
Design, synthesis and anticonvulsant-analgesic activity of new N-[(phenoxy)alkyl]- and N-[(phenoxy)ethoxyethyl]aminoalkanols†‡
Anna
Rapacz
a,
Anna M.
Waszkielewicz
 *b,
Katarzyna
Pańczyk
b,
Karolina
Pytka
a,
Paulina
Koczurkiewicz
c,
Kamil
Piska
c,
Elżbieta
Pękala
c,
Bogusława
Budziszewska
d,
Beata
Starek-Świechowicz
d and
Henryk
Marona
b
*b,
Katarzyna
Pańczyk
b,
Karolina
Pytka
a,
Paulina
Koczurkiewicz
c,
Kamil
Piska
c,
Elżbieta
Pękala
c,
Bogusława
Budziszewska
d,
Beata
Starek-Świechowicz
d and
Henryk
Marona
b
aDepartment of Pharmacodynamics, Faculty of Pharmacy, Jagiellonian University Medical College, Medyczna 9 Str., 30-688 Krakow, Poland
bDepartment of Bioorganic Chemistry, Faculty of Pharmacy, Jagiellonian University Medical College, Medyczna 9 Str., 30-688 Krakow, Poland. E-mail: awaszkie@cm-uj.krakow.pl
cDepartment of Pharmaceutical Biochemistry, Faculty of Pharmacy, Jagiellonian University Medical College, Medyczna 9 Str., 30-688 Krakow, Poland
dDepartment of Biochemical Toxicology, Faculty of Pharmacy, Jagiellonian University Medical College, Medyczna 9, 30-688 Krakow, Poland
First published on 11th November 2016
Abstract
New derivatives of N-[(phenoxy)alkyl]- and N-[(phenoxy)ethoxyethyl]aminoalkanols have been synthesized and evaluated for their anticonvulsant activity in maximal electroshock (MES), maximal electroshock seizure threshold (MEST), and pentylenetetrazol (PTZ) tests. Their neurotoxicity was evaluated via rotarod and chimney tests. The compounds exhibiting the most beneficial activity and protection indices were evaluated for analgesic activity using the formalin test for neurogenic pain. They were also evaluated for their influence on cytotoxic activity using in vitro cellular models (HepG2 and CRL-2534 cell lines). Experiments performed using MTT and neutral red cytotoxicity assays showed that all evaluated compounds were safe for normal, glial cells (astrocytes) and did not induce hepatotoxic effects. Based on the results from the in vitro studies, the safety of the evaluated compounds was inferred. The most promising compound in this research was 1-{2-[2-(2,3-dimethylphenoxy)ethoxy]ethyl}piperidin-3-ol hydrochloride. Additionally, in silico metabolism prediction for the compound has been performed.
Introduction
Epilepsy is a set of neurological disorders characterized by hypersynchronous discharges of neurons and expressing itself as recurrent seizures. The disease affects approximately 1% of the human world population, influencing physical, social, and economic aspects of life. Approximately 35% of seizures are considered pharmacoresistant, and this, when added to the toxicity of antiepileptic drugs and their interactions, is a great premise for the search of novel and safe therapies.1Premises for the search of anticonvulsants among aminoalkanols include the activity of known antiarrhythmic drugs, such as propranolol and mexiletine, in the maximal electroshock (MES) test; propranolol is also useful in the treatment of seizures in children.2–4 Moreover, it has been proven that mexiletine exhibits analgesic properties in neurogenic pain, since it significantly and dose-dependently reduces the durations of the first and second phases of the formalin test.5 The mechanism of action in this case, although mexiletine is a non-selective voltage-gated sodium channel blocker, may possibly rely on interaction with δ1 opioid receptors.6
Aroxyalkyl and aroxyethoxyethyl derivatives of aminoalkanols as potential anticonvulsant agents have been a subject of our interest for many years. Among these, we have synthesized and evaluated appropriate derivatives of 4-methylphenol, 2,6-dimethylphenol,7–9 4-chloro-3-methylphenol, and 2-chloro-5-methylphenol,10 as well as 4-chloro-2-methylphenol.11 The most active derivatives in the MES test include structures with substitution in the phenyl ring at positions 2,6-(CH3)2 of the phenyl group, and aminoalkanols in configuration 1,2 (derivatives of colamine), with the linker being ethylene or ethoxyethylene. However, in the case of hydroxyxanthone derivatives as analogs of the studied group of compounds modified by the aroxyl moiety, the propylene linker proved more beneficial.12
Fig. 1 presents structures, activity in MES (mice, ip), and neurotoxicity of reference drugs (mexiletine, propranolol) and compounds: (S)-(+)-2N-[(2,6-dimethylphenoxy)ethyl]aminobutan-1-ol hydrochloride (I),7 (D,L)-trans-2N-[(2,6-dimethylphenoxy)ethyl]aminocyclohexan-1-ol (II),9 and (R)-2-[(2,6-dimethylphenoxy)ethyl]aminopropan-1-ol (III). The last one showed activity in MES, and also in audiogenic seizures and in female mouse electroshock or 6 Hz seizures.11 The ethoxyethyl analog of III—(R)-2-[(2,6-dimethylphenoxy)ethoxyethyl]aminopropan-1-ol—in the form of hydrochloride (IV) also exerts activity in MES.12 The mechanism of action of III and IV is probably binding to σ, 5-HT1A receptors, as well as 5-HT transporters. Compound IV additionally binds to 5-HT2B receptors.13,14 It also exhibits analgesic properties in the formalin test. Fig. 1 also presents compounds V–VII,13,14 since they were found to be active in the MES and/or 6 Hz tests. These compounds differ structurally from the most active ones, but they also possess beneficial properties and can serve as additional references. Especially, VII contains piperidin-4-ol, where nitrogen is contained in the heterocyclic aliphatic ring instead of the secondary amine group, and such a moiety has not been explored sufficiently.
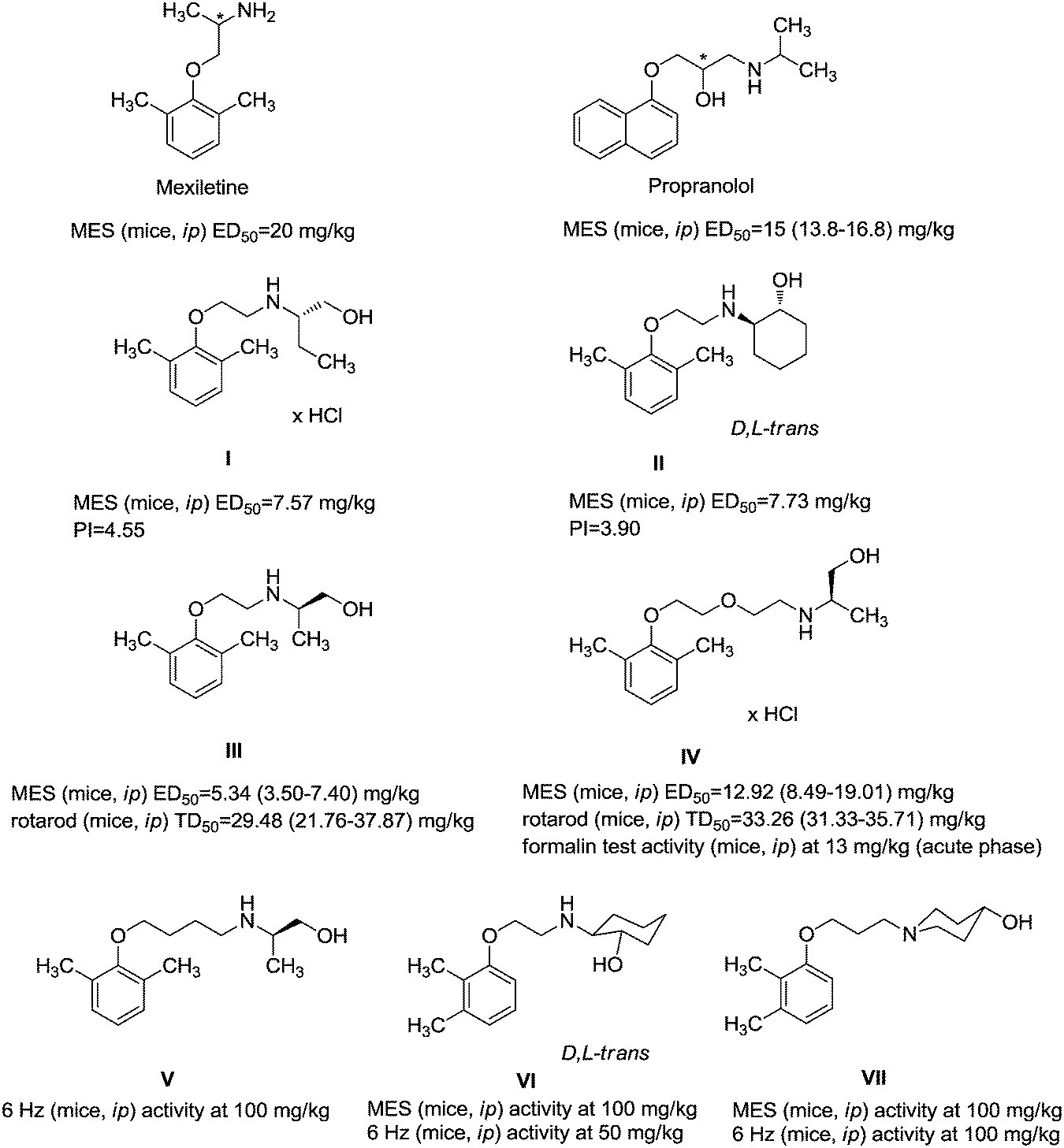 | ||
| Fig. 1 Chemical structures and anticonvulsant activity of reference compounds.2–4,9,13,14 | ||
The purpose of the present study is continuation of our former research. We planned new structures in terms of various substitutions in the phenyl ring (2,3-, 2,4-, 2,6-dimethyl, 2,4,6-trimethyl-, 2-chloro-5-methyl-, 2-chloro-6-methyl), and aminoalkanols (so far beneficial: 2-aminopropan-1-ol, 1-aminopropan-2-ol, 2-aminobutan-1-ol, 1-aminobutan-2-ol, as racemates and when appropriate – enantiomers, D,L-trans-2-aminocyclohexan-1-ol, or 4-aminocyclohexan-1-ol, 2-amino1-phenylethan-1-ol, as well as piperidin-3- or -4-ol), as well as varying the length and type of the linker between the two moieties (2–4 methylene units, possibly containing oxygen in the middle and thus forming an ether moiety).
Results and discussions
Chemistry
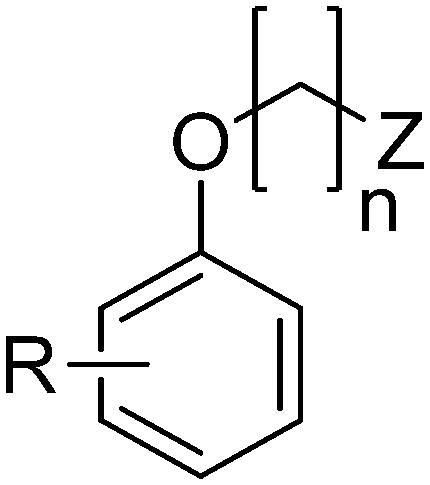
The title compounds consist of a group of phenoxyethyl, phenoxypropyl, phenoxybutyl, and (phenoxyethoxy)ethyl derivatives of chiral or achiral aminoalkanols. Their chemical structures are presented in Table 1. The structures of all compounds were preliminarily examined using the Molinspiration online toolkit,15 in order to estimate their physicochemical parameters, including octanol–water partition coefficient (milog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P), molecular weight, and topological polar surface area (TPSA) (Table 1). None of the estimated values exhibited derogations of the rules for “drug-like” compounds.16,17
P), molecular weight, and topological polar surface area (TPSA) (Table 1). None of the estimated values exhibited derogations of the rules for “drug-like” compounds.16,17
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Compd. | R | n | Z | Configuration | milogP |
Molecular weight | TPSA (Å2) |
| 1 | 2,4-(CH3)2 | 2 |
|
D,L-trans | 3.36 | 263.38 | 41.49 |
| 2 | 2,6-(CH3)2 | 4 |
|
R,S | 2.50 | 251.37 | 41.49 |
| 3 |
|
R,S | 3.35 | 265.40 | 41.49 | ||
| 4 |
