DOI:
10.1039/C6MB00730A
(Paper)
Mol. BioSyst., 2017,
13, 193-207
Conformational states of HAMP domains interacting with sensory rhodopsin membrane systems: an integrated all-atom and coarse-grained molecular dynamics simulation approach†
Received
24th October 2016
, Accepted 11th November 2016
First published on 11th November 2016
Abstract
Understanding the downstream signaling mechanism of sensory rhodopsin and its cognate transducer complex (srII–htrII) has long been a challenge in the field of photoreceptor research. Here, an integration of all-atom and coarse-grained (CG) molecular dynamics (MD) simulations in different srII–htrII complex states is carried out. It is shown that the cytoplasmic four-helix HAMP dimer gives rise to a gear-box model interaction with discrete hydrophobic packing in Natronomonas pharaonis (Np). Structural analysis in all-atom and CG-MD reveals a stable conformational state in the physiological environment (323 K and 1.15 M salt). Comparative analysis in the ground and intermediate state conformations reveals substantial inter-HAMP interactions in the intermediate state with uniform clockwise (+10° to +30°) and counterclockwise (−20° to −40°) rotations in the α1 helix and the α2 helix of the monomer, respectively. Low temperature and low salt environments (283 K and 0.15 M) significantly affect srII–htrII binding affinity in both states with unusual helix bending. The distinguished control cable, knob-into-holes packing and piston-like movements in HAMP helices are found in the intermediate state complex. The N-terminal htrII (159 residues) coupled with srII yields a binding energy (ΔGbind) of −309.22, −436.53 and −331.11 kJ mol−1 in the MM/PBSA calculation for the NphtrII homodimer, the NpsrII–htrII ground state conformation and the NpsrII–htrII intermediate state conformation, respectively. Only the HAMP1 domain shows a very low ΔGbind value (−21.03 kJ mol−1) for the ground state in comparison to that for the intermediate state (−54.68 kJ mol−1). The structural analysis highlights the key residues that include Y199srII, T189srII, E43htrII, T86htrII, M100htrII, E116htrII, E126htrII and S130htrII for complex stabilization and signal transduction.
Introduction
Bacterial and archaeal photoreceptors mediate phototaxis and modulate cell motility. This action assists microorganisms in coping with diverse environmental niches.1 The phototaxis movement is associated with a complex signaling mechanism that involves sensory and transducer molecular interaction. These molecules are characterized by transmembrane (TM) protein domains that arbitrate the structural changes to stimulate phototaxis. The haloarchaeum Natronomonas pharaonis (Np) induces the phototaxis movement through sensory rhodopsin (srII) and its cognate transducer (htrII) interaction.2–5 The srII and htrII are composed of seven and two TM domains, respectively. The membrane-bound htrII further contains two cytoplasmic HAMP domains (HAMP1 and HAMP2) and a methyl-accepting transducer.6,7 The all-trans to 13-cis retinal photoisomerization exerts conformational changes in srII. These signals are transferred to the tightly coupled htrII molecule through the TM–TM interface.8 The four-helix HAMP domain that connects the htrII TM domain further propagates the signal and triggers other downstream signaling molecules.9,10
Several mechanisms of srII–htrII (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) activation have been proposed, and these mechanisms rely on four-bundle helical switching of the HAMP domain.11–13 The conformational alteration of the HAMP domain has also been described in several model studies in other species.12,13 In addition, truncation of HAMP restricts the heterodimer complex mediated proton pumping. This condition suggests the crucial involvement of HAMP in sensory-transducer tight binding and proton pumping.9,14 Despite their significance, the structural changes in HAMP and its antagonistic effects on the NpsrII–htrII complex are still elusive. Previous studies in srII homologous systems proposed various models, such as scissor-like motions, dynamic helix destabilization and helical shift motion, and gear-box model, among others, for HAMP relay signal transduction.15–18 Moreover, four TM-helix motions with respect to the membrane plane have been proposed. These motions are: translation in the plane of the membrane, perpendicular to the membrane (piston motion), and rotations along the axis parallel and perpendicular to the membrane.16,17
:2) activation have been proposed, and these mechanisms rely on four-bundle helical switching of the HAMP domain.11–13 The conformational alteration of the HAMP domain has also been described in several model studies in other species.12,13 In addition, truncation of HAMP restricts the heterodimer complex mediated proton pumping. This condition suggests the crucial involvement of HAMP in sensory-transducer tight binding and proton pumping.9,14 Despite their significance, the structural changes in HAMP and its antagonistic effects on the NpsrII–htrII complex are still elusive. Previous studies in srII homologous systems proposed various models, such as scissor-like motions, dynamic helix destabilization and helical shift motion, and gear-box model, among others, for HAMP relay signal transduction.15–18 Moreover, four TM-helix motions with respect to the membrane plane have been proposed. These motions are: translation in the plane of the membrane, perpendicular to the membrane (piston motion), and rotations along the axis parallel and perpendicular to the membrane.16,17
Although a substantial effort has been exerted, the signaling mechanism is poorly characterized in the NpsrII–htrII complex. The structural inadequacy of HAMP confines such exploration in NpsrII–htrII systems and warrants investigation. The difficulties and limitations of experimental techniques (NMR/X-ray crystallography) in studying such large membrane-bound systems have attracted computational biologists to conduct structural inspection. A computational study reported two distinct HAMP states for NphtrII (residues 85–133) and suggested that without HAMP flanking segments, the results may be misleading.13 Moreover, experimental studies have shown that the HAMP conformational states are environmentally (pH and temperature) dependent.19 Thus, a computational study that combines the HAMP flanking region under different environmental conditions will be useful in exploring the srII–htrII signaling mechanism.
