
T- and B-cell immunosuppressive activity of novel α-santonin analogs with humoral and cellular immune response in Balb/c mice†‡
Nisar A.
Dangroo
a,
Jasvinder
Singh
bc,
Nidhi
Gupta
a,
Shashank
Singh
bc,
Anapurna
Kaul
b,
Mohmmed A.
Khuroo
d and
Payare L.
Sangwan
*ac
aBioorganic Chemistry Division, Canal Road, Jammu-180001, India
bCancer Pharmacology Division, CSIR-Indian Institute of Integrative Medicine, Canal Road, Jammu-180001, India
cAcademy of Scientific and Innovative Research (AcSIR), CSIR-IIIM Campus, Jammu, India. E-mail: plsangwan@iiim.ac.in; Fax: +91 191 2586333; Tel: +91 191 2585006-13 Extn. 371
dUniversity of Kashmir, Hazaratbal Road, Srinagar, India
First published on 11th November 2016
Abstract
In continuation of our endeavours to synthesize immunosuppressive agents from α-santonin, we report herein the design and synthesis of a new series of α-santonin derived O-aryl/aliphatic ether, ester and amide analogs and the evaluation of their immunosuppressive activities. The in vitro studies led to several analogs with significant immunosuppressive effects by inhibiting ConA and LPS stimulated T- and B-cell proliferation in a dose dependent manner. The more significant compounds 4d, 4e, 4f, 4h, 6a and 6b displayed potent inhibitory activity on the mitogen-induced T- and B-cell proliferation in comparison to α-santonin 1. Compound 4e displayed stupendous in vitro immunosuppressive effects with ∼80% suppression of B and ∼75% suppression of T lymphocyte proliferation, respectively. The in vivo investigation on BALB/c mice revealed that non-cytotoxic compound 4e suppresses both humoral and cellular immunity.
1. Introduction
Natural products (NPs) have proved to be promising sources of leads for drug discovery, and in fact, one-third of the top-selling drugs in the world are either NPs or NP-derived.1,2 In the field of immunology, the discovery of cyclosporin A from the fungus Cylindrocarpon lucidum revolutionised organ transplantation.3 Since then, NPs have played a dominant role in the development of most efficient immunosuppressive drugs, including cortisol, tacrolimus, rapamycin, rituximab, everolimus, as well as a number of NPs and their analogs, which are under clinical trials.4 The T lymphocytes undoubtedly play a key role in the initiation of an immune response in transplant rejection and other autoimmune diseases. T-cell proliferation results from activation by antigen-presenting cells (APCs) in combination with the major histocompatibility complex class II and B7 complex. This mechanism results in the activation of calcineurin, which leads to the production of interleukin-2 (IL2). Autocrine stimulus by interleukin-2 results in T-cell proliferation.5 Immunosuppressive drugs, for instance cyclosporin A, inhibit the abnormal activation and proliferation of T lymphocytes and the immune system associated with organ transplantation and other cell-mediated autoimmune diseases.6 Side effects associated with these drugs, along with their high cost, have led the need for the investigation of alternative drugs, especially those belonging to the traditional system of medicine, with better safety profiles.The plant Artemisia laciniata (asteraceae family) is usually found at altitudes from 2700 to 6300 m in Himalayan regions and, in India, it is widely distributed in Jammu and Kashmir, Leh, Ladakh, and Uttar Pradesh.7 The plant has been traditionally used to treat jaundice, gall bladder, high fever and blood purification.8 α-Santonin, a major chemical constituent of the plant, possesses several biological activities, including anticancer, antifungal and anti-inflammatory activities.9–11 We have explored the immunosuppressive activity of α-santonin derived triazoles in our previous communications.12,13 These findings indicate that α-santonin symbolizes an ideal scaffold for the synthesis of diverse analogues for exploration of their structure activity relationship (SAR). Acetyl α-desmotroposantonin 2 exhibited more potent anti-proliferation effects against lymphocytes compared to the parent molecule,12 and this indicated that compound 2 could be a useful scaffold for the synthesis of new chemical entities (NCEs) to understand SAR for the immunosuppressive activity. Therefore, in continuation of our research interest for the design and synthesis of potent NCEs through the structural modification of NPs,12–15 compound 2 was used as a starting material, and a series of new α-santonin derived O-aryl/aliphatic ether, ester and amide analogs were synthesized for the evaluation of the effect of carbon chain length on immunosuppressive activity in the present study. Several analogs displayed potent immunosuppressive activity and compound 4e, showing stupendous in vitro activity, was further investigated for in vivo activity in BALB/c mice models for cellular immune response and sheep red blood cell (SRBC) induced humoral antibody production. Both in vitro and in vivo results demonstrate the immunosuppressive activity of compound 4e.
2. Results and discussion
2.1. Chemistry
α-Santonin 1 was isolated from the dichloromethane–methanol extract of the aerial parts of Artemisia laciniata. Compounds 2 and 3 were synthesised as reported in our previous communication.12 Briefly, NP santonin 1 was subjected to the dienone phenone type reaction using Ac2O/H2SO4 to get acetyl α-desmotroposantonin 2, which on deacetylation in NH3: MeOH, furnished α-desmotroposantonin 3. The ester analogs 4a–e were prepared by treating 3 with the appropriate anhydride in dry dichloromethane (DCM) in the presence of pyridine at room temperature, while analogs 4f–h were synthesized by treatment of 3 with the appropriate acid chloride in dry dichloromethane in good to excellent yields (Scheme 1).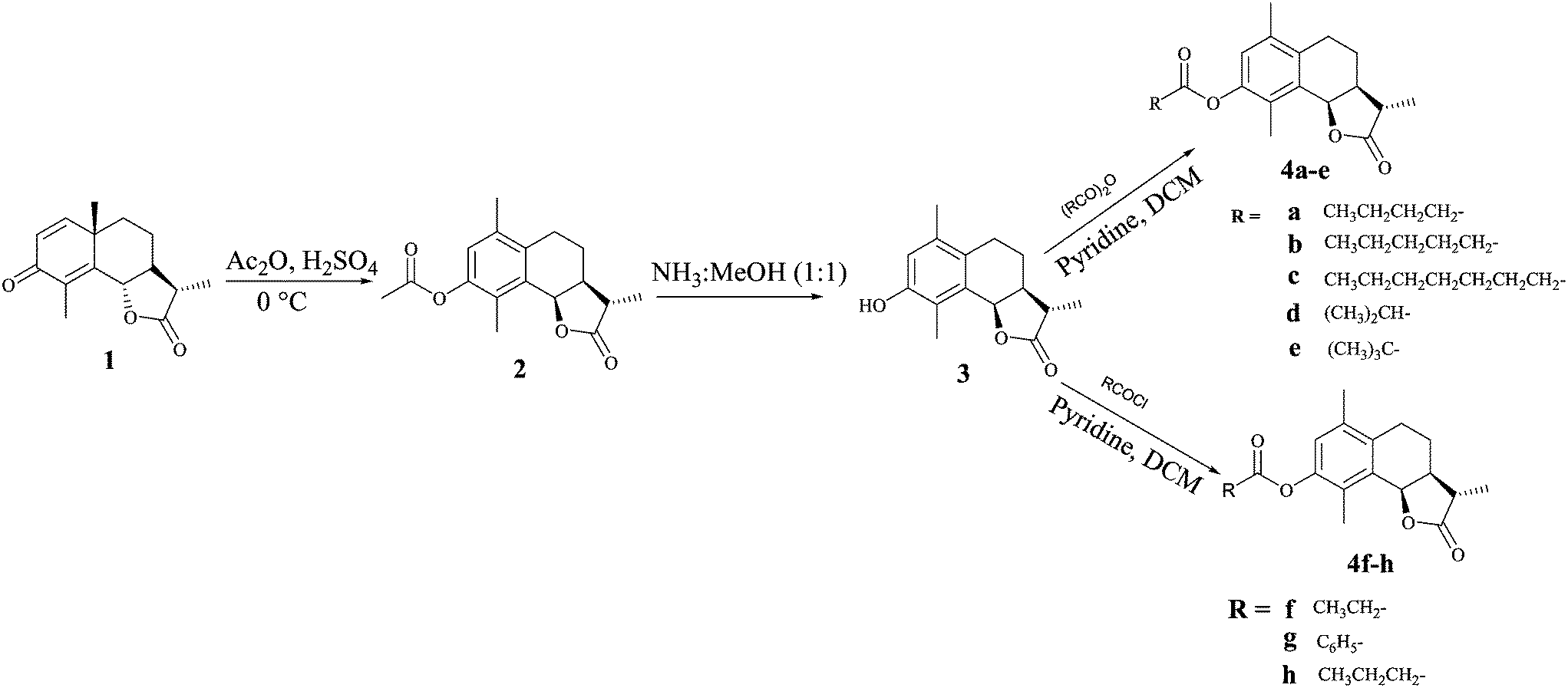 | ||
| Scheme 1 Synthetic route for compounds 4a–h. | ||
Compound 5 was prepared by reacting 3 with succinic anhydride in dry DCM, by employing different bases like NaHCO3, K2CO3, pyridine, Et3N, dimethyl amino pyridine (DMAP) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU). However, the best results were obtained in DMAP. Compound 5, on reaction with SOCl2 in dry DCM under reflux resulted in the in situ generation of the acid chloride, followed by the addition of the appropriate amine, afforded compounds 6a–c (Scheme 2).
