
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDetermination and reduction of translocator protein (TSPO) ligand rs6971 discrimination†
Renee
Sokias‡
a,
Eryn L.
Werry‡
bc,
Sook W.
Chua‡
d,
Tristan A.
Reekie
a,
Lenka
Munoz
e,
Erick C. N.
Wong
c,
Lars M.
Ittner
d and
Michael
Kassiou
*a
aSchool of Chemistry, The University of Sydney, NSW 2006, Australia. E-mail: michael.kassiou@sydney.edu.au
bFaculty of Health Sciences, The University of Sydney, NSW 2006, Australia
cSchool of Medical Sciences (Pharmacology), Bosch Institute, The University of Sydney, NSW 2006, Australia
dDementia Research Unit, School of Medical Sciences, University of New South Wales, NSW 2052, Australia
eSchool of Medical Sciences (Pathology) and Charles Perkins Centre, The University of Sydney, NSW 2006, Australia
First published on 15th November 2016
Abstract
The 18 kDa translocator protein (TSPO) is a target for development of diagnostic imaging agents for glioblastoma and neuroinflammation. Clinical translation of TSPO imaging agents has been hindered by the presence of a polymorphism, rs6971, which causes a non-conservative substitution of alanine for threonine at amino acid residue 147 (TSPO A147T). Disclosed brain-permeant second-generation TSPO ligands bind TSPO A147T with reduced affinity compared to the wild type protein (TSPO WT). Efforts to develop a TSPO ligand that binds TSPO WT and TSPO A147T with similarly high affinity have been hampered by a lack of knowledge about how ligand structure differentially influences interaction with the two forms of TSPO. To gain insight, we have established human embryonic kidney cell lines stably over-expressing human TSPO WT and TSPO A147T, and tested how modifications of a novel N-alkylated carbazole scaffold influence affinity to both TSPO isoforms. Most of the new analogues developed in this study showed high affinity to TSPO WT and a 5–6-fold lower affinity to TSPO A147T. Addition of electron-withdrawing substituents yielded analogues with highest affinity for TSPO A147T without decreasing affinity for TSPO WT. This knowledge can be used to inform further development of non-discriminating TSPO ligands for use as diagnostic markers for glioblastoma and neuroinflammation irrespective of rs6971.
Introduction
The translocator protein (TSPO) is an 18 kDa outer mitochondrial membrane protein.1 Endogenous ligands include protoporphyrin IX and cholesterol,1 and early in vitro work demonstrated a role for TSPO in heme synthesis, cholesterol translocation and steroidogenesis.2,3 More recent in vivo knockout studies, however, have failed to find robust changes in heme metabolism, mitochondrial cholesterol transport and serum pregnenolone levels.4,5 More concordant evidence links the TSPO to a role in proliferation and apoptosis, particularly in cancer cells. Upregulation of TSPO in C6 glioblastoma cells enhances cell proliferation while cell lines with decreased TSPO levels show lower proliferation.6 Intriguingly, numerous chemotherapeutic agents that induce glioblastoma apoptosis require intact TSPO to exert their effects, and direct ligation of the TSPO both in vitro and in vivo can induce apoptosis and anti-proliferative effects in cancer cells.6Clinical interest in TSPO ligands stems partly from the observation that TSPO is expressed at low levels in the healthy brain, but appears at up to 15-times higher levels on glioblastoma cells.7–9 Levels of TSPO in glioblastoma tumours correlate with severity and clinical outcome.7–9 Furthermore, TSPO is upregulated on activated glia during neuroinflammation.10–13 In addition to pre-clinical data,11,14–17 a number of small clinical trials have demonstrated the utility of TSPO imaging in brain tumours and neuroinflammatory disorders. For example, a 2-fold higher level of radioactivity was found in glioma tissue compared to non-malignant brain tissue in patients injected with a radiolabelled TSPO ligand18 and quantification of the radiolabelled TSPO ligand (123)I-CLINDE using SPECT proved useful for predicting glioblastoma progression.19 Increased TSPO ligand PET signal was found in the brains of patients with neuroinflammatory disorders such as HIV, presymptomatic Huntington's disease, temporal lobe epilepsy, mild cognitive impairment and Alzheimer's disease.17,20–22 Furthermore, injection of the immunogen lipopolysaccharide into healthy humans resulted in a 30–60% increase in brain signal of the TSPO ligand [(11)C]PBR28, concurrent with increased blood levels of inflammatory cytokines.23 These suggest the TSPO shows good clinical promise as an imaging marker for glioblastoma and neuroinflammation.
The clinical translation of TSPO imaging agents has been hindered by variation in the affinity of TSPO ligands across human subjects. Almost every known brain-permeable second-generation TSPO ligand binds TSPO with high affinity in some healthy subjects (high affinity binders; HABs) but low affinity in other healthy subjects (low affinity binders; LABs). For example the affinities of DPA-713, XBD-173 and PBR-28 show a 4-, 13- and 55-fold reduction, respectively, in brain tissue or lymphocytes from LABs compared to HABs.24,25 This reduction in affinity is attributed to the single nucleotide polymorphism, rs6971, present in 30% of the Caucasian population and at lower levels in African American, Han Chinese and Japanese subjects.26 The rs6971 polymorphism results in a non-conservative substitution of alanine for threonine at amino acid residue 147 (TSPO A147T) which falls in the fifth transmembrane domain, within a potential ligand binding site.27 The crystal structure of the bacterial homolog of TSPO A147T (34% homology) suggests this mutation moves the second and fifth transmembrane domains closer together than in the wild type TSPO (TSPO WT).27 This has a clinical impact, as administration of radiolabelled versions of DPA-713 and PBR-28 to rs6971 homo- or heterozygotes produces a lower brain PET signal than in wild type homozygotes.25 This necessitates subjects to be genotyped for their rs6971 status and that neuroimaging results be interpreted in light of their genotype.26
A more ideal situation would involve development of a ligand that could bind with equally high affinity to both TSPO WT and A147T. While the affinity of the first-generation TSPO ligand PK 11195 is not affected by the presence of the polymorphism,25 PK 11195 has highly variable kinetics and poor in vivo bioavailability.28,29 Development of non-discriminating ligands based on high affinity brain-permeable second generation TSPO ligands has been hindered by a paucity of knowledge about how ligand structure influences discrimination between the TSPO WT and A147T. To this end, we have established human embryonic kidney (293T) cell lines over-expressing human TSPO WT and A147T to allow for high throughput assessment of TSPO ligand discrimination. Using these isogenic cell lines, we explored how modifications of a novel N-alkylated carbazole scaffold influence affinity at both TSPO forms, generating new insight into the different binding requirements of TSPO WT and A147T. The carbazole scaffold was chosen as a simple and commercially available analogue to known nitrogen-containing tricyclic TSPO ligands such as SSR-180,575, XBD-173 and DPA-713. The carbazole building block could be readily N-substituted with acetamides and functionalised through cross-coupling to explore the requirements for TSPO WT and A147T selectivity.