To understand this, we studied the consequence of the NpsrII–htrII complex with HAMP1 conformational variations in the ground and signaling states. The effect of salt concentration and temperature on structural stabilization was also investigated. All-atom molecular dynamics (MD) and coarse-grained (CG) MD simulations were conducted to investigate the signaling mechanism in srII–htrII complex states. Essential MD calculations and comparisons were performed to quantify the helix movements and rotation in the active and inactive srII–htrII conformation. Binding free energy was computed in homodimer (1:1) and heterotetramer (2:2) srII/htrII systems. Comparative energetic analysis in wild and mutant varieties was performed using the GMXAPBS tool20 that combines GROMACS21 and Adaptive Poisson–Boltzmann Solver (APBS).22 The key amino acid residues involved in active and inactive srII–htrII complex formations were highlighted.
Experimental
Structural modeling
The 3-D coordinates of the HAMP1 domain were assigned using the NMR structure of Af1503 HAMP (PDB ID: 2L7H)17 as the template. The extended HAMP1 structure (137–159) was obtained from the I-TASSER server. The HAMP1 (1–159) was modeled combining the Af1503 HAMP and I-TASSER model structure in MODELLER v9.12.23 The lowest discrete optimized protein energy (DOPE) model was initially energy minimized and equilibrated to fix conformational problems associated with the modelled structure. The htrII linker region from N. pharaonis (PDB ID: 2RM8) was identified as the top template in the threading server for the connecting helix modeling in I-TASSER. Referring to the previous methods,24 the concatenation of the experimental structure (ground: PDB ID 1H2S and intermediate: PDB ID 2F95) of the NpsrII/htrII TM domains, HAMP1 model (84–159), and extended HAMP1 helix (137–159) was performed. The NpsrII–htrII complexes were iteratively energy minimized to ward off steric clashes and bumps. The refined models were validated at different web servers as described elsewhere.25 The homodimer and heteromer complex coordinates were achieved by target-template superposition using a Discovery Studio Visualizer (DSV 3.5, Accelarys).
All-atom MD simulations
The MD simulation systems were prepared by embedding proteins in different lipid bilayers. 1,2-Dimyristoyl-sn-glycero-3-phosphocholine (DMPC) lipid bilayers in an aqueous environment were used in the MD simulation study. The pre-equilibrated DMPC lipid bilayer (128 lipids) was obtained from the Stockholm lipids database (http://people.su.se/˜jjm/Stockholm_Lipids/Downloads.html) and replicated to generate big systems (256 and 512 lipids). The protein was oriented in the membrane using the g_membed program26 followed by solvation in simple point charge water models. The z-axis of the MD systems was rotated and extended to add more solvents to provide a sufficient solvent environment to the cytoplasmic domain. The Amber99SB*-ILDN-RETK force field27,28 with experimental retinal topology extension29 was used for complex MD simulations. Salt concentrations (NaCl) ranging from 0.15 mM to 1.15 mM were used for the different systems, as listed in Table 1. The systems were energy minimized using the steepest descent method. Equilibration of the energy-minimized MD systems were performed using a short NVT of 0.2 ns followed by a 1 ns NPT ensemble.30 In all MD systems, a minimum distance of ∼30 Å was maintained between the surface of the protein and the simulation box. Nonbonded interactions were calculated using a cutoff radius of 12 Å for both short-range electrostatics and van der Waals interactions. Long-range interactions were calculated by applying the particle mesh Ewald method.31 The equilibrated systems were allowed to undergo unrestrained MD simulations. A total of ∼1.4 μs all-atom MD simulations were carried out and are listed in Table 1. The production MD run was carried above the phase transition temperature of DMPC, 323 K for protein, and at 1 bar atmospheric pressure. To investigate the effect of pH and temperature on the HAMP1 structure, MD simulations were conducted at 323 K and 1.15 NaCl (physiological) and 283 K and 0.15 M NaCl concentrations (nonphysiological). The stability and helix packing of different-sized HAMP1 monomers and dimers (i.e., 84–136 and 84–159) were investigated using Gromos53a632 and Amber99SB-ILDN force fields (Table 1).27,28 Visualization of MD trajectories was conducted using VMD,33 and structural interpretations were performed with PyMOL (academic license) and DSV 3.5. All graphs were drawn using QtGrace (http://qtgrace.sourceforge.net/) and Microsoft Excel (academic license). The protein–protein interaction maps were generated using LigPlot+ (academic license, http://www.ebi.ac.uk/thornton-srv/software/LigPlus/). Parallel computation was performed using an SGI UV 3000 system equipped with GROMACS 4.5.5 and 5.0.1 at the Institute for Protein Research, Osaka University, Japan.
