
Design and synthesis of novel selective estrogen receptor degradation inducers based on the diphenylheptane skeleton†‡
Takashi
Misawa
*a,
Takuma
Fujisato
a,
Yasunari
Kanda
a,
Nobumichi
Ohoka
a,
Takuji
Shoda
a,
Momoko
Yorioka
a,
Makoto
Makishima
b,
Yuko
Sekino
a,
Mikihiko
Naito
a,
Yosuke
Demizu
a and
Masaaki
Kurihara
*a
aNational Institute of Health Sciences Setagaya, Tokyo, 158-8501, Japan. E-mail: misawa@nihs.go.jp; masaaki@nihs.go.jp
bNihon University Itabashi, Tokyo, 173-8610, Japan
First published on 8th December 2016
Abstract
Estrogen receptors (ERs) are a family of nuclear receptors (NRs) that regulate physiological effects such as reproduction and bone homeostasis. It has been reported that approximately 70% of human breast cancers are hormone-dependent and ERα-positive. Recently, novel anti-breast cancer drugs based on different mechanisms of action have received significant attention. In this article, we have designed and synthesized a selective ER degradation inducer based on the diphenylheptane skeleton. Western blotting analysis revealed that PBP-NC10 degraded ERα through the ubiquitin–proteasome system. We also performed computational docking analysis to predict the binding mode of PBP-NC10 to ERα.
Introduction
Estrogen receptor (ER), a member of the nuclear receptor (NR) family of transcription factors, relates to several biological functions such as reproduction and bone physiology.1 The binding of 17β-estradiol (E2) and its metabolites, estrone and estriol, to the ER ligand binding domain (LBD) induces the folding of Helix 12 (H12) and the subsequent interaction with co-activators along the hydrophobic groove, resulting in ER-mediated target gene transcriptions.2 The ER is divided into two subtypes, ERα and ERβ, which are encoded by distinct genes but are highly conserved in their LBD.3 It is well known that approximately 70% of