Results and discussion
Synthesis of compounds
The carbazole acetamide derivatives were synthesised according to the synthetic scheme shown in Fig. 1. Briefly, the synthetic route follows a three-step pathway. Regioselective bromination of the commercially available 9H-carbazole (1) with N-bromosuccinimide provided the 9H-carbazole 2. Subsequent NH-deprotonation of 1 or 2 with sodium hydride, followed by the addition of the alkylating agents gave the required carbazole acetamide derivatives, 3a–b and 4a–4b. A palladium-catalysed Suzuki–Miyaura cross-coupling reaction with the required boronic acid derivative gave the corresponding freely rotating phenyl group 5a–5c.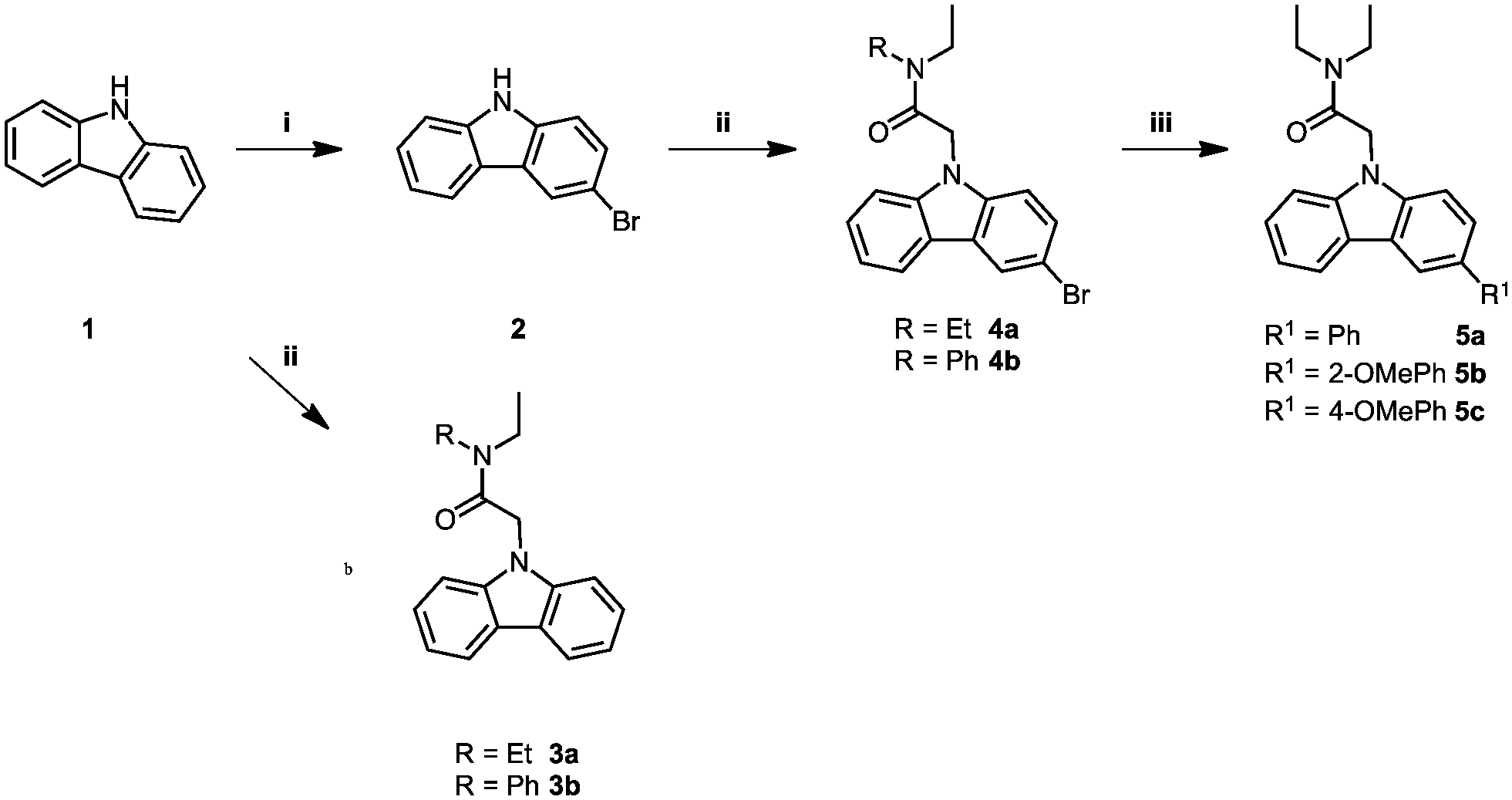 | ||
| Fig. 1 Synthesis of carbazole acetamide derivatives. Conditions and reagents: (i) NBS, DMF, dark, 0 °C – RT, 8 h; (ii) corresponding alkylating agent, NaH, THF, 0 °C – RT, 1 h; (iii) boronic acid derivative, [Pd(PPh3)4], Cs2CO3, toluene, 95 °C, argon, 24 h. | ||
Validation of cell lines
In order to study differential binding of compounds to TSPO WT and A147T, we established novel cell lines, each stably expressing one of the TSPO variants (Fig. 2). Cell lines were derived from clonally selected 293T cells that have been transduced with lentiviruses, which either encode human TSPO WT or A147T for stable integration into their genome. Western blotting of protein extracts showed comparable expression levels of human TSPO WT and A147T in the respective cell lines, but interestingly, lower total levels of TSPO compared to endogenous TSPO in non-transfected controls (Fig. 2A). Furthermore, confocal microscopy demonstrated that both human TSPO WT and A147T localized virtually exclusively to mitochondria, without changes to mitochondrial patterns (Fig. 2B). Hence, we have established two new cell lines expressing human TSPO WT and A147T, suitable for pharmacological assessment of compounds.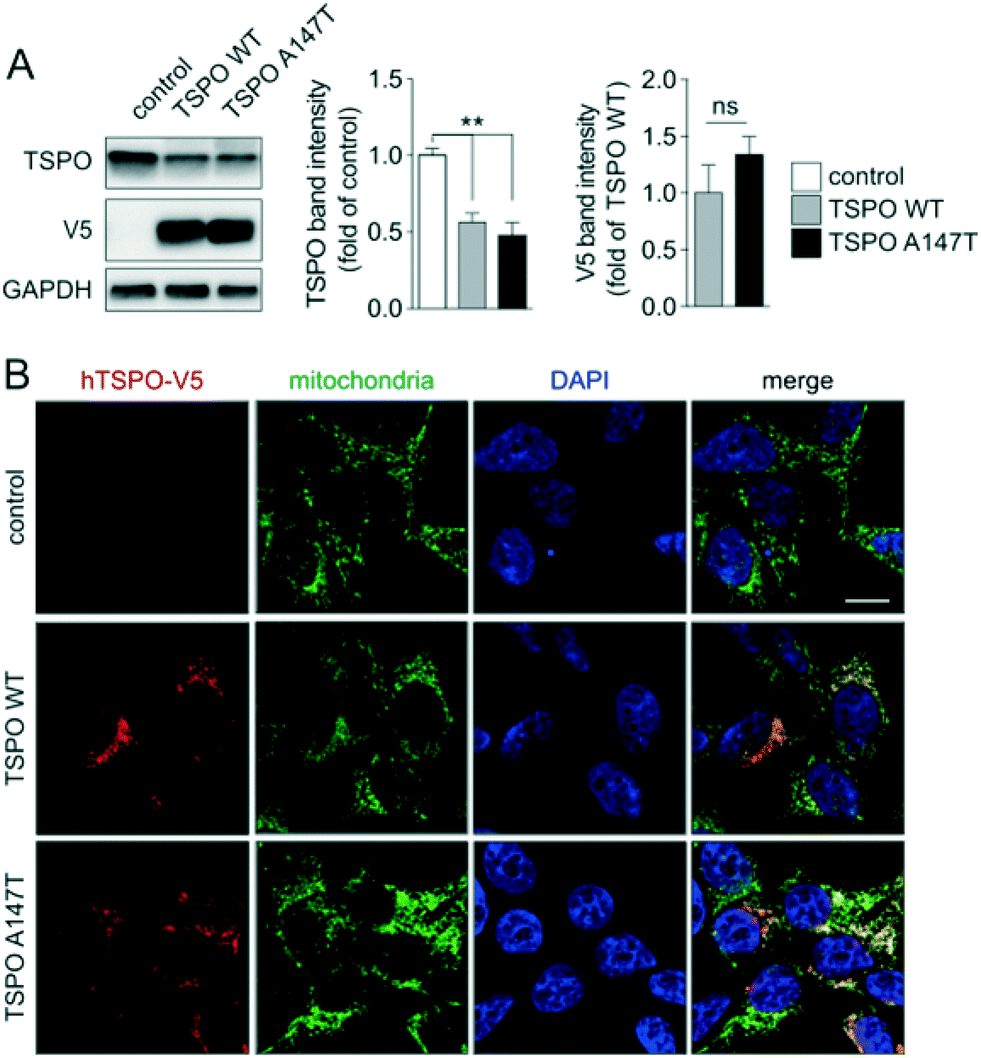 | ||
| Fig. 2 TSPO WT or A147T-expressing cells. (A) Western blotting and quantification (3 independent blots) of stably expressing 293T cells and (B) confocal microscopy together with staining for V5-tagged TSPO WT or A147T and the mitochondrial marker porin. Merges confirm mitochondrial localization of expressed TSPO variants. Scale bar, 10 μm. | ||
As this is the first description of cell lines engineered to over-express the TSPO WT or A147T, we examined the affinity of TSPO ligands PK 11195, DPA-713 and XBD-173, which have been well characterised in human tissue from low and high affinity TSPO binders. In saturation radioligand binding experiments using cell membranes from 293T cells over-expressing TSPO WT and A147T, Kd values of 13.7 ± 5.9 nM and 13.4 ± 2.2 nM, respectively, were determined for [3H]-PK 11195. Equal affinity of PK 11195 towards TSPO WT and A147T was recapitulated in competition radioligand binding assays (Table 1), mirroring the lack of sensitivity of PK 11195 to the mutated TSPO in human brain tissue.30 Although the affinity of PK 11195 to human TSPO seen in this study is lower than that commonly reported at the rat TSPO (0.7–18 nM),31 it aligns almost exactly with that reported in human brain tissue (28.5–30.6 nM),30 adding to reports of species differences in binding of other scaffolds to the TSPO.32,33
| Compound | TSPO WT Ki (nM) | TSPO A147T Ki (nM) | TSPO A147T Ki/TSPO WT Ki |
|---|---|---|---|
| PK 11195 | 29.2 ± 7.3 | 36.0 ± 6.8 | 1.2 |
| DPA-713 | 19.5 ± 3.5 | 98.8 ± 2.3 | 5.1 |
| XBD-173 | 6.4 ± 1.4 | 31.3 ± 3.6 | 4.9 |
| 3a | 30.1 ± 3.6 | 176.6 ± 41.6 | 5.9 |
| 3b | 91.6 ± 0.9 | 459.6 ± 83.0 | 5.0 |
| 4a | 31.5 ± 5.4 | 160.9 ± 20.6 | 5.1 |
| 4b | 54.3 ± 8.4 | 123.4 ± 16.3 | 2.3 |
| 5a | 31.6 ± 1.9 | 159.8 ± 31.0 | 5.1 |
| 5b | 25.4 ± 0.2 | 137.3 ± 20.9 | 5.4 |
| 5c | 28.9 ± 3.6 | 93.4 ± 14.6 | 3.2 |
These novel cell lines also recapitulate the drop-off in affinity of DPA-713 and XBD-173 at human brain tissue from LABs versus HABs (Table 1).24,25 DPA-713 produced a 5-fold drop off in affinity at TSPO A147T compared to TSPO WT, which is similar to the 4.4-fold loss in affinity in human brain tissue from LAB's compared to HAB's.25 XBD-173 showed a 5-fold loss in affinity at TSPO A147T, and while this is less than the 12.6-fold loss in affinity at LAB brain tissue compared to HAB brain tissue, the affinity of XBD-173 at LABs (30.3 nM) is almost identical to the affinity at TSPO A17T in this study (31.3 nM). The demonstration that the pharmacodynamic characteristics of both TSPO A147T-discriminating and non-discriminating ligands reported on endogenous human cells are mirrored in these novel cell lines verifies their validity for use as a high throughput model for assessment of polymorphism discrimination by TSPO ligands.