Table 1 Summary of the all-atom and coarse-grained MD simulation parameters
|
Simulation system
|
Force field
|
Lipid number
|
Solvent
|
Na
+
|
Cl
−
|
Time (ns)
|
|
All-atom MD systems
|
|
NpHAMP1 |
OPLS/AA |
— |
24369 |
479 |
469 |
100 |
|
NpHAMP1 |
Amber99sb-ildn |
— |
24369 |
479 |
469 |
100 |
|
NpHAMP1 |
Gromos53a6 |
— |
24076 |
471 |
461 |
100 |
| HAMP1-dimer (physiological) |
Amber99sb-ildn |
— |
7624 |
172 |
158 |
100 |
| HAMP1-dimer (physiological) |
Amber99sb-ildn |
— |
7541 |
24 |
10 |
100 |
|
NpHtrII-dimer (84–159, physiological) |
Amber99sb-ildn |
— |
12792 |
410 |
390 |
100 |
|
NpHtrII-dimer (84–159, nonphysiological) |
Amber99sb-ildn |
— |
12664 |
28 |
18 |
100 |
|
NpHtrII-dimer (23–159, physiological) |
Amber99sb-ildn |
256 |
18289 |
668 |
650 |
100 |
|
NpHtrII-dimer (23–159, nonphysiological) |
Amber99sb-ildn |
256 |
16114 |
32 |
14 |
100 |
| Ground state (physiological) |
Amber99sb-star-ildn-RETK |
512 |
28898 |
1032 |
1012 |
100 |
| Ground state (nonphysiological) |
Amber99sb-star-ildn-RETK |
512 |
30658 |
152 |
132 |
100 |
| Intermediate state (physiological) |
Amber99sb-star-ildn-RETK |
512 |
28893 |
1036 |
1012 |
100 |
| Intermediate state (nonphysiological) |
Amber99sb-star-ildn-RETK |
512 |
30653 |
156 |
132 |
100 |
| PDB ID: 2F95 |
Amber99sb-star-ildn-RETK |
128 |
5289 |
124 |
122 |
30 |
| PDB ID: 2l7H |
Amber99sb-ildn |
— |
9849 |
29 |
28 |
100 |
|
|
|
Coarse-grained MD systems
|
| Ground state (physiological) |
Martini |
512 |
10570 |
1013 |
993 |
2000 |
| Ground state (nonphysiological) |
Martini |
512 |
10570 |
1013 |
993 |
2000 |
| Intermediate state (physiological) |
Martini |
512 |
11932 |
1144 |
1120 |
2000 |
| Intermediate state (nonphysiological) |
Martini |
512 |
11932 |
1144 |
1120 |
2000 |
| Mutant |
Martini |
256 |
7827 |
88 |
85 |
10000 |
Coarse-grained MD simulations
Coarse-grained MD simulations were performed for all complexes using the MARTINI v2.2 force field34 in GROMACS. A 4:1 mapping of (non-hydrogen) atoms onto CG particles was considered for all CG approaches. The methods of CG-MD were adopted from previous studies.35,36 A large box extended in the z-dimension was used for the protein–lipid complexes in different states. Standard CG beads for proteins and lipids were considered. All systems were equilibrated for 200 ns followed by 2 μs long MD simulations. A mutant system of NpsrII–htrII (PDB ID: 1H2S)3 was generated by selective alanine mutation at Y199srII and F28htrII and was simulated for 10 μs to investigate the possible molecular disintegration.37 The temperature and salt concentrations were the same as for the all-atom MD simulations.
Conformational analysis from the MD simulations
The structural motions during the MD simulation in the NpsrII, NphtrII, and HAMP1 helices were studied by performing principal component analysis (PCA) and anisotropic network model (ANM) analyses derived from the top eigenvector. The protein dynamics and sequence analysis (ProDy)38 program was used to measure the structural displacements and elasticity, and was created using the Normal Mode Wizard extension.
Evaluation of binding free energy
Intermolecular binding free energy between different molecules in the NpsrII–htrII complexes was calculated using the MM/PBSA method.39 The shell-script files combining GROMACS and APBS commands were executed in different complex MD systems. Based on a single-trajectory system, 500 snapshots were collected at equal intervals for different MD systems, and binding energy (BE) computation was used in each snapshot. Our previous report40 described the BE parameters and methods in detail. Different molecular combinations were utilized to compute the intermolecular BE and were interpreted.
In silico mutagenesis
To explore the crucial amino acid residues in terms of their binding energy contribution in the NpsrII–htrII complex, alanine mutant complexes were generated by selective alanine scanning. Binding free energy for these mutants was calculated from the 500 snapshots using the same method described earlier, and the different BE components in the mutant and wild-type NpsrII–htrII complexes were correlated.