human breast cancers are hormone-dependent and ERα-positive.4 Therefore, several ERα-mediated transcriptional inhibitors such as tamoxifen (ERα antagonist)5 and raloxifene6 are widely used for ERα-positive breast cancer therapy (Fig. 1a).7 Tamoxifen is metabolized to 4-hydroxytamoxifen (4-OHT) with a much higher binding affinity for ERα, and 4-OHT exerts its anti-estrogenic actions.8 However, it has been problematic that long-term administration of tamoxifen might cause drug resistance in breast cancer cells and also increases the risk factors for uterine cancer because of its agonistic effects on the uterus. Therefore, novel anti-breast cancer drugs based on different mechanisms of action have received significant attention. Recently, several methods to induce targeted protein degradation using small molecules have been reported such as PROTAC,9 SNIPER,10 and hydrophobic tag systems.11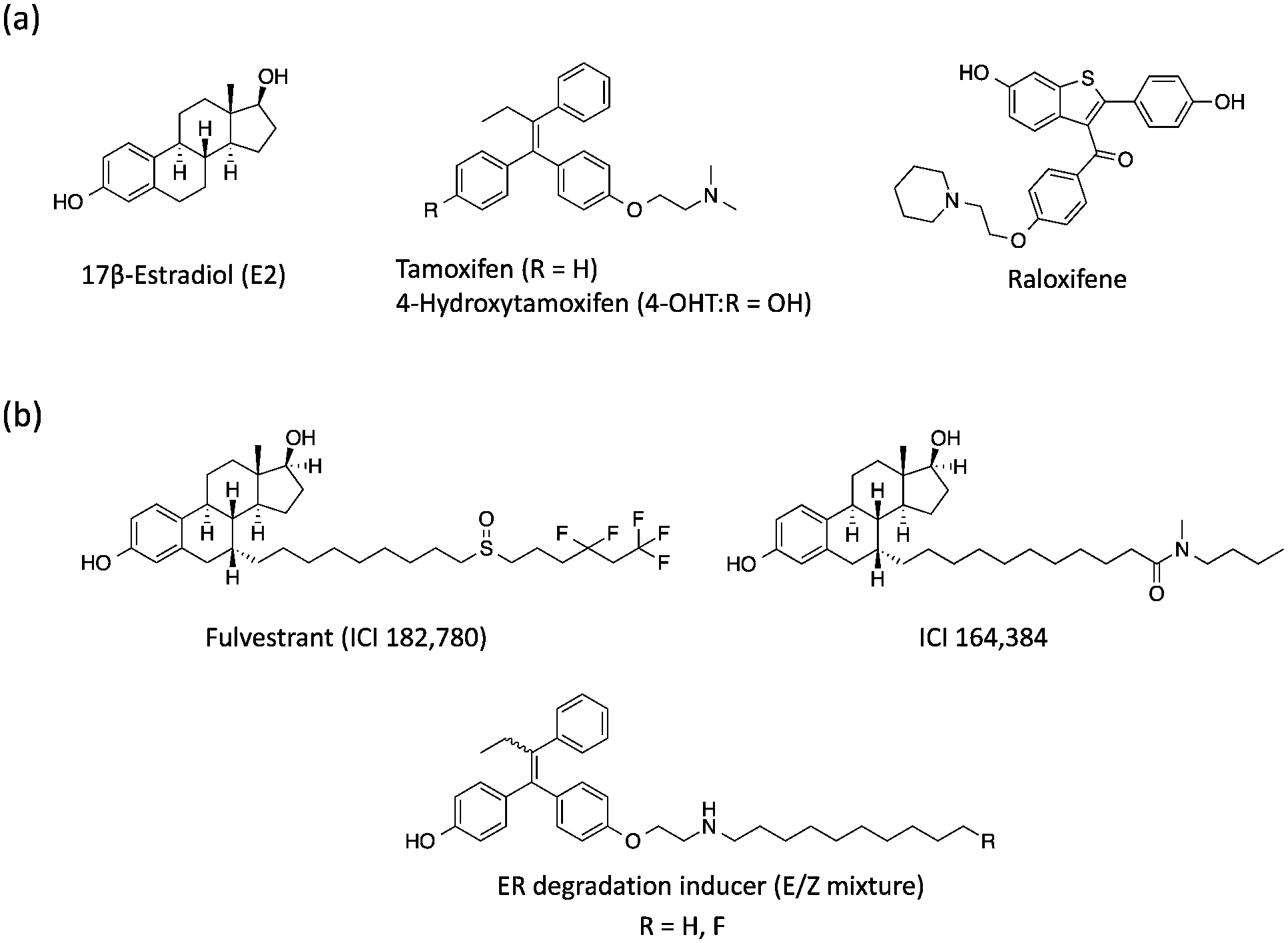 | ||
| Fig. 1 (a) Chemical structures of 17β-estradiol, anti-estrogenic compounds and (b) selective estrogen receptor downregulators. | ||
Among these, selective ER downregulators (SERDs) have been discussed as anti-breast cancer drugs. Fulvestrant (ICI182,780) and ICI164,384 (Fig. 1b), which are E2 derivatives bearing a long alkyl chain on the 7α-position of E2, are representative SERDs and are capable of inducing ER degradation through the ubiquitin–proteasome system (UPS).12 Based on the X-ray structure analysis of ICI164,384 bound to the ER-LBD, the ER-degradation mechanism of ICI164,384 is speculated to involve binding of the long alkyl chain of ICI164,384 to the hydrophobic groove of ER, sterically preventing H12 folding in ER and the co-activator interaction with ER, resulting in destabilization of ER and subsequent degradation.13