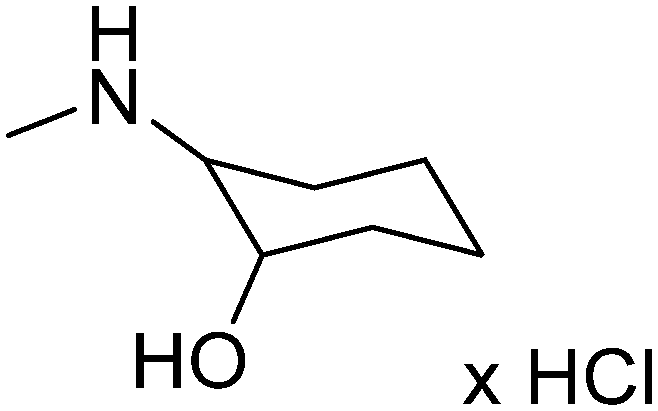
|
D,L-trans | 3.88 | 291.44 | 41.49 | ||
| 5 |
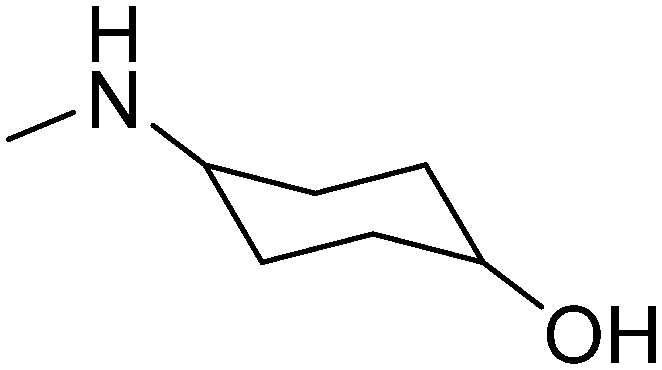
|
trans | 3.41 | 291.44 | 41.49 | ||
| 6 | 2,4,6-(CH3)3 | 3 |
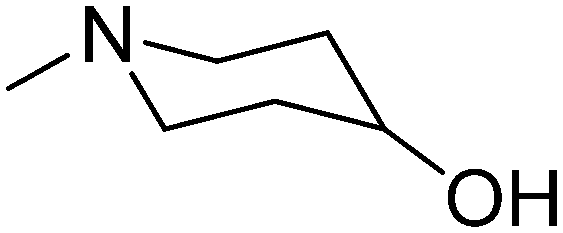
|
2.52 | 277.41 | 32.70 | |
| 7 | 2-Cl, 5-CH3 | 2 |
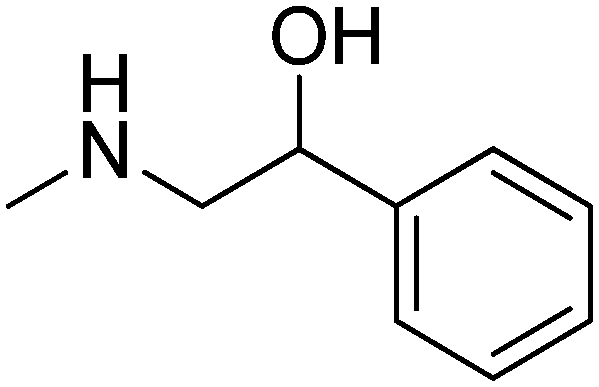
|
R,S | 3.30 | 305.81 | 41.49 |
| 8 |
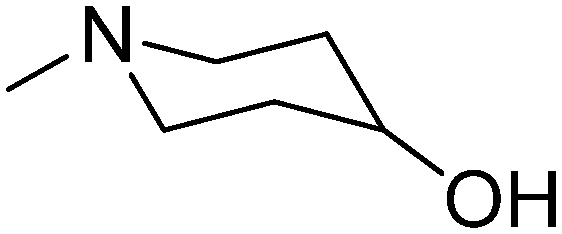
|
2.10 | 269.77 | 32.70 | |||
| 9 |

|
trans | 3.12 | 283.80 | 41.49 | ||
| 10 | 4 |
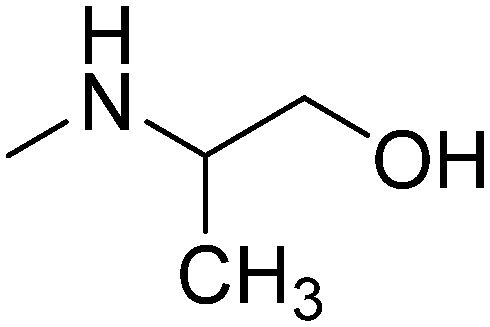
|
R,S | 3.07 | 271.79 | 41.49 | |
| 11 |
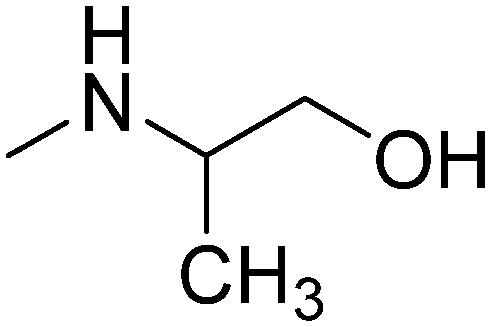
|
R-(−) | 3.07 | 271.79 | 41.49 | ||
| 12 |
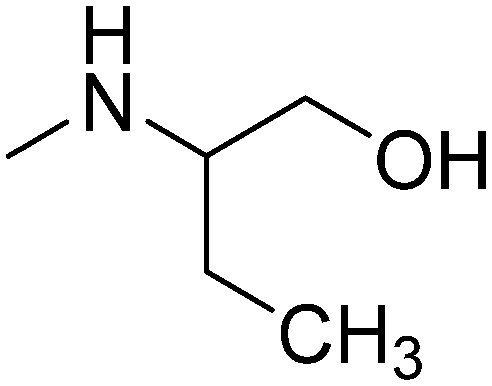
|
R,S | 3.61 | 285.81 | 41.49 | ||
| 13 |
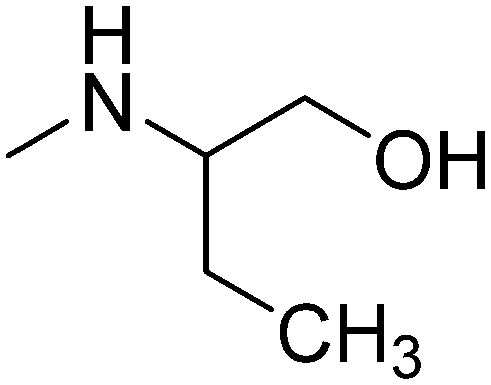
|
R-(−) | 3.61 | 285.81 | 41.49 | ||
| 14 |
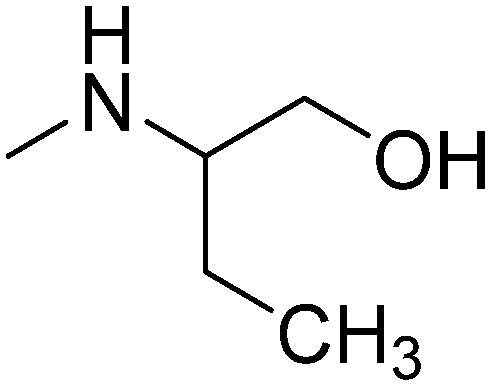
|
S-(+) | 3.61 | 285.81 | 41.49 | ||
| 15 | 2-Cl, 6-CH3 |
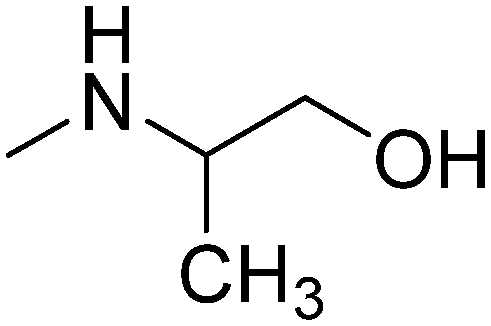
|
R,S | 3.05 | 271.79 | 41.49 | |
| 16 |
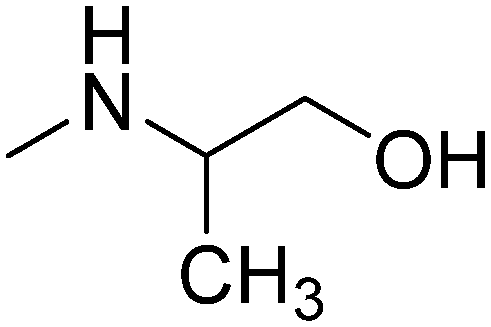
|
S-(+) | 3.05 | 271.79 | 41.49 | ||
| 17 |
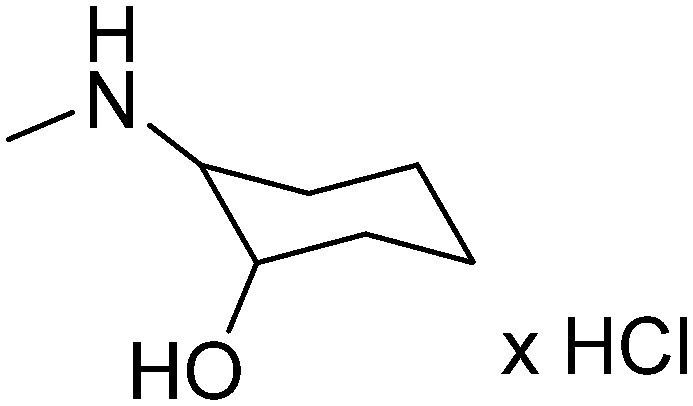
|
D,L-trans | 4.11 | 311.85 | 41.49 | ||
| 18 |
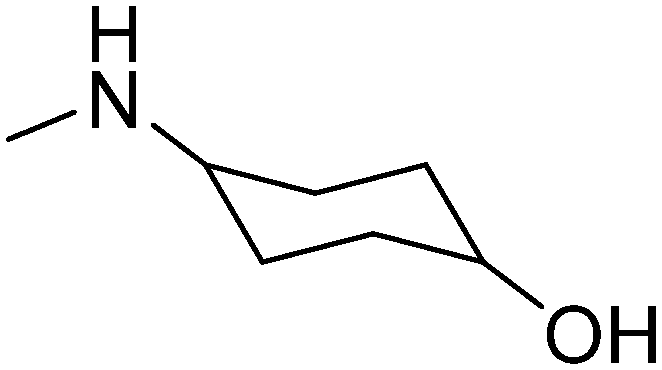
|
trans | 3.64 | 311.85 | 41.49 | ||
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Compd. | R | Z | Configuration | milogP |
Molecular mass | TPSA (Å2) | |
| 19 | 2,3-(CH3)2 |
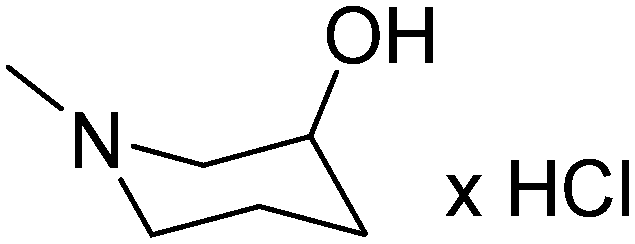
|
2.47 | 293.41 | 41.93 | ||
| 20 |
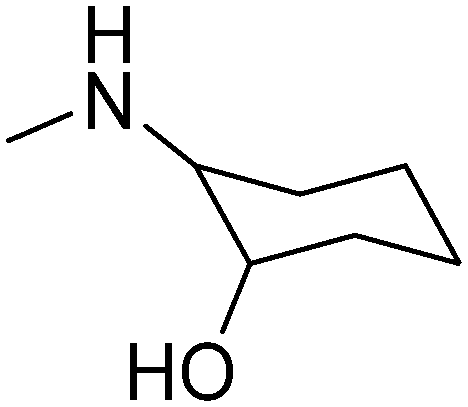
|
D,L-trans | 3.23 | 307.43 | 50.72 | ||
| 21 | 2,4-(CH3)2 |
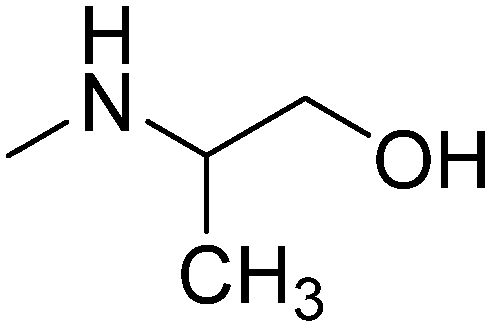
|
R,S | 2.19 | 267.37 | 50.72 | |
| 22 |
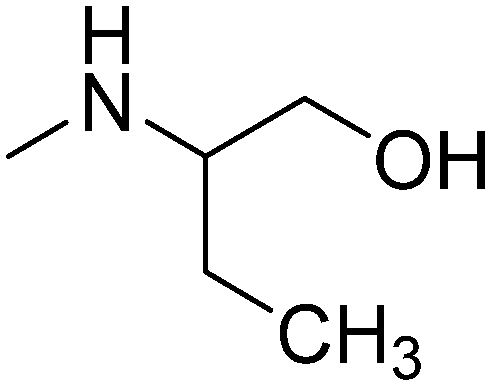
|
R-(−) | 2.73 | 281.40 | 50.72 | ||
| 23 | 2,6-(CH3)2 |
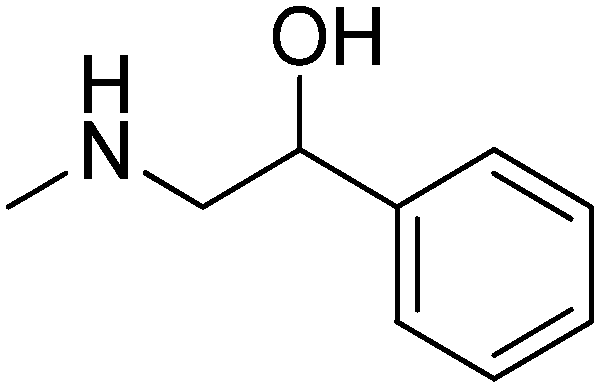
|
R,S | 2.93 | 329.44 | 50.72 | |
| 24 | 2-Cl, 5-CH3 |
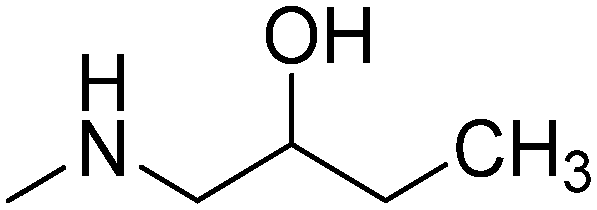
|
R,S | 2.61 | 301.81 | 50.72 | |
| 25 |
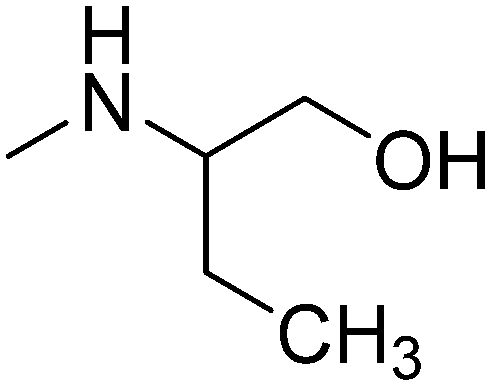
|
R,S | 2.96 | 301.81 | 50.72 | ||
| 26 |
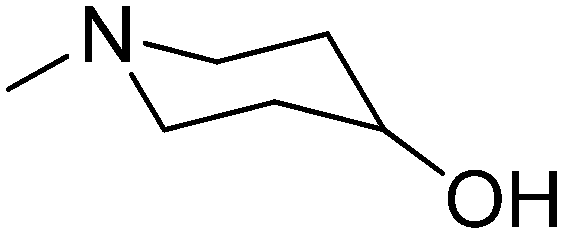
|
2.00 | 313.82 | 41.93 | |||
| 27 | 2-Cl, 6-CH3 |
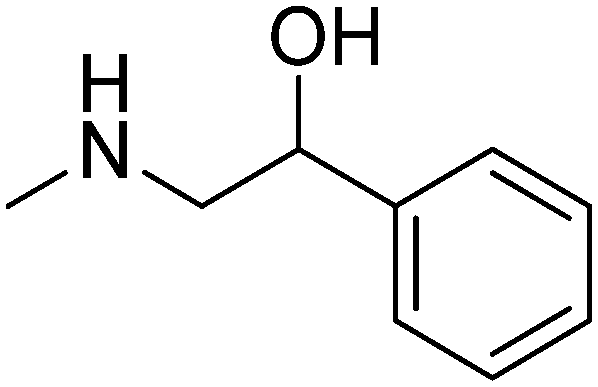
|
R,S | 3.19 | 349.86 | 50.72 | |
| 28 |
|
R-(−) | 3.19 | 349.86 | 50.72 | ||
All compounds were achieved by means of multistep chemical synthesis (Scheme 1), according to formerly published procedures.7,18,19 The first step constituted obtaining substituted phenoxyalkyl halide (chloride or bromide) or phenoxyethoxyethyl bromide, which were subsequently used in the N-alkylation of the appropriate aminoalkanol for achievement of each title compound in the form of a base. Compounds 4, 17, and 19 were achieved as salts by saturation with gaseous HCl in acetone, while other compounds were finally characterized as bases.
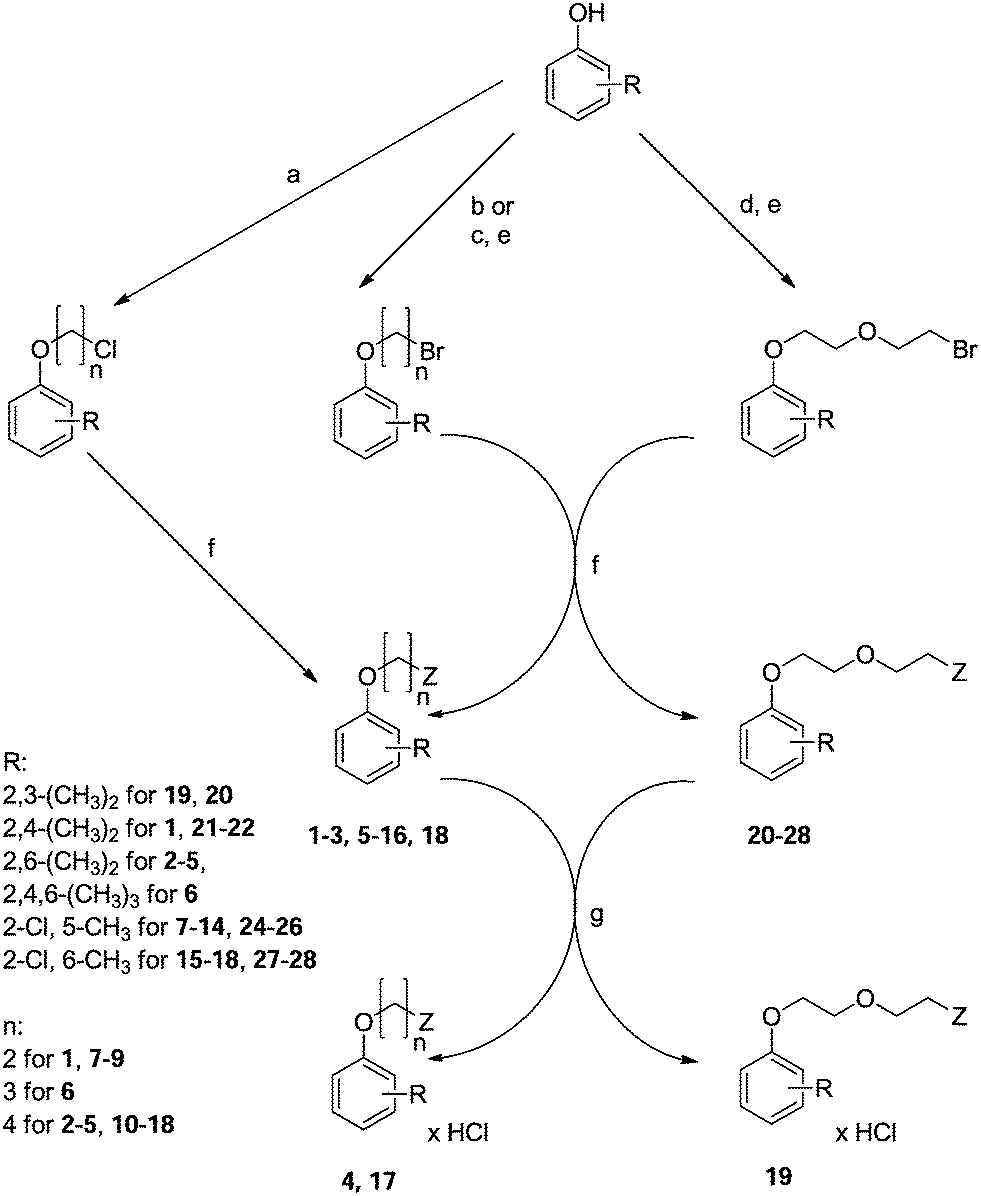 | ||
| Scheme 1 Synthesis of compounds 1–28 (Table 1). Reagents: (a) Br–(CH2)n–Cl, K2CO3, TEBA, acetone; (b) Br–(CH2)n–Br, NaOH; (c) Cl–(CH2)n–OH, K2CO3, acetone/EtOH; (d) Cl–CH2–CH2–O–CH2–CH2–OH, K2CO3, acetone/EtOH; (e) PBr3; (f) Z (aminoalkanol) as in Table 1, K2CO3, toluene or TEA, DMF; (g) HClgas, acetone. | ||
Pharmacology
The title compounds have been subject to preliminary screening of anticonvulsant activity via the maximal electroshock seizure (MES) test. Among the derivatives of N-[(phenoxy)alkyl]aminoalkanols 1–18, the most active compounds are 1: D,L-trans-2-[2-(2,4-dimethylphenoxy)ethyl]aminocyclohexan-1-ol, which is a 2,4-dimethyl analog of reference compound II, 2: (R,S)-1-[4-(2,6-dimethylphenoxy)butyl]aminopropan-2-ol, 6: 1-[3-(2,4,6-trimethylphenoxy)propyl]piperidin-4-ol, 15: (R,S)-2-[4-(2-chloro-6-methylphenoxy)butyl]aminopropan-1-ol, and 18: D,L-trans-4-[4-(2-chloro-6-methylphenoxy)butyl]aminocyclohexan-1-ol. Among these, the most favourable ED50 and protection indices PI are observed for 6 and 15 (Table 2).| Compd. | Dose (mg kg−1) | MESa | Lethality in MES | Rotarodb | ED50 (confidence interval) TD50 (confidence interval) (mg kg−1) | PIc |
|---|---|---|---|---|---|---|
| MES – maximal electroshock seizure test. a No. of animals protected/no. of animals tested. b TD50 (mg kg−1) or number of mice in which motor impairment was observed/number of mice tested (10 rpm). c PI = TD50/ED50. | ||||||
| Control | — | 0/6 | 5/6 | — | ||
| 1 | 100 | 6/6 | 0/6 | ED50 = 31.80 (24.12–41.93) TD50 = 76.78 (63.19–93.29) | 2.41 | |
| 60 | 3/6 | 1/6 | ||||
| 30 | 4/6 | 1/6 | ||||
| 10 | 0/6 | 6/6 | ||||
| 2 | 100 | 4/4 | 0/4 | ED50 = 27.11 (22.38–32.83) TD50 = 43.29 (34.19–54.81) | 1.60 | |
| 30 | 4/6 | 0/6 | ||||
| 25 | 2/6 | 0/6 | ||||
| 20 | 1/6 | 1/6 | ||||
| 3 | 30 | 4/6 | 2/6 | — | ||
| 4 | 30 | 4/6 | 1/6 | — | ||
| 5 | 100 | 5/6 | 6/6 | — | ||
| 60 | 0/6 | 3/6 | ||||
| 6 | 100 | 6/6 | 0/6 | ED50 = 20.87 (12.56–34.69) TD50 = 81.95 (70.45–95.32) | 3.93 | |
| 30 | 4/6 | 1/6 | ||||
| 20 | 3/6 | 1/6 | ||||
| 10 | 1/6 | 3/6 | ||||
| 7 | 100 | 1/4 | 1/4 | 2/4 | ||
| 8 | 100 | 4/4 | 0/4 | 4/4 | ||
| 9 | 100 | — | 3/4 | TOX | ||
| 10 | 100 | 4/4 | 0/4 | 4/4 | ||
| 30 | 0/4 | 0/4 | 0/4 | |||
| 11 | 100 | 4/4 | 0/4 | 4/4 | ||
| 30 | 0/4 | 0/4 | 0/4 | |||
| 12 | 100 | 4/4 | 0/4 | 4/4 | ||
| 30 | 1/4 | 0/4 | 0/4 | |||
| 13 | 100 | 4/4 | 0/4 | 4/4 | ||
| 30 | 1/4 | 0/4 | 0/4 | |||
| 14 | 100 | 4/4 | 0/4 | 4/4 | ||
| 30 | 0/4 | 0/4 | 1/4 | |||
| 15 | 100 | 4/4 | 0/4 | ED50 = 19.06 (13.84–26.25) TD50 = 45.66 (35.84–58.17) | 2.40 | |
| 30 | 5/6 | 1/6 | ||||
| 20 | 3/6 | 0/6 | ||||
| 15 | 2/6 | 0/6 | ||||
| 16 | 100 | — | 3/4 | TOX | ||
| 17 | 100 | — | 4/4 | TOX | ||
| 18 | 100 | 6/6 | 0/6 | ED50 = 41.11 (32.54–51.94) TD50 = 49.99 (44.75–55.84) | 1.22 | |
| 60 | 3/6 | 3/6 | ||||
| 30 | 2/6 | 1/6 | ||||
| 19 | 100 | 6/6 | 0/6 | ED50 = 68.71 (63.95–73.82) TD50 = 121.64 (116.53–126.99) | 1.77 | |
| 80 | 5/6 | 0/6 | ||||
| 60 | 4/6 | 0/6 | ||||
| 30 | 2/6 | 1/6 | ||||
| 20 | 100 | 6/6 | 0/6 | ED50 = 32.87 (26.21–41.21) TD50 = 47.39 (38.69–58.04) | 1.44 | |
| 60 | 5/6 | 0/6 | ||||
| 30 | 3/6 | 3/6 | ||||
| 10 | 0/6 | 1/6 | ||||
| 21 | 100 | 6/6 | 0/6 | ED50 = 27.11 (22.38–32.83) TD50 = 108.91 (101.81–116.50) | 4.02 | |
| 30 | 4/6 | 0/6 | ||||
| 25 | 2/6 | 0/6 | ||||
| 20 | 1/6 | 1/6 | ||||
| 22 | 100 | 4/4 | 0/4 | 3/4 | ||
| 30 | 0/4 | 0/4 | 0/4 | |||
| 23 | 100 | 4/4 | 0/4 | ED50 = 13.03 (11.58–14.65) TD50 = 36.00 (31.43–41.22) | 2.76 | |
| 20 | 6/6 | 0/6 | ||||
| 15 | 2/6 | 0/6 | ||||
| 10 | 1/6 | 0/6 | ||||
| 24 | 100 | 4/4 | 0/4 | 4/4 | ||
| 30 | 0/4 | 0/4 | 1/4 | |||
| 25 | 100 | 4/4 | 0/4 | 4/4 | ||
| 30 | 1/4 | 0/4 | 0/4 | |||
| 26 | 100 | 4/4 | 0/4 | 3/4 | ||
| 30 | 0/4 | 2/4 | 0/4 | |||
| 27 | 100 | 6/6 | 0/6 | ED50 = 23.94 (21.29–26.92) TD50 = 103.04 (67.57–157.14) | 4.30 | |
| 30 | 5/6 | 0/6 | ||||
| 25 | 2/6 | 0/6 | ||||
| 20 | 1/6 | 0/6 | ||||
| 28 | 100 | — | 2/4 | TOX | ||
| Phenytoin | ED50 = 6.65 (5.42–8.16) TD50 = 68.73 (60.79–77.71) | 10.34 | ||||
| Lacosamide | ED50 = 10.40 (9.43–11.47) TD50 = 42.52 (40.93–44.18) 15 min. after administration TD50 = 46.20 (44.48–48.00) 30 min. after administration | 4.09 | ||||
| 4.44 | ||||||
Among the N-[(phenoxyethoxy)ethyl]aminoalkanols, ED50 values were derived for 19–21, 23, and 27: 1-{2-[2-(2,3-dimethylphenoxy)ethoxy]ethyl}piperidin-3-ol hydrochloride (19), D,L-trans-2-{2-[2-(2,3-dimethylphenoxy)ethoxy]ethyl}aminocyclohexan-1-ol (20), and (R,S)-2-{2-[2-(2,4-dimethylphenoxy)ethoxy]ethyl}aminopropan-1-ol (21), as well as (R,S)-2-{2-[2-(2,6-dimethylphenoxy)ethoxy]ethyl}amino-1-phenylethan-1-ol (23), and (R,S)-2-{2-[2-(2-chloro-6-methylphenoxy)ethoxy]ethyl}amino-1-phenylethan-1-ol (27).