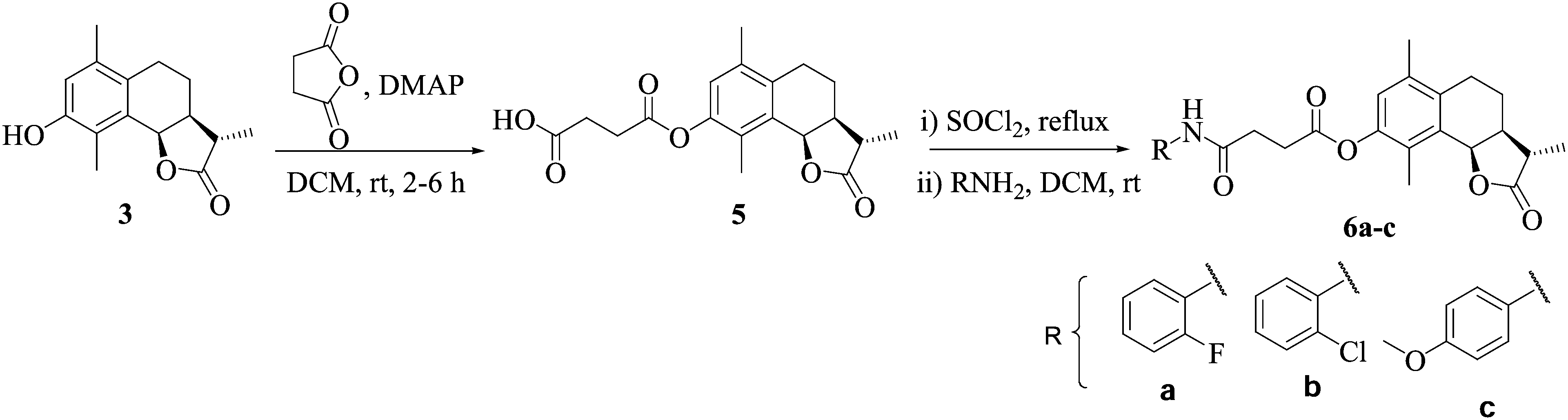 | ||
| Scheme 2 Synthetic route for compounds 6a–c. | ||
Ether analogs were synthesised by the treatment of 3 with different alkyl halides in DCM in the presence of pyridine at room temperature to afford 7a–g (Scheme 3). All the analogs were characterized by 1H NMR, 13C NMR and HRESIMS.
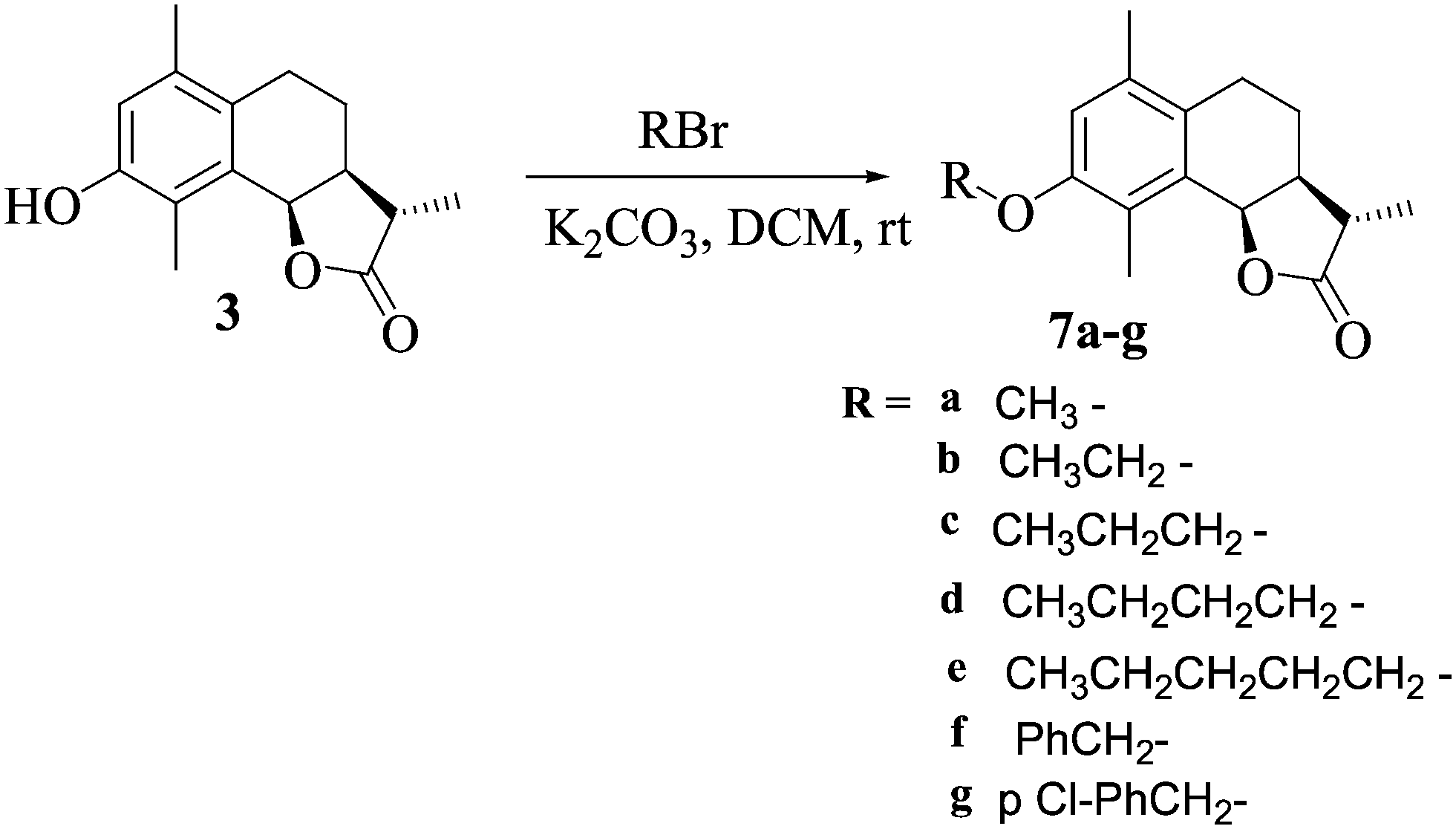 | ||
| Scheme 3 Synthetic route for compounds 7a–g. | ||
2.2. Biological activity
| Compounds | Conc. (μM) | Cytotoxicity | LPS mean ± SE | LPS-induced B-cell proliferation rate (%) | Con-A mean ± SE | ConA-induced T-cell proliferation rate (%) |
|---|---|---|---|---|---|---|
| ↑ indicates immune stimulant agents and ↓ indicates immunosuppressive agents. + indicates cytotoxic and − indicates non-cytotoxic. Results are shown as means of three readings and standard error by Student's ‘t’ test (±S.E.) for three separate experiments, conducted in triplicate at three different 1, 10 and 100 μM concentrations. | ||||||
| The numbers with bold values represent those compounds that have displayed potent immunosuppressive activity. | ||||||
| Blank | − | 0.06 ± 0.00 | − | 0.04 ± 0.00 | − | |
| Control | − | 1.13 ± 0.12 | − | 0.60 ± 0.01 | − | |
| 1 | 100 | − | 0.49 ± 0.35 | 33.25↓ | 0.21 ± 0.02 | 8.16↓ |
| 10 | − | 0.50 ± 0.02 | 27.12↓ | 1.10 ± 0.45 | 3.55↑ | |
| 1 | − | 1.23 ± 0.76 | 19.00↓ | 0.80 ± 0.03 | 35.36↓ | |
| 2 | 100 | + | 0.38 ± 0.04 | 67.99↓ | 0.64 ± 0.19 | 63.00↓ |
| 10 | − | 0.73 ± 0.25 | 44.27↓ | 0.30 ± 0.09 | 45.24↓ | |
| 1 | − | 0.50 ± 0.04 | 51.83↓ | 0.16 ± 0.01 | 39.72↓ | |
| 3 | 100 | + | 0.50 ± 0.02 | 38.25↑ | 0.21 ± 0.00 | 58.13↑ |
| 10 | + | 0.91 ± 0.02 | 24.87↓ | 0.25 ± 0.00 | 27.35↓ | |
| 1 | + | 0.89 ± 0.02 | 23.28↓ | 0.32 ± 0.08 | 22.05↓ | |
| 4a | 100 | + | 0.90 ± 0.12 | 6.25↑ | 0.91 ± 0.16 | 28.16↑ |
| 10 | − | 0.49 ± 0.04 | 48.95↓ | 0.50 ± 0.31 | 29.57↓ | |
| 1 | − | 0.94 ± 0.18 | 2.08↓ | 0.15 ± 0.00 | 43.23↓ | |
| 4b | 100 | + | 1.03 ± 0.10 | 7.29↑ | 1.25 ± 0.07 | 78.87↓ |
| 10 | + | 0.71 ± 0.01 | 26.04↓ | 1.70 ± 0.29 | 46.05↓ | |
| 1 | − | 0.74 ± 0.33 | 22.91↓ | 0.19 ± 0.01 | 39.43↓ | |
| 4c | 100 | + | 1.51 ± 0.38 | 57.29↓ | 0.74 ± 0.14 | 4.22↓ |
| 10 | − | 1.47 ± 0.36 | 53.12↑ | 1.49 ± 0.53 | 43.12↓ | |
| 1 | − | 0.60 ± 0.27 | 37.5↓ | 0.21 ± 0.00 | 50.42↓ | |
| 4d | 100 | − | 1.46 ± 0.06 | 52.08↓ | 1.23 ± 0.09 | 73.23↓ |
| 10 | − | 1.16 ± 0.34 | 33.33↓ | 0.16 ± 0.00 | 61.46↓ | |
| 1 | − | 0.64 ± 0.30 | 20.83↑ | 0.14 ± 0.01 | 53.28↓ | |
| 4e | 100 | − | 0.22 ± 0.00 | 80.53↓ | 0.15 ± 0.01 | 75.00↓ |
| 10 | − | 0.38 ± 0.13 | 66.00↓ | 0.25 ± 0.01 | 58.11↓ | |
| 1 | − | 0.58 ± 0.33 | 48.67↓ | 0.39 ± 0.15 | 35.11↓ | |
| 4f | 100 | + | 0.34 ± 0.15 | 69.00↓ | 0.35 ± 0.16 | 41.00↓ |
| 10 | + | 0.64 ± 0.08 | 43.00↓ | 0.40 ± 0.18 | 33.00↓ | |
| 1 | + | 0.90 ± 0.18 | 20.00↓ | 0.53 ± 0.19 | 11.66↓ | |
| 4g | 100 | − | 0.79 ± 0.07 | 17.70↓ | 0.88 ± 0.16 | 23.94↑ |
| 10 | − | 0.54 ± 0.02 | 43.75↓ | 0.43 ± 0.20 | 39.43↓ | |
| 1 | − | 0.78 ± 0.01 | 18.75↓ | 0.20 ± 0.01 | 71.83↓ | |
| 4h | 100 | − | 0.29 ± 0.14 | 69.79↓ | 0.15 ± 0.00 | 71.87↓ |
| 10 | − | 0.62 ± 015 | 35.41↓ | 0.37 ± 0.16 | 38.00↓ | |
| 1 | − | 0.84 ± 0.04 | 12.50↓ | 0.57 ± 0.28 | 28.50↓ | |
| 5 | 100 | + | 1.01 ± 0.11 | 5.20↓ | 1.29 ± 0.08 | 61.69↓ |
| 10 | + | 0.64 ± 0.06 | 42.70↓ | 0.21 ± 0.00 | 53.42↓ | |
| 1 | − | 0.55 ± 0.02 | 33.33↓ | 0.21 ± 0.01 | 29.42↓ | |
| 6a | 100 | − | 0.25 ± 0.01 | 77.87↓ | 0.20 ± 0.12 | 66.66↓ |