Assessment of affinity
We characterised the affinity of a new TPSO ligand chemotype based on an N-alkylated carbazole scaffold in 293T cells over-expressing TSPO WT and A147T (Table 1). 3a bound TSPO WT with an affinity comparable to that of PK 11195. As has been reported for most known second generation TSPO ligands,24,253a showed a 6× loss of affinity at TSPO A147T, similar to the drop off seen in this study with DPA-713 and XBD-173. Changing the N,N-diethylamide portion of the scaffold to N,N-benzylethylamide (3b) reduced affinity at both TSPO WT and A147T, although the reduction in TSPO A147T affinity (2.6×) was marginally less than at TSPO WT (3×) resulting in a slightly reduced level of discrimination between TSPO WT and A147T (5-fold discrimination) when compared to 3a.Incorporating a bromine atom at the 3 carbon to produce 4a led to a slight increase in affinity for TSPO A147T without impacting affinity at TSPO WT, resulting in a small improvement in discrimination (5-fold discrimination) compared to 3a. Changing the N,N-diethylamide portion of the brominated scaffold 4a to N,N-benzylethylamide (4b) again resulted in loss of affinity at TSPO WT (4b has 1.6× lower affinity than 4a), although the loss of affinity was not as severe as seen with the non-brominated carbazole derivatives (3b has 3× lower affinity than 3a). In contrast to the effect seen in the non-brominated carbazoles (3avs.3b), the N,N-benzylethylamide substitution in the brominated carbazole (4b) improved affinity 1.3× at TSPO A147T. As a result, compared to 3a, 4b showed a reduction in discrimination between TSPO WT and A147T (2.3-fold discrimination). Together, the loss in affinity for TSPO WT when the N,N-diethylamide portion of the scaffold is replaced with N,N-benzylethylamide suggests that bulk or hydrophobicity in this portion of the scaffold may interfere with interactions with key amino acids in the TSPO WT binding pocket. The observation that bromination of the N,N-benzylethylamide scaffold reverses the loss in affinity at TSPO A147T suggests that even though the extra bulk or hydrophobicity may impart steric hindrance, extra substitution of the carbazole through bromination, may allow the molecule to reach the key amino acids in the binding pocket.
The impact of substitution at the 3 carbon of the 3aN,N-diethylamide carbazole was further explored by introduction of a freely rotating phenyl in the absence (5a) or presence of a methoxy group at the 2′ (5b) or 4′ (5c) position on the phenyl ring. These derivatives retained the high affinity for TSPO WT seen with 3a, but showed increased affinity for TSPO A147T. This was particularly pronounced for 5c, with a 2× higher affinity for TSPO A147T compared to 3a resulting in an improved reduction of discrimination (3.2-fold discrimination). The fifth transmembrane segment of a bacterial homolog of TSPO A147T is more angled than that of the wild type homolog.27 This fifth transmembrane segment contains key binding amino acids, and it could be predicted that the angling of this segment makes the key binding amino acids more difficult to access. Addition of substituents at carbon 3 of the carbazole may allow the molecule to transduce the required distance to access these amino acids. In addition, the substitution of alanine for threonine seen at amino acid residue 147 of TSPO A147T occurs at a key binding amino acid.27,34 Alanine is a hydrophobic amino acid, while threonine is a polar amino acid, indicating that a ligand with polar substituents compared to hydrophobic substituents will interact more ideally at this amino acid in TSPO A147T, perhaps explaining the benefit imparted to carbazole affinity by methoxy substitutions. Given these substitutions at carbon 3 of the carbazole were tolerated by TSPO WT, and improved affinity for TSPOA147T, further exploration of substitutions to carbon 3 of N-diethylamide carbazoles may aid in the development of high affinity non-discriminating TSPO ligands.