ED50s of compounds have been presented with confidence intervals of active doses. In the cases of 1–2, 6, 15, 20–21, and 27, these CIs are largely covered, which suggests similar pharmacological profiles. This is not surprising, given such structural similarity, since we can observe that all these compounds contain one methyl substituent in position 2 of the phenyl ring and a second substituent – methyl in positions 4 or 6 or chlorine in position 6. An additional methyl (in 2,4,6-trimethyl derivative 6) does not appear to influence the activity. Within the aminoalkanol moieties, we can observe derivatives of colamine. In all cases, the linker used represents all tested possibilities – ethylene, propylene, and tetramethylene, as well as ethoxyethylene group.
It is worth commenting on the protection indices (PI) of the active compounds, since it is not only their activity that influences the behavior of the animals, but also the neurotoxicity observed in the rotarod test and quantified by TD50 (Table 2). The most promising ratio between TD50 and ED50 was observed for compound 6, followed by 1, 15, 21, 23, and finally 19; this ratio, although not the highest, is still comparable to that of valproic acid (1.7).4
In the MEST test, two compounds, 19 and 27, caused a significantly elevated electroconvulsive threshold (ECT) in comparison to vehicle-treated mice. Compound 19 at a dose of 30 mg kg−1 increased the ECT by 75% (p < 0.001), whereas compound 27 given at a dose of 10 mg kg−1 increased it by 57% (p < 0.001). Compounds 1 and 6 only slightly elevated the ECT (by 24 and 12%, respectively). Compounds 20 and 21 were not able to elevate the ECT at the tested doses. Reference drugs phenytoin (2.5 mg kg−1) and lacosamide (10 mg kg−1) increased the ECT by 23% (p < 0.001) and above 25 mA, respectively (Table 3).
| Compd. | Dose (mg kg−1) | CS50 (confidence interval) (mA)a | Effect (%) |
|---|---|---|---|
| a Statistical analysis: unpaired t test **p < 0.01, ***p < 0.001, ****p < 0.0001; – lack of effect. | |||
| Control | — | — | — |
| 1 | 20 | 9.33 (7.65–11.38) | 23.90 |
| Control: 7.53 (6.45–8.79) | |||
| 6 | 10 | 9.15 (7.76–10.78) | 12.55 |
| Control: 8.13 (7.41–8.92) | |||
| 19 | 30 | 13.18 (12.12–14.33)*** | 75.03 |
| Control: 7.53 (6.45–8.79) | |||
| 20 | 20 | 7.01 (6.26–16.72) | — |
| Control: 7.53 (6.45–8.79) | |||
| 21 | 10 | 6.76 (3.77–12.11) | — |
| Control: 8.13 (7.41–8.92) | |||
| 27 | 10 | 11.09 (6.14–19.20)*** | 56.64 |
| Control: 7.08 (6.48–7.73) | |||
| Phenytoin | 2.5 | 7.46 (6.66–8.34)** | 23.31 |
| Control: 6.05 (5.20–7.03) | |||
| Lacosamide | 10 | >25**** | >413.22 |
| Control: 6.05 (5.20–7.03) | |||
The scPTZ test employs chemically induced clonic seizures and is related to human generalized absence seizures. The most beneficial activity in the scPTZ seizures was observed in the case of compound 21, which, at a dose of 100 mg kg−1, significantly prolonged the latency time to the first seizure episode by 117% (p < 0.05). Moreover, compound 1 delayed the onset of seizures by 58%, but this result was not statistically significant. Ethosuximide – the reference drug – delayed the onset of seizures at a dose of 100 mg kg−1 by 140% (p < 0.001), and at a dose of 50 mg kg−1 by 124% (p < 0.01) (Table 4).
| Compound | Dose (mg kg−1) | Latency of tonic seizures ± SEM (s)a | Effect (%) |
|---|---|---|---|
| a Statistical analysis: univariate ANOVA and Dunnett's post hoc test; *p < 0.05, **p < 0.01, ***p < 0.001; − lack of effect. | |||
| Control | — | — | — |
| 1 | 100 | 921 ± 361.9 | 57.71 |
| Control: 584 ± 145.8 | |||
| 30 | 576.4 ± 73.69 | 2.42 | |
| Control: 562.8 ± 83.17 | |||
| 6 | 100 | 275.4 ± 45.14 | — |
| Control: 584 ± 145.8 | |||
| 19 | 100 | 564.8 ± 197 | — |
| Control: 584 ± 145.8 | |||
| 20 | 80 | 596 ± 224 | 2.05 |
| Control: 584 ± 145.8 | |||
| 21 | 100 | 1269 ± 220.1* | 117.23 |
| Control: 584 ± 145.8 | |||
| 30 | 575.8 ± 66.22 | 2.31 | |
| Control: 562.8 ± 83.17 | |||
| 27 | 100 | 219.6 ± 12.77 | — |
| Control: 584 ± 145.8 | |||
| Ethosuximide | 100 | 1470 ± 166.2*** | 139.06 |
| Control: 614.9 ± 61.92 | |||
| 50 | 1377 ± 181.5** | 123.94 | |
| Control: 614.9 ± 61.92 | |||
Antinociceptive activity was evaluated via the formalin test, which is typically used as a tonic model of nociception. For each selected compound, the respective ED50 value from the MES test was used to establish its analgesic activity. In the first (neurogenic) phase of the test, five compounds (1, 6, 19, 20, and 27) only slightly reduced the duration of the licking response by 9–27%. In the second (late) phase of the formalin test, four compounds (1, 19, 20, and 21) revealed a prominent antinociceptive activity. They significantly reduced the nocifensive response by 46–83%. The precise results are summarized in Table 5.
| Compd. | Dose (mg kg−1) | Time of paw licking ± SEM (s) (phase I)a | Time of paw licking ± SEM (s) (phase II)a |
|---|---|---|---|
| a Statistical analysis: unpaired t test; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. | |||
| Control | — | 43.89 ± 6.64 | 79.89 ± 11.45 |
| 1 | 32 | 35.75 ± 5.86 (18.55% of effect) | 43 ± 10.99 (46.16% of effect)* |
| 6 | 21 | 32 ± 9.32 (27.01% of effect) | 76 ± 126.8 (4.87% of effect) |
| 19 | 69 | 37.86 ± 7.08 (13.74% of effect) | 28.29 ± 14.05 (64.59% of effect)* |
| 20 | 33 | 32.71 ± 8.32 (25.47% of effect) | 23.86 ± 8.33 (70.13% of effect)** |
| 21 | 27 | 67.25 ± 13.04 (lack of effect) | 13.25 ± 2.7 (83.41% of effect)*** |
| 27 | 24 | 40.13 ± 4.29 (8.57% of effect) | 72.63 ± 15.17 (8.80% of effect) |
Among the compounds active in MES, compounds 19–21 (ethoxyethyl derivatives) were the most active. All three are dimethylphenol derivatives (2,3 in the cases of 19 and 20 and 2,4 in the case of 21). They are also derivatives of piperidin-3-ol, D,L-trans-2-aminocyclohexan-1-ol, or R,S-2-aminopropan-1-ol, respectively. Compound 1 is a derivative of 2,4-dimethylphenol and D,L-trans-2-aminocyclohexan-1-ol with the shortest linker tested – ethylene.
Among the tested compounds, 21 and 27 at doses equal to the respective ED50 values obtained in the MES test significantly reduced the number of crossings registered with photoresistor actometers by 69 (p < 0.01), and 44% (p < 0.05), respectively. The results indicated that these compounds possess sedative properties. The other tested compounds 1, 6, 19, and 20 did not significantly influence locomotor activity in mice. None of the tested compounds affected motor coordination in the chimney test (Table 6).
| Compd. | Spontaneous activity | Chimney test | |||
|---|---|---|---|---|---|
| Dose (mg kg−1) | Activity ± SEMa | Effect (%) | Time of escaping from the chimney ± SEM (s) | Animals exhibiting loss of coordination (%) | |
| a Statistical analysis: unpaired t test; *p < 0.05, **p < 0.01. | |||||
| Control | — | 1402 ± 201.2 | — | 8.87 ± 1.79 | — |
| 1 | 32 | 1287 ± 295.5 | 8.20 | 4.75 ± 0.67 | 0 |
| 6 | 21 | 1125 ± 238.8 | 19.76 | 6.5 ± 1.68 | 0 |
| 19 | 69 | 1192 ± 213.0 | 14.98 | 4.75 ± 1.76 | 0 |
| 20 | 33 | 1354 ± 299.5 | 3.42 | 4.13 ± 0.64 | 0 |
| 21 | 27 | 519.5 ± 137.5** | 68.92 | 6.0 ± 2.35 | 0 |
| 27 | 24 | 779.7 ± 175.4* | 44.39 | 3.5 ± 0.65 | 0 |
Taking into consideration the structural aspects of the studied compounds, substitution in the phenyl ring with methyl moieties in positions 2,4, 2,6, and 2,4,6, as well as 2-chloro-6-methyl substitution, are confirmed to be beneficial for anticonvulsant activity. Analgesic activity was the most promising in the case of 2,3- or (2,4-dimethylphenoxy)ethoxyethyl derivatives (compounds 19–21), and this suggests that the ethoxyethyl linker is beneficial with respect to the anticonvulsant and analgesic activity. In case of 2,4-dimethyl and 2-chloro-6-methyl derivatives, sedative properties have been observed (i.e. (R,S)-2-{2-[2-(2,4-dimethylphenoxy)ethoxy]ethyl}aminopropan-1-ol (21) and (R,S)-2-{2-[2-(2-chloro-6-methylphenoxy)ethoxy]ethyl}amino-1-phenylethan-1-ol (27), respectively).
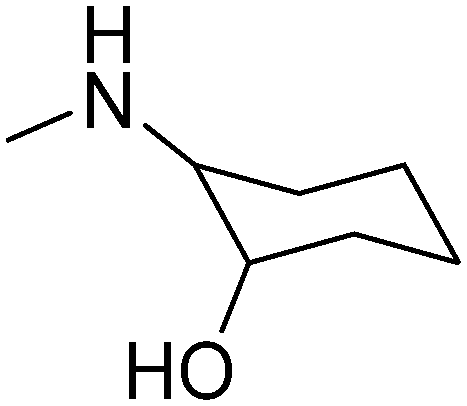
The most beneficial in vivo pharmacological profile was observed for four compounds (1, 6, 19, and 21). It is worth mentioning that compound 1 is a D,L-trans-2-aminocyclohexan-1-ol derivative similar to reference compound II, which may contribute to the activity. Compound 21 is a derivative of R,S-2-aminopropan-1-ol, similar to III and IV, which are R enantiomer derivatives. Compounds 6 and 19 are derivatives of piperidin-3- or −4-ol, respectively. These aminoalkanols were also present in the structures of other active compounds discovered within our former research, including reference compound VII.14
Analysis of cytotoxic activity of compounds 1, 6, 19, and 21 using in vitro cellular models
Data on the hepatotoxicity of some antiepileptic drugs (e.g. valproic acid,20,21 pregabalin22,23) became the premise for the determination of the potential hepatotoxicity of selected compounds possessing anticonvulsant activity. Compounds 1, 6, 19, and 21 were evaluated for potential cytotoxicity against human cancer cells (HepG2). Results obtained from two independent viability tests investigating different mechanisms of cell-mediated cytotoxicity clearly show that none of the analyzed compounds show a cytotoxic effect on hepatoma cells (Fig. 2 and 4). Performed analyses proved that the tested compounds do not affect cellular metabolism (MTT) and do not have an influence on endosomal activity – an important physiological process occurring in cells (neutral red uptake). What is more, they are completely safe with respect to normal cells [astrocytes (glial cells)] derived from the nervous system (Fig. 5). In addition, the cytotoxicity effect of a chemotherapeutic agent, doxorubicin (anthracycline antibiotic), on HepG2 cells was evaluated as a positive control (Fig. 3). Doxorubicin was dosed at the same concentration as the evaluated compounds (1, 6, 19, and 21) and exhibited a strong cytotoxic effect on HepG2 cells.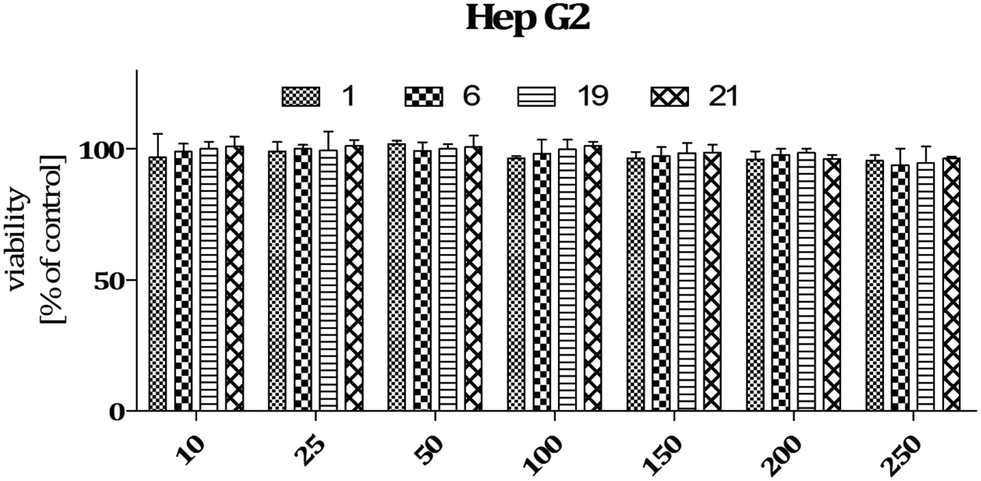 | ||
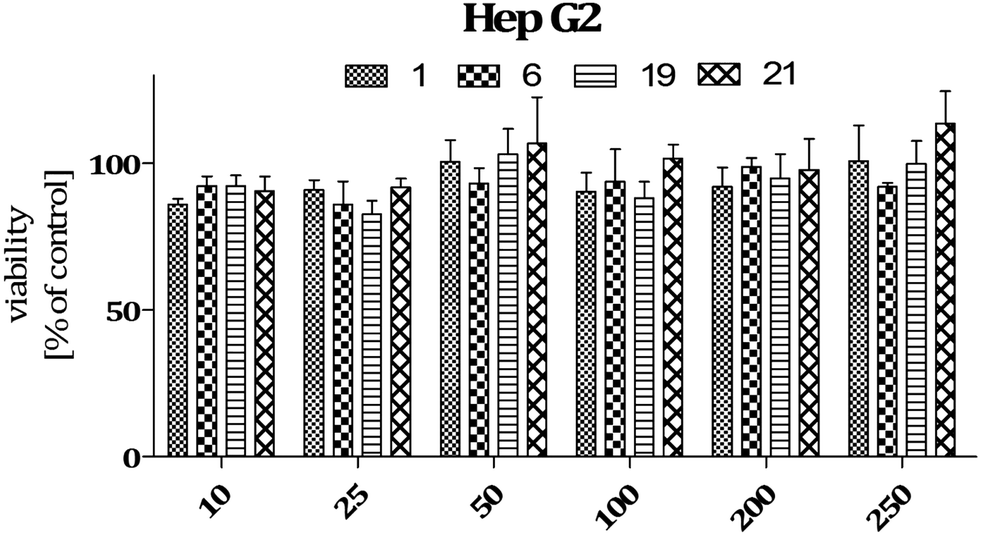
| Fig. 2 Viability of HepG2 cells incubated in the presence of selected compounds in the concentration range 10–250 μM. The graph shows results from MTT assay expressed as a percentage of the control conditions ± SEM. Three independent experiments were performed. | ||
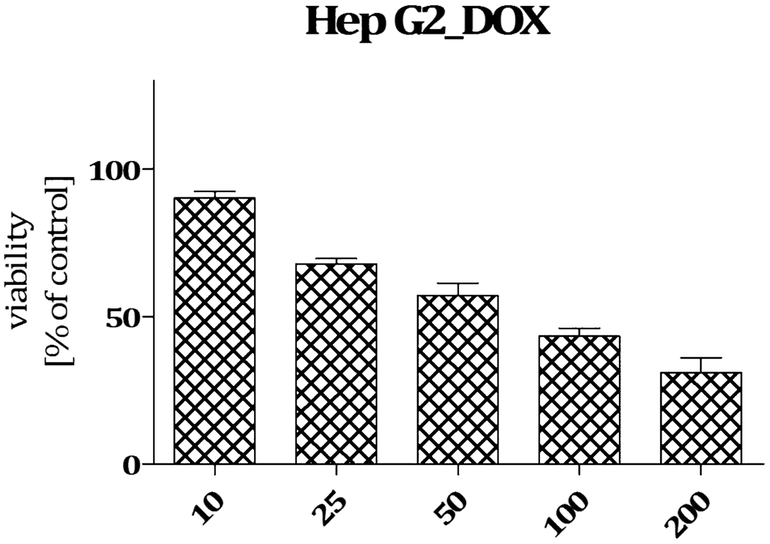 | ||
| Fig. 3 Viability of HepG2 cells incubated in the presence of doxorubicin (DOX) in the concentration range 10–200 μM. The graph shows results from MTT assay expressed as a percentage of the control conditions ± SEM. Three independent experiments were performed. | ||
 | ||
| Fig. 4 Viability of HepG2 cells incubated in the presence of selected compounds in the concentration range 10–250 μM. The graph shows results from neutral red assay expressed as a percentage of the control conditions ± SEM. Three independent experiments were performed. | ||
 | ||
| Fig. 5 Viability of CRL-2541 cells (astrocytes) incubated in the presence of selected compounds in the concentration range 10–250 μM. The graph shows results from MTT assay expressed as a percentage of the control conditions ± SEM. Three independent experiments were performed. | ||
The potential cytotoxic effect of compound 19 was examined by LDH release and MTT reduction assays. Exposure of SH-SY5Y cells to compound 19 at concentrations from 10−8 to 10−4 M for 72 hours did not change the LDH-release (Fig. 6). The test compound at concentrations from 10−8 to 10−4 M also had no significant effect on MTT reduction, although an increasing trend was observed (Fig. 7). Addition of hydrogen peroxide alone significantly damaged cells by increasing above twofold the release of LDH and attenuated MTT reduction by approx. 20% (Fig. 8 and 9). In the highest examined concentration, 10−5 M, compound 19 statistically significantly potentiated the damaging effects of hydrogen peroxide on LDH release, while at lower concentrations it had no effect on this parameter. At concentrations of 10−6 and 10−5 M, the tested compound significantly potentiated the H2O2-induced decrease in MTT reduction. Triton, used in all conducted assays as a positive control, strongly increased LDH release and decreased MTT reduction.