| 10 | − | 0.63 ± 0.36 | 44.00↓ | 0.30 ± 0.10 | 41.66↓ | |
| 1 | − | 0.89 ± 0.24 | 21.00↓ | 0.50 ± 0.12 | 16.66↓ | |
| 6b | 100 | − | 0.36 ± 0.00 | 69.00↓ | 0.21 ± 0.11 | 56.00↓ |
| 10 | − | 0.41 ± 0.13 | 51.00↓ | 0.29 ± 0.32 | 45.00↓ | |
| 1 | − | 0.63 ± 0.27 | 27.69↓ | 0.49 ± 0.21 | 13.00↓ | |
| 6c | 100 | + | 0.26 ± 0.00 | 68.00↓ | 0.25 ± 0.01 | 58.00↓ |
| 10 | − | 0.54 ± 0.17 | 52.00↓ | 0.39 ± 0.07 | 35.00↓ | |
| 1 | − | 0.93 ± 0.17 | 17.69↓ | 0.59 ± 0.20 | 1.00↓ | |
| 7a | 100 | + | 0.89 ± 0.13 | 21.00↓ | 1.30 ± 0.29 | 16.12↓ |
| 10 | + | 0.91 ± 0.02 | 19.10↓ | 1.34 ± 0.15 | 23.25↓ | |
| 1 | + | 0.65 ± 0.21 | 42.00↓ | 0.90 ± 0.32 | 50.00↓ | |
| 7b | 100 | + | 0.61 ± 0.03 | 46.00↓ | 0.70 ± 0.16 | 16.00↑ |
| 10 | + | 1.15 ± 0.31 | 1.00↓ | 0.57 ± 0.16 | 5.00↓ | |
| 1 | − | 0.33 ± 0.11 | 35.00↓ | 0.77 ± 0.05 | 28.00↓ | |
| 7c | 100 | + | 0.64 ± 0.08 | 43.00↓ | 0.82 ± 0.19 | 36.00↑ |
| 10 | + | 0.34 ± 0.15 | 49.00↓ | 0.35 ± 0.16 | 41.00↓ | |
| 1 | − | 0.37 ± 0.18 | 47.00↓ | 0.40 ± 0.18 | 33.00↓ | |
| 7d | 100 | − | 1.11 ± 0.05 | 39.01↓ | 1.28 ± 0.16 | 33.82↓ |
| 10 | − | 1.25 ± 0.10 | 33.31↓ | 0.85 ± 0.07 | 51.00↓ | |
| 1 | − | 1.38 ± 0.09 | 25.17↓ | 0.77 ± 0.14 | 32.05↓ | |
| 7e | 100 | + | 1.70 ± 0.05 | 9.59↓ | 0.96 ± 0.13 | 29.41↓ |
| 10 | + | 1.23 ± 0.05 | 32.41↓ | 0.91 ± 0.09 | 33.08↓ | |
| 1 | + | 1.32 ± 0.09 | 29.47↓ | 1.25 ± 0.18 | 11.08↓ | |
| 7g | 100 | + | 1.46 ± 0.27 | 29.00↑ | 0.24 ± 0.01 | 16.00↑ |
| 10 | − | 0.29 ± 0.05 | 74.00↓ | 1.30 ± 0.57 | 60.00↓ | |
| 1 | − | 0.52 ± 0.28 | 53.00↓ | 0.25 ± 0.00 | 58.00↓ | |
| 7f | 100 | + | 1.45 ± 0.14 | 28.00↓ | 0.93 ± 0.04 | 45.00↓ |
| 10 | − | 0.71 ± 0.47 | 37.00↓ | 0.40 ± 0.11 | 33.00↓ | |
| 1 | − | 0.71 ± 0.47 | 37.00↓ | 0.51 ± 0.28 | 15.00↓ | |
Compound 2, with aromatized ring A, displayed better inhibition against both the mitogens than the parent compound 1. Compound 4c displayed 57% inhibition against LPS induced B-cell proliferation at 100 μM in the ester series (4a–h), while 4b and 4h were found to be more active against Con A and displayed 39–78% and 28–78% suppression of T-cells in the tested dose range, respectively. Non-cytotoxic compound 4d showed potent and uniform suppression of T-cell proliferation, with 53% inhibition at 1 μM and finally attaining ∼73% inhibition at 100 μM. Compound 4e was found to be non-toxic and proved most potent among the ester analogs, with ∼80% suppression of B-cells and 75% inhibition of T lymphocyte proliferation. Compound 4f displayed a uniform suppression and reached up to 69% against LPS induced B-cell and 41% against Con A induced T-cell proliferation at 100 μM, but was cytotoxic. Compound 4g was non cytotoxic, but could not prove potency, while cytotoxic compound 5 displayed ∼61% inhibition of T-cells at 100 μM.
Amide analogs (6a–c) were found to be non-cytotoxic and displayed uniform inhibition against both the mitogens, and among them, 6a showed potent inhibition, reaching up to 77% at 100 μM concentration, against LPS induced B lymphocytes, and 66% against Con-A induced T-cell proliferation. Compound 6b showed 76% suppression of B lymphocytes and 58% suppression of T lymphocytes at 100 μM concentration. Among ethers, compound 7g displayed 74% inhibition of B and 66% inhibition of T lymphocytes without cytotoxicity at 10 μM, but the compound was found to be a stimulant, as well as cytotoxic at higher doses. The rest of the ether analogs displayed less than 50% inhibition along with cytotoxicity and were found less active compared to ester and amide derivatives.