Functional characterisation
Pro-apoptotic and anti-proliferative actions are two of the best characterised functions of TSPO ligands.6 To explore if TSPO WT and TSPO A147T affinity discrimination was reflected in functional outcomes, we examined the effect of these novel TSPO ligands on the viability and proliferation of 293T cells over-expressing TSPO WT and TSPO A147T. Due to limited solubility of 3a, 3b, 4b, 4c and 5a in aqueous cell culture media, only the functional effect of 3b and 5a could be assessed. 5a reduced viability and proliferation with greater potency than PK 11195 and XBD-173 (Fig. 3; Tables 2 and 3). DPA-713 and 3b impaired viability at 100 μM, but the effect was weak (IC50 > 100 μM) and these compounds did not significantly affect proliferation, even at 100 μM.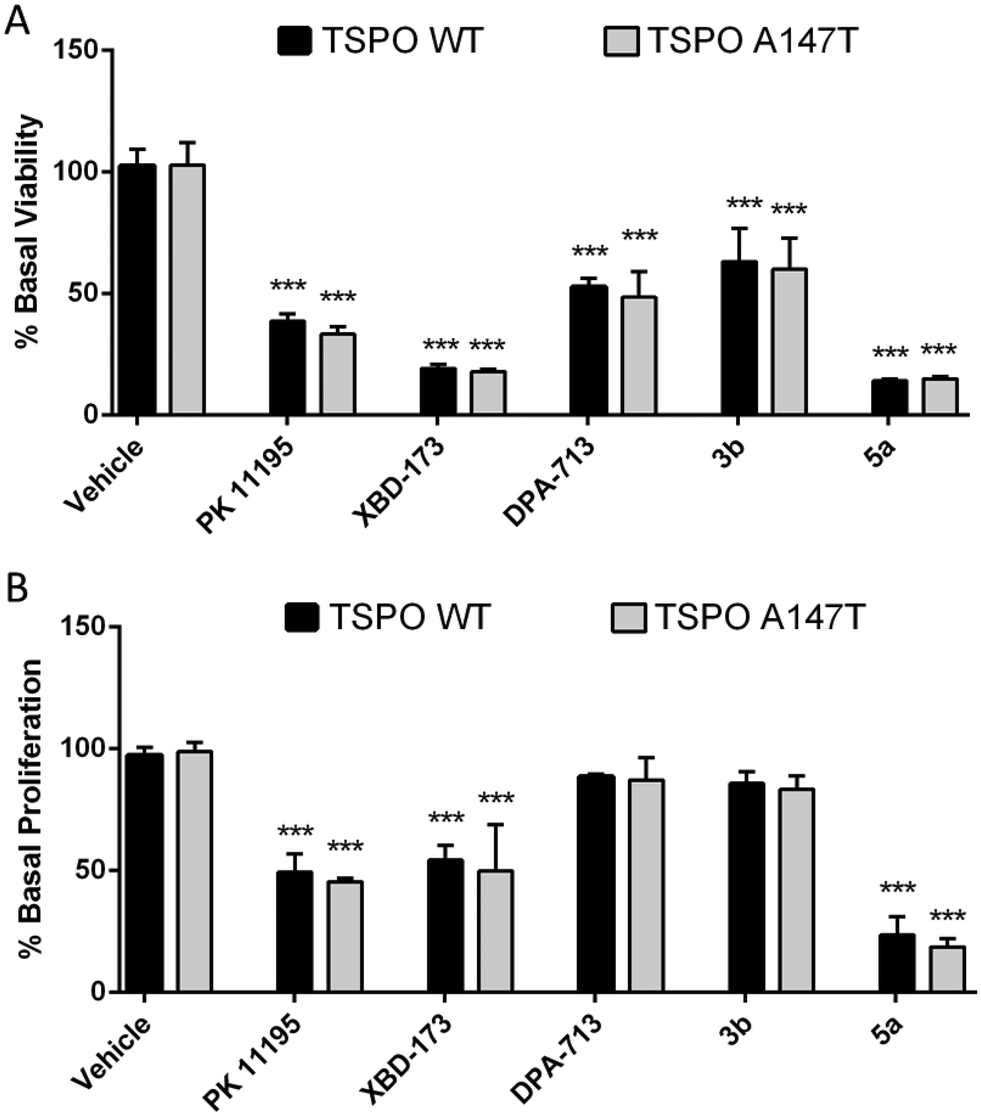 | ||
| Fig. 3 The effect of carbazole derivatives on (A) viability and (B) proliferation of TSPO WT and A147T over-expressing 293T cells. Both cell lines were treated with drugs (100 μM) for 48 h. Cell viability was determined with CellTiter Blue and proliferation was assessed with a BrdU ELISA. Values represent mean ± SEM (n ≥ 3, one-way ANOVA followed by Dunnett's multiple comparison test, ***p < 0.001 compared to vehicle-treated cells). | ||
| Compound | TSPO WT IC50 (μM) | TSPO A147T IC50 (μM) | TSPO A147T IC50/TSPO WT IC50 |
|---|---|---|---|
| PK 11195 | 67.4 ± 10.5 | 69.15 ± 2.6 | 1.0 |
| DPA-713 | >100 | >100 | n/a |
| XBD-173 | 29.5 ± 3.5 | 26.2 ± 3.3 | 0.9 |
| 3b | >100 | >100 | n/a |
| 5a | 23.7 ± 1.0 | 22.9 ± 1.1 | 1.0 |
| Compound | TSPO WT IC50 (μM) | TSPO A147T IC50 (μM) | TSPO A147T IC50/TSPO WT IC50 |
|---|---|---|---|
| PK 11195 | >100 | >100 | n/a |
| DPA-713 | >100 | >100 | n/a |
| XBD-173 | >100 | >100 | n/a |
| 3b | >100 | >100 | n/a |
| 5a | 50.5 ± 5.0 | 47.1 ± 1.2 | 0.9 |
Interestingly, binding sensitivity to TSPO A147T was not reflected in functional assays (Fig. 2; Tables 2 and 3). XBD-173 and 5a showed equal viability-reducing and anti-proliferative potency at both cell lines despite their 5- to 6-fold drop in affinity to TSPO A147T. Further, the potencies of these compounds are several orders of magnitude less than their TSPO affinities. This could suggest that the functional effects of these ligands on viability and proliferation are not mediated by TSPO. However, the anti-proliferative and viability-reducing effects of PK 11195 can be mitigated by TSPO silencing,35 despite the fact that its potency for these functions is several orders of magnitude lower than its affinity for TSPO.35–37 Also, a 4-phenyl quinazoline-2-carboxamide with high affinity for the TSPO (Ki = 0.5 nM) decreased U343 glioblastoma cell viability at micromolar concentrations without off-target activation of G-proteins or inhibiting thirteen protein kinases linked to glioblastoma.38 These observations suggest that the common disjunction between TSPO affinity and potency does not necessitate non-specificity. More recently, the duration of time in which a TSPO ligand interacts with the TSPO, as indexed by residence time, has been shown to be a more informative predictor of TSPO potency or efficacy than affinity. A longer residence time correlates with greater functional effects, including more potent viability-impairment.39,40 Hence, it may be that while the polymorphism affects affinity, this sensitivity may not be reflected in functional assays as the polymorphism may not affect residence time of the ligands.