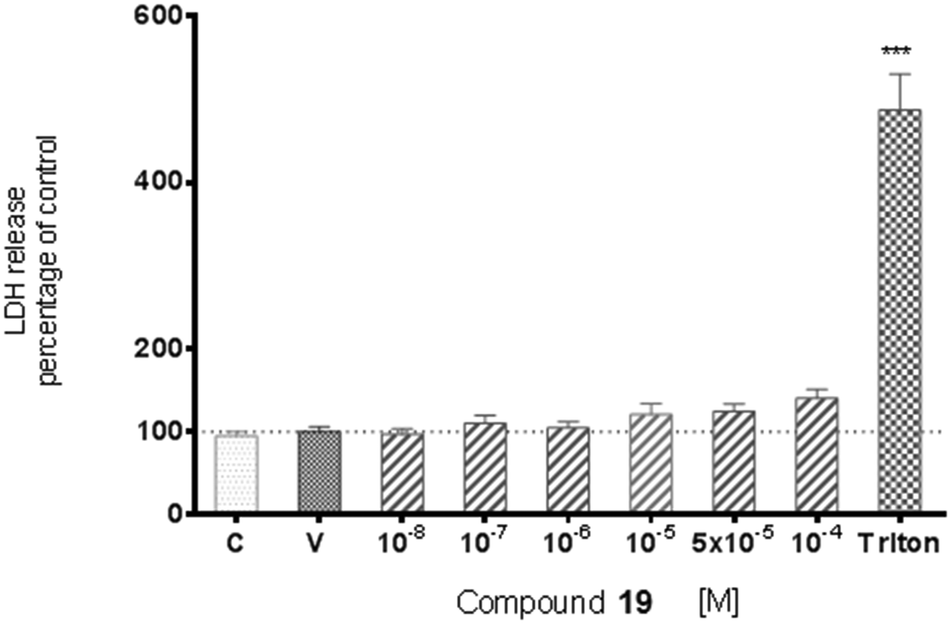 | ||
| Fig. 6 The effect of compound 19 on basal lactate dehydrogenase (LDH) release from SH-SY5Y cells. Results are shown as a percentage of control cells incubated with vehicle alone. The significance of differences between the means was evaluated by Dunnett's test following a one-way analysis of variance; ***p < 0.001 vs. culture with vehicle. C-culture without vehicle, V-culture with appropriate vehicle. | ||
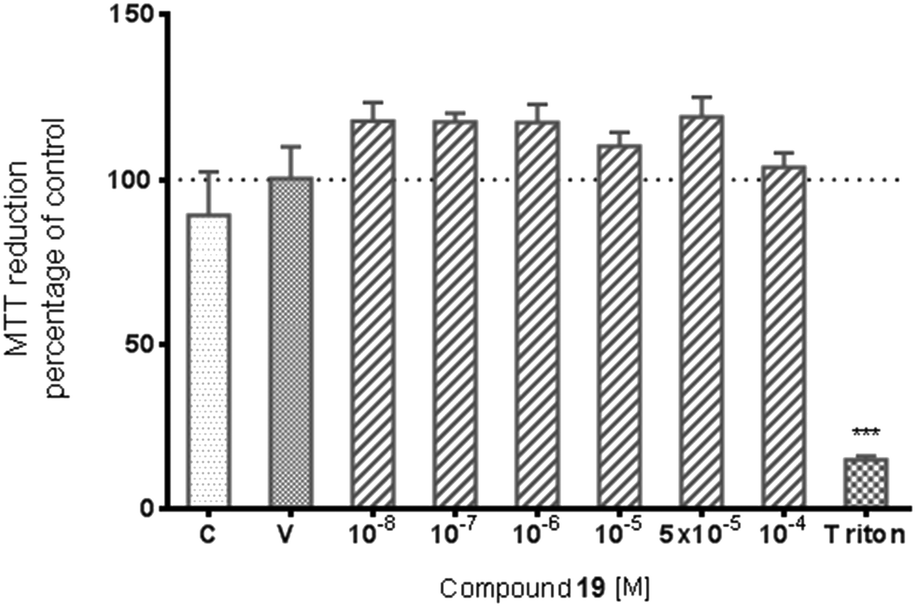 | ||
| Fig. 7 The effect of compound 19 on basal MTT reduction in SH-SY5Y cells. Results are shown as a percentage of control cells incubated with vehicle alone. The significance of differences between the means was evaluated by Dunnett's test following a one-way analysis of variance; ***p < 0.001 vs. culture with vehicle. C-culture without vehicle, V-culture with appropriate vehicle. | ||
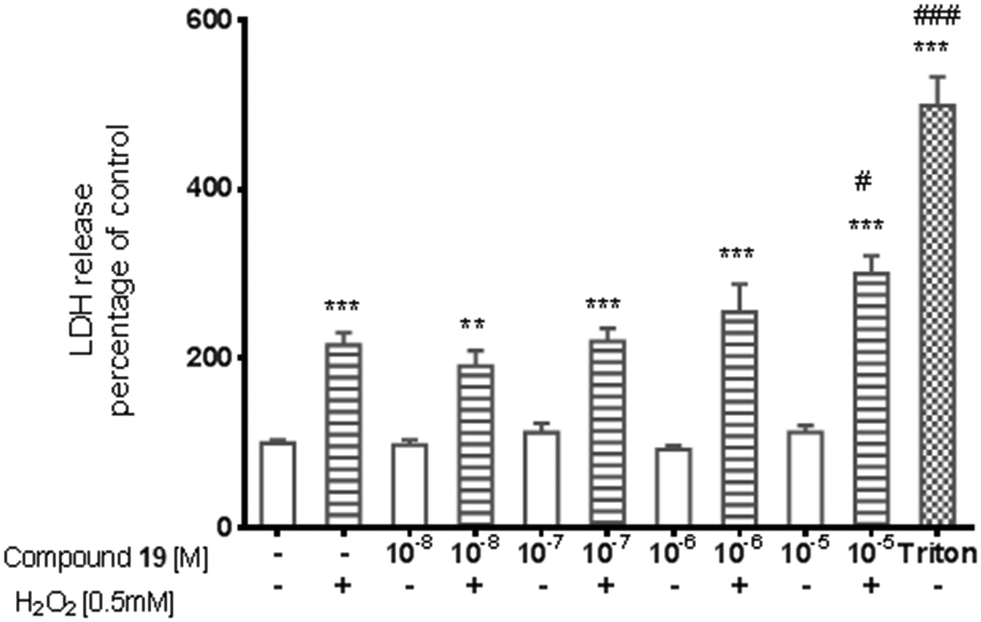 | ||
| Fig. 8 The effect of compound 19 on H2O2-induced lactate dehydrogenase (LDH) release from SH-SY5Y cells. The significance of differences between the means was evaluated by Dunnett's test following a one-way analysis of variance; **p < 0.01; ***p < 0.001 vs. culture with vehicle only; #p < 0.05; ###p < 0.001 vs. cells treated with H2O2. | ||
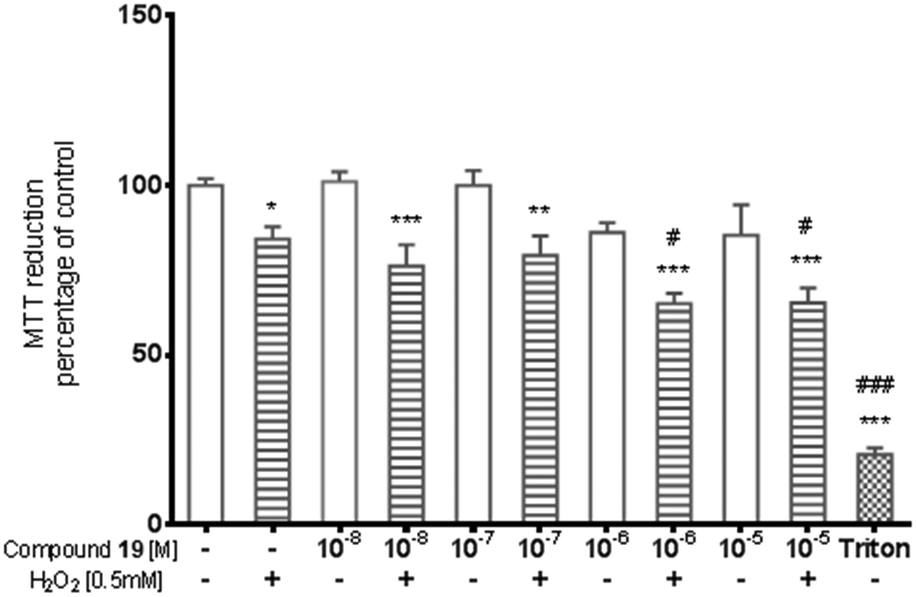 | ||
| Fig. 9 The effect of compound 19 on H2O2-induced MTT reduction in SH-SY5Y cells. The significance of differences between the means was evaluated by Dunnett's test following a one-way analysis of variance; *p < 0.05; **p < 0.01; p < 0.001 vs. culture with vehicle only; #p < 0.05; ###p < 0.001 vs. cells treated with H2O2. | ||
The potential cytotoxic activity of compound 19 was also tested in SH-SY5Y cell culture. This cell line is of human origin, possesses a dopaminergic phenotype, and is one of the most broadly used models for screening of the putative neurotoxic and neuroprotective activity of a wide range of chemicals.24,25 Similarly, addition of hydrogen peroxide to cell medium, as the source of detrimental reactive oxygen species, is a well-validated and frequently used model for oxidative stress. Moreover, both in the pathogenesis of neurodegenerative diseases and in the mechanism of action of most neurotoxic substances, oxidative stress plays a major role. The present study showed that the test compound at concentrations up to 10−4 M did not decrease the SH-SY5Y cell viability, assessed by MTT reduction assay, and did not produce a cytotoxic effect, defined by LDH release. Considering the possible use of this compound in the future in clinics, its lack of adverse action on the integrity of membranes and viability of cells with neuronal phenotypes is important. Neuroprotection is a favorable effect of antiepileptic drugs, because seizures can lead to nerve cell damage. However, compound 19, in the model used in the present study, did not reduce hydrogen-peroxide-induced cell damage. This should be noted. Nevertheless, the potential neurotoxic/neuroprotective action of compound 19 was determined only in one experimental in vitro model and reflected the short-term (72 hour) exposure of SH-SY5Y cells to a test substance. Therefore, further more detailed studies in that regard will be useful.
In vitro cellular models are commonly used to determine the cytotoxicity of tested compounds at an early stage of the drug evaluation process. By applying several methods, which are based on different mechanisms of action, we can accurately determine how the tested substance affects individual cells and thus predict its potential side effects.
Analyses performed using two different cytotoxicity assays show that the tested compounds (1, 6, 19, and 21) are safe. They do not induce hepatotoxicity (as do many anti-epileptic drugs) and are also safe for normal cells derived from the nervous system (astrocytes). Compound 19 does not affect the metabolic activity of cells, which was proven using MTT assay (Fig. 7 and 9). In addition, neutral red assay showed that the tested compound did not interfere with the endosomal activity of cells.
In silico prediction of metabolism
Metabolism of compound 19 was predicted using the MetaSite program. The premise for the study is the possibility of further research regarding these compounds, facilitated by in silico methods. The MetaSite program was chosen for simulation due to its high probability of finding correct metabolites in further research: in about 85% of cases, the method predicts the correct site of metabolism within the first two choices in the ranking list.26,27The most probable results for compound 19 are presented in Fig. 10, and they show that the two main sites of metabolism are the methyl group in position 3 of the phenyl ring (reactions 1–3), as well as the methylene in position 6 of piperidinol (reactions 4–5). Reactions 1–3 on the methyl group constitute aliphatic hydroxylation as well as carbonylation (oxidation), resulting in the formation of alcohol, aldehyde, or carboxylic acid, respectively. Reactions 4 and 5 constitute N-dealkylation (oxidation), resulting in opening of the piperidinol ring and formation of aldehyde or carboxylic acid, respectively. All metabolites are less lipophilic (all exert lower logP) than the parent compound. Such results were expected, since they are typical for liver metabolism. The obtained structures will be useful for future research regarding 19 and the group of N-[(phenoxyethoxy)ethyl]aminoalkanol derivatives.
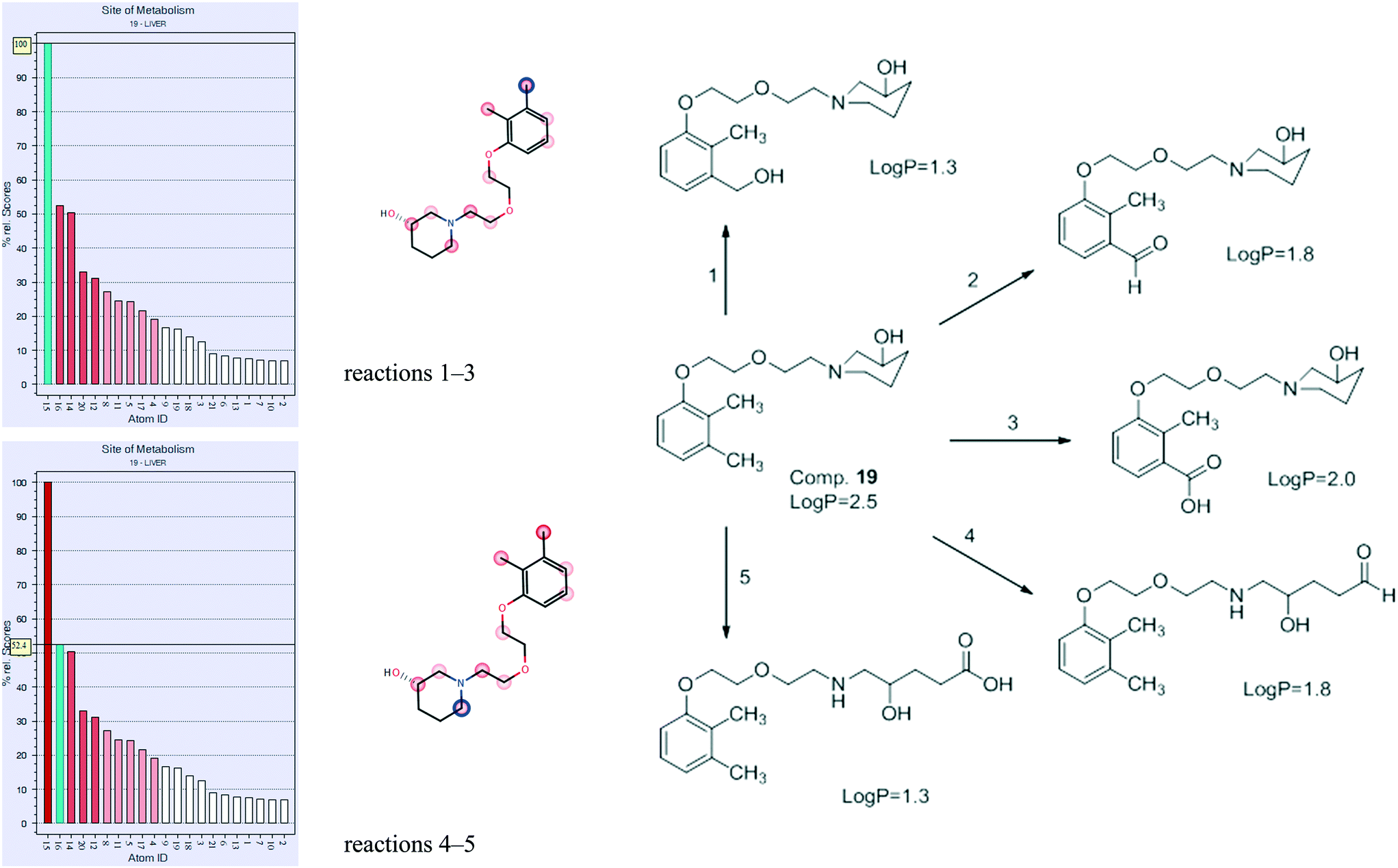 | ||
| Fig. 10 The main results of in silico metabolism prediction for compound 19. | ||
Conclusion
The presented research shows that there exist premises for the search of an anticonvulsant as well as analgesic compounds among the derivatives of N-[(phenoxy)alkyl]- or N-[(phenoxy)ethoxyethyl]aminoalkanols. The results are consistent with our former research, indicating the possibility of finding active compounds among derivatives of 2,6-dimethyl, and also 2,3-dimethyl and 2,4-dimethyl, as well as 2,4,6-trimethylphenol, among aminoalkanols – derivatives of 2-aminopropan-1-ol, and D,L-trans-2-aminocyclohexan-1-ol, as well as piperidin-3- or -4-ol. Both moieties are linked in the structures of active compounds by ethylene, propylene, tetramethylene, or ethoxyethyl. The linker varies among active compounds, which suggests a premise for further investigation of its effect on the activity.The most promising compound in this research was 1-{2-[2-(2,3-dimethylphenoxy)ethoxy]ethyl}piperidin-3-ol hydrochloride (19), and this compound was subject to additional research on astrocytes as well as the in silico prediction of metabolism, which confirms that this compound may be of interest in terms of further research. The presented results are intended to facilitate further research regarding this compound and the group of N-[(phenoxyethoxy)ethyl]aminoalkanol derivatives.