Previously, we reported that 4-OHT derivatives bearing long alkyl chains (Fig. 1b) induced ER degradation similar to fulvestrant.14 However, these synthesized compounds were obtained and assayed as a mixture of geometric isomer E and Z forms. It has been reported that the triphenylethylene skeleton of 4-OHT is easily isomerized from the active Z form to an inactive E form in solution.15 Therefore, these findings raise the possibilities that its isomerization decreases the ER-antagonistic and ER-downregulating activities. On the other hand, Hashimoto and Ishikawa et al. recently reported that the diphenylalkane skeleton acted as a steroid skeleton mimic and could be a key to the development of several NR ligands.16 Among these, the ER antagonist PBP based on the diphenylheptane skeleton has potent anti-estrogenic activity with an IC50 value of 4.5 nM from a reporter gene assay.17 Moreover, the diphenylalkane skeleton has an advantage of good chemical stability and for short-step synthesis over the triphenylethylene skeleton of 4-OHT. Herein, we described the development of PBP derivatives bearing several long alkyl chains on the hydroxy group (Fig. 2), and evaluated their biological properties such as ER degradation, binding affinity, transcriptional activity and anti-proliferation activity.
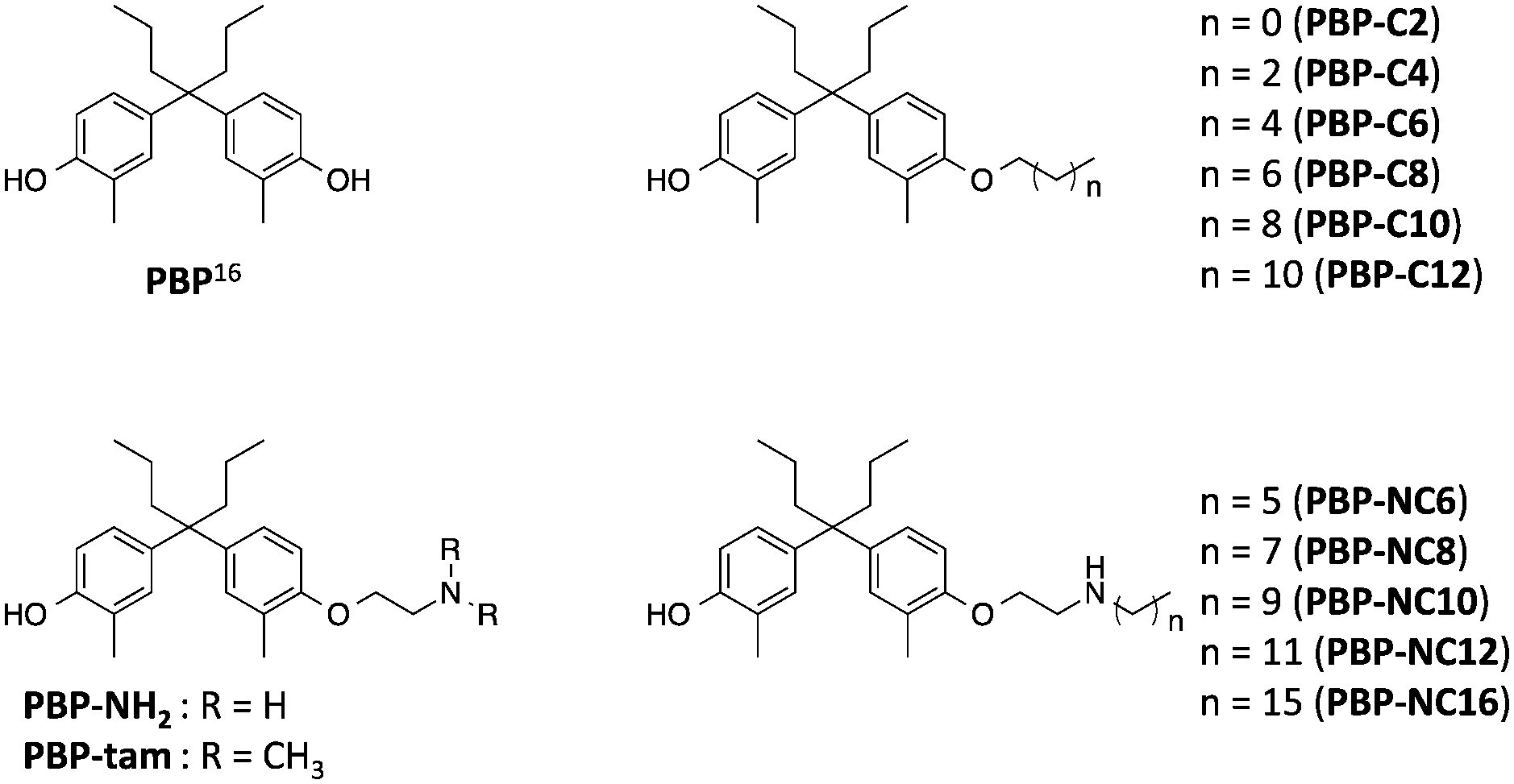 | ||
| Fig. 2 Chemical structures of the diphenylheptane skeleton (PBP) and designed compounds PBP-NH2, PBP-tam, PBP-C2 to PBP-C12, and PBP-NC6 to PBP-NC16 in this study. | ||
Results and discussion
The synthetic routes to the designed compounds PBP-C2–C12, PBP-NC6–NC16, PBP-NH2, and PBP-tam are shown in Scheme 1. 4-Heptanone and o-cresol were combined under acidic conditions to give the diphenylheptane skeleton (PBP, 3). Alkylation of the 4-hydroxy group of PBP with the corresponding 1-bromoalkanes afforded alkylated products PBP-C2–C12. Compounds PBP-NC6–NC16 were synthesized by mono-alkylation of PBP with 