It was evident from the cell proliferation results that ester and amide analogs display better inhibition activity, compared to ether analogs, which indicates that carbonyl groups present in these derivatives play an important role. Among the ester analogs, as the carbon chain length increases, the activity increases in compounds containing chains of up to four carbons (4h), and then decreases rapidly as the carbon chain increases. Further, isobutyrate (4d) and trimethyl acetate (4e) analogs proved more potent compared to straight chain analogs. The compound 6a, bearing an ortho-F group in the amide moiety, exhibited potent activity among the amide analogs.
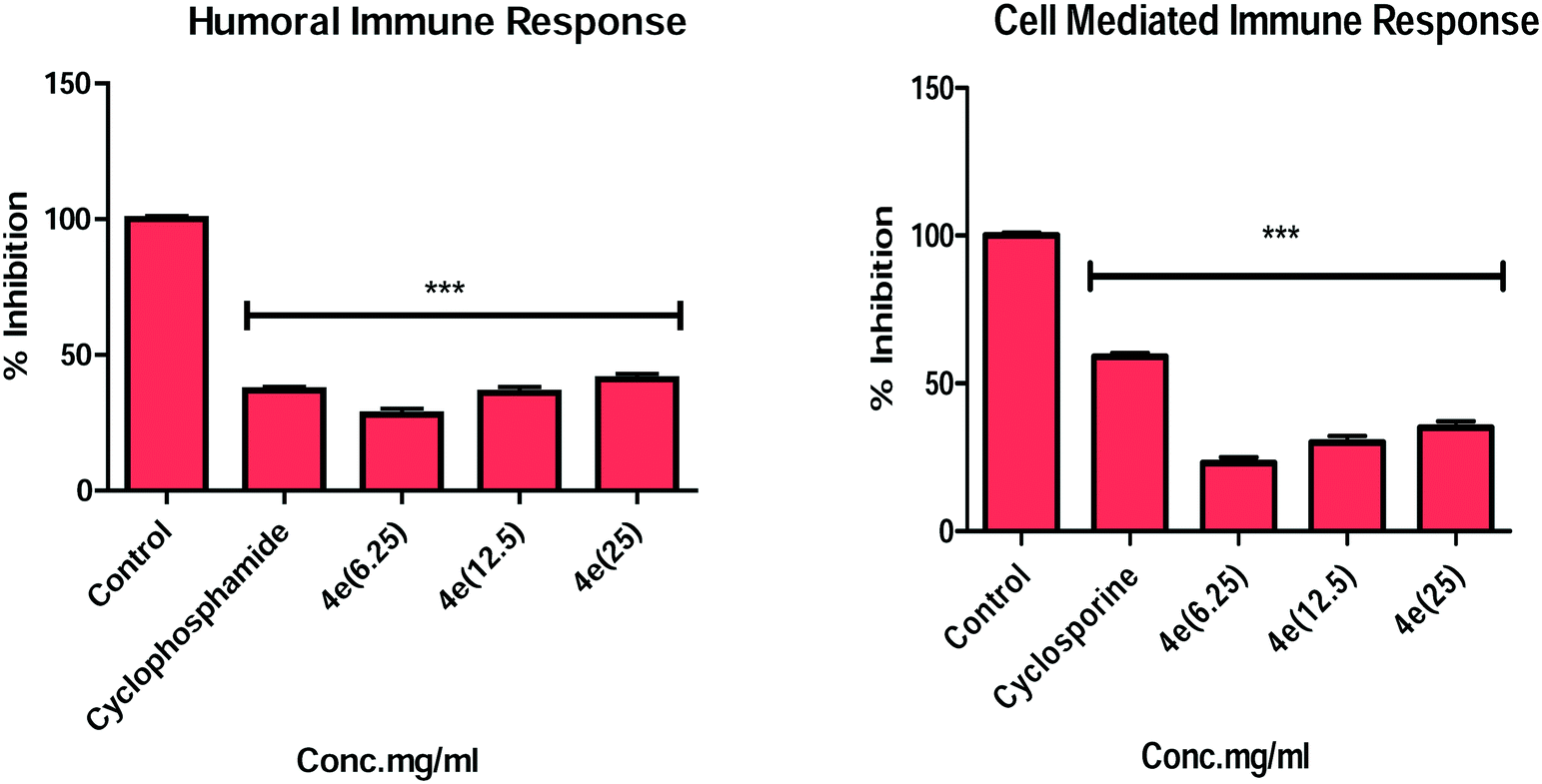 | ||
| Fig. 1 Effect of compound 4e on humoral immunity and DTH response. Data are expressed as means ± SE of five observations. | ||
3. Conclusion
In summary, a series of new α-santonin derived O-aryl/aliphatic ether, ester and amide derivatives were synthesised. All the compounds were investigated for their in vitro immunosuppressive effect on T- and B-lymphocytes of BALB/c mice. Many analogs showed strong inhibition against Con-A and LPS stimulated T-cell and B-cell proliferation in a dose dependent manner. The screening data revealed that the presence of carbonyl groups and appropriate aliphatic side chains play important factors toward contributing to the high immunosuppressive potency in these analogs. More interestingly, compounds 4c, 4d, 4e and 4h exhibited potent in vitro activity and the compound 4e revealed the most immunosuppressive effects. Further, compound 4e was investigated for in vivo delayed-type hypersensitivity (DTH) reaction and antibody production in BALB/c mice models. The in vitro and in vivo immunosuppressive activity suggested that compound 4e suppressed both humoral and cell mediated immunity.4. Experimental
4.1. Chemistry
All the reagents and solvents for executing the present work were obtained from Sigma Aldrich. Dry DCM was prepared by refluxing freshly distilled DCM over CaH2 (5% w/v) for a few hours and then re-distilling it. Dry DCM was stored over 4 Å molecular sieves for future use. Pyridine was dried over KOH for one week, then distilled and stored over 4 Å molecular sieves. All the chemical reactions were monitored by TLC on 0.25 mm silica gel 60 F254 plates (E. Merck) using 2% ceric ammonium sulphate solution as a spraying reagent for the detection of the spots. Purification of compounds was carried out either by crystallization or column chromatography using silica gel 60–120 mesh as the stationary phase. 1H NMR and 13C NMR spectra were recorded on Bruker DPX 400 and DPX 500 instruments using CDCl3 or CD3OD as the solvents, with TMS as the internal standard. The chemical shifts were expressed in δ ppm and coupling constants in Hertz. High resolution mass spectra (HRESIMS) were recorded on an Agilent Technologies 6540 instrument.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) extract of the aerial parts of Artemisia laciniata by column chromatography over silica gel 60–120 mesh, and further purified by crystallization.12
:1 mixture of NH3:MeOH to furnish α-desmotroposantonin 3 with 98% yield, mp: 197 °C, HRESIMS m/z calcd for C15H18O3 [M + H]+ 247.13287, found 247.13195.