Conclusions
We have established isogenic cell lines stably over-expressing TSPO WT and TSPO A147T, and used this model to explore how structural variants of a novel N-alkylated carbazole scaffold affect affinity to TSPO WT and TSPO A147T. Through substitutions at the carbazole and the N,N-diethylamide portion of the molecule, we improved the discrimination of the scaffold from 6-fold (3a) to 2.3-fold (4b). TSPO WT was not as sensitive to substitution at the carbazole as TSPO A147T, and addition of electron-donating groups to the 3-position of the carbazole produced the biggest improvement in TSPO A147T affinity without imparting loss in affinity at TSPO WT (5c). This knowledge can be used to inform further development of TSPO ligands with equally high affinity at TSPO WT and A147T for ultimate use as diagnostic markers for glioblastoma and neuroinflammation.Experimental section
Synthesis
Elemental microanalysis was obtained from the Department of Chemistry and Biomolecular Sciences at Macquarie University, Australia. Low- and high-resolution mass spectra were obtained through electron ionisation (ESI). Low-resolution mass spectra were performed on a Finnigan LCQ mass spectrometer. High-resolution mass spectra were performed on a Bruker 7T Apex Qe Fourier Transform Ion Cyclotron resonance mass spectrometer equipped with an Apollo II ESI/APCI/MALDI Dual source.
Procedure for the preparation of 3-bromo-9H-carbazole (2). N-Bromosuccinimide (23 g, 0.13 mol, 1.10 equiv.) was added in parts to a solution of the 9H-carbazole (20 g, 0.12 mol, 1.00 equiv.) in DMF (60 mL) at 0 °C. The reaction was carried out under dark conditions at room temperature for 8 hours. The reaction mixture was poured into ice water and the precipitate was filtered, washed with water and dried under vacuum. Purification by recrystallisation with ethanol gave 2 (23 g, 0.094 mmol, 76%) as a colourless crystalline solid; Rf 0.88 (ethyl acetate/hexane, 3
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 v/v); m.p 195.4–196.9 °C; 1H NMR (300 MHz, CDCl3) δ = 8.21–8.04 (3H, m), 7.54–7.46 (3H, m), 7.35–7.28 (2H, m); 13C NMR (75 MHz, CDCl3) δ = 138.2, 128.64, 126.58, 123.17, 122.48, 122.21, 121.27, 120.51, 119.93, 112.25, 112.03, 110.79; IR (νmax/cm−1): 3401, 1563, 1434, 1053, 809, 744, 566; MS (+ESI) 248/246 [(M + H)+ 35/100%] (found (+ESI): [M + H]+, 247.9893/245.9913. C12H9BrN+ requires 247.9892/245.9913).
:7 v/v); m.p 195.4–196.9 °C; 1H NMR (300 MHz, CDCl3) δ = 8.21–8.04 (3H, m), 7.54–7.46 (3H, m), 7.35–7.28 (2H, m); 13C NMR (75 MHz, CDCl3) δ = 138.2, 128.64, 126.58, 123.17, 122.48, 122.21, 121.27, 120.51, 119.93, 112.25, 112.03, 110.79; IR (νmax/cm−1): 3401, 1563, 1434, 1053, 809, 744, 566; MS (+ESI) 248/246 [(M + H)+ 35/100%] (found (+ESI): [M + H]+, 247.9893/245.9913. C12H9BrN+ requires 247.9892/245.9913).
The required alkylating agent (1.50 equiv.) was added at 0 °C and the mixture was warmed to room temperature and stirred for an additional 1 hour. The reaction mixture was poured into ice water and neutralised with aqueous hydrochloric acid (1 M). The organic layer was collected and the aqueous layer was extracted with ethyl acetate (3×). The organic layers were combined, dried over anhydrous MgSO4, filtered, and the solvent was removed in vacuo. Purification by flash chromatography on silica gave the desired product.
2-(9H-Carbazol-9-yl)-N,N-diethylacetamide (3a). General procedure A was followed with 9H-carbazole (300 mg, 1.79 mmol, 1.00 equiv.) and 2-bromo-N,N-diethylacetamide (369 μL, 2.69 mmol, 1.50 equiv.). Purification by flash chromatography (silica, ethyl acetate/hexane, 3
:7 v/v) gave 3a (460 mg, 1.64 mmol, 90%) as a colourless powder. Rf 0.43 (ethyl acetate/hexane, 3:7 v/v); m.p 138.7–139.9 °C; 1H NMR (300 MHz, CDCl3) δ = 8.10–8.07 (2H, d), 7.46–7.20 (6H, m), 5.02 (2H, s), 3.40–3.34 (4H, q, J = 6.45), 1.28–1.05 (6H, t, J = 6.45); 13C NMR (75 MHz, CDCl3) δ = 166.29, 140.72, 125.90, 123.17, 120.38, 119.38, 108.52, 45.50, 41.63, 14.39, 14.21; IR (νmax/cm−1) 2970, 1653, 1464, 1248, 799; MS (+ESI) 583 [(2M + Na)+ 17%], 303 [(M + Na)+ 45%], 281 [(M + H)+ 100%]; found: C, 78.29; H, 6.52; N, 8.11. Calc for C18H20N2O: C, 78.11; H, 6.59; N, 8.11%.
N-Benzyl-2-(9H-carbazol-9-yl)-N-ethylacetamide (3b). General procedure A was followed with 9H-carbazole (300 mg, 1.79 mmol, 1.00 equiv.) N-benzyl-2-bromo-N-ethylacetamide (530 μL, 2.69 mmol, 1.50 equiv.). Purification by flash chromatography (ethyl acetate/hexane, 1
:4 v/v) gave 3b (530 mg, 1.55 mmol, 86%) as a colourless powder. The 1H NMR spectrum of this amide shows the presence of two different rotamers in a 1:1 ratio. For the sake of simplification, the NMR data for only one rotamer is reported. Rf 0.70 (ethyl acetate/hexane, 3:7 v/v); m.p 144.2–145.6 °C; 1H NMR (300 MHz, CDCl3) δ = 8.11–8.02 (2H, m), 7.46–7.16 (11H, m), 4.99 (2H, s), 4.62 (2H, s), 3.37–3.34 (2H, q), 1.06–1.02 (3H, t); 13C NMR (75 MHz, CDCl3) δ = 169.10, 140.75, 128.62, 128.29, 125.95, 125.95, 121.75, 120.47, 119.49, 115.42, 108.46, 49.83, 48.67, 43.11, 13.25; IR (νmax/cm−1) 3402, 1658, 1536, 1452, 799; MS (+ESI) 365 [(M + Na)+ 40%], 343 [(M + H)+ 100%]; found: C, 80.55; H, 6.49; N, 8.19. Calc for C23H22N2O: C, 80.67; H, 6.48; N, 8.18%.
2-(3-Bromo-9H-carbazole-9-yl)-N,N-diethylacetamide (4a). General procedure A was followed with 2 (2.00 g, 8.13 mmol, 1.00 equiv.) and 2-bromo-N,N-diethylacetamide (1.70 mL, 12.19 mmol, 1.50 equiv.). Purification by flash chromatography (ethyl acetate/hexane, 3
:7 v/v) gave 4a (2.72 g, 8.13 mmol, 93%) as a colourless powder. Rf 0.38 (ethyl acetate/hexane, 3:7 v/v); m.p 133.8–135.2 °C; 1H NMR (300 MHz, CDCl3) δ = 8.17 (1H, s) 8.03–8.00 (1H, m), 7.51–7.43 (1H, m), 7.33–7.16 (4H, m), 4.95 (2H, s), 3.40–3.35 (4H, q, J = 7.05), 1.42–1.10 (6H, t, J = 7.05); 13C NMR (75 MHz, CDCl3) δ = 165.8, 141.03, 139.46, 128.51, 126.65, 124.94, 123.13, 122.15, 120.63, 119.83, 112.25, 110.08, 108.65, 45.34, 41.62, 12.95; IR (νmax/cm−1) 2925, 1642, 1426, 1268, 739, 564; MS (+ESI) 741/739 [(2 M + Na)+ 5/20%], 383/381 [(M + Na)+ 40/100%]; found: C, 60.26; H, 5.10; N, 7.88. Calc for C18H19BrN2O: C, 60.18; H, 5.33; N, 7.80%.