1,2-dichloroethane and subsequent alkylation of alkylamines with compound 4. After alkylation of PBP with benzyl (2-bromoethyl)carbamate, the Cbz group was removed using Pd(OH)2 under H2 atmosphere to give PBP-NH2. PBP-tam was obtained by the treatment of PBP with 2-chloro-N,N-dimethylethylamine.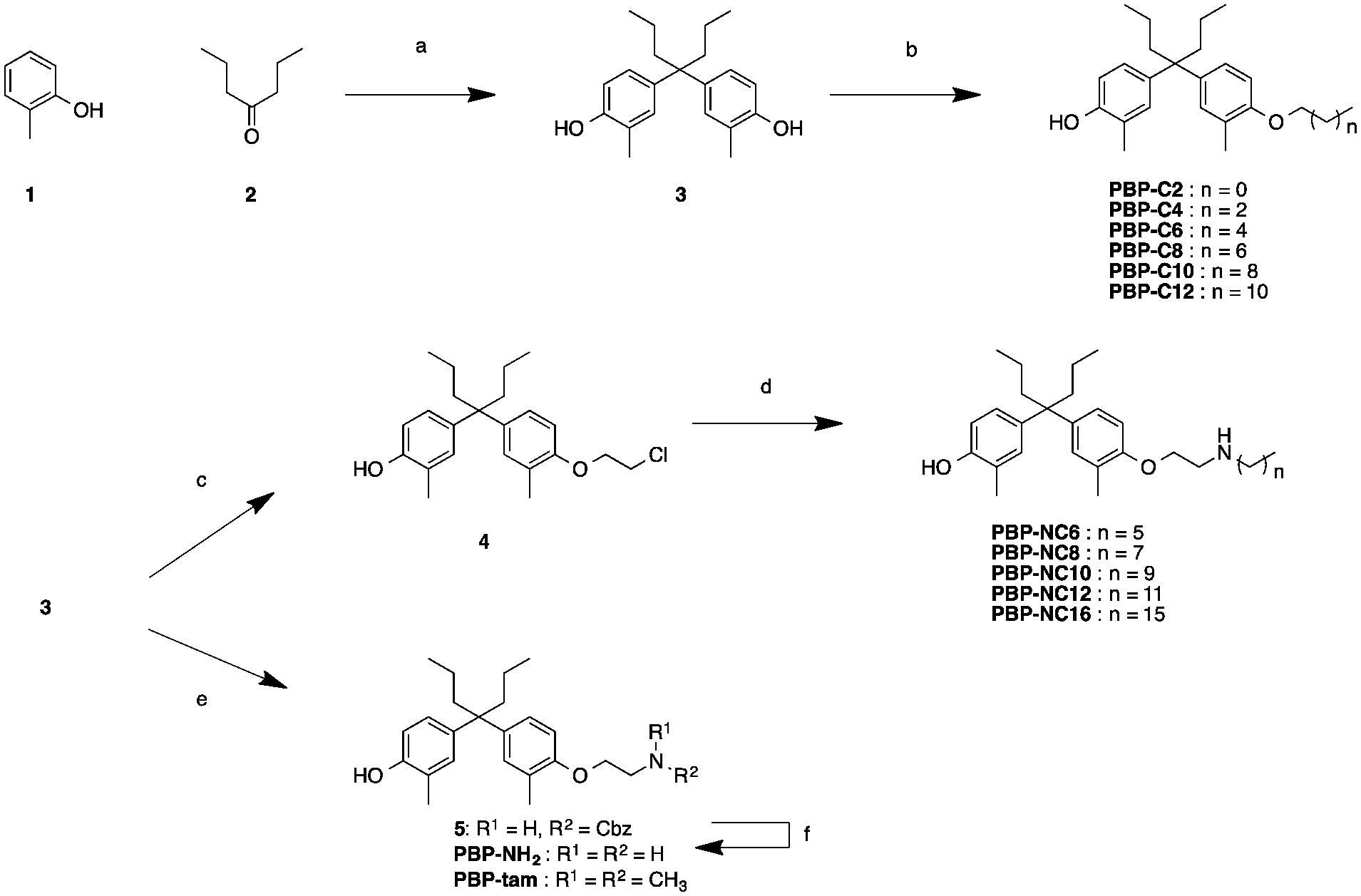 | ||
| Scheme 1 Synthesis of compounds PBP-NH2, PBP-tam, PBP-C2 to PBP-C12, and PBP-NC6 to PBP-NC16. Reagents and conditions: (a) MsOH, rt, 3 days, 59%; (b) R-I (R = ethyl, n-hexyl, n-octyl, n-decyl, n-dodecyl), K2CO3, MeCN, 60–100 °C, 16–47%; (c) 1,2-dichloroethane, K2CO3, MeCN, 48 h, 19%; (d) R-NH2 (R = n-hexyl, n-octyl, n-decyl, n-dodecyl, n-hexadecyl), MeCN, microwave, 120 °C, 4 h, 20–42%; (e) benzyl (2-bromoethyl)carbamate or 2-chloro-N,N-dimethylethylamine Cs2CO3, NaI, DMF, 100 °C, 21 h, 6%; (f) Pd(OH)2, H2, MeOH, rt, 26 h, 25%. | ||
First, we investigated the effects of the synthesized compounds on ERα-downregulating activity by western blotting analysis using MCF-7 breast cancer cells. The potency of the ERα-downregulating activity is described as the ratio of ERα to standard proteins. As shown in Fig. 3, ERα downregulation was observed by treatment with PBP-NC8, PBP-NC10, and PBP-NC12 with amino alkyl chains (ERα downregulation ratio at 10 μM; PBP-NC8: 69%, PBP-NC10: 32%, and PBP-NC12: 68%). In particular, the ERα expression level was dose-dependently reduced in the presence of PBP-NC10 (ERα downregulation ratio at 1 μM: PBP-NC10: 58%). On the other hand, PBP-C2–C12, with simple alkyl chains, PBP-NC6 and PBP-NC16, with short or long alkyl chains, and PBP-tam, with a dimethylamino group, did not show ERα-downregulating activity (Fig. 3a and b). These results indicated that the nitrogen atom and the optimal length of the alkyl chain of PBP-NC derivatives play a pivotal role in the ERα-downregulating activity.