Results and discussion
Structural analysis
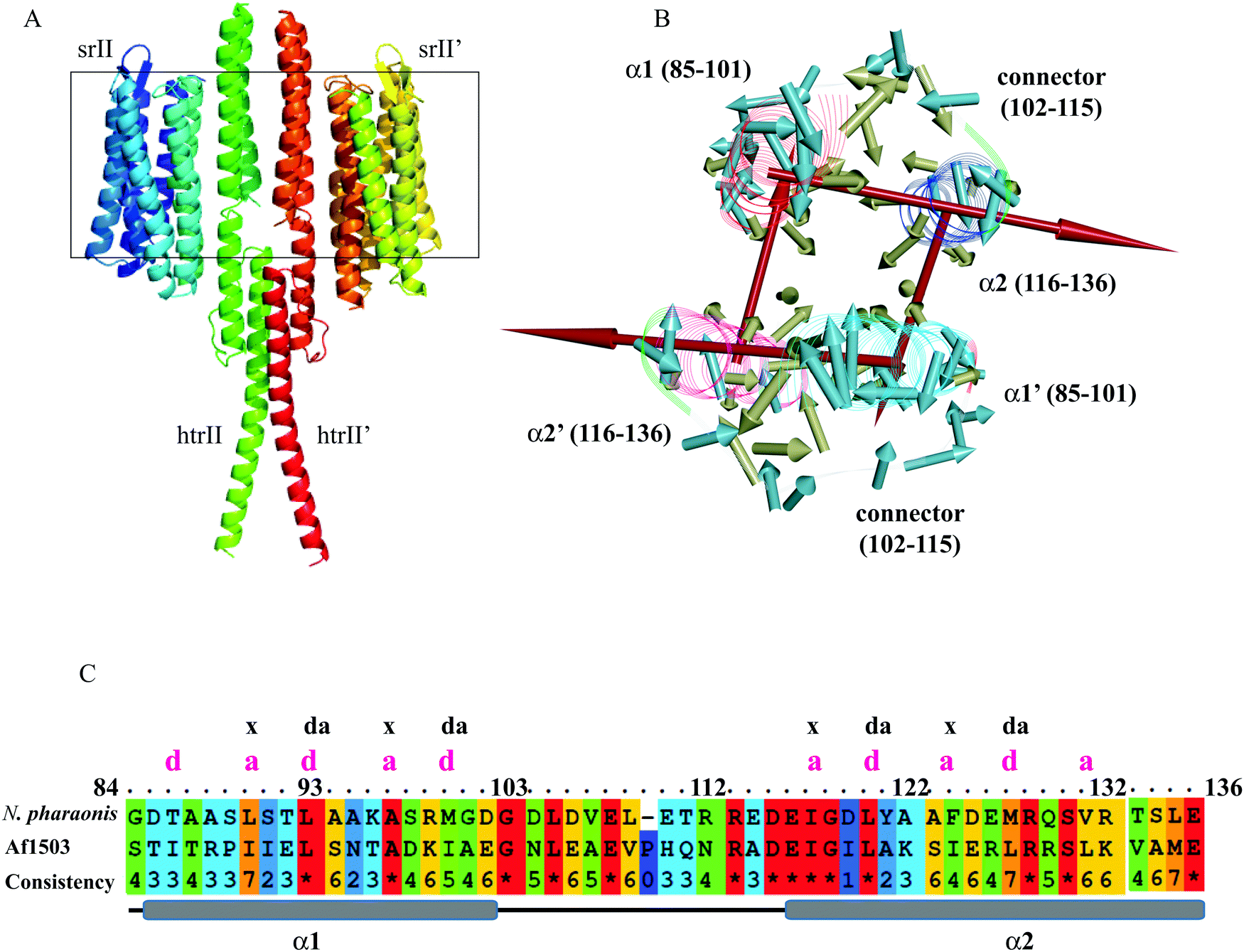
The experimental structures of the ground and intermediate states were patched with the cytoplasmic domain model (Fig. 1A), as described in “Materials and methods.” The HAMP1 model (84–136) domain presented a double helix (α1 and α2) conformation connected by a right-handed 14-residue long loop (Fig. 1B and C). The α1 was oriented in a fashion that depicted offset by one helical turn with respect to α2. The loop connecting α1 and α2 exhibited a distance of ∼24 Å between the sharp reverse turns at the helix ends. The facing terminal peaks of both helices were separated by ∼9 Å. The sharp reverse turns were contributed by the N-terminus conserved Gly101 and Asp115 residues. The C-terminal domain (136–159) showed continuation of the α2 helix with a unique crossing angle as observed in other multi-HAMP proteins.10,11 The I-TASSER model of the C-terminal domain was built using the top templates that include the HtrII linker region from N. pharaonis (PDB ID: 2RM8), the crystal structure of de novo designed helical assembly protein (PDB ID: 3S0R) and the solution structure of a polypeptide containing four heptad repeats from a merozoite surface antigen of P. falciparum (PDB ID: 1PSM) with an estimated TM-score of 0.67 ± 0.13, which measures the global structural similarity between the query and template proteins. The structural report of the HAMP1 model depicted reliable scores with respect to the chosen template structure in all validation servers (Table S1, ESI†) and can be considered for further structural exercises. The Ramachandran plot analysis showed a 100% amino acid distribution pattern in the allowed regions similar to the template. The compatibility of the HAMP1 model structure with its amino acid sequence accessed using verify3D showed a positive score for all residues with a higher average 3D-1D score >0.2 of 63.29 in comparison to the score of the template structure (2L7H). Similarly, the MolProbity analysis revealed the absence of bad backbone bonds and angle and Cβ deviations. Other validation servers, as listed in Table S1 (ESI†), also presented a reliable validation score as compared to the template structure.