:1) extract of the aerial parts of Artemisia laciniata by column chromatography over silica gel 60–120 mesh, and further purified by crystallization.12
:1 mixture of NH3:MeOH to furnish α-desmotroposantonin 3 with 98% yield, mp: 197 °C, HRESIMS m/z calcd for C15H18O3 [M + H]+ 247.13287, found 247.13195.
4.1.4.1. α-Desmotroposantonin pentanoate (4a). White solid; mp: 128 °C; 1H NMR (400 MHz, CDCl3): δ 6.84 (s, 1H, Ar–H), 5.58 (d, J = 6.4 Hz, 1H, ArCHO–), 2.71–2.78 (m, 1H, –CHCHCH3), 2.55–2.61 (m, 2H, ArCH2CH2–), 2.51–2.60 (m, 2H, –CH2CH2CO), 2.36–2.49 (m, 1H, –CHCHCH2–), 2.21 and 2.19 (s, 3H each, 2× Ar–CH3), 1.93–1.96 and 1.69–1.75 (m, 1H each, –CH2CH2CH–), 1.93–1.96 (m, 2H, –CH2CH2CO), 1.42–1.45 (m, 2H, –CH3CH2CH2), 1.40 (d, J = 7.0 Hz, 3H, CH3CH–), 0.98 (t, J = 7.4 Hz, 3H, CH3CH2–); 13C NMR (100 MHz, CDCl3): δ 179.37, 172.25, 147.45, 134.78, 133.87, 131.27, 129.05, 123.93, 75.51, 41.68, 40.40, 34.23, 31.35, 24.76, 24.02, 22.34, 19.53, 14.45, 13.91, 12.09. HRESIMS m/z calcd for C20H26O4 [M + H]+ 331.1904, found 331.1904.
4.1.4.2. α-Desmotroposantonin hexanoate (4b). White solid; mp: 132 °C; 1H NMR (400 MHz, CDCl3): δ 6.84 (s, 1H, Ar–H), 5.58 (d, J = 6.4 Hz, 1H, ArCHO–), 2.71–2.78 (m, 1H, –CHCHCH3), 2.51–2.59 (m, 2H each, ArCH2CH2– and –OCOCH2CH2–), 2.39–2.43 (m, 1H, –CHCHCH2–), 2.21 and 2.20 (s, 3H each, 2× Ar–CH3), 1.91–1.96 and 1.72–1.75 (m, 1H each, –CH2CH2CH–), 1.71–1.73 (m, 2H, CH3CH2CH2–), 1.40 (d, J = 7.0 Hz, 3H, CH3CH–), 0.90–0.95 (t, J = 7.4 Hz, 3H, –CH2CH3); 13C NMR (100 MHz, CDCl3): δ 179.37, 172.25, 147.45, 134.78, 133.87, 131.27, 129.05, 123.93, 75.51, 41.68, 40.40, 34.23, 31.35, 24.76, 24.02, 23.38, 22.34, 19.53, 14.45, 13.91, 12.09. HRESIMS m/z calcd for C21H28O4 [M + H]+ 345.2060, found 345.2077.
4.1.4.3. α-Desmotroposantonin octanoate (4c). White solid; mp: 135 °C; 1H NMR (400 MHz, CDCl3): δ 6.85 (s, 1H, Ar–H), 5.58 (d, J = 6.4 Hz, 1H, ArCHO–), 2.71–2.77 (m, 1H, –CHCHCH3), 2.50–2.59 (m, 2H each, ArCH2CH2– and –OCOCH2CH2–), 2.39–2.44 (m, 1H, –CHCHCH2–), 2.21 and 2.20 (s, 3H each, 2× Ar–CH3), 1.91–1.97 and 1.72–1.75 (m, 1H each, –CH2CH2CH–), 1.40 (d, J = 7.0 Hz, 3H, CH3CH–), 1.22–1.46 (m, 10H, –CH2(CH2)5CH3), 0.90 (t, J = 7.4 Hz, 3H, –CH2CH3); 13C NMR (100 MHz, CDCl3): δ 179.37, 172.37, 147.50, 134.73, 133.87, 131.27, 129.06, 123.93, 75.47, 41.68, 40.40, 34.26, 31.65, 29.14, 28.91, 25.06, 24.01, 23.36, 22.59, 19.53, 14.45, 14.05, 12.09. HRESIMS m/z calcd for C23H32O4 [M + H]+ 373.2373, found 373.2406.
4.1.4.4. α-Desmotroposantonin isobutyrate (4d). White solid; mp: 121 °C; 1H NMR (400 MHz, CDCl3): δ 6.83 (s, 1H, Ar–H), 5.58 (d, J = 6.4 Hz, 1H, ArCHO–), 2.81–2.89 (m, 1H, –COCH(CH3)2), 2.71–2.78 (m, 1H, –CHCHCH3), 2.55–2.61 (m, 2H, ArCH2CH2–), 2.36–2.49 (m, 1H, –CHCHCH2–), 2.20 and 2.17 (s, 3H each, 2× Ar–CH3), 1.93–1.96 and 1.69–1.75 (m, 1H each, –CH2CH2CH–), 1.39 (d, J = 7.0 Hz, 3H, CH3CH–), 1.31 (d, J = 6.4 Hz, 6H, CH3CHCH3); 13C NMR (100 MHz, CDCl3): δ 179.37, 175.25, 147.45, 134.78, 133.87, 131.27, 129.05, 123.24, 75.51, 41.68, 40.40, 34.15, 23.97, 23.40, 19.51, 19.10, 19.03, 14.47, 12.0, HRESIMS m/z calcd for C19H24O4 [M + H]+ 317.1747, found 317.1759.