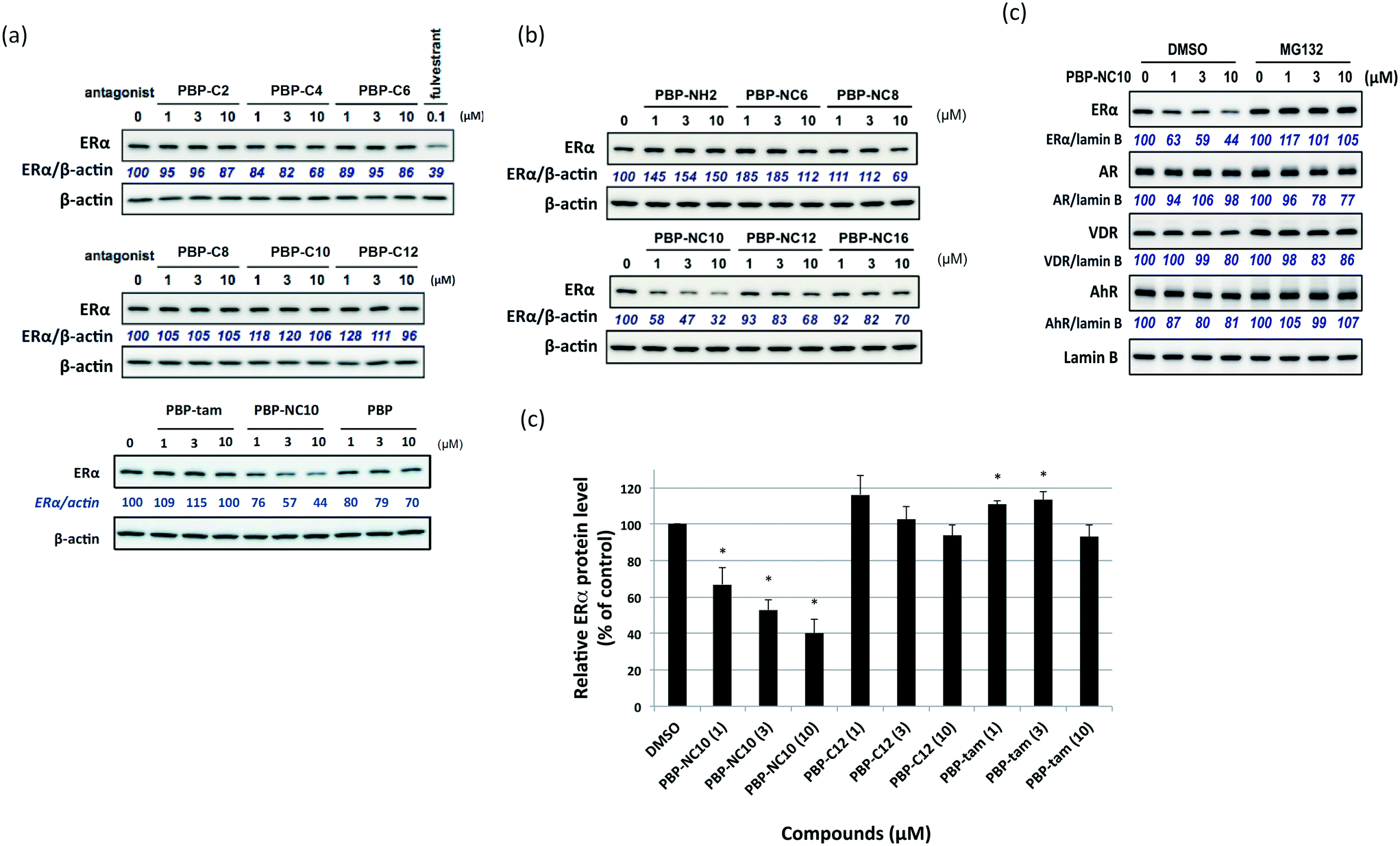 | ||
| Fig. 3 (a and b) Western blotting analysis of estrogen receptor (ER) α levels in MCF-7 cells after 6 h of incubation with the synthesized compounds. (c) Quantification of the levels of ERα is provided in a histogram. Data are expressed as means ± S.E. of three independent experiments (*P < 0.05 versus control). | ||
Next, we investigated the selectivity of PBP-NC10 over other NRs such as the androgen receptor (AR), vitamin D receptor (VDR), and aryl hydrocarbon receptor (AhR) because the diphenylalkane skeleton could act as several NR ligands.16 Moreover, we also performed the western blotting analysis in the presence of proteasome inhibitor MG132 to investigate whether ERα downregulation by PBP-NC10 was mediated by UPS as hypothesized. As shown in Fig. 4, ERα was downregulated by treatment with PBP-NC10 in a dose-dependent manner, whereas the other NR expression levels were unaffected. Furthermore, reductions in the expression levels of ERα induced by PBP-NC10 were suppressed by MG132, indicating that PBP-NC10 induced proteasomal degradation.
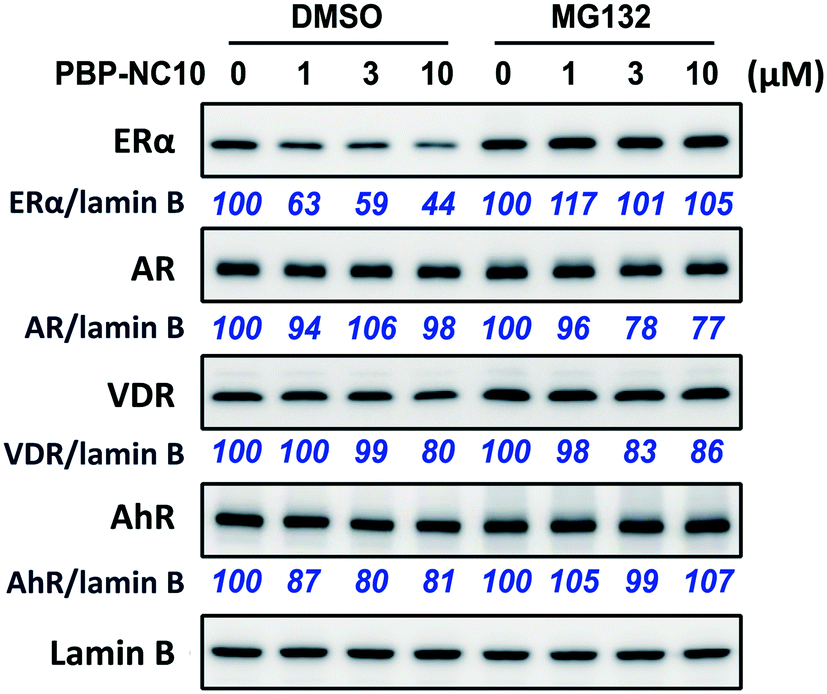 | ||
| Fig. 4 The effects of PBP-NC10 on nuclear receptor levels in MCF-7 cells with or without proteasome inhibitor MG132. ER, estrogen receptor; AR, androgen receptor; VDR, vitamin D receptor; AhR, aryl hydrocarbon receptor. | ||
Next, we investigated the effects of the synthesized compounds on the binding affinity and transcriptional activity against ERα. To evaluate the ability of the compounds to bind to ERα, a fluorescence polarization (FP)-based competitive binding assay was conducted using a PolarScreen nuclear receptor competitor assay kit (Life Technologies).14 As reported previously, 4-OHT and PBP strongly bound to ERα with IC50 values of 5.6 nM and 7.3 nM, respectively. Table 1 shows that PBP-C2 to PBP-C8 exhibited moderate binding affinity to ERα (IC50 = 40–400 nM), whereas PBP-C10 and PBP-C12 did not bind to ERα. These data indicated that the introduction of longer alkyl chains rather than an n-octyl group lowered the binding affinity. On the other hand, PBP-NC8 to PBP-NC12 showed pronounced ERα binding affinity (IC50 values of 2.4 nM for PBP-NC8, 4.9 nM for PBP-NC10, and 4.9 nM for PBP-NC12), which was similar to that of 4-OHT (IC50 value of 5.6 nM) in the FP assay. Moreover, PBP-tam exhibited lower binding affinity to ERα compared with PBP-NH2 and PBP-NC derivatives (PBP-tam: IC50 = 313 nM, PBP-NH2: IC50 = 78 nM). These results revealed that the hydrogen atom on the nitrogen atom of PBP-NC derivatives was important for binding to ERα.