Experimental section
Chemistry
Most of the reagents and solvents were commercially available materials of reagent grade and were purchased from Alfa Aesar (purchased from Chemat, Gdansk, Poland), Sigma-Aldrich (purchased from Sigma-Aldrich, Poznan, Poland) or Merck Sp. z o.o. (Warsaw, Poland). D,L-trans-2-aminocyclohexan-1-ol (1, 4, 17, 20) was synthesized according to a previously published procedure from cyclohexene oxide.28Synthesis of the title compounds
The synthesis was performed according to previously published procedures.7,18,19 The aim of the first step of the synthesis was to obtain the appropriate phenoxyalkyl (for achievement of 1–18) or phenoxyethoxyethyl halide (for achievement of 19–28), which was synthesized by three methods.The first method resulted in obtaining phenoxyalkyl chlorides by heating the appropriately substituted phenol with 1-bromo-3-chloropropane or 1-bromo-4-chlorobutane in acetone in the presence of K2CO3 and TEBA.
Another method constituted achieving substituted phenoxyalkyl and phenoxyethoxyethyl bromides in two steps. At first, the appropriate phenol was heated with chloroalkanol (2-chloroethan-1-ol, 3-chloropropan-1-ol, 4-chlorobutan-1-ol), or 2-(2-chloroethoxy)ethan-1-ol in a mixture of acetone and ethanol in the presence of K2CO3. The crude products were subsequently subject to bromination with the use of PBr3.
Substituted phenoxybutyl bromides were alternatively achieved by heating the appropriate phenol with 1,4-dibromobutan19 in the presence of NaOH to limit the number of synthesis steps. However, this method was the least efficient.
In order to obtain the final products 1–28, aminolysis of the achieved halides (0.03 mole) with the appropriate aminoalkanols (0.025 mole) was performed. The reaction was carried out in toluene, in the presence of K2CO3 as a proton acceptor or in DMF in the presence of TEA. Both methods produce comparable yields. Compounds 4, 17, and 19 were subsequently converted into hydrochlorides, by means of saturation with gaseous HCl in acetone solution, and purified by recrystallization from a mixture of acetone and MeOH (3:1, v/v). Other obtained amines were purified either by recrystallization from n-hexane or by column chromatography (compounds 20–28, silica gel, eluting with CH2Cl2/MeOH 70:30, v/v).
Melting points (mp) were determined by means of a Büchi SMP-20 apparatus (Büchi Labortechnik, Flawil, Switzerland) and were uncorrected. Analytical TLC was carried out on precoated plates (silica gel, 60F-254 Merck), and spots were visualized with UV light.
The 1H and 13C NMR spectra for compounds 19 and 27 were obtained with a Bruker AVANCE III 600 spectrometer (Bruker, Karlsruhe, Germany) (600.2 MHz for 1H and 150.94 MHz for 13C) at the Faculty of Chemistry, Jagiellonian University in Krakow. The 1H NMR spectra for other compounds and 13C NMR spectra for compounds 9, 11, 14, 18 were recorded at the Faculty of Pharmacy, Jagiellonian University Medical College (Krakow, Poland) with a Varian Mercury-VX 300 NMR spectrometer at 29 °C. Chemical shifts were referenced against solvent lock signal. Standard Varian pulse sequences were used for 2D experiments. The 1H and 13C NMR spectra for compounds 4, 17, and 19 were recorded in DMSO-d6 and for other compounds in CDCl3. The results are presented in the following format: chemical shift δ (ppm), multiplicity, number of protons, J values in Hertz (Hz), protons' position. Multiplicity abbreviations: s (singlet), br s (broad singlet), d (doublet), dd (doublet of doublets), dt (doublet of triplets), dtd (doublet of triplets of doublets), t (triplet), tt (triplet of triplets), m (multiplet).
The IR spectra of compounds 1, 6, and 21 were recorded on a Jasco FT/IR 410 spectrometer (KBr pellets).
The purity of the obtained compounds was confirmed by LCMS. The LCMS system consisted of a Waters Acquity UPLC, coupled to a Waters TQD mass spectrometer (electrospray ionization mode ESI-tandem quadrupole). All the analyses were carried out using an Acquity UPLC BEH C18, 1.7 lm, 2.1 × 100 mm column. A flow rate of 0.3 mL min−1 and a gradient of 5–95% B over 10 min and then 100% B over 2 min was used. Eluent A: water/0.1% HCO2H; eluent B: acetonitrile/0.1% HCO2H. LCMS data were obtained by scanning the first quadrupole in 0.5 s in a mass range from 50 to 1000 Da; eight scans were summed to produce the final spectrum. Measurement of optical rotation ([α]20.0589) for compounds 11, 13, 14, 16, 22, and 28 was carried out using Jasco 2000.
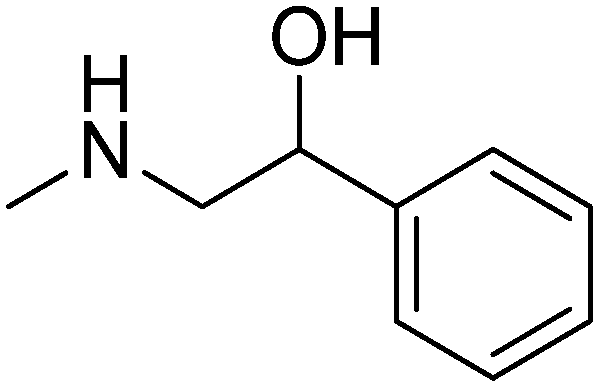
:1); NMR 1H (300 MHz, CDCl3): δ 6.91–6.96 (m, 2H, Ar-H3, Ar-H5), 6.72 (d, 1H, J = 7.9 Hz, Ar-H6), 4.03 (t, 2H, Ar–O–CH2–), 3.14–3.23 (m, 2H, CHH–NH–, OH), 2.83–2.89 (m, 1H, –CHH–NH–), 2.24–2.26 (m, 1H, >CH–NH–), 2.25 (s, 3H, Ar–CH3), 2.19 (s, 3H, Ar–CH3), 0.97–2.15 (m, 8H, cyclohex); IR (KBr, νmax cm−1): 3318, 3270, 3064, 2928, 2856, 1629, 1451, 1351, 1110, 1068, 967, 894, 884, 857, 661; LCMS [M + H]+m/z calcd for C16H25O2N 264.19, found 264.28, 99.27%.
:1); NMR 1H (300 MHz, CDCl3): δ 6.99 (d, 1H, J = 7.2 Hz, Ar-H4), 6.58–6.70 (m, 2H, Ar-H3, Ar-H5), 4.12 (dt, 1H, J = 6.3 Hz, J = 3.3 Hz, –CH–CH3), 3.96 (t, 2H, J = 5.8 Hz, Ar–O–CH2–), 2.86–3.00 (m, 2H, –CH2–CH2–NH–), 2.68 (dd, 2H, J = 12.1 Hz, J = 9.9 Hz, –CH2–CH–), 2.30 (s, 6H, Ar–CH3), 1.79–2.00 (m, 4H, –CH2–CH2–CH2–NH–), 1.42 (d, 1H, J = 6.3 Hz, –OH), 1.16–1.28 (m, 3H, –CH–CH3); LCMS [M + H]+m/z calcd for C15H25O2N 252.19, found 252.21, 100.00%.
:1); NMR 1H (300 MHz, CDCl3): δ 6.98–7.02 (m, 2H, Ar-H3, Ar-H5), 6.87–6.94 (m, 1H, Ar-H4), 3.77 (t, 2H, J = 6.4 Hz, Ar–O–CH2–), 3.62 (dd, 1H, J = 10.5 Hz, J = 4.1 Hz, –CHH–OH), 3.28 (dd, 1H, J = 10.5 Hz, J = 6.4 Hz, –CHH–OH), 2.73–2.83 (m, 1H, –NH–CH<), 2.51–2.69 (m, 2H, –CH2–NH–), 2.27 (s, 6H, Ar–(CH3)2), 1.81–1.92 (m, 2H, Ar–O–CH2–CH2–), 1.66–1.77 (m, 2H, –CH2–CH2–NH–), 1.35–1.57 (m, 2H, –CH2–CH3), 0.92 (t, 3H, J = 7.3 Hz, 3H, –CH2–CH3); LCMS [M + H]+m/z calcd for C16H27O2N 266.21, found 266.28, 100.00%.
:1); NMR 1H (300 MHz, DMSO-d6): δ 8.39–8.81 (m, 2H, NH2+), 6.98–7.02 (m, 2H, Ar-H3, Ar-H5), 6.85–6.92 (m, 1H, Ar-H4), 5.50 (d, 1H, J = 5.3 Hz, OH), 3.72 (t, 2H, J = 5.9 Hz, Ar–O–CH2–), 3.48 (br s, 1H, >CH–OH), 3.01 (br s, 2H, –CH2–NH–), 2.70–2.82 (m, 1H, >CH–NH–), 2.20 (s, 6H, Ar–(CH3)2), 1.59–2.09 (m, 8H, pip), 1.19 (d, 4H, J = 8.2 Hz, –CH2–CH2–CH2–CH2–NH–); LCMS [M + H]+m/z calcd for C18H29O2N 292.22, found 292.33, 99.39%.
:1); NMR 1H (300 MHz, CDCl3): δ 6.97–7.02 (m, 2H, Ar-H3, Ar-H5), 6.87–6.93 (m, 1H, Ar-H4), 3.76 (t, 2H, J = 6.4 Hz, Ar–O–CH2–), 3.56–3.67 (m, 1H, >CH–OH), 2.66–2.74 (m, 2H, –CH2–NH–), 2.40–2.51 (m, 1H, >CH–NH–), 2.26 (s, 6H, Ar–CH3), 1.89–2.03 (m, 4H, –CH2–CH2–CH2–NH–), 1.77–1.89 (m, 2H, pip), 1.63–1.75 (m, 2H, pip), 1.08–1.38 (m, 4H, pip); LCMS [M + H]+m/z calcd for C18H29O2N 292.22, found 292.33, 98.88%.
:1); NMR 1H (300 MHz, CDCl3): δ 6.81 (s, 2H, Ar–H), 3.77 (t, 2H, J = 6.2, Ar–O–CH2–), 3.62–3.74 (m, 1H, CH–OH), 2.76–2.88 (m, 2H, pip), 2.58 (t, 2H, J = 7.2, 2H, –CH2–N<), 2.23 (s, 9H, Ar–(CH3)3), 2.12–2.22 (m, 2H, pip), 1.87–2.00 (m, 4H, pip, –CH2–CH2–CH2–), 1.58–1.68 (m, 2H, pip); IR (KBr, νmax cm−1): 3393, 3230, 2937, 2859, 2786, 2740, 2299, 1682, 1486, 1381, 1218, 1149, 1057, 979, 852, 784, 720, 648, 586, 431, 418; LCMS [M + H]+m/z calcd for C17H27O2N 278.21, found 278.24, 98.18%.
:1); NMR 1H (300 MHz, CDCl3): δ 7.27–7.42 (m, 5H, CH–Ar), 7.23 (d, 1H, J = 8.0 Hz, H-4), 6.66–6.81 (m, 2H, H-3, H-6), 4.75 (dd, 1H, J = 9.0 Hz, J = 3.6 Hz, CH–Ar), 4.04–4.22 (m, 2H, –O–CH2–CH2), 3.08–3.13 (m, 2H, –O–CH2–CH2), 3.04 (dd, 1H, J = 12.3 Hz, J = 3.6 Hz, –NH–CHH–CH–), 2.80 (dd, 1H, J = 12.1 Hz, J = 9.0 Hz, –NH–CHH–CH–), 2.33 (s, 3H, Ar–CH3), 1.41–2.00 (m, 1H, NH); LCMS [M + H]+m/z calcd for C17H20O2NCl 306.12, found 306.10, 99.39%.
:1); NMR 1H (300 MHz, CDCl3): δ 7.20 (d, 1H, J = 7.95 Hz, J = 1.4 Hz, Ar-H3), 6.66–6.75 (m, 2H, Ar-H4, Ar-H6), 4.13 (t, 2H, J = 5.90 Hz, Ar–O–CH2), 3.70 (tt, 1H, J = 8.88 Hz, J = 4.33 Hz, >CH–OH), 2.88–2.96 (m, 2H, pip-H2,6), 2.85 (t, 2H, J = 5.90 Hz, –CH2–N<), 2.32–2.39 (m, 2H, pip-H2,6), 2.30 (s, 3H, Ar–CH3), 1.86–1.97 (m, 2H, pip-H3,5), 1.53–1.67 (m, 2H, pip-H3,5); LCMS [M + H]+m/z calcd for C14H20O2NCl 270.12, found 270.20, 98.89%.
:3); NMR 1H (300 MHz, CDCl3): δ 7.21 (d, 1H, J = 8.0 Hz, Ar–H), 6.68–6.75 (m, 2H, Ar–H), 4.11 (t, 2H, J = 5.3 Hz, 2H, –O–CH2–CH2–), 3.54–3.70 (m, 1H, CH–OH), 3.04 (t, 2H, J = 5.3 Hz, –CH2–NH–), 2.53 (tt, 1H, J = 10.5 Hz, J = 3.5 Hz, –NH–CH), 2.31 (s, 3H, Ar–CH3), 1.99 (dd, 4H, J = 8.7 Hz, J = 2.1 Hz, (CH2)2 cyclohex), 1.59 (br s, 1H, NH), 1.11–1.40 (m, 4H, (CH2)2 cyclohex); 13C (75 MHz, CDCl3): 153.91 (Ar-C1), 137.90 (Ar-C5), 129.72 (Ar-C3), 122.31 (Ar-C4), 119.93 (Ar-C2), 114.76 (Ar-C6), 70.52 (>CH–OH), 68.90 (Ar–O–CH2–), 55.62 (–NH–CH<), 46.08 (–CH2–NH–), 33.96 (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) (CH2)2–OH), 31.22 ((CH2)2–NH), 21.33 (Ar–CH3); LCMS [M + H]+m/z calcd for C15H22O2NCl 284.14, found 284.17, 98.22%.
:1); NMR 1H (300 MHz, CDCl3): δ 7.22 (d, 1H, J = 8.0 Hz, Ar-H4), 6.59–6.80 (m, 2H, Ar-H3, Ar-H6), 4.04 (t, 2H, J = 6.3 Hz, –O–CH2–), 3.59 (dd, 1H, J = 10.5 Hz, J = 4.1 Hz, –CH2–OH), 3.24 (dd, 1H, J = 10.5 Hz, J = 6.9 Hz, –CH2–OH), 2.73–2.93 (m, 2H, –CH2–NH–CH–), 2.62 (dt, 1H, J = 11.3 Hz, J = 7.1 Hz, –CH2–NH–CH–), 2.32 (s, 3H, Ar–CH3), 1.82–2.02 (m, 2H, –O–CH2–CH2–), 1.70 (quin, 2H, J = 7.4 Hz, –CH2–CH2–NH–), 1.58 (br s, 1H, NH), 1.07 (d, 3H, J = 6.4 Hz, –CH–CH3); LCMS [M + H]+m/z calcd for C14H22O2N 272.14, found 272.26, 99.60%.
:1); NMR 1H (300 MHz, CDCl3): δ 7.21 (d, 1H, J = 8.0 Hz, Ar-4H), 6.72 (s, 1H, Ar-6H), 6.66–6.71 (m, 1H, Ar-3H), 4.02 (t, 2H, J = 6.3 Hz, –O–CH2–), 3.58 (dd, 1H, J = 10.4 Hz, J = 4.2 Hz, –CH–CHH–OH), 3.23 (dd, 1H, J = 10.4 Hz, J = 7.1 Hz, –CH–CHH–OH), 2.74–2.86 (m, 2H, –CH2–NH–), 2.61 (dt, 1H, J = 11.5, J = 7.2, –CH<), 2.31 (s, 3H, Ar–CH3), 1.84–1.94 (m, 2H, –O–CH2–CH2–), 1.64–1.75 (m, 2H, –CH2–CH2–NH–), 1.06 (d, 3H, J = 6.4 Hz, –CH–CH3); 13C (75 MHz, CDCl3): δ 154.10 (Ar-C1), 137.79 (Ar-C5), 129.73 (Ar-C3), 121.87 (Ar-C4), 119.75 (Ar-C2), 114.30 (Ar-C6), 68.79 (>CH–), 65.47 (Ar–O–CH2–), 54.26 (>CH–CH2–OH), 46.56 (–CH2–NH–), 27.16 (–CH2–CH2–NH–), 26.96 (Ar–O–CH2–CH2–), 21.35 (Ar–CH3), 17.38 (>CH–CH3); LCMS [M + H]+m/z calcd for C14H22O2NCl 272.14, found 272.08, 99.04%; [α]20.0589 = −16.73°.
:1); NMR 1H (300 MHz, CDCl3): δ 7.21 (d, 1H, J = 8.2 Hz, Ar-H3), 6.66–6.74 (m, 2H, Ar-H4, Ar-H6), 4.02 (t, 2H, J = 6.2 Hz, Ar–O–CH2–), 3.62 (dd, 1H, J = 10.5 Hz, J = 4.1 Hz, –CHH–OH), 3.27 (dd, 1H, J = 10.5 Hz, J = 6.4 Hz, –CHH–OH), 2.60–2.83 (m, 2H, –CH2–NH–), 2.50–2.58 (m, 1H, –NH–CH<), 2.32 (s, 3H, Ar–CH3), 1.83–1.95 (m, 2H, Ar–O–CH2–CH2–), 1.63–1.75 (m, 2H, –CH2–CH2–NH–), 1.34–1.57 (m, 2H, –CH2–CH3), 0.88–0.96 (m, 3H, –CH2–CH3); LCMS [M + H]+m/z calcd for C15H24O2NCl 286.15, found 286.17, 100.00%.
:1); NMR 1H (300 MHz, CDCl3): δ 7.21 (d, 1H, J = 8.0, Ar-H3), 6.66–6.74 (m, 2H, Ar-H4, Ar-H6), 4.02 (t, 2H, J = 6.2 Hz, Ar–O–CH2–), 3.64 (dd, 1H, J = 10.6 Hz, J = 4.0 Hz, –CHH–OH), 3.30 (dd, 1H, J = 10.6 Hz, J = 6.5 Hz, –CHH–OH), 2.63–2.87 (m, 2H, –CH2–NH–), 2.52–2.63 (m, 1H, –CH<), 2.31 (s, 3H, Ar–CH3), 2.09 (br s, 2H, NH, OH), 1.84–1.96 (m, 2H, –O–CH2–CH2–), 1.66–1.77 (m, 2H, –CH2–CH2–NH–), 1.36–1.60 (m, 2H, –CH2–CH3), 0.92 (t, 3H, J = 7.4 Hz, –CH2–CH3); LCMS [M + H]+m/z calcd for C15H24O2NCl 286.15, found 286.35, 100.00%; [α]20.0589 = −11.98°.
:1); NMR 1H (300 MHz, CDCl3): δ 7.21 (d, 1H, J = 8.0 Hz, Ar-3H), 6.72 (s, 1H, Ar-6H), 6.67–6.71 (m, 1H, Ar-4H), 4.02 (t, 2H, J = 6.2 Hz, –O–CH2–), 3.62 (dd, 1H, J = 10.5 Hz, J = 4.1 Hz, CH–CHH–OH), 3.27 (dd, 1H, J = 10.5 Hz, J = 6.4 Hz, CH–CHH–OH), 2.72–2.83 (m, 1H, –NH–CH), 2.50–2.68 (m, 2H, –CH2–NH), 2.31 (s, 3H, Ar–CH3), 1.84–1.94 (m, 2H, –O–CH2–CH2–), 1.69 (dt, 2H, J = 14.9 Hz, J = 7.2 Hz, –CH2–CH2–NH), 1.34–1.57 (m, 2H, –CH2–CH3), 0.91 (t, 3H, J = 7.4 Hz, –CH2–CH3); 13C (75 MHz, CDCl3): δ 156.17 (Ar-C1), 137.79 (Ar–C5), 129.74 (Ar-C3), 121.90 (Ar-C4), 119.78 (Ar-C2), 114.34 (Ar-C6), 68.81 (Ar–O–CH2–), 62.54 (–NH–CH<), 60.19 (>CH–CH2–), 46.40 (–CH2–NH–), 27.23 (–CH2–CH2–NH–), 26.95 (Ar–O–CH2–CH2–), 24.47 (–CH2–CH3), 21.32 (Ar–CH3), 10.44 (–CH2–CH3); LCMS [M + H]+m/z calcd for C15H24O2NCl 286.15, found 286.10, 99.63%; [α]20.0589 = +12.49°.