4.1.4.5. α-Desmotroposantonin trimethyl acetate (4e). White solid; mp: 127 °C; 1H NMR (400 MHz, CDCl3): δ 6.94 (s, 1H, Ar–H), 5.59 (d, J = 6.4 Hz, 1H, ArCHO–), 2.70–2.78 (m, 1H, –CHCHCH3), 2.55–2.61 (m, 2H, ArCH2CH2–), 2.51–2.60 (m, 2H, –CH2CH2CO), 2.36–2.49 (m, 1H, –CHCHCH2–), 2.26 and 2.22 (s, 3H each, 2× Ar–CH3), 1.93–1.96 and 1.69–1.75 (m, 1H each, –CH2CH2CH–), 1.93–1.96 (m, 2H, –CH2CH2CO), 1.42–1.45 (m, 2H, –CH3CH2CH2), 1.55 (s, 9H, C(CH3)3), 1.40 (d, J = 7.0 Hz, 3H, CH3CH–); 13C NMR (100 MHz, CDCl3): δ 179.37, 167.73, 147.69, 134.78, 134.05, 131.20, 129.20, 123.93, 75.46, 41.68, 40.35, 38.04, 27.68 (3× CH3), 23.76, 23.27, 19.17, 13.91, 13.75. HRESIMS m/z calcd for C20H26O4 [M + H]+ 331.1904, found 331.1887.
4.1.5.1. α-Desmotroposantonin propanoate (4f). White solid; mp: 132 °C; 1H NMR (400 MHz, CDCl3): δ 6.84 (s, 1H, Ar–H), 5.58 (d, J = 6.4 Hz, 1H, ArCHO–), 2.71–2.78 (m, 1H, –CHCHCH3), 2.51–2.59 (m, 2H, ArCH2CH2–), 2.55–2.61 (m, 2H, –OCOCH2CH3), 2.39–2.43 (m, 1H, –CHCHCH2–), 2.22 and 2.20 (s, 3H each, 2× Ar–CH3), 1.93–1.96 and 1.72–1.75 (m, 1H each, –CH2CH2CH–), 1.40 (d, J = 7.0 Hz, 3H, CH3CH–), 1.30–1.35 (t, J = 7.4 Hz, 3H, –CH2CH3); 13C NMR (100 MHz, CDCl3): δ 179.37, 173.25, 147.45, 134.78, 133.87, 131.27, 129.05, 123.93, 75.51, 41.68, 40.40, 27.21, 24.01, 23.38, 19.53, 14.45, 12.09, 9.29. HRESIMS m/z calcd for C18H22O4 [M + H]+ 303.1591, found 303.1596.
4.1.5.2. α-Desmotroposantonin benzoate (4g). White solid; mp: 142 °C; 1H NMR (400 MHz, CDCl3): δ 8.21–8.23 (m, 2H, 2× Ar1–H), 7.60–7.67 (m, 1H, Ar1–H), 7.46–7.55 (m, 2H, 2× Ar1–H), 6.99 (s, 1H, Ar–H), 5.62 (d, J = 6.4 Hz, 1H, ArCHO–), 2.74–2.81 (m, 1H, –CHCHCH3), 2.53–2.61 (m, 2H, ArCH2CH2–), 2.42–2.48 (m, 1H, –CHCHCH2–), 2.27 and 2.25 (s, 3H each, 2× Ar–CH3), 1.93–1.99 and 1.74–1.81 (m, 1H each, –CH2CH2CH–), 1.40 (d, J = 7.0 Hz, 3H, CH3CH–); 13C NMR (100 MHz, CDCl3): δ 179.37, 165.25, 147.45, 134.78, 134.14, 133.87, 131.27, 130.17, 129.31(2× CH), 129.05, 128 (2× CH), 124.03, 75.51, 41.68, 40.40, 24.01, 23.38, 19.53, 14.45, 12.09. HRESIMS m/z calcd for C22H22O4 [M + H]+ 351.1591, found 351.1597.
4.1.5.3. α-Desmotroposantonin butanoate (4h). White solid; mp: 144 °C; 1H NMR (400 MHz, CDCl3): δ 6.84 (s, 1H, Ar–H), 5.58 (d, J = 6.4 Hz, 1H, ArCHO–), 2.70–2.77 (m, 1H, –CHCHCH3), 2.51–2.59 (m, 2H each, ArCH2CH2– and –OCOCH2CH2–), 2.39–2.43 (m, 1H, –CHCHCH2–), 2.21 and 2.20 (s, 3H each, 2× Ar–CH3), 1.91–1.96 and 1.72–1.75 (m, 1H each, –CH2CH2CH–), 1.71–1.73 (m, 2H, CH3CH2CH2–), 1.40 (d, J = 7.0 Hz, 3H, CH3CH–), 1.08 (t, J = 7.4 Hz, 3H, –CH2CH3); 13C NMR (100 MHz, CDCl3): δ 179.37, 172.25, 147.45, 134.78, 133.87, 131.27, 129.05, 123.93, 75.51, 41.68, 40.40, 36.11, 24.01, 23.38, 19.53, 18.60, 14.45, 13.76, 12.09. HRESIMS m/z calcd for C19H24O4 [M + H]+ 317.1747, found 317.1779.
4.1.7.1. 4-α-Desmotroposantoninyl-(2-fluorophenyl)-4-oxobutanmide (6a). White solid; mp: 123 °C; 1H NMR (400 MHz, CDCl3): δ 8.02 (s, 1H, NH), 7.27 (m, 4H, 4× Ar1–H), 6.90 (s, 1H, Ar–H), 5.65 (d, J = 6.4 Hz, 1H, ArCHO–), 3.01–3.04 (m, 2H, –NHCOCH2CH2), 2.93–2.97 (m, 2H, –CH2CH2COO), 2.73–2.79 (m, 1H, –CHCHCH3), 2.59–2.65 (m, 2H, ArCH2CH2–), 2.36–2.49 (m, 1H, –CHCHCH2–), 2.25 and 2.24 (s, 3H each, 2× Ar–CH3), 1.91–1.95 and 1.63–1.71 (m, 1H each, –CH2CH2CH–), 1.41 (d, J = 6.4 Hz, 3H, CH3CH–); 13C NMR (100 MHz, CDCl3): δ 180.56, 171.87, 169.52, 147.30, 134.94, 134.22, 130.96, 129.88, 128.92, 124.78, 123.82, 121.24, 117.55, 115.12, 114.96, 75.99, 41.76, 40.61, 31.45, 28.50, 23.89, 23.26, 19.29, 14.19, 11.86. HRESIMS m/z calcd for C25H26FNO5 [M + H]+ 440.1868, found 440.1808.
4.1.7.2. 4-α-Desmotroposantoninyl-(2-chlorophenyl)-4-oxobutanmide (6b). White solid; mp: 142 °C; 1H NMR (400 MHz, CDCl3): δ 8.35 (s, 1H, NH), 7.36 (d, J = 8.2 Hz, 1H, Ar1–H), 7.26–7.29 (m, 2H, 2× Ar1–H), 7.03–7.06 (m, 1H, Ar1–H), 6.88 (s, 1H, Ar–H), 5.57 (d, J = 6.4 Hz, 1H, ArCHO–), 3.04–3.07 (m, 2H, –NHCOCH2CH2), 2.94–2.97 (m, 2H, –CH2CH2COO), 2.71–2.78 (m, 1H, –CHCHCH3), 2.55–2.61 (m, 2H, ArCH2CH2–), 2.36–2.49 (m, 1H, –CHCHCH2–), 2.25 and 2.23 (s, 3H each, 2× Ar–CH3), 1.93–1.96 and 1.69–1.75 (m, 1H each, –CH2CH2CH–), 1.40 (d, J = 6.4 Hz, 3H, CH3CH–); 13C NMR (100 MHz, CDCl3): δ 179.45, 171.44, 169.52, 147.45, 134.86, 134.46, 134.16, 131.27, 130.82 129.88, 129.07, 127.72, 124.78, 123.82, 121.24, 75.51, 41.62, 40.44, 32.18, 29.32, 24.02, 23.37, 19.56, 14.49, 12.17. HRESIMS m/z calcd for C25H26ClNO5 [M + H]+ 456.1572, found 456.1589.