Furthermore, the ERα transcriptional activity was evaluated using a reporter gene assay with CMX-GAL4N-hERα as the recombinant receptor gene, TK-MH100x4-LUC as a reporter gene, and the CMX-β-galactosidase gene for normalization, as reported previously.18 Human Embryonic Kidney 293 (HEK293) cells were incubated with the synthesized compounds in the absence or presence of E2 (0.3 nM). After incubation, the cells were assayed for reporter gene and β-galactosidase activities. As shown in Fig. 5, it was demonstrated that PBP-NC10 exhibited potent ERα antagonistic activity, whereas PBP-C12, which has the same length of alkyl chain as PBP-NC10, showed slight ERα antagonistic activity (ERα antagonistic activity: PBP-C12, 61% inhibition at 10 μM; PBP-NC10, IC50 = 45 nM).
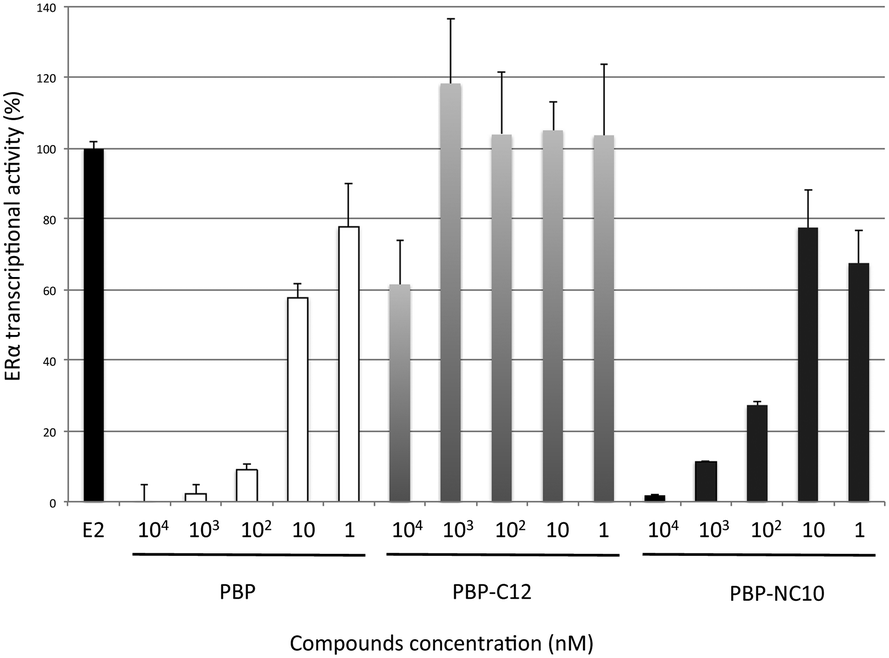 | ||
| Fig. 5 ERα antagonistic activity of PBP, PBP-C12 and PBP-NC10 in HEK293 cells. Data are expressed as means ± S.E. of three independent experiments. | ||
Finally, we performed a computational docking analysis to predict the binding mode of PBP-NC10 to ERα as follows; the X-ray co-crystal structure of ERα bound to 4-OHT was obtained from the Protein Data Bank (PDB: 3ERT) and used in docking studies.19 Docking models of PBP-NC10 bound to ERα were constructed via a conformational search analysis using Molecular Operating Environment (MOE). MMFF94S was used as a force field. In the modeled structures, the amino group of PBP-NC10 interacted with the side-chain carboxyl group of Glu351 and the alkyl chain of PBP-NC10 was included in the hydrophobic groove composed of hydrophobic amino acids such as Leu354, Met357, ILe358, Leu379, and Trp383 of ERα. Therefore, these results indicated that the amino group of PBP-NC10 played an important role in leading the alkyl chain to the hydrophobic groove. Furthermore, the results of the docking analysis suggested that the length of the hydrophobic groove interacting with the long alkyl chain was the best around a decyl group on the amino group of PBP-NC10 (Fig. 6).