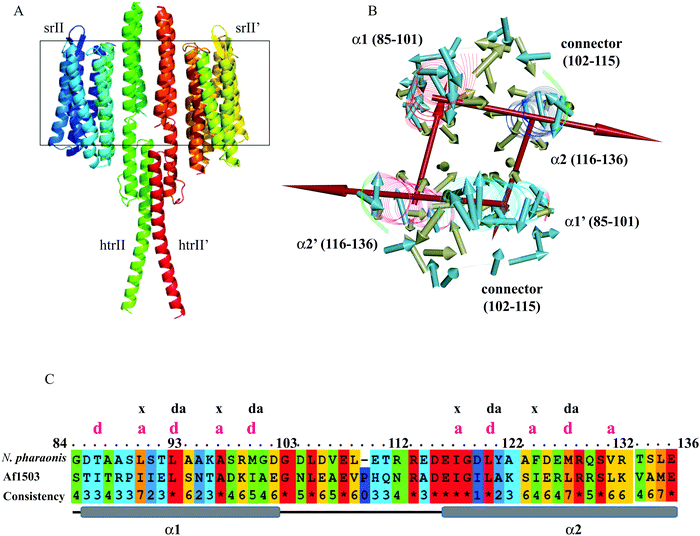 |
| | Fig. 1 (A) Illustration of the model conformation of the NpsrII–htrII complex. The transmembrane region is shown inside the rectangular box. (B) The hydrophobic moments in the HAMP1 homodimer were constructed using Kyte and Doolittle hydrophobicity values in DSV 3.5. The vectors are shown to originate from the Cα atom and averaged to indicate the direction. The hydrophilic and hydrophobic residues in the direction of the vector are in the opposite and same direction of the side chain, respectively. Both HAMP1 molecules in the homodimer are labeled as α1/α2 and α1′/α2′. The pale yellow, pale blue and red colored arrows represent the hydrophobic moment vector for hydrophobic and hydrophilic residues, and the sum of all residues, respectively. (C) The HAMP sequence alignment between N. pharaonis and Af1503 is shown. The conserved residues are marked as “*”, and the heptad sequence repeat (a–b–c–d–e–f–g) with position “a” and “d” having hydrophobic residues in both α1 and α2 helices are labeled. | |
Hydrophobic movements in HAMP
The NpHAMP1 helices exhibited a heptad repeat pattern “a–b–c–d–e–f–g” (where “a” and “d” represent hydrophobic residues) that is a characteristic feature of a coiled-coil conformation.17 The conservation of hydrophobic residues in the heptad repeats (positions “a” and “d”) showed a small variation in the Np and Af1503 HAMP domains (Fig. 1C). As mentioned in the previous reports,10,13 the strong heptad sequence in NphtrII suggests a knob-into-holes packing for cytoplasmic signal propagation. As shown in Fig. 1B, the hydrophobic moment analysis of different residues in the NpHAMP1 model presents a substantial intra and intermolecular interaction. The vectors in both chains were created from the origin of the Cα atoms using the hydrophobicity scale proposed by Kyte and Doolittle.41 The side chains of the first hydrophobic residue (position “a”) at the heptad motif of α1 were oriented toward the α2 helix (intra), whereas the second residue (position “d”) pointed towards the α2′ helix (inter) in the HAMP1 dimer. This movement indicated a tendency for α1–α2 (intra) and α1–α2′ (inter) interactions. The sum of the hydrophobic moments for all residues represented a ring similar to an interhelical interaction pattern (Fig. 1). These movements showed a cyclic interconnection and rotation in the four-bundle structure where the α1/α1′ and α2/α2′ were uniformly interconnected.