:1); NMR 1H: δ 7.20 (dd, 1H, J = 7.9 Hz, J = 1.5 Hz, Ar-H3), 7.04–7.09 (m, 1H, Ar-H5), 6.89–6.96 (m, 1H, Ar-H4), 3.92 (t, 2H, J = 6.4 Hz, Ar–O–CH2–), 3.58 (dd, 1H, J = 10.5 Hz, J = 4.1 Hz, –CHH–OH), 3.23 (dd, 1H, J = 10.5 Hz, J = 7.0 Hz, –CHH–OH), 2.74–2.87 (m, 2H, –CH2–NH–), 2.62 (dt, 1H, J = 11.3 Hz, J = 7.3 Hz, –NH–CH<), 2.30 (s, 3H, Ar–CH3), 1.83–1.94 (m, 2H, Ar–O–CH2–CH2–), 1.67–1.78 (m, 2H, –CH2–CH2–NH–), 1.06 (d, 3H, J = 6.4 Hz, >CH–CH3); LCMS [M + H]+m/z calcd for C14H22O2NCl 272.14, found 272.21, 100.00%.
:1); NMR 1H (300 MHz, CDCl3): δ 7.19 (dd, 1H, J = 8.1 Hz, J = 1.4 Hz, Ar-H3), 7.06 (dd, 1H, J = 7.6 Hz, J = 0.6 Hz, Ar-H5), 6.93 (t, 1H, J = 7.7 Hz, Ar-H4), 3.92 (t, 2H, J = 6.2 Hz, Ar–O–CH2–), 3.61 (dd, 1H, J = 10.6 Hz, J = 4.0 Hz, –CHH–OH), 3.29 (dd, 1H, J = 10.6 Hz, J = 7.1 Hz, –CHH–OH), 2.77–2.92 (m, 2H, –CH2–NH–), 2.66 (dt, 1H, J = 11.3 Hz, J = 7.1 Hz, –NH–CH<), 2.30 (s, 3H, Ar–CH3), 2.03 (br s, 2H, NH, OH), 1.84–1.95 (m, 2H, –O–CH2–CH2–), 1.72–1.83 (m, 2H, –CH2–CH2–NH–), 1.09 (d, 3H, J = 6.4 Hz, >CH–CH3); LCMS [M + H]+m/z calcd for C14H22O2NCl 272.14, found 272.32, 98.32%; [α]20.0589 = +15.61°.
:1); NMR 1H (300 MHz, CDCl3): δ 7.19 (dd, 1H, J = 8.0 Hz, J = 1.3 Hz, Ar-H3), 7.06 (dd, 1H, J = 7.6 Hz, J = 0.9 Hz, Ar-H5), 6.92 (t, 1H, J = 7.7 Hz, Ar-H4), 3.91 (t, 2H, J = 6.4 Hz, Ar–O–CH2), 3.62 (tt, 1H, J = 10.5 Hz, J = 4.0 Hz, >CH–OH), 2.73 (t, 2H, J = 1.0 Hz, –CH2–NH–), 2.45 (tt, 1H, J = 10.7 Hz, J = 3.7 Hz, –NH–CH<), 2.29 (s, 3H, Ar–CH3), 1.90–2.01 (m, 4H, cyclohex), 1.81–1.89 (m, 2H, Ar–O–CH2–CH2–), 1.65–1.77 (m, 2H, cyclohex), 1.23–1.39 (m, 3H, –CH2–CH2–NH–), 1.07–1.21 (m, 2H, cyclohex); 13C (75 MHz, CDCl3): δ 153.43 (Ar–C1), 133.29 (Ar-C6), 129.50 (Ar-C5), 127.94 (Ar-C3), 124.41 (Ar-C4), 72.57 (>CH–OH), 70.60 (Ar–O–CH2–), 56.05 (–NH–CH<), 47.22 (–CH2–NH–), 34.07 ((CH2)2CH–OH), 31.36 ((CH2)2CH–NH–), 28.12 (Ar–O–CH2–CH2–), 27.13 (–CH2–CH2–NH–), 16.47 (Ar–CH3); LCMS [M + H]+m/z calcd for C17H26O2NCl 312.17, found 312.09, 100.00%.
:1); NMR 1H (600.2 MHz, DMSO-d6): δ 10.67 (br s, 1H, NH+), 7.00 (dd, 1H, J = 8.2 Hz, J = 7.4 Hz, Ar-H5), 6.78 (d, 1H, J = 8.2 Hz, Ar-H6), 6.75 (d, 1H, J = 7.4 Hz, Ar-H4), 4.79 (br s, 1H, OH), 4.10 (ddd, 2H, J = 6.1 Hz, J = 4.7 Hz, J = 3.3 Hz, Ar–O–CH2–), 3.91 (t, 2H, J = 5.2 Hz, –O–CH2–CH2–NH–), 3.88 (br s, 1H, >CH–OH), 3.80 (ddd, 2H, J = 6.1 Hz, J = 4.7 Hz, J = 3.3 Hz, –O–CH2–CH2–O–), 3.65 (br s, 2H, pip), 3.41 (br s, 2H, pip), 3.22 (t, 2H, J = 5.2 Hz, –O–CH2–CH2–NH–), 2.21 (s, 3H, Ar–CH3 (3)), 2.09 (s, 3H, Ar–CH3 (2)), 1.98 (br s, 2H, pip), 1.74 (br s, 2H, pip); 13C (150.94 MHz, DMSO-d6): δ 157.01 (Ar-C1), 137.82 (Ar-C3), 126.33 (Ar-C5), 125.14 (Ar-C2), 122.88 (Ar-C4), 110.52 (Ar-C6), 69.80 (Ar–O–CH2–CH2–O–), 68.22 (Ar–O–CH2–CH2–O–), 65.55 (–O–CH2–CH2–NH+–), 64.67 (CH–OH), 55.50 (–O–CH2–CH2–NH+–), 51.32 (piperidine-C2), 49.26 (piperidine-C6), 31.78 (piperidine-C4), 29,98 (piperidine-C5), 20.00 (Ar–CH3 (3)), 11.86 (Ar–CH3 (2)); LCMS [M + H]+m/z calcd for C17H27O3N 294.20, found 294.21, 100.00%.
:1); NMR 1H (300 MHz, CDCl3): δ 7.00–7.06 (m, 1H, Ar-H3), 6.78 (d, 1H, J = 7.6 Hz, Ar-H4), 6.71 (d, 1H, J = 8.2 Hz, Ar-H2), 4.07–4.14 (m, 2H, Ar–O–CH2–), 3.80–3.86 (m, 2H, Ar–O–CH2–CH2–), 3.67 (dd, 2H, J = 5.9 Hz, J = 4.7 Hz, –CH2–O–CH2–), 3.08–3.20 (m, 1H, >CH–OH), 3.03 (dt, 1H, J = 12.2 Hz, J = 5.9 Hz, –CHH–NH–), 2.65 (dt, 1H, J = 12.3 Hz, J = 4.7 Hz, –CHH–NH–), 2.27 (s, 3H, Ar–CH3 (3)), 2.18–2.25 (m, 1H, >CH–NH–), 2.16 (s, 3H, Ar-CH3 (2)), 1.97–2.11 (m, 2H, pip), 1.66–1.77 (m, 2H, pip), 1.32–1.48 (m, 1H, NH), 1.18–1.31 (m, 3H, pip), 0.86–1.04 (m, 1H, pip); LCMS [M + H]+m/z calcd for C18H28O3N 308.22, found 308.35, 98.56%.
:1); NMR 1H (300 MHz, CDCl3): δ 6.90–6.96 (m, 2H, Ar-H3, Ar-H5), 6.72 (d, 1H, J = 7.6 Hz, Ar-H2), 4.07–4.12 (m, 2H, Ar–O–CH2–), 3.80–3.84 (m, 2H, Ar–O–CH2–CH2–), 3.64–3.69 (m, 2H, –CH2–O–CH2–), 3.56 (dd, 1H, J = 10.5 Hz, J = 4.1 Hz, –CHH–OH), 3.21–3.28 (m, 1H, –CHH–OH), 2.88–2.97 (m, 1H, >CH–NH–), 2.67–2.81 (m, 2H, –CH2–NH–), 2.25 (s, 3H, Ar–CH3(4)), 2.20 (s, 3H, Ar–CH3(2)), 1.05 (d, 3H, J = 6.4 Hz, >CH–CH3); IR (KBr, νmax cm−1): 3434, 3300, 3126, 2961, 2924, 2879, 1612, 1506, 1459, 1379, 1256, 1226, 1132, 1066, 1028, 952, 801, 556, 538; LCMS [M + H]+m/z calcd for C15H25O3N 268.19, found 268.40, 100.00%.
:1); NMR 1H (300 MHz, CDCl3): δ 6.88–6.98 (m, 2H, Ar-H3, Ar-H5), 6.72 (d, 1H, J = 8.0 Hz, Ar-H6), 4.06–4.12 (m, 2H, Ar–O–CH2–), 3.80–3.85 (m, 2H, Ar–O–CH2–CH2–), 3.65–3.70 (m, 2H, –CH2–O–CH2–), 3.58–3.64 (m, 1H, >CH–CHH–OH), 3.31 (dd, 1H, J = 10.8 Hz, J = 6.4 Hz, >CH–CHH–OH), 2.85–2.98 (m, 1H, –CHH–NH–), 2.70–2.80 (m, 1H, –CHH–NH–), 2.52–2.62 (m, 1H, –CH<), 2.25 (s, 3H, Ar–CH3 (para)), 2.20 (s, 3H, Ar–CH3 (ortho)), 2.09–2.17 (m, 1H, NH), 1.39–1.53 (m, 2H, –CH2–CH3), 0.91 (t, 3H, J = 7.4 Hz, –CH2–CH3); LCMS [M + H]+m/z calcd for C16H27O3N 282.20, found 282.43, 100.00%; [α]20.0589= −9.64°.
:3); NMR 1H (300 MHz, CDCl3): δ 7.27–7.38 (m, 5H, >CH–Ar), 6.97–7.03 (m, 2H, Ar-H3, Ar-H5), 6.88–6.95 (m, 1H, Ar-H4), 4.81 (dd, 1H, J = 9.2 Hz, J = 3.3 Hz, >CH–Ar), 3.91–3.96 (m, 2H, Ar–O–CH2–), 3.78–3.84 (m, 2H, Ar–O–CH2–CH2–O–), 3.74 (t, 2H, J = 5.1 Hz, –CH2–CH2–NH–), 2.93–3.05 (m, 3H, –CH2–CH2–NH–), 2.79 (dd, 2H, J = 12.1 Hz, J = 9.5 Hz, –NH–CH2–CH<), 2.27 (s, 6H, Ar–(CH3)2); LCMS [M + H]+m/z calcd for C20H27O3N 330.20, found 330.42, 97.22%.
: 1); NMR 1H (300 MHz, CDCl3): δ 7.23 (d, 1H, J = 8.0 Hz, Ar-H4), 6.76 (s, 1H, Ar-H6), 6.72 (dd, 1H, J = 8.1 Hz, J = 1.2 Hz, Ar-H3), 4.18 (t, 2H, J = 1.0 Hz, Ar–O–CH2–), 3.82–3.95 (m, 2H, Ar–O–CH2–CH2–), 3.70 (t, 2H, J = 5.1 Hz, –CH2–O–CH2–), 3.51 (dtd, 1H, J = 9.3 Hz, J = 6.3 Hz, J = 3.1 Hz, –CH2–CH–OH), 2.79–2.90 (m, 2H, –CH2–CH2–NH–), 2.72–2.79 (m, 1H, –NH–CHH–CH–), 2.43 (dd, 1H, J = 12.1 Hz, J = 9.5 Hz, –NH–CHH–CH–), 2.32 (s, 3H, Ar–CH3), 1.64 (br s, 2H, NH, OH), 1.35–1.53 (m, 2H, –CH–CH2–CH3), 0.96 (t, 3H, J = 7.6 Hz, –CH–CH2–CH3); LCMS [M + H]+m/z calcd for C15H24O3NCl 302.15, found 302.12, 99.51%.
:1); NMR 1H (300 MHz, CDCl3): δ 7.21 (d, 1H, J = 8.0 Hz, Ar-H3), 6.69–6.78 (m, 2H, Ar-H4, Ar-H6), 4.15–4.20 (m, 2H, Ar–O–CH2–), 3.90 (dd, 2H, J = 5.6 Hz, J = 4.1 Hz, Ar–O–CH2–CH2–), 3.82 (t, 2H, J = 5.1 Hz, –CH2–CH2–NH–), 3.72 (dd, 1H, J = 11.5 Hz, J = 3.6 Hz, –CHH–OH), 3.47 (dd, 1H, J = 11.5 Hz, J = 6.7 Hz, –CHH–OH), 3.00–3.11 (m, 1H, –NH–CH<), 2.86–2.97 (m, 2H, –CH2–NH–), 2.70–2.78 (m, 1H, NH), 2.31 (s, 3H, Ar–CH3), 1.50–1.63 (m, 2H, –CH2–CH3), 0.88–0.97 (m, 3H, –CH2–CH3); LCMS [M + H]+m/z calcd for C15H24O3NCl 302.15, found 302.18, 98.85%.
:1); NMR 1H (300 MHz, CDCl3): δ 7.21 (d, 1H, J = 8.0 Hz, Ar-3H), 6.68–6.76 (m, 2H, Ar-H4, Ar-H6), 4.16 (t, 2H, J = 4.4 Hz, –O–CH2–), 3.84–3.90 (m, 3H, –CH2–O–, –CH–OH), 3.01 (br s, 2H, –CH2–CH2–N–), 2.83 (br s, 2H, –CH2–N–), 2.31 (s, 3H, Ar–CH3), 2.11 (br s, 2H, –N–CH2–), 1.72 (br s, 4H, –N–CH2–, –CH2–CH–OH), 1.47 (br s, 2H, –CH2–CH–OH); LCMS [M + H]+m/z calcd for C16H24O3NCl 314.15, found 314.15, 99.05%.
:1); NMR 1H (600.2 MHz, CDCl3): δ 7.32–7.35 (m, 2H, Ar-H2′, Ar-H6′), 7.29–7.31 (m, 2H, Ar-H3′, Ar-H5′) 7.26–7.28 (m, 1H, Ar-H3), 7.19–7.23 (m, 1H, Ar-H4′), 7.16–7.19 (m, 1H, Ar-H5), 7.03 (dd, 1H, J = 7.8 Hz, J = 7.6 Hz, Ar-H4), 5.23 (d, 1H, J = 4.4 Hz, OH), 4.58–4.62 (m, 1H, >CH–OH), 4.00 (t, 2H, J = 4.8 Hz, Ar–O–CH2–), 3.70 (t, 2H, J = 4.8 Hz, Ar–O–CH2–CH2–), 3.53 (t, 2H, J = 5.5 Hz, –O–CH2–CH2–NH–), 2.71 (t, 2H, J = 5.5 Hz, –O–CH2–CH2–NH–), 2.61–2.68 (m, 1H, –NH–CH2–CH<), 2.26 (s, 3H, Ar–CH3 (6)), 1.78 (br s, 1H, NH); 13C (150.94 MHz, CDCl3): δ 152.79 (Ar-C1), 144.55 (Ar-C1′), 133.30 (Ar-C6), 129.84 (Ar-C5), 127.81 (Ar-C3′, Ar-C5′), 127.67 (Ar-C3), 126.65 (Ar-C4′), 126.63 (Ar-C2), 125.82 (Ar-C6′, Ar-C2′), 124.79 (Ar-C4), 71.84 (Ar–O–CH2–), 71.55 (>CH–OH), 70.29 (–O–CH2–CH2–NH–), 69.36 (Ar–O–CH2–CH2–), 57.59 (–NH–CH2–CH<), 48.56 (–O–CH2–CH2–NH–), 15.93 (Ar–CH3); LCMS [M + H]+m/z calcd for C19H24O3NCl 350.15, found 350.17, 100.00%.
:1); NMR 1H (300 MHz, CDCl3): δ 7.27–7.40 (m, 5H, >CH–Ar), 7.19 (d, 1H, J = 7.7 Hz, Ar-H3), 7.03–7.08 (m, 1H, Ar-H5), 6.90–6.97 (m, 1H, Ar-H4), 4.81 (dd, 1H, J = 9.5 Hz, J = 3.6 Hz, >CH-), 4.06–4.11 (m, 2H, Ar–O–CH2–), 3.81–3.86 (m, 2H, Ar–O–CH2–CH2–), 3.74 (t, 2H, J = 5.0 Hz, –O–CH2–CH2–NH–), 2.97–3.05 (m, 2H, –O–CH2–CH2–NH–), 2.90–2.96 (m, 1H, NH), 2.79 (dd, 2H, J = 12.2 Hz, J = 9.1 Hz, –NH–CH2–CH<), 2.30 (s, 3H, Ar–CH3); LCMS [M + H]+m/z calcd for C19H24O3NCl 350.15, found 350.37, 97.94%; [α]20.0589 = −22.29°.
(CH2)2–OH), 31.22 ((CH2)2–NH), 21.33 (Ar–CH3); LCMS [M + H]+m/z calcd for C15H22O2NCl 284.14, found 284.17, 98.22%.
:1); NMR 1H (300 MHz, CDCl3): δ 7.22 (d, 1H, J = 8.0 Hz, Ar-H4), 6.59–6.80 (m, 2H, Ar-H3, Ar-H6), 4.04 (t, 2H, J = 6.3 Hz, –O–CH2–), 3.59 (dd, 1H, J = 10.5 Hz, J = 4.1 Hz, –CH2–OH), 3.24 (dd, 1H, J = 10.5 Hz, J = 6.9 Hz, –CH2–OH), 2.73–2.93 (m, 2H, –CH2–NH–CH–), 2.62 (dt, 1H, J = 11.3 Hz, J = 7.1 Hz, –CH2–NH–CH–), 2.32 (s, 3H, Ar–CH3), 1.82–2.02 (m, 2H, –O–CH2–CH2–), 1.70 (quin, 2H, J = 7.4 Hz, –CH2–CH2–NH–), 1.58 (br s, 1H, NH), 1.07 (d, 3H, J = 6.4 Hz, –CH–CH3); LCMS [M + H]+m/z calcd for C14H22O2N 272.14, found 272.26, 99.60%.