N-Benzyl-2-(3-bromo-9H-carbazol-9-yl)-N-ethylacetamide (4b). General procedure A was followed with 2 (1.00 g, 4.06 mmol, 1.00 equiv.) and N-benzyl-2-bromo-N-ethylacetamide (1.20 mL, 6.09 mL, 1.50 equiv.). Purification by flash chromatography (ethyl acetate/hexane, 1
:4 v/v) gave 4b (1.38 mg, 3.28 mmol, 81%) as a colourless powder. The 1H NMR spectrum of this amide shows the presence of two different rotamers in a 1:1 ratio. For the sake of simplification, the NMR data for only one rotamer is reported. Rf 0.60 (ethyl acetate/hexane, 3:7 v/v); m.p 121.9–123.44 °C; 1H NMR (300 MHz, CDCl3) δ = 8.08–8.01 (1H, m), 7.94–7.85 (1H, m), 7.40–7.01 (9H, m), 6.89–6.86 (1H, m), 4.77 (2H, s), 4.52 (2H, s), 3.43–3.24 (2H, q), 1.14–0.98 (3H, t); 13C NMR (75 MHz, CDCl3) δ = 166.81, 141.07, 139.47, 129.20, 128.67, 128.30, 126.35, 125.92, 124.94, 123.17, 123.03, 122.14, 120.66, 119.75, 110.03, 108.65, 108.47, 50.12, 44.98, 42.41, 13.71; IR (νmax/cm−1) 1645, 1450, 1263, 817, 718, 588; MS (+ESI) 865/863 [(2M + Na)+ 10/25%], 445/443 [(M + Na)+ 35/100%]; found: C, 65.24; H, 5.14; N, 6.41. Calc for C23H21BrN2O: C, 65.57; H, 5.02; N, 6.65%.
N,N-Diethyl-2-(3-phenyl-9H-carbazole-9-yl)acetamide (5a). General procedure B was followed using 4a (200 mg, 0.579 mmol, 1.00 equiv.) and phenylboronic acid (102 mg, 0.835 mmol, 1.50 equiv.). Purification by flash chromatography (ethyl acetate/hexane, 3
:7 v/v) gave 5a (137 mg, 0.384 mmol, 69%) as a colourless solid. Rf 0.38 (ethyl acetate/hexane, 3:7 v/v); m.p 94.3–95.6 °C; 1H NMR (300 MHz, CDCl3) δ = 8.22–8.21 (1H, m), 8.06–8.00 (1H, m), 7.63–7.59 (2H, m), 7.41–7.15 (8H, m), 4.95 (2H, s), 3.37–3.27 (4H, q, J = 7.04), 1.08–0.97 (6H, t, J = 7.05); 13C NMR (75 MHz, CDCl3) δ = 166.20, 142.14, 141.21, 140.30, 132.96, 127.32, 126.11, 125.92, 125.50, 123.71, 123.35, 120.49, 119.56, 119.41, 118.98, 108.78, 108.64, 45.55, 41.65, 12.97; IR (νmax/cm−1) 2969, 1650, 1217, 768, 573; MS (+ESI) 735 [(2M + Na)+ 20%], 379 [(M + Na)+ 100%], 357 [(M + H)+ 10%]; found: C, 80.82; H, 6.74; N, 7.89. Calc for C24H24N2O: C, 80.87; H, 6.79; N, 7.86%.
N,N-Diethyl-2-(3-(2-methoxyphenyl)-9H-carbazol-9-yl)acetamide (5b). General procedure B was followed using 4a (200 mg, 0.557 mmol, 1.00 equiv.) and 2-methoxyphenylboronic acid (127 mg, 0.835 mmol, 1.50 equiv.). Purification by flash chromatography (ethyl acetate/hexane, 3
:7 v/v) gave 5b as a colourless powder (156 mg, 0.404 mmol, 73%). Rf 0.32 (ethyl acetate/hexane, 3:7 v/v) m.p 148.1–150.6 °C; 1H NMR (300 MHz, CDCl3) δ = 8.22 (1H, s), 8.01–8.08 (1H, m), 7.64–7.61 (1H, m), 7.46–7.20 (7H, m), 7.08–6.84 (1H, m), 5.03 (2H, s), 3.82 (3H, s), 3.43–3.39 (4H, q, J = 6.90), 1.16–1.10 (6H, q, J = 6.90); 13C NMR (75 MHz, CDCl3) δ = 166.31, 156.67, 141.09, 139.93, 131.61, 131.31, 129.91, 128.02, 127.86, 125.85, 123.47, 123.13, 121.41, 120.89, 120.46, 119.39, 111.39, 108.54, 107.95, 55.67, 45.62, 41.44, 14.20; IR (νmax/cm−1) 2963, 1638, 1463, 1219, 1022, 749; MS (+ESI) 795 [(2M + Na)+ 25%], 409 [(M + Na)+ 100%], 387 [(M + H)+ 15%]; found: C, 77.72; H, 6.88; N, 7.21. Calc for C25H26N2O2: C, 77.69; H, 6.78; N, 7.25%.
N,N-Diethyl-2-(3-(4-methoxyphenyl)-9H-carbazole-9-yl)acetamide (5c). General procedure B was followed using 4a (500 mg, 1.39 mmol, 1.00 equiv.) and 4-methoxyphenylboronic acid (317 mg, 2.086 mmol, 1.50 equiv.). Purification by flash chromatography (ethyl acetate/hexane, 3
:7 v/v) gave 5c as a colourless powder (392 mg, 1.014 mmol, 73%). Rf 0.28 (ethyl acetate/hexane, 3:7 v/v); m.p 119.8–121.2 °C; 1H NMR (300 MHz, CDCl3) δ = 8.26 (1H, s), 8.16–8.13 (1H, m), 7.68–7.63 (3H, m), 7.51–7.25 (4H, m), 7.05–7.02 (2H, m), 5.10 (2H, s), 4.90 (3H, s), 3.46–3.42 (4H, t, J = 7.05), 1.19–1.12 (6H, t, J = 7.05). 13C NMR (75 MHz, CDCl3) δ = 166.26, 158.60, 141.17, 139.94, 134.80, 132.66, 128.28, 126.05, 125.25, 123.69, 123.34, 120.48, 119.48, 118.53, 114.21, 108.73, 108.60, 55.40, 45.62, 41.66, 12.98; IR (νmax/cm−1) 2932, 1658, 1476, 1260, 1216, 1033, 750, 524; MS (+ESI) 795 [(2 M + Na)+ 25%], 409 [(M + Na)+ 100%], 387 [(M + H)+ 15%]; found: C, 77.60; H, 6.85; N, 7.18. Calc for C25H26N2O2: C, 77.69; H, 6.78; N, 7.25%.