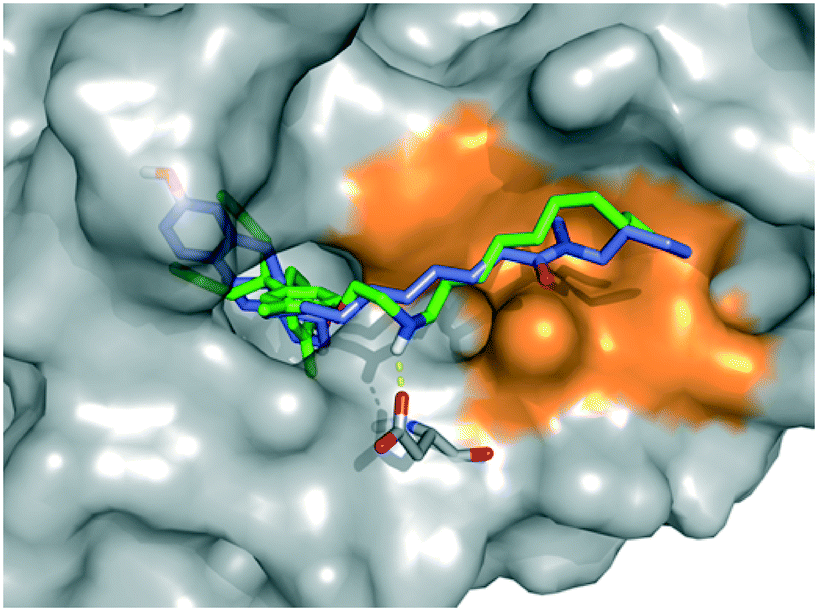 | ||
| Fig. 6 Computational model of the estrogen receptor (ER) α ligand binding domain and PBP-NC10, ICI164,384. The hydrophobic region is colored orange, PBP-NC10 is colored green, and ICI164,384 is colored blue. | ||
Conclusions
We have developed a novel selective estrogen degradation inducer PBP-NC10, which could be useful as a lead structure in the development of compounds with more potent and selective ER degradation-inducing activities. We also revealed that the amino group and optimal length of the long alkyl chain of PBP-NC10 are essential for exerting its ERα-downregulating activity. Furthermore, a docking study was performed and predicted that the interaction between the hydrogen atom on the amino group of PBP-NC10 and the carboxylic acid of Glu351 of ERα leads the long alkyl chain to bind to the hydrophobic groove of ERα.Experimental section
Chemistry
4-Hydroxytamoxifen was purchased from Fluka. All other reagents and solvents were purchased from Sigma-Aldrich, Wako Pure Chemical and Tokyo Chemical Industry, and were used without purification. Analytical TLC was carried out using Merck silica gel 60 F254 pre-coated plates and visualized using a 254 nm UV lamp, phosphomolybdic acid, p-anisaldehyde or ninhydrin staining. Column chromatography was performed with silica gel (spherical, neutral) purchased from Kanto Chemical. The microwave reactions were carried out using a Biotage Initiator. 1H and 13C NMR spectra were obtained on a Varian AS 400 Mercury spectrometer (400 MHz for 1H and 100 MHz for 13C). FT-IR spectra were recorded on a JASCO FT/IR-4100 spectrometer at 1 cm−1 resolution, with an average of 128 scans used for the solution (CDCl3) method and a 0.1 mm path length for NaCl cells. Chemical shifts are expressed as ppm downfield from a solvent residual peak or internal standard tetramethylsilane (TMS). High-resolution mass spectra were obtained on a Shimadzu IT-TOF MS equipped with an electrospray ionization source.Biology
Acknowledgements
This work was supported in part by a grant for the Research on Development of New Drugs from Japan Agency for Medical Research and Development (AMED; M. N.) and a Grant-in-Aid for Young Scientists (B) from the Japan Society for the Promotion of Science (T. M.).Notes and references
- (a) S. Ogawa, V. Eng, J. Taylor, B. D. Lubahn, S. K. Korach and W. D. Pfaff, Endocrinologia, 1998, 139, 5070 CAS; (b) Y. Imai, M. Y. Youn, S. Kondoh, T. Nakamura, T. Kouzmenko, T. Matsumoto, I. Takeda, K. Takaoka and S. Kato, Ann. N. Y. Acad. Sci., 2009, 1173, E31 CrossRef CAS PubMed.
- J. M. Hall and D. P. McDonnell, Mol. Interventions, 2005, 5, 343 CrossRef PubMed.
- (a) G. G. Kuiper, E. Enmark, M. Pelto-Huikko, S. Nilsson and J. A. Gustafsson, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 5925 CrossRef CAS PubMed; (b) C. Thomas and J. Gusrafsson, Nat. Rev. Cancer, 2011, 11, 597 CrossRef CAS PubMed.