:1); NMR 1H (300 MHz, CDCl3): δ 7.21 (d, 1H, J = 8.0 Hz, Ar-4H), 6.72 (s, 1H, Ar-6H), 6.66–6.71 (m, 1H, Ar-3H), 4.02 (t, 2H, J = 6.3 Hz, –O–CH2–), 3.58 (dd, 1H, J = 10.4 Hz, J = 4.2 Hz, –CH–CHH–OH), 3.23 (dd, 1H, J = 10.4 Hz, J = 7.1 Hz, –CH–CHH–OH), 2.74–2.86 (m, 2H, –CH2–NH–), 2.61 (dt, 1H, J = 11.5, J = 7.2, –CH<), 2.31 (s, 3H, Ar–CH3), 1.84–1.94 (m, 2H, –O–CH2–CH2–), 1.64–1.75 (m, 2H, –CH2–CH2–NH–), 1.06 (d, 3H, J = 6.4 Hz, –CH–CH3); 13C (75 MHz, CDCl3): δ 154.10 (Ar-C1), 137.79 (Ar-C5), 129.73 (Ar-C3), 121.87 (Ar-C4), 119.75 (Ar-C2), 114.30 (Ar-C6), 68.79 (>CH–), 65.47 (Ar–O–CH2–), 54.26 (>CH–CH2–OH), 46.56 (–CH2–NH–), 27.16 (–CH2–CH2–NH–), 26.96 (Ar–O–CH2–CH2–), 21.35 (Ar–CH3), 17.38 (>CH–CH3); LCMS [M + H]+m/z calcd for C14H22O2NCl 272.14, found 272.08, 99.04%; [α]20.0589 = −16.73°.
:1); NMR 1H (300 MHz, CDCl3): δ 7.21 (d, 1H, J = 8.2 Hz, Ar-H3), 6.66–6.74 (m, 2H, Ar-H4, Ar-H6), 4.02 (t, 2H, J = 6.2 Hz, Ar–O–CH2–), 3.62 (dd, 1H, J = 10.5 Hz, J = 4.1 Hz, –CHH–OH), 3.27 (dd, 1H, J = 10.5 Hz, J = 6.4 Hz, –CHH–OH), 2.60–2.83 (m, 2H, –CH2–NH–), 2.50–2.58 (m, 1H, –NH–CH<), 2.32 (s, 3H, Ar–CH3), 1.83–1.95 (m, 2H, Ar–O–CH2–CH2–), 1.63–1.75 (m, 2H, –CH2–CH2–NH–), 1.34–1.57 (m, 2H, –CH2–CH3), 0.88–0.96 (m, 3H, –CH2–CH3); LCMS [M + H]+m/z calcd for C15H24O2NCl 286.15, found 286.17, 100.00%.
:1); NMR 1H (300 MHz, CDCl3): δ 7.21 (d, 1H, J = 8.0, Ar-H3), 6.66–6.74 (m, 2H, Ar-H4, Ar-H6), 4.02 (t, 2H, J = 6.2 Hz, Ar–O–CH2–), 3.64 (dd, 1H, J = 10.6 Hz, J = 4.0 Hz, –CHH–OH), 3.30 (dd, 1H, J = 10.6 Hz, J = 6.5 Hz, –CHH–OH), 2.63–2.87 (m, 2H, –CH2–NH–), 2.52–2.63 (m, 1H, –CH<), 2.31 (s, 3H, Ar–CH3), 2.09 (br s, 2H, NH, OH), 1.84–1.96 (m, 2H, –O–CH2–CH2–), 1.66–1.77 (m, 2H, –CH2–CH2–NH–), 1.36–1.60 (m, 2H, –CH2–CH3), 0.92 (t, 3H, J = 7.4 Hz, –CH2–CH3); LCMS [M + H]+m/z calcd for C15H24O2NCl 286.15, found 286.35, 100.00%; [α]20.0589 = −11.98°.
:1); NMR 1H (300 MHz, CDCl3): δ 7.21 (d, 1H, J = 8.0 Hz, Ar-3H), 6.72 (s, 1H, Ar-6H), 6.67–6.71 (m, 1H, Ar-4H), 4.02 (t, 2H, J = 6.2 Hz, –O–CH2–), 3.62 (dd, 1H, J = 10.5 Hz, J = 4.1 Hz, CH–CHH–OH), 3.27 (dd, 1H, J = 10.5 Hz, J = 6.4 Hz, CH–CHH–OH), 2.72–2.83 (m, 1H, –NH–CH), 2.50–2.68 (m, 2H, –CH2–NH), 2.31 (s, 3H, Ar–CH3), 1.84–1.94 (m, 2H, –O–CH2–CH2–), 1.69 (dt, 2H, J = 14.9 Hz, J = 7.2 Hz, –CH2–CH2–NH), 1.34–1.57 (m, 2H, –CH2–CH3), 0.91 (t, 3H, J = 7.4 Hz, –CH2–CH3); 13C (75 MHz, CDCl3): δ 156.17 (Ar-C1), 137.79 (Ar–C5), 129.74 (Ar-C3), 121.90 (Ar-C4), 119.78 (Ar-C2), 114.34 (Ar-C6), 68.81 (Ar–O–CH2–), 62.54 (–NH–CH<), 60.19 (>CH–CH2–), 46.40 (–CH2–NH–), 27.23 (–CH2–CH2–NH–), 26.95 (Ar–O–CH2–CH2–), 24.47 (–CH2–CH3), 21.32 (Ar–CH3), 10.44 (–CH2–CH3); LCMS [M + H]+m/z calcd for C15H24O2NCl 286.15, found 286.10, 99.63%; [α]20.0589 = +12.49°.
:1); NMR 1H: δ 7.20 (dd, 1H, J = 7.9 Hz, J = 1.5 Hz, Ar-H3), 7.04–7.09 (m, 1H, Ar-H5), 6.89–6.96 (m, 1H, Ar-H4), 3.92 (t, 2H, J = 6.4 Hz, Ar–O–CH2–), 3.58 (dd, 1H, J = 10.5 Hz, J = 4.1 Hz, –CHH–OH), 3.23 (dd, 1H, J = 10.5 Hz, J = 7.0 Hz, –CHH–OH), 2.74–2.87 (m, 2H, –CH2–NH–), 2.62 (dt, 1H, J = 11.3 Hz, J = 7.3 Hz, –NH–CH<), 2.30 (s, 3H, Ar–CH3), 1.83–1.94 (m, 2H, Ar–O–CH2–CH2–), 1.67–1.78 (m, 2H, –CH2–CH2–NH–), 1.06 (d, 3H, J = 6.4 Hz, >CH–CH3); LCMS [M + H]+m/z calcd for C14H22O2NCl 272.14, found 272.21, 100.00%.
:1); NMR 1H (300 MHz, CDCl3): δ 7.19 (dd, 1H, J = 8.1 Hz, J = 1.4 Hz, Ar-H3), 7.06 (dd, 1H, J = 7.6 Hz, J = 0.6 Hz, Ar-H5), 6.93 (t, 1H, J = 7.7 Hz, Ar-H4), 3.92 (t, 2H, J = 6.2 Hz, Ar–O–CH2–), 3.61 (dd, 1H, J = 10.6 Hz, J = 4.0 Hz, –CHH–OH), 3.29 (dd, 1H, J = 10.6 Hz, J = 7.1 Hz, –CHH–OH), 2.77–2.92 (m, 2H, –CH2–NH–), 2.66 (dt, 1H, J = 11.3 Hz, J = 7.1 Hz, –NH–CH<), 2.30 (s, 3H, Ar–CH3), 2.03 (br s, 2H, NH, OH), 1.84–1.95 (m, 2H, –O–CH2–CH2–), 1.72–1.83 (m, 2H, –CH2–CH2–NH–), 1.09 (d, 3H, J = 6.4 Hz, >CH–CH3); LCMS [M + H]+m/z calcd for C14H22O2NCl 272.14, found 272.32, 98.32%; [α]20.0589 = +15.61°.
:1); NMR 1H (300 MHz, CDCl3): δ 7.19 (dd, 1H, J = 8.0 Hz, J = 1.3 Hz, Ar-H3), 7.06 (dd, 1H, J = 7.6 Hz, J = 0.9 Hz, Ar-H5), 6.92 (t, 1H, J = 7.7 Hz, Ar-H4), 3.91 (t, 2H, J = 6.4 Hz, Ar–O–CH2), 3.62 (tt, 1H, J = 10.5 Hz, J = 4.0 Hz, >CH–OH), 2.73 (t, 2H, J = 1.0 Hz, –CH2–NH–), 2.45 (tt, 1H, J = 10.7 Hz, J = 3.7 Hz, –NH–CH<), 2.29 (s, 3H, Ar–CH3), 1.90–2.01 (m, 4H, cyclohex), 1.81–1.89 (m, 2H, Ar–O–CH2–CH2–), 1.65–1.77 (m, 2H, cyclohex), 1.23–1.39 (m, 3H, –CH2–CH2–NH–), 1.07–1.21 (m, 2H, cyclohex); 13C (75 MHz, CDCl3): δ 153.43 (Ar–C1), 133.29 (Ar-C6), 129.50 (Ar-C5), 127.94 (Ar-C3), 124.41 (Ar-C4), 72.57 (>CH–OH), 70.60 (Ar–O–CH2–), 56.05 (–NH–CH<), 47.22 (–CH2–NH–), 34.07 ((CH2)2CH–OH), 31.36 ((CH2)2CH–NH–), 28.12 (Ar–O–CH2–CH2–), 27.13 (–CH2–CH2–NH–), 16.47 (Ar–CH3); LCMS [M + H]+m/z calcd for C17H26O2NCl 312.17, found 312.09, 100.00%.
:1); NMR 1H (600.2 MHz, DMSO-d6): δ 10.67 (br s, 1H, NH+), 7.00 (dd, 1H, J = 8.2 Hz, J = 7.4 Hz, Ar-H5), 6.78 (d, 1H, J = 8.2 Hz, Ar-H6), 6.75 (d, 1H, J = 7.4 Hz, Ar-H4), 4.79 (br s, 1H, OH), 4.10 (ddd, 2H, J = 6.1 Hz, J = 4.7 Hz, J = 3.3 Hz, Ar–O–CH2–), 3.91 (t, 2H, J = 5.2 Hz, –O–CH2–CH2–NH–), 3.88 (br s, 1H, >CH–OH), 3.80 (ddd, 2H, J = 6.1 Hz, J = 4.7 Hz, J = 3.3 Hz, –O–CH2–CH2–O–), 3.65 (br s, 2H, pip), 3.41 (br s, 2H, pip), 3.22 (t, 2H, J = 5.2 Hz, –O–CH2–CH2–NH–), 2.21 (s, 3H, Ar–CH3 (3)), 2.09 (s, 3H, Ar–CH3 (2)), 1.98 (br s, 2H, pip), 1.74 (br s, 2H, pip); 13C (150.94 MHz, DMSO-d6): δ 157.01 (Ar-C1), 137.82 (Ar-C3), 126.33 (Ar-C5), 125.14 (Ar-C2), 122.88 (Ar-C4), 110.52 (Ar-C6), 69.80 (Ar–O–CH2–CH2–O–), 68.22 (Ar–O–CH2–CH2–O–), 65.55 (–O–CH2–CH2–NH+–), 64.67 (CH–OH), 55.50 (–O–CH2–CH2–NH+–), 51.32 (piperidine-C2), 49.26 (piperidine-C6), 31.78 (piperidine-C4), 29,98 (piperidine-C5), 20.00 (Ar–CH3 (3)), 11.86 (Ar–CH3 (2)); LCMS [M + H]+m/z calcd for C17H27O3N 294.20, found 294.21, 100.00%.
:1); NMR 1H (300 MHz, CDCl3): δ 7.00–7.06 (m, 1H, Ar-H3), 6.78 (d, 1H, J = 7.6 Hz, Ar-H4), 6.71 (d, 1H, J = 8.2 Hz, Ar-H2), 4.07–4.14 (m, 2H, Ar–O–CH2–), 3.80–3.86 (m, 2H, Ar–O–CH2–CH2–), 3.67 (dd, 2H, J = 5.9 Hz, J = 4.7 Hz, –CH2–O–CH2–), 3.08–3.20 (m, 1H, >CH–OH), 3.03 (dt, 1H, J = 12.2 Hz, J = 5.9 Hz, –CHH–NH–), 2.65 (dt, 1H, J = 12.3 Hz, J = 4.7 Hz, –CHH–NH–), 2.27 (s, 3H, Ar–CH3 (3)), 2.18–2.25 (m, 1H, >CH–NH–), 2.16 (s, 3H, Ar-CH3 (2)), 1.97–2.11 (m, 2H, pip), 1.66–1.77 (m, 2H, pip), 1.32–1.48 (m, 1H, NH), 1.18–1.31 (m, 3H, pip), 0.86–1.04 (m, 1H, pip); LCMS [M + H]+m/z calcd for C18H28O3N 308.22, found 308.35, 98.56%.
:1); NMR 1H (300 MHz, CDCl3): δ 6.90–6.96 (m, 2H, Ar-H3, Ar-H5), 6.72 (d, 1H, J = 7.6 Hz, Ar-H2), 4.07–4.12 (m, 2H, Ar–O–CH2–), 3.80–3.84 (m, 2H, Ar–O–CH2–CH2–), 3.64–3.69 (m, 2H, –CH2–O–CH2–), 3.56 (dd, 1H, J = 10.5 Hz, J = 4.1 Hz, –CHH–OH), 3.21–3.28 (m, 1H, –CHH–OH), 2.88–2.97 (m, 1H, >CH–NH–), 2.67–2.81 (m, 2H, –CH2–NH–), 2.25 (s, 3H, Ar–CH3(4)), 2.20 (s, 3H, Ar–CH3(2)), 1.05 (d, 3H, J = 6.4 Hz, >CH–CH3); IR (KBr, νmax cm−1): 3434, 3300, 3126, 2961, 2924, 2879, 1612, 1506, 1459, 1379, 1256, 1226, 1132, 1066, 1028, 952, 801, 556, 538; LCMS [M + H]+m/z calcd for C15H25O3N 268.19, found 268.40, 100.00%.
:1); NMR 1H (300 MHz, CDCl3): δ 6.88–6.98 (m, 2H, Ar-H3, Ar-H5), 6.72 (d, 1H, J = 8.0 Hz, Ar-H6), 4.06–4.12 (m, 2H, Ar–O–CH2–), 3.80–3.85 (m, 2H, Ar–O–CH2–CH2–), 3.65–3.70 (m, 2H, –CH2–O–CH2–), 3.58–3.64 (m, 1H, >CH–CHH–OH), 3.31 (dd, 1H, J = 10.8 Hz, J = 6.4 Hz, >CH–CHH–OH), 2.85–2.98 (m, 1H, –CHH–NH–), 2.70–2.80 (m, 1H, –CHH–NH–), 2.52–2.62 (m, 1H, –CH<), 2.25 (s, 3H, Ar–CH3 (para)), 2.20 (s, 3H, Ar–CH3 (ortho)), 2.09–2.17 (m, 1H, NH), 1.39–1.53 (m, 2H, –CH2–CH3), 0.91 (t, 3H, J = 7.4 Hz, –CH2–CH3); LCMS [M + H]+m/z calcd for C16H27O3N 282.20, found 282.43, 100.00%; [α]20.0589= −9.64°.
:3); NMR 1H (300 MHz, CDCl3): δ 7.27–7.38 (m, 5H, >CH–Ar), 6.97–7.03 (m, 2H, Ar-H3, Ar-H5), 6.88–6.95 (m, 1H, Ar-H4), 4.81 (dd, 1H, J = 9.2 Hz, J = 3.3 Hz, >CH–Ar), 3.91–3.96 (m, 2H, Ar–O–CH2–), 3.78–3.84 (m, 2H, Ar–O–CH2–CH2–O–), 3.74 (t, 2H, J = 5.1 Hz, –CH2–CH2–NH–), 2.93–3.05 (m, 3H, –CH2–CH2–NH–), 2.79 (dd, 2H, J = 12.1 Hz, J = 9.5 Hz, –NH–CH2–CH<), 2.27 (s, 6H, Ar–(CH3)2); LCMS [M + H]+m/z calcd for C20H27O3N 330.20, found 330.42, 97.22%.
: 1); NMR 1H (300 MHz, CDCl3): δ 7.23 (d, 1H, J = 8.0 Hz, Ar-H4), 6.76 (s, 1H, Ar-H6), 6.72 (dd, 1H, J = 8.1 Hz, J = 1.2 Hz, Ar-H3), 4.18 (t, 2H, J = 1.0 Hz, Ar–O–CH2–), 3.82–3.95 (m, 2H, Ar–O–CH2–CH2–), 3.70 (t, 2H, J = 5.1 Hz, –CH2–O–CH2–), 3.51 (dtd, 1H, J = 9.3 Hz, J = 6.3 Hz, J = 3.1 Hz, –CH2–CH–OH), 2.79–2.90 (m, 2H, –CH2–CH2–NH–), 2.72–2.79 (m, 1H, –NH–CHH–CH–), 2.43 (dd, 1H, J = 12.1 Hz, J = 9.5 Hz, –NH–CHH–CH–), 2.32 (s, 3H, Ar–CH3), 1.64 (br s, 2H, NH, OH), 1.35–1.53 (m, 2H, –CH–CH2–CH3), 0.96 (t, 3H, J = 7.6 Hz, –CH–CH2–CH3); LCMS [M + H]+m/z calcd for C15H24O3NCl 302.15, found 302.12, 99.51%.
:1); NMR 1H (300 MHz, CDCl3): δ 7.21 (d, 1H, J = 8.0 Hz, Ar-H3), 6.69–6.78 (m, 2H, Ar-H4, Ar-H6), 4.15–4.20 (m, 2H, Ar–O–CH2–), 3.90 (dd, 2H, J = 5.6 Hz, J = 4.1 Hz, Ar–O–CH2–CH2–), 3.82 (t, 2H, J = 5.1 Hz, –CH2–CH2–NH–), 3.72 (dd, 1H, J = 11.5 Hz, J = 3.6 Hz, –CHH–OH), 3.47 (dd, 1H, J = 11.5 Hz, J = 6.7 Hz, –CHH–OH), 3.00–3.11 (m, 1H, –NH–CH<), 2.86–2.97 (m, 2H, –CH2–NH–), 2.70–2.78 (m, 1H, NH), 2.31 (s, 3H, Ar–CH3), 1.50–1.63 (m, 2H, –CH2–CH3), 0.88–0.97 (m, 3H, –CH2–CH3); LCMS [M + H]+m/z calcd for C15H24O3NCl 302.15, found 302.18, 98.85%.
:1); NMR 1H (300 MHz, CDCl3): δ 7.21 (d, 1H, J = 8.0 Hz, Ar-3H), 6.68–6.76 (m, 2H, Ar-H4, Ar-H6), 4.16 (t, 2H, J = 4.4 Hz, –O–CH2–), 3.84–3.90 (m, 3H, –CH2–O–, –CH–OH), 3.01 (br s, 2H, –CH2–CH2–N–), 2.83 (br s, 2H, –CH2–N–), 2.31 (s, 3H, Ar–CH3), 2.11 (br s, 2H, –N–CH2–), 1.72 (br s, 4H, –N–CH2–, –CH2–CH–OH), 1.47 (br s, 2H, –CH2–CH–OH); LCMS [M + H]+m/z calcd for C16H24O3NCl 314.15, found 314.15, 99.05%.