Biological methods
Lentivirus-producing constructs were generated using the Gateway® cloning system (Life Technologies) according to the manufacturer's instructions. Constructs were first cloned into the pENTR™/SD/D-TOPO® entry vector and subsequently into the destination vector, pLVX-V5 (ref. 41) using Gateway® LR Clonase II® enzyme. All clones were sequenced to verify the correct insertion and sequence. TSPO WT cDNA was amplified by PCR with the 5′ oligonucleotide forward primer 5′-ATGGCCCCGCCCTGGGTGC-3′ and reverse primer 5′-CTCTGGCAGCCGCCGTCCC-3′. The A147T mutant was produced as previously described using the QuikChange Site-directed Mutagenesis Kit protocol (Agilent) with the forward primer 5′-CTGGCCTGGCTGGCCTTCACGACCACACTCAACTACTGC-3′ and reverse primer 5′-GCAGTAGTTGAGTGTGGTCGTGAAGGCCAGCCAGGCCAG-3′.42 Lentivirus production for generation of stable TSPO WT or A147T-expressing cell lines was conducted as previously described.43 293T cells were cultured in DMEM containing 10% fetal bovine serum, 100 U ml−1 penicillin/streptomycin and 2 mM glutamine. Seven days after infection with lentiviruses, cells were selected for expression using puromycin.
000g at 4 °C for 15 min. The obtained pellet was re-suspended in 10 ml Tris-HCl (50 mM; pH 7.4) and centrifuged at 48000g for 15 min at 4 °C. The pellet was resuspended in 10 ml of Tris-HCl (50 mM; pH 7.4) and spun at 48000g for 15 min, at 4 °C. The resulting pellet was resuspended in 6 ml Tris-HCl (50 mM; pH 7.4) and total protein concentration was determined using the BCA protein assay (Pierce Biotechnology Inc., Rockford, IL) according to the manufacturer's protocol.
:100; Roche Molecular Diagnostics Indianapolis, IN) at 37 °C for 4 h. Detection of BrdU incorporated into DNA of proliferating cells was performed with a BrdU ELISA (Roche Molecular Diagnostics) following the manufacturer's instructions. The reaction was stopped with 1 M sulfuric acid and the absorbance was measured with a POLARstar Omega plate reader (BMG Labtech) at 450 nm. Data were normalised to vehicle controls and IC50 values calculated by non-linear regression analysis using Graphpad Prism 6.0 (Graphpad). IC50 values represent mean ± SEM from 3 independent experiments. Statistical significance was assessed with a one-way ANOVA with Dunnett's multiple comparison's test. p < 0.05 was considered statistically significant.
Acknowledgements
We wish to acknowledge Dr Donna Lai and Dr Sheng Hua for assistance with use of the Bosch Molecular Biology Facility's equipment. Work performed was supported in part by the European Union's Seventh Framework Programme [FP7/2007-2013] INMiND (Grant agreement No. HEALTH-F2-2011-278850).References
- V. Papadopoulos, M. Baraldi, T. R. Guilarte, T. B. Knudsen, J. J. Lacapere, P. Lindemann, M. D. Norenberg, D. Nutt, A. Weizman, M. R. Zhang and M. Gavish, Trends Pharmacol. Sci., 2006, 27, 402–409 CrossRef CAS PubMed.
- V. Papadopoulos, H. Amri, N. Boujrad, C. Cascio, M. Culty, M. Garnier, M. Hardwick, H. Li, B. Vidic, A. S. Brown, J. L. Reversa, J. M. Bernassau and K. Drieu, Steroids, 1997, 62, 21–28 CrossRef CAS PubMed.
- S. Taketani, H. Kohno, T. Furukawa and R. Tokunaga, J. Biochem., 1995, 117, 875–880 CAS.
- R. B. Banati, R. J. Middleton, R. Chan, C. R. Hatty, W. Wai-Ying Kam, C. Quin, M. B. Graeber, A. Parmar, D. Zahra, P. Callaghan, S. Fok, N. R. Howell, M. Gregoire, A. Szabo, T. Pham, E. Davis and G. J. Liu, Nat. Commun., 2014, 5, 5452 CrossRef PubMed.
- A. H. Zhao, L. N. Tu, C. Mukai, M. P. Sirivelu, V. V. Pillai, K. Morohaku, R. Cohen and V. Selvaraj, J. Biol. Chem., 2016, 291, 1591–1603 CrossRef CAS PubMed.
- E. L. Werry, M. L. Barron and M. Kassiou, Biochem. Soc. Trans., 2015, 43, 531–536 CrossRef CAS PubMed.
- H. Miettinen, J. Kononen, H. Haapasalo, P. Helen, P. Sallinen, T. Harjuntausta, H. Helin and H. Alho, Cancer Res., 1995, 55, 2691–2695 CAS.
- N. Miyazawa, E. Hamel and M. Diksic, J. Neuro-Oncol., 1998, 38, 19–26 CrossRef CAS PubMed.
- E. Vlodavsky and J. F. Soustiel, J. Neuro-Oncol., 2007, 81, 1–7 CrossRef PubMed.
- M. Wang, X. Wang, L. Zhao, W. Ma, I. R. Rodriguez, R. N. Fariss and W. T. Wong, J. Neurosci., 2014, 34, 3793–3806 CrossRef CAS PubMed.
- A. M. Dickens, S. Vainio, P. Marjamaki, J. Johansson, P. Lehtiniemi, J. Rokka, J. Rinne, O. Solin, M. Haaparanta-Solin, P. A. Jones, W. Trigg, D. C. Anthony and L. Airas, J. Nucl. Med., 2014, 55, 466–472 CrossRef CAS PubMed.
- S. Lavisse, M. Guillermier, A. S. Herard, F. Petit, M. Delahaye, N. Van Camp, L. Ben Haim, V. Lebon, P. Remy, F. Dolle, T. Delzescaux, G. Bonvento, P. Hantraye and C. Escartin, J. Neurosci., 2012, 32, 10809–10818 CrossRef CAS PubMed.
- J. Maeda, M. Higuchi, M. Inaji, B. Ji, E. Haneda, T. Okauchi, M. R. Zhang, K. Suzuki and T. Suhara, Brain Res., 2007, 1157, 100–111 CrossRef CAS PubMed.
- Y. Y. Cheung, M. L. Nickels, D. Tang, J. R. Buck and H. C. Manning, Bioorg. Med. Chem. Lett., 2014, 24, 4466–4471 CrossRef CAS PubMed.
- D. Tang, M. L. Nickels, M. N. Tantawy, J. R. Buck and H. C. Manning, Mol. Imaging Biol., 2014, 16, 813–820 CrossRef PubMed.
- B. Gulyas, B. Makkai, P. Kasa, K. Gulya, L. Bakota, S. Varszegi, Z. Beliczai, J. Andersson, L. Csiba, A. Thiele, T. Dyrks, T. Suhara, K. Suzuki, M. Higuchi and C. Halldin, Neurochem. Int., 2009, 54, 28–36 CrossRef CAS PubMed.
- B. Janssen, D. J. Vugts, U. Funke, G. T. Molenaar, P. S. Kruijer, B. N. van Berckel, A. A. Lammertsma and A. D. Windhorst, Biochim. Biophys. Acta, 2016, 1862, 425–441 CrossRef CAS PubMed.
- S. Pappata, P. Cornu, Y. Samson, C. Prenant, J. Benavides, B. Scatton, C. Crouzel, J. J. Hauw and A. Syrota, J. Nucl. Med., 1991, 32, 1608–1610 CAS.
- P. Jensen, L. Feng, I. Law, C. Svarer, G. M. Knudsen, J. D. Mikkelsen, R. de Nijs, V. A. Larsen, A. Dyssegaard, G. Thomsen, W. Fischer, D. Guilloteau and L. H. Pinborg, J. Nucl. Med., 2015, 56, 1386–1390 CrossRef CAS PubMed.