HAMP1 conformational variability
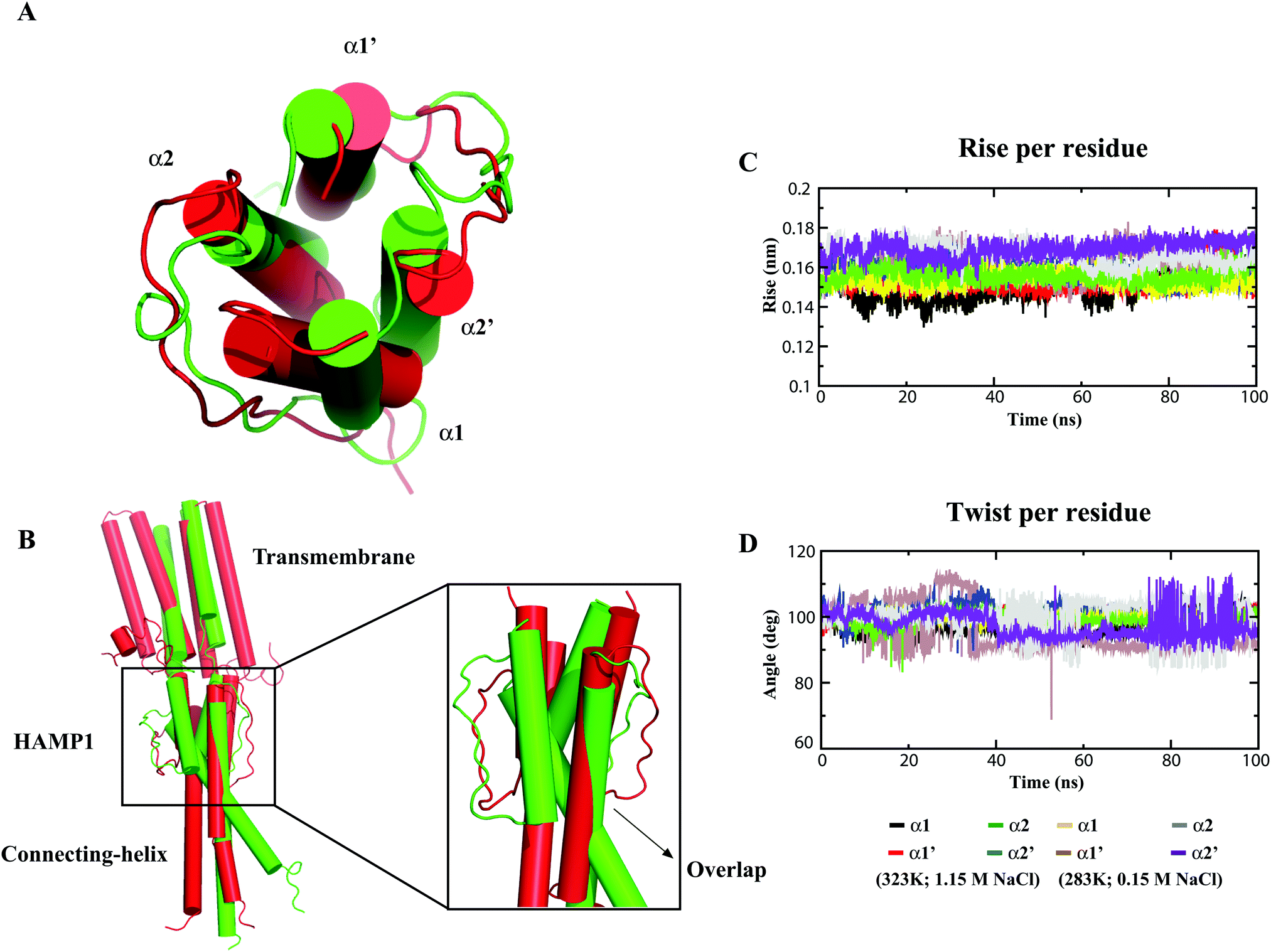
The MD calculation of the HAMP1 domain in the monomeric form showed a disorder conformation (see ESI,† Fig. S1) with significant helicity loss and unusual misfolding in the three force fields (Table 1) during 100 ns. Likewise, 100 ns MD simulation analysis of the experimental solved template structure (2L7H) also revealed a loss in helicity and misfolding at the end of the simulation (see the ESI,† Fig. S1). By contrast, the HAMP1 dimer presented a stable conformation under physiological conditions (see the ESI,† Fig. S1). This result is in agreement with previous MD results carried out at 310 K and 1 M salt concentration.13 Under 283 K and 0.15 M salt conditions (nonphysiological), the helical properties were restrained and a low RMSD was calculated (see the ESI,† Fig. S1). However, a relatively less ideal helix with a helix rise higher than 0.15 nm and a twist smaller than 100° was revealed. A well-oriented stable helix conformation was observed for both the HAMP1 (84–136) and truncated NphtrII (23–159) systems under physiological conditions (Fig. 2A). In contrast, in a nonphysiological environment, HAMP1 helix overlapping was identified (Fig. 2B) with a significant helix tilt (average tilt: α1 = 2.3° and α2 = 5.90°). MD analysis showed that the conformational stability of HAMP1 is environmentally dependent for the extremely haloalkaliphilic archaeon. This result is in agreement with the experimental results.19
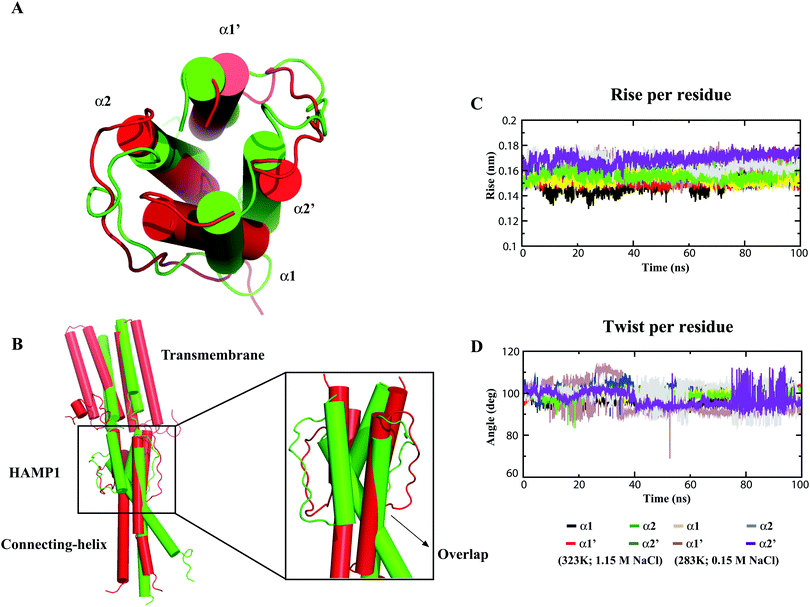 |
| | Fig. 2 Conformational analysis of the HAMP1 homodimer at different temperatures and salt environments after 100 ns all-atom MD simulations. (A) NphtrII-HAMP1 (84–136) and (B) NphtrII (23–159). The protein is shown as a cartoon drawn in PyMOL and the HAMP1 conformations are shown in red and green for high and low environmental conditions, respectively. (C) The helix rise per residue in the HAMP1 helices (ideal helix = ∼0.15 nm), and (D) the helix twist per residue (ideal helix = ∼100°) in HAMP1 during 100 ns. The color legends for the four different HAMP1 helices are shown below the graph. | |