- (a) P. Walter, S. Green, G. Greene, A. Krust, J. M. Bornert, J. M. Jeltsch, A. Staub, E. Jensen, G. Scrase and M. Waterfield, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 7889 CrossRef CAS PubMed; (b) F. Holst, P. R. Stahl, C. Ruiz, O. Hellwinkel, Z. Jehan, M. Wendland, A. Lebeau, L. Terracciano, K. Al-Kuraya, F. Jänicke, G. Sauter and R. Simon, Nat. Genet., 2007, 39, 655 CrossRef CAS PubMed.
- V. C. Jordan, Br. J. Pharmacol., 2006, 147, 269 CrossRef PubMed.
- T. A. Grese, S. Cho, D. R. Finley, A. G. Godfrey, C. D. Jones, C. W. Lugar, M. J. Martin, K. Matsumoto, L. D. Pennington, M. A. Winter, M. D. Adrian, H. W. Cole, D. E. Magee, D. L. Phillips, E. R. Rowley, L. L. Short, A. L. Glasebrook and H. U. Bryant, J. Med. Chem., 1997, 40, 146 CrossRef CAS PubMed.
- A. Howell, J. Cuzick, M. Baum, A. Buzdar, M. Dowsett, J. F. Forbes, G. Hoctin-Boes, J. Houghton, G. Y. Locker and J. S. Tobias, Lancet, 2005, 365, 60 CrossRef CAS.
- (a) M. D. Johnson, H. Zuo, K. H. Lee, J. P. Trebley, J. M. Rae, R. V. Weatherman, Z. Desta, D. A. Flockhart and T. C. Skaar, Breast Cancer Res. Treat., 2004, 85, 151 CrossRef CAS PubMed; (b) P. Y. Maximov, C. B. Myers, R. F. Curpan, J. S. Lewis-Wambi and V. C. Jordan, J. Med. Chem., 2010, 53, 3273 CrossRef CAS PubMed.
- J. S. Schneekloth Jr., F. N. Fonseca, M. Koldobskiy, A. Mandal, R. Deshaies, K. Sakamoto and C. M. Crews, J. Am. Chem. Soc., 2004, 126, 3748 CrossRef PubMed.
- Y. Itoh, M. Ishikawa, M. Naito and Y. Hashimoto, J. Am. Chem. Soc., 2010, 132, 5820–5826 CrossRef CAS PubMed.
- J. L. Gustafson, T. K. Neklesa, C. S. Cox, A. G. Roth, D. L. Buckley, H. S. Tae, T. B. Sundberg, D. B. Stagg, J. Hines, D. P. McDonnell, J. D. Norris and C. M. Crews, Angew. Chem., Int. Ed., 2015, 54, 9659 CrossRef CAS PubMed.
- (a) A. E. Wakeling, M. Dukes and J. Bowler, Cancer Res., 1991, 51, 3867 CAS; (b) W. L. Yeh, K. Shioda, K. R. Coser, D. Rivizzigno, K. R. McSweeney and T. Shioda, PLoS One, 2013, 8, e60889 CAS.
- A. C. Pike, A. M. Brzozowski, J. Walton, R. E. Hubbard, A. G. Thorsell, Y. L. Li, J. A. Gustafsson and M. Carlquist, Structure, 2001, 9, 145 CrossRef CAS PubMed.
- T. Shoda, M. Kato, R. Harada, T. Fujisato, K. Okuhira, Y. Demizu, H. Inoue, M. Naito and M. Kurihara, Bioorg. Med. Chem., 2015, 23, 3091 CrossRef CAS PubMed.
- (a) R. McCague, M. Jarman, O. T. Leung, A. B. Foster, G. Leclercq and S. Stoessel, J. Steroid Biochem., 1988, 31, 545 CrossRef CAS; (b) B. S. Katzenellenbogen, M. J. Norman, R. L. Eckert, S. W. Peltz and W. F. Mangel, Cancer Res., 1984, 44, 112 CAS.
- (a) K. Maruyama, T. Noguchi-Yachide, K. Sugita, Y. Hashimoto and M. Ishikawa, Bioorg. Med. Chem. Lett., 2010, 20, 6661 CrossRef CAS PubMed; (b) M. Kainuma, J. Kasuga, S. Hosoda, K. Wakabayashi, A. Tanatani, K. Nagasawa, H. Miyachi, M. Makishima and Y. Hashimoto, Bioorg. Med. Chem. Lett., 2006, 16, 3213 CrossRef CAS PubMed.
- K. Maruyama, M. Nakamura, S. Tomoshige, K. Sugita, M. Makishima, Y. Hashimoto and M. Ishikawa, Bioorg. Med. Chem. Lett., 2013, 23, 4031 CrossRef CAS PubMed.
- T. Noguchi-Yachide, K. Sugita and Y. Hashimoto, Heterocycles, 2011, 83, 2137 CrossRef CAS.
- A. K. Shiau, D. Barstad, P. M. Loria, L. Cheng, P. J. Kushner, D. A. Agard and G. L. Greene, Cell, 1998, 95, 927 CrossRef CAS PubMed.
Footnotes |
| † The authors declare no competing interests. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c6md00553e |
| This journal is © The Royal Society of Chemistry 2017 |