- J. H. Vera, Q. Guo, J. H. Cole, A. Boasso, L. Greathead, P. Kelleher, E. A. Rabiner, N. Kalk, C. Bishop, R. N. Gunn, P. M. Matthews and A. Winston, Neurology, 2016, 86, 1425–1432 CrossRef CAS PubMed.
- M. Politis, N. Lahiri, F. Niccolini, P. Su, K. Wu, P. Giannetti, R. I. Scahill, F. E. Turkheimer, S. J. Tabrizi and P. Piccini, Neurobiol. Dis., 2015, 83, 115–121 CrossRef CAS PubMed.
- L. D. Gershen, P. Zanotti-Fregonara, I. H. Dustin, J. S. Liow, J. Hirvonen, W. C. Kreisl, K. J. Jenko, S. K. Inati, M. Fujita, C. L. Morse, C. Brouwer, J. S. Hong, V. W. Pike, S. S. Zoghbi, R. B. Innis and W. H. Theodore, JAMA Neurol., 2015, 72, 882–888 CrossRef PubMed.
- C. M. Sandiego, J. D. Gallezot, B. Pittman, N. Nabulsi, K. Lim, S. F. Lin, D. Matuskey, J. Y. Lee, K. C. O'Connor, Y. Huang, R. E. Carson, J. Hannestad and K. P. Cosgrove, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 12468–12473 CrossRef CAS PubMed.
- D. R. Owen, A. J. Lewis, R. Reynolds, R. Rupprecht, D. Eser, M. R. Wilkins, I. Bennacef, D. J. Nutt and C. A. Parker, Synapse, 2011, 65, 257–259 CrossRef CAS PubMed.
- D. R. Owen, R. N. Gunn, E. A. Rabiner, I. Bennacef, M. Fujita, W. C. Kreisl, R. B. Innis, V. W. Pike, R. Reynolds, P. M. Matthews and C. A. Parker, J. Nucl. Med., 2011, 52, 24–32 CrossRef CAS PubMed.
- D. R. Owen, A. J. Yeo, R. N. Gunn, K. Song, G. Wadsworth, A. Lewis, C. Rhodes, D. J. Pulford, I. Bennacef, C. A. Parker, P. L. StJean, L. R. Cardon, V. E. Mooser, P. M. Matthews, E. A. Rabiner and J. P. Rubio, J. Cereb. Blood Flow Metab., 2012, 32, 1–5 CrossRef CAS PubMed.
- F. Li, J. Liu, Y. Zheng, R. M. Garavito and S. Ferguson-Miller, Science, 2015, 347, 555–558 CrossRef CAS PubMed.
- C. J. Endres, M. G. Pomper, M. James, O. Uzuner, D. A. Hammoud, C. C. Watkins, A. Reynolds, J. Hilton, R. F. Dannals and M. Kassiou, J. Nucl. Med., 2009, 50, 1276–1282 CrossRef CAS PubMed.
- F. Chauveau, H. Boutin, N. Van Camp, F. Dolle and B. Tavitian, Eur. J. Nucl. Med. Mol. Imaging, 2008, 35, 2304–2319 CrossRef PubMed.
- D. R. Owen, O. W. Howell, S. P. Tang, L. A. Wells, I. Bennacef, M. Bergstrom, R. N. Gunn, E. A. Rabiner, M. R. Wilkins, R. Reynolds, P. M. Matthews and C. A. Parker, J. Cereb. Blood Flow Metab., 2010, 30, 1608–1618 CrossRef PubMed.
- A. M. Scarf, K. M. Auman and M. Kassiou, Curr. Mol. Med., 2012, 12, 387–397 CAS.
- R. Narlawar, E. L. Werry, A. M. Scarf, R. Hanani, S. W. Chua, V. A. King, M. L. Barron, R. N. Martins, L. M. Ittner, L. M. Rendina and M. Kassiou, J. Med. Chem., 2015, 58, 8743–8749 CrossRef CAS PubMed.
- A. M. Scarf, C. Luus, E. Da Pozzo, S. Selleri, C. Guarino, C. Martini, L. M. Ittner and M. Kassiou, Curr. Mol. Med., 2012, 12, 488–493 CAS.
- L. Jaremko, M. Jaremko, K. Giller, S. Becker and M. Zweckstetter, Science, 2014, 343, 1363–1366 CrossRef CAS PubMed.
- B. Chelli, A. Salvetti, E. Da Pozzo, M. Rechichi, F. Spinetti, L. Rossi, B. Costa, A. Lena, G. Rainaldi, F. Scatena, R. Vanacore, V. Gremigni and C. Martini, J. Cell. Biochem., 2008, 105, 712–723 CrossRef CAS PubMed.
- W. Kugler, L. Veenman, Y. Shandalov, S. Leschiner, I. Spanier, M. Lakomek and M. Gavish, Cell. Oncol., 2008, 30, 435–450 CAS.
- D. M. Zisterer, N. Hance, G. Campiani, A. Garofalo, V. Nacci and D. C. Williams, Biochem. Pharmacol., 1998, 55, 397–403 CrossRef CAS PubMed.
- S. Castellano, S. Taliani, M. Viviano, C. Milite, E. Da Pozzo, B. Costa, E. Barresi, A. Bruno, S. Cosconati, L. Marinelli, G. Greco, E. Novellino, G. Sbardella, F. Da Settimo and C. Martini, J. Med. Chem., 2014, 57, 2413–2428 CrossRef CAS PubMed.
- B. Costa, E. Da Pozzo, C. Giacomelli, E. Barresi, S. Taliani, F. Da Settimo and C. Martini, Sci. Rep., 2016, 6, 18164 CrossRef CAS PubMed.
- B. Costa, E. Da Pozzo, C. Giacomelli, S. Taliani, S. Bendinelli, E. Barresi, F. Da Settimo and C. Martini, Apoptosis, 2015, 20, 383–398 CrossRef CAS PubMed.
- N. Krupka, P. Strappe, J. Gotz and L. M. Ittner, Plasmid, 2010, 63, 155–160 CrossRef CAS PubMed.
- D. Koller, L. M. Ittner, R. Muff, K. Husmann, J. A. Fischer and W. Born, J. Biol. Chem., 2004, 279, 20387–20391 CrossRef CAS PubMed.
- Y. D. Ke, J. Dramiga, U. Schutz, J. J. Kril, L. M. Ittner, H. Schroder and J. Gotz, PLoS One, 2012, 7, e35678 CAS.
- L. M. Ittner, D. Koller, R. Muff, J. A. Fischer and W. Born, Biochemistry, 2005, 44, 5749–5754 CrossRef CAS PubMed.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J. Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- V. Chandrakanthan, A. Yeola, J. C. Kwan, R. A. Oliver, Q. Qiao, Y. C. Kang, P. Zarzour, D. Beck, L. Boelen, A. Unnikrishnan, J. E. Villanueva, A. C. Nunez, K. Knezevic, C. Palu, R. Nasrallah, M. Carnell, A. Macmillan, R. Whan, Y. Yu, P. Hardy, S. T. Grey, A. Gladbach, F. Delerue, L. Ittner, R. Mobbs, C. R. Walkley, L. E. Purton, R. L. Ward, J. W. Wong, L. B. Hesson, W. Walsh and J. E. Pimanda, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E2306–2315 CrossRef CAS PubMed.
Footnotes |
| † The authors declare no competing interests. |
| ‡ Joint first authors. |
| This journal is © The Royal Society of Chemistry 2017 |