4.1.7.3. 4-α-Desmotroposantoninyl-(4-methoxyphenyl)-4-oxobutanmide (6c). White solid; mp: 147 °C; 1H NMR (400 MHz, CDCl3): δ 7.05 (s, 1H, NH), 7.37 (d, J = 8.4 Hz, 2× Ar1–H), 6.86 (s, 1H, Ar–H), 6.83 (d, J = 8.4 Hz, 2× Ar1–H), 5.55 (d, J = 6.0 Hz, 1H, ArCHO–), 3.78 (s, 3H, OCH3), 3.01–3.04 (m, 2H, –NHCOCH2CH2), 2.71–2.75 (m, 2H, –CH2CH2COO), 2.65–2.70 (m, 1H, –CHCHCH3), 2.50–2.61 (m, 2H, ArCH2CH2–), 2.39–2.41 (m, 1H, –CHCHCH2–), 2.19 and 2.18 (s, 3H each, 2× Ar–CH3), 1.91–1.95 and 1.69–1.74 (m, 1H each, –CH2CH2CH–), 1.38 (d, J = 6.4 Hz, 3H, CH3CH–); 13C NMR (125 MHz, CDCl3) δ 179.56, 171.93, 169.30, 156.34, 147.34, 134.92, 134.18, 131.25, 130.95, 129.04, 123.82, 121.77 (2× CH), 114.07 (2× CH), 75.51, 55.48, 41.61, 40.47, 31.74, 29.46, 24.02, 23.36, 19.56, 14.50, 12.18. HRESIMS m/z calcd for C26H29NO6 [M + H]+ 452.2093, found 452.2068.
4.1.8.1. α-Desmotroposantonin methyl ether (7a). White solid; mp: 141 °C; 1H NMR (400 MHz, CDCl3): δ 6.75 (s, 1H, Ar–H), 5.63 (d, J = 6.4 Hz, 1H, ArCHO–), 3.81 (s, 3H, OCH3), 2.70–2.75 (m, 1H, –CHCHCH3), 2.55–2.61 (m, 2H, ArCH2CH2–), 2.37–2.49 (m, 1H, –CHCHCH2–), 2.25 and 2.23 (s, 3H each, 2× Ar–CH3), 1.85–1.93 and 1.69–1.75 (m, 1H each, –CH2CH2CH–), 1.38 (d, J = 7.0 Hz, 3H, CH3CH–); 13C NMR (100 MHz, CDCl3): δ 179.55, 155.67, 134.12, 130.74, 127.43, 125.58, 113.35, 75.83, 55.8, 41.86, 40.54, 23.70, 23.65, 20.04, 14.53, 11.52. HRESIMS m/z calcd for C16H20O3 [M + H]+ 261.1485, found 261.1491.
4.1.8.2. α-Desmotroposantonin ethyl ether (7b). White solid; mp: 113 °C; 1H NMR (400 MHz, CDCl3): δ 6.71 (s, 1H, Ar–H), 5.60 (d, J = 6.4 Hz, 1H, ArCHO–), 3.99 (q, 2H, J = 5.6 Hz, –OCH2CH3), 2.66–2.71 (m, 1H, –CHCHCH3), 2.47–2.54 (m, 2H, ArCH2CH2–), 2.37–2.39 (m, 1H, –CHCHCH2–), 2.27 and 2.21 (s, 3H each, 2× Ar–CH3), 1.87–1.89 and 1.68–1.71 (m, 1H each, –CH2CH2CH–), 1.36–1.41 (m, 3H each, CH3CH–OCH2CH3); 13C NMR (100 MHz, CDCl3): δ 179.74, 155.20, 134.00, 130.71, 127.79, 125.97, 114.76, 75.90, 64.23, 41.87, 40.59, 23.73, 23.70, 19.99, 15.08, 14.53, 11.57. HRESIMS m/z calcd for C17H22O3 [M + H]+ 275.1642, found 275.1638.
4.1.8.3. α-Desmotroposantonin propyl ether (7c). White solid; mp: 128 °C; 1H NMR (400 MHz, CDCl3): δ 6.72 (s, 1H, Ar–H), 5.60 (d, J = 6.4 Hz, 1H, ArCHO–), 3.89 (t, 2H, J = 6.4 Hz, OCH2CH2–), 2.66–2.72(m, 1H, –CHCHCH3), 2.45–2.55 (m, 2H, ArCH2CH2–), 2.36–2.42 (m, 1H, –CHCHCH2–), 2.27 and 2.21 (s, 3H each, 2× Ar–CH3), 1.87–1.92 and 1.68–1.75 (m, 1H each, –CH2CH2CH–), 1.80 (q, 2H, J = 6.0 Hz, –CH2CH3), 1.38 (d, J = 7.6 Hz, 3H, CH3CH–), 1.04 (t, J = 7.2 Hz, 3H, CH3CH2–); 13C NMR (100 MHz, CDCl3): δ 179.77, 155.30, 134.02, 130.69, 127.67, 125.86, 114.57, 75.91, 70.13, 41.85, 40.59, 23.73, 23.69, 22.82, 19.98, 14.52, 11.57, 10.73. HRESIMS m/z calcd for C18H24O3 [M + H]+ 289.1789, found 289.1812.
4.1.8.4. α-Desmotroposantonin butyl ether (7d). White solid; mp: 118 °C; 1H NMR (400 MHz, CDCl3) δ 6.72 (s, 1H, Ar–H), 5.61 (d, J = 6.4 Hz, 1H, ArCHO–), 3.93 (t, J = 7.8 Hz, 2H, –CH2CH2O), 2.67–2.72 (m, 1H, –CHCHCH3), 2.46–2.57 (m, 2H, ArCH2CH2–), 2.36–2.42 (m, 1H, –CHCHCH2–), 2.27 and 2.22 (s, 3H each, 2× Ar–CH3), 1.86–1.94 and 1.67–1.75 (m, 1H each, –CH2CH2CH–), 1.76–1.82 (m, 2H, –CH2CH2CH2O), 1.42–1.45 (m, 2H, CH3CH2CH2–), 1.38 (d, J = 7.0 Hz, 3H, CH3CH–), 0.97 (t, J = 7.8 Hz, 3H, CH3CH2–); 13C NMR (125 MHz, CDCl3): δ 179.70, 155.49, 133.96, 130.69, 127.77, 125.75, 114.65, 75.90, 68.26, 41.86, 40.49, 31.36, 23.55, 23.52, 19.93, 19.31, 14.43, 13.71, 11.45. HRESIMS m/z calcd for C19H26O3 [M + H]+ 303.1955, found 303.1981.
4.1.8.5. α-Desmotroposantonin pentyl ether (7e). White solid; mp: 131 °C; 1H NMR (400 MHz, CDCl3) δ 6.72 (s, 1H, Ar–H), 5.62 (d, J = 6.4 Hz, 1H, ArCHO–), 3.81 (t, J = 7.8 Hz, 2H, –CH2CH2O), 2.67–2.75 (m, 1H, –CHCHCH3), 2.52–2.57 (m, 2H, ArCH2CH2–), 2.37–2.42 (m, 1H, –CHCHCH2–), 2.28 and 2.22 (s, 3H each, 2× Ar–CH3), 1.85–1.90 and 1.67–1.75 (m, 1H each, –CH2CH2CH–), 1.76–1.82 (m, 2H, –CH2CH2CH2O), 1.42–1.45 (m, 2H, –CH2CH2CH2O), 1.38 (d, J = 7.0 Hz, 3H, CH3CH–), 0.93 (t, J = 7.8 Hz, 3H, CH3CH2CH2–); 13C NMR (100 MHz, CDCl3): δ 179.74, 155.35, 134.00, 130.69, 127.63, 125.90, 114.54, 75.79, 68.60, 41.87, 40.55, 29.17, 28.36, 23.73, 232.69, 22.47, 20.00, 14.53, 14.10, 11.56. HRESIMS m/z calcd for C20H28O3 [M + H]+ 317.2011, found 317.2087.
