deshpande, Bioorg. Med. Chem. Lett., 2008, 18, 2043–2047 CrossRef CAS PubMed.
- N. S. Vatmurge, B. G. Hazra, V. S. Pore, F. Shirazi, M. V. Deshpande, S. Kadreppa, S. Chattopadhyay and R. G. Gonnade, Org. Biomol. Chem., 2008, 6, 3823–3830 Search PubMed.
- A. H. Kategaonkar, P. V. Shinde, A. H. Kategaonkar, S. K. Pasale, B. B. Shingate and M. S. Shingare, Eur. J. Med. Chem., 2010, 45, 3142–3146 CrossRef CAS PubMed.
- R. V. Somu, H. Boshoff, C. Qiao, E. M. Bennett, C. E. Barry and C. C. Aldrich, J. Med. Chem., 2006, 49, 31–34 CrossRef CAS PubMed.
- M. S. Costa, N. Boechat, E. A. Rangel, C. Da Silva Fde, A. M. De Souza, C. R. Rodrigues, H. C. Castro, I. N. Junior, M. C. Lourenco, S. M. Wardell and V. F. Ferreira, Bioorg. Med. Chem., 2006, 14, 8644–8653 CrossRef CAS PubMed.
- R. P. Tripathi, A. K. Yadav, A. Ajay, S. S. Bisht, V. Chaturvedi and S. K. Sinha, Eur. J. Med. Chem., 2010, 45, 142–148 CrossRef CAS PubMed.
- M. Whiting, J. C. Tripp, Y. C. Lin, W. Lindstrom, A. J. Olson, J. H. Elder, K. B. Sharpless and V. V. Fokin, J. Med. Chem., 2006, 49, 7697–7710 CrossRef CAS PubMed.
- Y. Saito, V. Escuret, D. Durantel, F. Zoulim, R. F. Schinazi and L. A. Agrofoglio, Bioorg. Med. Chem., 2003, 11, 3633–3639 CrossRef CAS PubMed.
- D. K. Mohapatra, P. K. Maity, M. Shabab and M. I. Khan, Bioorg. Med. Chem. Lett., 2009, 19, 5241–5245 CrossRef CAS PubMed.
- C. A. Lipinski, Drug Discovery Today: Technol., 2004, 1, 337–341 CrossRef CAS PubMed.
- D. F. Veber, S. R. Johnson, H. Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, J. Med. Chem., 2002, 45, 2615–2623 CrossRef CAS PubMed.
Footnotes |
| † The authors declare no competing interests. |
| ‡ Electronic supplementary information (ESI) available: Supplementary data provided as a separate electronic file. That file can then be transformed into PDF format and submitted along with the manuscript and graphic files to the appropriate editorial office. See DOI: 10.1039/c6md00479b |
| This journal is © The Royal Society of Chemistry 2017 |