The α1 and α2 helix stability analysis also exhibited a comparatively high-order ideal helix property (ideal helix rise: 0.15 nm; ideal helix twist: 100°) in NphtrII only under physiological conditions (Fig. 2C and D). On the other hand, although a comparatively stable NphtrII dimer conformation was revealed at physiological temperature and pH, a significant tilt in TM domains (average tilt: TM1 = 6.47°; TM2 = 7.44°; TM1′ = 2.63°, and TM2′ = 2.96°) was noticed. This condition correlates with an analogous report that showed weak homodimerization as compared with the heterodimerization in the NpsrII–htrII complex using FRET analysis.42 To quantify the NphtrII homodimer binding efficacy, the free BE calculation was conducted. The results yielded an approximately similar binding energy value under physiological (−156.62 kJ mol−1) and nonphysiological (−148.20 kJ mol−1) conditions in the HAMP1 (84–136) dimer (Table 2). However, the truncated NphtrII dimer (23–159) presented a comparatively high binding energy (i.e., −309.22 kJ mol−1 and −130.36 kJ mol−1 for the physiological and nonphysiological MD systems, respectively). The computed BE value under physiological conditions is in agreement with the FRET results (KD ∼ 16 μM).42 In contrast, a comparatively small binding energy obtained from the nonphysiological simulated system indicated homodimer instability. The different BE contributions are listed in Table 2. To further investigate NphtrII and HAMP1 conformational stability, we studied heterodimeric systems in both active and inactive states.
Table 2 Binding free energy (kJ mol−1) calculations using the MM/PBSA method in the NpsrII–htrII and HAMP1 complex systems
| Systems |
Polar contribution |
Nonpolar contribution |
| ΔGbinda |
ΔGcoulb |
ΔGpsc |
ΔGpolard |
ΔGvdwe |
ΔGnpsf |
ΔGnonpolarg |
|
Binding free energy.
Coulombic term.
Polar solvation terms.
Polar solvation energy.
van der Waals energy.
Nonpolar solvation energy.
Nonpolar solvation terms. Standard errors are presented in parentheses, TM – transmembrane.
|
| srII–htrII (ground state) |
−436.53 (9.68) |
−2176.14 (11.63) |
2282.99 (11.60) |
106.85 |
−478.34 (8.50) |
−65.03 (0.20) |
−543.38 |
| srII–htrII (intermediate state) |
−331.11 (14.07) |
−1317.73 (17.75) |
1488.41 (19.06) |
160.68 |
−430.41 (13.57) |
−61.38 (0.34) |
−491.80 |
|
2F95 (TM-only) |
−211.85 (7.80) |
−745.81 (8.41) |
806.62 (8.75) |
60.85 |
−235.75 (8.01) |
−36.95 (0.12) |
−272.70 |
| HAMP1-dimer (physiological) |
−156.62 (3.37) |