:1); NMR 1H (600.2 MHz, CDCl3): δ 7.32–7.35 (m, 2H, Ar-H2′, Ar-H6′), 7.29–7.31 (m, 2H, Ar-H3′, Ar-H5′) 7.26–7.28 (m, 1H, Ar-H3), 7.19–7.23 (m, 1H, Ar-H4′), 7.16–7.19 (m, 1H, Ar-H5), 7.03 (dd, 1H, J = 7.8 Hz, J = 7.6 Hz, Ar-H4), 5.23 (d, 1H, J = 4.4 Hz, OH), 4.58–4.62 (m, 1H, >CH–OH), 4.00 (t, 2H, J = 4.8 Hz, Ar–O–CH2–), 3.70 (t, 2H, J = 4.8 Hz, Ar–O–CH2–CH2–), 3.53 (t, 2H, J = 5.5 Hz, –O–CH2–CH2–NH–), 2.71 (t, 2H, J = 5.5 Hz, –O–CH2–CH2–NH–), 2.61–2.68 (m, 1H, –NH–CH2–CH<), 2.26 (s, 3H, Ar–CH3 (6)), 1.78 (br s, 1H, NH); 13C (150.94 MHz, CDCl3): δ 152.79 (Ar-C1), 144.55 (Ar-C1′), 133.30 (Ar-C6), 129.84 (Ar-C5), 127.81 (Ar-C3′, Ar-C5′), 127.67 (Ar-C3), 126.65 (Ar-C4′), 126.63 (Ar-C2), 125.82 (Ar-C6′, Ar-C2′), 124.79 (Ar-C4), 71.84 (Ar–O–CH2–), 71.55 (>CH–OH), 70.29 (–O–CH2–CH2–NH–), 69.36 (Ar–O–CH2–CH2–), 57.59 (–NH–CH2–CH<), 48.56 (–O–CH2–CH2–NH–), 15.93 (Ar–CH3); LCMS [M + H]+m/z calcd for C19H24O3NCl 350.15, found 350.17, 100.00%.
:1); NMR 1H (300 MHz, CDCl3): δ 7.27–7.40 (m, 5H, >CH–Ar), 7.19 (d, 1H, J = 7.7 Hz, Ar-H3), 7.03–7.08 (m, 1H, Ar-H5), 6.90–6.97 (m, 1H, Ar-H4), 4.81 (dd, 1H, J = 9.5 Hz, J = 3.6 Hz, >CH-), 4.06–4.11 (m, 2H, Ar–O–CH2–), 3.81–3.86 (m, 2H, Ar–O–CH2–CH2–), 3.74 (t, 2H, J = 5.0 Hz, –O–CH2–CH2–NH–), 2.97–3.05 (m, 2H, –O–CH2–CH2–NH–), 2.90–2.96 (m, 1H, NH), 2.79 (dd, 2H, J = 12.2 Hz, J = 9.1 Hz, –NH–CH2–CH<), 2.30 (s, 3H, Ar–CH3); LCMS [M + H]+m/z calcd for C19H24O3NCl 350.15, found 350.37, 97.94%; [α]20.0589 = −22.29°.
Pharmacology
The MES test was performed according to a procedure described by Löscher et al.29 Briefly, the mice received an electrical stimulus of sufficient intensity (25 mA, 500 V, 50 Hz, 0.2 s) delivered via auricular electrodes by an electroshock generator (Rodent Shocker, Type 221, Hugo Sachs, Germany) to induce maximal seizures. The endpoint was the tonic extension of the hind limbs. In vehicle-treated mice, the procedure caused immediate hind-limb tonic extension. Mice not displaying hind-limb tonic extension were considered to be protected from seizures. For selected compounds exhibiting potent anticonvulsant activity, the ED50 was evaluated, defined as the dose of a drug protecting 50% of animals against seizures in the MES test. At least three groups of animals were injected with various doses of tested compounds and ED50 values were calculated with 95% confidence intervals by probit analysis.30Minimal motor impairment was established in mice by a standard rotarod procedure.31 Mice were trained to balance on an accelerating rotarod (Rotarod apparatus Panlab/Harvard Apparatus, LE 8200, Spain). Proper experimentation was conducted at least 24 h after the training trial. On the test day, mice were intraperitoneally (ip) pretreated with the test compound and tested on a rotarod revolving at 10 rotations per minute. Neurotoxicity was indicated by the inability of the animal to maintain equilibrium on the rod for one minute. For the selected, most potent compounds, the neurotoxic effect was expressed as a TD50 value, representing the doses at which the compound resulted in minimal motor impairment in 50% of the animals in the rotarod test. At least three groups of animals were injected with various doses of tested compounds and TD50 values were calculated with 95% confidence intervals by probit analysis.30
In the maximal electroshock seizure threshold test (MEST test), electroconvulsions were produced by alternating current (duration of the stimulus: 0.2 s; 50 Hz) delivered via auricular electrodes by an electroshock generator (Rodent Shocker, Type 221, Hugo Sachs, Germany), according to the procedure previously described.32 Full tonic extension of both hind limbs was taken as the endpoint. Mice not displaying hind-limb tonic extension were considered protected from seizure. The current intensity was established according to an ‘up-and-down’ method previously described by Socała et al.33 The current intensity was lowered or raised by 0.06-log intervals depending on whether the previously stimulated animal did or did not exert tonic hind-limb extension, respectively. The convulsive threshold was evaluated as CS50, defined as current strength (in mA) required to induce tonic hind-limb extension in 50% of the animals tested, using the log-probit method.30
ScPTZ-induced seizures were performed by subcutaneous injection of pentylenetetrazole (PTZ, 85 mg kg−1). This produced clonic convulsions lasting for at least 5 s, with accompanying loss of the righting reflex. PTZ was administered 0.5 h after injections of tested compounds and observations were carried out for 30 min. In the control groups, the first episodes of clonic convulsions were recorded between 4–16 min in the observation period. The absence of clonic convulsions within the observed time period was interpreted as the compound's ability to protect against PTZ-induced seizures.34
Antinociceptive activity in the formalin hind paw test was examined as previously described.35,36 Twenty microliters of a 5% formalin solution were injected intraplantarly (ipl) into the right hind paw of the mouse. Immediately after formalin injection, the animals were placed individually into glass beakers and were observed for the next 30 min. Time (in s) spent on licking or biting the injected hind paw in selected intervals, 0–5 and 15–30 min after injection was measured in each experimental group and was an indicator of nociceptive behavior. In mice, an intraplantar injection of diluted formalin produces a biphasic nocifensive behavioral response (i.e., licking or biting the injected hind paw). The acute nociceptive phase lasts for the first 5 min, whereas the second inflammatory phase occurs between 15 and 30 min after formalin injection.
Locomotor activity was measured with photoresistor actometers (Ugo Basile, Italy) connected to a counter for the recording of light-beam interruptions. Mice were individually placed in plastic cages for a 30 min habituation period, and then the number of light-beam crossings was counted for a 30 min session. The cages were cleaned with 70% ethanol after each mouse.
The chimney test was performed as previously described.37 Previously trained and selected animals were placed in a 25 cm long and 2.5 cm in diameter, horizontally located tube, which was reversed in such a way that the mice were able to leave it only by climbing up backward until they reached the other end. The inability of mice to perform the test within 60 seconds indicated motor impairment.
The experimental protocol was approved by the First Local Ethics Committee on Animal Testing at the Jagiellonian University in Krakow (No 15/2014, 73/2014 and 108/2014) and was in accordance with the European Communities Council Directive of 24 November 1986 (86/609/EEC).
Cytotoxicity analysis
The SH-SY5Y neuroblastoma cell line was obtained from the American Type Culture Corporation (ATCC). Cells were cultured in Dulbecco's modified Eagle's medium (DMEM, Gibco-BRL) supplemented with 10% fetal bovine serum (FBS, Gibco-BRL), 100 units per mL of penicillin, and 100 μg mL−1 of streptomycin (Sigma Co.), and kept in a humidified atmosphere of 5% CO2/95% O2 at 37 °C.An astrocyte cell line was obtained from the American Type Culture Corporation (ATCC, CRL-2541), and human hepatoma HepG2 (Sigma Aldrich, 85011430) were used in the study. The cells were cultured under standard conditions (37 °C, 5% CO2), in DMEM low glucose medium (astrocytes) and MEM medium (hepatoma), all supplemented with 10% FBS and antibiotics.
The cytotoxic effect was determined with the use of both MTT (Cayman) and neutral red assay. Briefly, in order to perform MTT assay, the cells were seeded at a density of 1 × 104 in 96-well plates. Following overnight culture, the cells were then treated with increasing doses of compounds (1, 6, 19, 21) and incubated for 24 h. Following cell exposure to each drug for 24 h in 96-well plates, 10 μL MTT reagent was added to each well, and after 3 hours of incubation (37 °C, 5% CO2), the medium was aspirated and the formazan produced in the cells appeared as dark crystals in the bottom of the wells. Next, the crystal-dissolving solution (Cayman) was added to each well. Then, the optical density (OD) of each well was determined at 570 nm on a plate reader (BIOTEK). The number of metabolically active cells is directly proportional to the absorbance of the samples.
In case of the neutral red assay, HepG2 cells were seeded in 96-well plates (Corning) at a density of 5 × 104 cells per well. After 24 h, the culture medium was replaced with the same medium (control) or medium containing tested compounds at concentrations from 10 to 250 μM. The cells were incubated in the presence of compounds for 24 h, and then cell viability was determined by neutral red staining (Sigma Aldrich) according to a procedure described in the literature.38 The optical density (OD) of each well was determined at 570 nm on a plate reader (BIOTEK). The number of living cells was directly proportional to the absorbance of the samples.
One day before the experiment, the cells were seeded at 15000 cells per well in 96-well plates in the medium containing a reduced amount of serum (1% FBS). The SH-SY5Y cells were treated with compound 19 (dissolved in ddH2O) at concentrations of 10−8, 10−7, 10−6, 10−5, 5 × 10−5, and 10−4 M for 72 hours. In order to assess the effect of the test compound on oxidative stress-induced cell damage, compound 19 was added at concentrations of 10−8, 10−7, 10−6, and 10−5 M for 72 hours alone or with hydrogen peroxide (0.5 mM). H2O2 was added for the last 24 hours of culture, i.e. 48 hours after the addition of compound 19. The control cultures were supplemented with the same amount of an appropriate vehicle. For LDH release and MTT reduction assays as a positive control, triton was used.
Cytotoxicity was quantified by measuring the efflux of lactate dehydrogenase (LDH) into the culture media 72 h after treatment with compound 19 (alone or with hydrogen peroxide). LDH activity was determined in medium using a cytotoxicity detection kit (Roche Diagnostic GmbH). The amount of colored hydrazone, formed in the reaction of pyruvic acid with 2,4-dinitrophenylhydrazine, was inversely proportional to the LDH activity in the sample and could be quantified by measuring the absorbance at 490 nm. Data were normalized as a percent of the LDH released by vehicle-treated cells (100%), i.e. cells cultured in the absence of compound 19 and without H2O2.
The viability of SH-SY5Y cells was measured, as described previously,39 by determining the cellular reducing capacity, estimated through the extent of MTT reduction to the insoluble intracellular formazan, which depends on the activity of intracellular dehydrogenases and is independent of changes in the integrity of the plasma membrane. After 72 h culture with the test compound (alone or with hydrogen peroxide) the medium was removed and SH-SY5Y cells were incubated with MTT (3-[4,5-dimethylthiazol-2-yl]2,5-diphenyl tetrazolium bromide; 0.15 mg mL−1 in PBS) for 1 h at 37 °C. After incubation, when purple colored precipitates were visible under the microscope, the supernatants were aspirated, and the formazan precipitates were solubilized by the addition of 100 μL of dimethylsulfoxide (DMSO) per well. Absorbance was measured at 570 nm in a plate-reader (Multiscan, Labsystem). The results were expressed as a percentage of control cells incubated in the absence of compound 19 and H2O2.
Statistical analysis
The data were obtained in three independent experiments, each performed in 3–4 wells and were presented as a percentage of the control ± SEM. The significance of differences between the means was evaluated by Dunnett's test, following a one-way analysis of variance (ANOVA).Metabolism prediction
The MetaSite 5.1.1 Mass 3.2.1 computer program (Molecular Discovery, Ltd.) was used for metabolism prediction. Within the tools option of the program, a site of metabolism was chosen, then liver metabolism. In the case of a metabolite identification tool, the minimum mass was limited by 50 g mol−1 and redundant metabolites (below 30%) were ignored. All common CYP reactions were taken into consideration.Acknowledgements
The work was financed by the National Science Centre, Poland, decision on grant no DEC-2013/11/B/NZ7/04834 and DEC-2015/17/N/NZ7/00966.References
- J. W. Sander, Curr. Opin. Neurobiol., 2003, 16, 165 CrossRef.
- G. J. Alexander, L. M. Kopeloff, R. B. Alexander and N. Chatterjie, Neurobehav. Toxicol. Teratol., 1986, 8, 231 CAS.
- W. Fischer, Seizure, 2002, 11, 285 CrossRef CAS PubMed.
- K. K. Borowicz and M. Banach, Pharmacol. Rep., 2014, 66, 545 CrossRef CAS PubMed.
- H. Hitosugi, T. Kashiwazaki, M. Ohsawa and J. Kamei, Methods Find. Exp. Clin. Pharmacol., 1999, 21, 409 CrossRef CAS PubMed.
- J. Kamei, A. Saitoh and Y. Kasuya, Neurosci. Lett., 1995, 196, 169 CrossRef CAS PubMed.
- H. Marona and L. Antkiewicz-Michaluk, Acta Pol. Pharm., 1998, 55, 487 CAS.
- A. Waszkielewicz, N. Szkaradek, E. Pękala, F. Galzarano and H. Marona, Biomed. Chromatogr., 2010, 24, 1365 CrossRef CAS PubMed.
- E. Pękala, A. M. Waszkielewicz, E. Szneler, M. Walczak and H. Marona, Bioorg. Med. Chem., 2011, 19, 6927 CrossRef PubMed.
- H. Marona, A. M. Waszkielewicz and E. Szneler, Acta Pol. Pharm., 2005, 62, 345 CAS.
- A. M. Waszkielewicz, E. Szneler, M. Cegła and H. Marona, Lett. Drug Des. Discovery, 2013, 10, 35 CrossRef CAS.
- A. M. Waszkielewicz, A. Gunia-Krzyżak, B. Powroźnik, K. Słoczyńska, E. Pękala, M. Walczak, M. Bednarski, E. Żesławska, W. Nitek and H. Marona, Bioorg. Med. Chem., 2016, 24(8), 1793–1810 CrossRef CAS PubMed.
- A. M. Waszkielewicz, M. Cegła, E. Żesławska, W. Nitek, K. Słoczyńska and H. Marona, Bioorg. Med. Chem., 2015, 23, 4197 CrossRef CAS PubMed.
- A. M. Waszkielewicz, A. Gunia-Krzyżak, B. Powroźnik, K. Słoczyńska, E. Pękala, M. Walczak, M. Bednarski, E. Żesławska, W. Nitek and H. Marona, Bioorg. Med. Chem., 2016, 24, 1793 CrossRef CAS PubMed.
- Bratislava, Slovac Repub. Molinspiration Cheminformatics, http://www.molinspiration.com/services/properties.html, (accessed January 2015).
- D. V. Veber, S. R. Johnson, H.-Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, J. Med. Chem., 2002, 45, 2615 CrossRef CAS PubMed.
- C. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2001, 46, 3 CrossRef CAS PubMed.
- A. M. Waszkielewicz, K. Pytka, A. Rapacz, E. Wełna, M. Jarzyna, G. Satała, A. Bojarski, J. Sapa, P. Żmudzki, B. Filipek and H. Marona, Chem. Biol. Drug Des., 2015, 85, 326 CAS.
- J. Augstein, W. Austin, R. Boscott, S. Green and C. Worthing, J. Med. Chem., 1965, 8, 356 CrossRef CAS PubMed.
- M. M. Schmid, R. W. Freudenmann, F. Keller, B. J. Connemann, C. Hiemke, M. Gahr, W. Kratzer, M. Fuchs and C. Schönfeldt-Lecuona, Pharmacopsychiatry, 2013, 46, 63 CAS.
- R. M. Nanau and M. G. Neuman, Clin. Biochem., 2013, 46, 1323 CrossRef CAS PubMed.
- J. M. Sendra, T. T. Junyent and M. J. R. Pellicer, Ann. Pharmacother., 2011, 45, 32 CrossRef PubMed.
- S. Doğan, S. Ozberk and A. Yurci, Eur. J. Gastroenterol. Hepatol., 2011, 23, 628 CrossRef PubMed.
- A. Krishna, M. Biryukov, C. Trefois, P. M. A. Antony, R. Hussong, J. Lin, M. Heinäniemi, G. Glusman, S. Köglsberger, O. Boyd, B. H. J. van den Berg, D. Linke, D. Huang, K. Wang, L. Hood, A. Tholey, R. Schneider, D. J. Galas, R. Balling and P. May, BMC Genomics, 2014, 15, 1154 CrossRef PubMed.
- S. H. Kwon, J. A. Kim, S. I. Hong, Z. H. Jung, H. C. Kim, S. Y. Lee and C. G. Jang, Neurochem. Int., 2011, 58, 533 CrossRef CAS PubMed.
- G. Cruciani, E. Carosati, B. De Boeck, K. Ethirajulu, C. Mackie, T. Howe and R. Vianello, J. Med. Chem., 2005, 48, 6970 CrossRef CAS PubMed.
- I. Zamora, L. Afzelius and G. Cruciani, J. Med. Chem., 2003, 46, 2313 CrossRef CAS PubMed.
- P. Newman, in Optical resolution procedures for chemical compounds, Optical Resolution Information Center, Manhattan College Riverdale, New York, 1984, vol. 1, Amines Search PubMed.
- W. Löscher, C. P. Fassbender and B. Nolting, Epilepsy Res., 1991, 8, 79 CrossRef.
- J. Litchfield and F. Wilcoxon, J. Pharmacol. Exp. Ther., 1949, 96, 99 CAS.
- N. Dunham, T. Miya and L. Edwards, J. Pharmacol. Exp. Ther., 1957, 46, 64 CAS.
- A. Rapacz, S. Rybka, J. Obniska, K. Sałat, B. Powroźnik, E. Pękala and B. Filipek, Naunyn-Schmiedeberg's Arch. Pharmacol., 2016, 389, 339 CrossRef CAS PubMed.
- K. Socała, D. Nieoczym, M. Pieróg and P. Wlaź, J. Neural Transm., 2015, 122, 1239 CrossRef PubMed.
- G. Ferreri, A. Chimirri, E. Russo, R. Gitto, P. Gareri, A. De Sarro and G. De Sarro, Pharmacol., Biochem. Behav., 2004, 77, 85 CrossRef CAS.
- T. M. Laughlin, K. V. Tram, G. L. Wilcox and A. K. Birnbaum, J. Pharmacol. Exp. Ther., 2002, 302, 1168 CrossRef CAS PubMed.
- A. Rapacz, J. Obniska, B. Wiklik-Poudel, S. Rybka, K. Sałat and B. Filipek, Eur. J. Pharmacol., 2016, 781, 239 CrossRef CAS PubMed.
- J. R. Boissier, J. Tardy and J. C. Diverres, J. Exp. Med., 1960, 3, 81 CAS.
- G. Repetto, A. del Peso and J. L. Zurita, Nat. Protoc., 2008, 3, 1125 CrossRef CAS PubMed.
- M. Regulska, B. Pomierny, A. Basta-Kaim, A. Starek, M. Filip, W. Lasoń and B. Budziszewska, Pharmacol. Rep., 2010, 62, 1243 CrossRef CAS PubMed.
Footnotes |
| † The authors declare no competing interests. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c6md00537c |
| This journal is © The Royal Society of Chemistry 2017 |