4.1.8.6. α-Desmotroposantonin benzyl ether (7f). White solid; mp: 124 °C; 1H NMR (400 MHz, CDCl3): δ 7.30–7.45 (m, 5H, 5× Ar1–H), 6.80 (s, 1H, Ar–H), 5.62 (d, J = 6.4 Hz, 1H, ArCHO–), 5.05 (s, 2H, Ar1CH2O), 2.70–2.75 (m, 1H, –CHCHCH3), 2.55–2.61 (m, 2H, ArCH2CH2–), 2.37–2.49 (m, 1H, –CHCHCH2–), 2.25 and 2.21 (s, 3H each, 2× Ar–CH3), 1.93–1.96 and 1.69–1.75 (m, 1H each, –CH2CH2CH–), 1.40 (d, J = 7.0 Hz, 3H, CH3CH–); 13C NMR (100 MHz, CDCl3): δ 179.93, 137.57, 134.09, 130.90, 130.74, 128.68 (2× CH), 128.26, 127.84, 127.14 (2× CH), 126.90, 114.83, 75.74, 70.05, 41.85, 40.64, 23.77, 23.73, 20.07, 14.57, 11.83. HRESIMS m/z calcd for C22H24O3 [M + H]+ 337.1798, found 337.1807.
4.1.8.7. α-Desmotroposantonin p-chlorobenzyl ether (7g). White solid; mp: 139 °C; 1H NMR (400 MHz, CDCl3): δ 7.33–7.38 (m, 4H, 4× Ar1–H), 6.76 (s, 1H, Ar–H), 5.63 (d, J = 6.4 Hz, 1H, ArCHO–), 5.01 (s, 2H, Ar1CH2O), 2.70–2.75 (m, 1H, –CHCHCH3), 2.55–2.61 (m, 2H, ArCH2CH2–), 2.37–2.49 (m, 1H, –CHCHCH2–), 2.25 and 2.21 (s, 3H each, 2× Ar–CH3), 1.93–1.96 and 1.69–1.75 (m, 1H each, –CH2CH2CH–), 1.40 (d, J = 7.0 Hz, 3H, CH3CH–); 13C NMR (100 MHz, CDCl3): δ 179.37, 154.73, 136.02, 134.20, 133.51, 130.99, 129.20, 128.72 (2× CH), 128.48 (2× CH), 126.08, 114.86, 75.80, 69.70, 41.83, 40.36, 23.73, 23.68, 20.04, 14.54, 11.79 HRESIMS m/z calcd for C22H23ClO3 [M + H]+ 371.1408, found 371.1412.
4.2. Biology
Mitogens and reagents: concanavalin-A (Con-A), lipopolysaccharide (LPS) and 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide (MTT) were procured from Sigma Aldrich. RPMI (Roswell Park Memorial Institute) 1640 medium and fetal bovine serum (FBS) were purchased from Gibco BRL, Life Technologies (USA).
Acknowledgements
Council of Scientific and Industrial Research (CSIR), New Delhi is well acknowledged for funding support under 12th FYP Project BSC-0108. One of the authors (NAD) is greatly thankful to the CSIR-UGC, New Delhi for the award of senior research fellowship. The authors are also thankful to the staff of instrumentation division of the institute for recording the spectroscopic (NMR, HRESIMS) data.References
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2016, 79, 629–661 CrossRef CAS PubMed.
- G. M. Cragg, P. G. Grothaus and D. J. Newman, J. Nat. Prod., 2014, 77, 703–723 CrossRef CAS PubMed.
- H. Hackstein and A. W. Thomson, Nat. Rev. Immunol., 2004, 4, 24–35 CrossRef CAS PubMed.
- J. Mann, Nat. Prod. Rep., 2001, 18, 417–430 RSC.
- J. A. Kobashigawa and J. K. Patel, Nat. Clin. Pract. Cardiovasc. Med., 2006, 3, 203–212 CrossRef CAS PubMed.
- F. Issa, A. Schiopu and K. J. Wood, Expert Rev. Clin. Immunol., 2010, 6, 155–169 CrossRef CAS PubMed.
- P. Weyerstahl, H. Marschall-Weyerstahl, M. Schröder and V. K. Kaul, J. Essent. Oil Res., 1992, 4, 107–112 CrossRef CAS.
- S. Fahad and A. Bano, Pak. J. Bot., 2012, 44, 165–170 Search PubMed.
- J. Khazir, P. P. Singh, D. M. Reddy, I. Hyder, S. Shafi, S. D. Sawant, G. Chashoo, A. Mahajan, M. S. Alam, A. K. Saxena, S. Aravinda, B. D. Gupta and H. M. S. Kumar, Eur. J. Med. Chem., 2013, 63, 279–289 CrossRef CAS PubMed.
- B. Singh, J. Srivastava, R. Khosa and U. Singh, Folia Microbiol., 2001, 46, 137–142 CrossRef CAS.
- G. Blay, L. Cardona, B. Garcia and J. R. Pedro, Stud. Nat. Prod. Chem., 2000, 24, 53–129 CAS.
- N. A. Dangroo, J. Singh, A. A. Dar, N. Gupta, P. K. Chinthakindi, A. Kaul, M. A. Khuroo and P. L. Sangwan, Eur. J. Med. Chem., 2016, 120, 160–169 CrossRef CAS PubMed.
- P. K. Chinthakindi, P. L. Sangwan, S. Farooq, R. R. Aleti, A. Kaul, A. K. Saxena, Y. L. N. Murthy, R. A. Vishwakarma and S. Koul, Eur. J. Med. Chem., 2013, 60, 365–375 CrossRef CAS PubMed.
- R. Majeed, P. L. Sangwan, P. K. Chinthakindi, I. Khan, N. A. Dangroo, N. Thota, A. Hamid, P. R. Sharma, A. K. Saxena and S. Koul, Eur. J. Med. Chem., 2013, 63, 782–792 CrossRef CAS PubMed.
- I. Khan, S. K. Guru, S. K. Rath, P. K. Chinthakindi, B. Singh, S. Koul, S. Bhushan and P. L. Sangwan, Eur. J. Med. Chem., 2016, 108, 104–116 CrossRef CAS PubMed.
- N. Doherty, Agents Actions, 1981, 11, 237–242 CrossRef CAS PubMed.
- D. S. Nelson and P. Mildenhall, Aust. J. Exp. Biol. Med. Sci., 1968, 46, 33–49 CrossRef CAS PubMed.
- M. Jonsson, M. Jestoi, A. V. Nathanail, U.-M. Kokkonen, M. Anttila, P. Koivisto, P. Karhunen and K. Peltonen, Food Chem. Toxicol., 2013, 53, 27–32 CrossRef CAS PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- K. Kobayashi, K. Kaneda and T. Kasama, Microsc. Res. Tech., 2001, 53, 241–245 CrossRef CAS PubMed.
- F. Malik, J. Singh, A. Khajuria, K. A. Suri, N. K. Satti, S. Singh, M. K. Kaul, A. Kumar, A. Bhatia and G. N. Qazi, Life Sci., 2007, 80, 1525–1538 CrossRef CAS PubMed.
- A. Gupta, A. Khajuria, J. Singh, K. L. Bedi, N. K. Satti, P. Dutt, K. A. Suri, O. P. Suri and G. N. Qazi, Int. Immunopharmacol., 2006, 6, 1543–1549 CrossRef CAS PubMed.
Footnotes |
| † The authors declare no competing interests. |
| ‡ Electronic supplementary information (ESI) available: Spectral data (1H NMR and 13C NMR) and HRESIMS of all compounds are provided in the supporting information. See DOI: 10.1039/c6md00527f |
| This journal is © The Royal Society of Chemistry 2017 |