2162.24 (17.32) |
−1874.92 (18.09) |
287.32 |
−396.98 (2.65) |
−46.96 (0.14) |
−443.94 |
| HAMP1-dimer (nonphysiological) |
−148.20 (2.82) |
1807.30 (14.43) |
−1506.06 (15.48) |
301.24 |
−399.58 (3.09) |
−49.86 (0.20) |
−449.44 |
| htrII-dimer (truncated, physiological) |
−309.22 (8.65) |
2736.87 (19.01) |
−2170.06 (17.82) |
566.81 |
−784.71 (7.18) |
−91.32 (0.17) |
−876.03 |
| htrII-dimer (truncated, nonphysiological) |
−130.36 (6.20) |
6030.58 (15.84) |
−5444.80 (18.53) |
585.78 |
−625.42 (6.36) |
−90.72 (0.38) |
−716.14 |
Conformational states of HAMP1 in ground and intermediate states
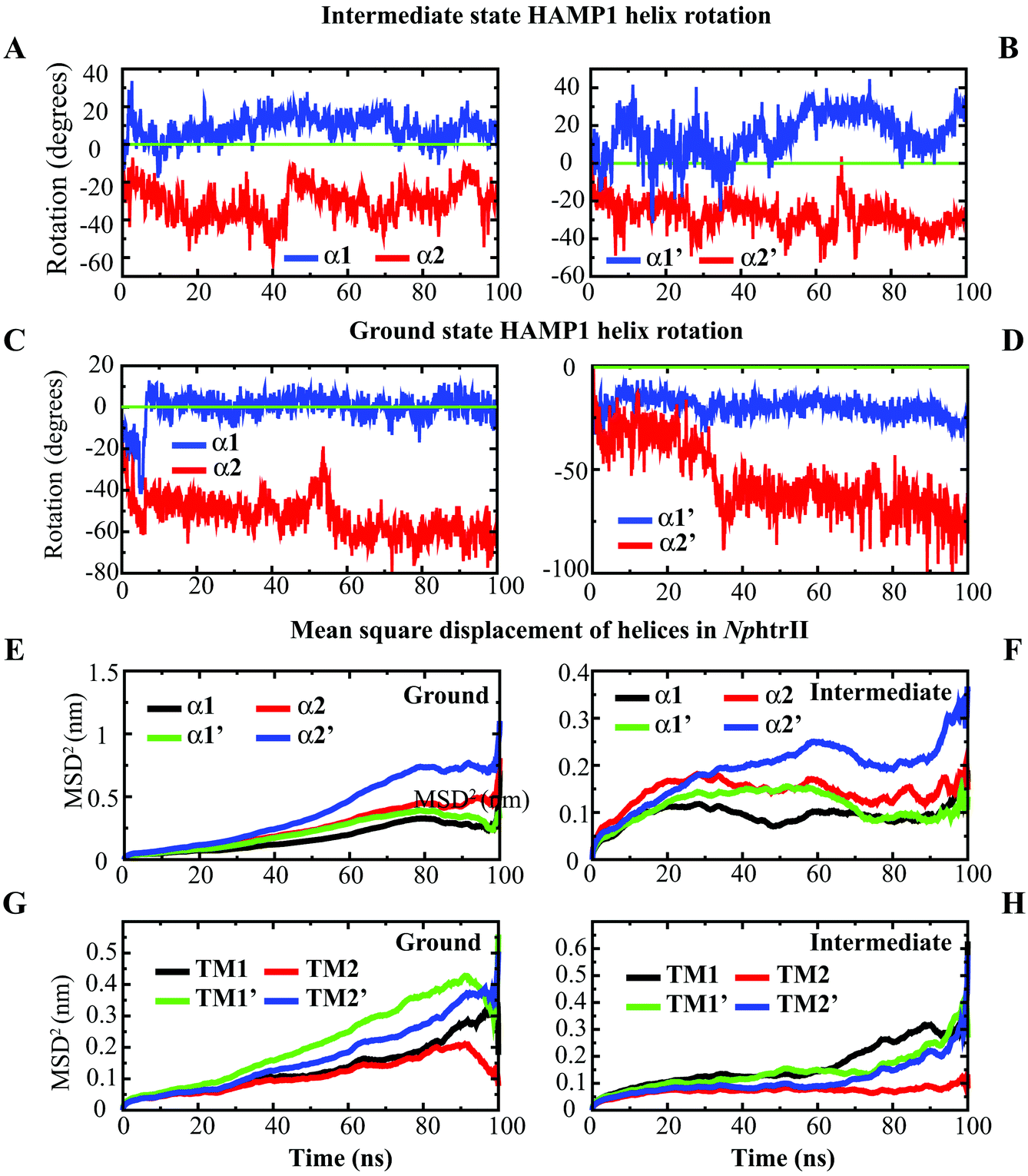
The distinct states of the HAMP domain in both transmembrane and soluble proteins have been studied extensively in the last few years to explore their biological consequences.11,13,16,18,43 These states also vary from proteins to proteins in terms of helix rotation, shift, and intra or interhelical interaction pattern. In multi-HAMP-domain proteins, the distinguishable characteristics of HAMP are prominent. The canonical HAMP that connects the TM domain is of more interest as far as photo-isomerized signal transduction is concerned. Comparison of helix rotation and displacement in the NpsrII–htrII (2:2) complex at the ground and intermediate states revealed a distinguishable helix rotation pattern during the 100 ns period (Fig. 3). The intermediate conformation exhibited opposite α1 and α2 helix rotations (i.e., clockwise and counterclockwise in both NphtrII molecules) (Fig. 3A and B). The NMR analysis and gear-box model studies in Af1503 HAMP also reported a ±26° rotation in all four helices in an alternative fashion.17 The intermediate state NphtrII–HAMP1 helices showed an opposite rotation (intramolecular) varying from ∼+10° to +30° and −20° to −40° in clockwise and counterclockwise directions, respectively (Fig. 3A and B). On the other hand, the ground state conformation presented a comparatively larger rotation (>50° in the negative axis) for α2 helices with an irregular α1 helix rotation (Fig. 3C and D). In addition, an increase in the rotational degrees of TM2 (NphtrII) was observed in the intermediate state conformation and indicated its influence on the HAMP1 rotation (see the ESI,† Fig. S2A). The TM2 domain of both molecules rotated in a similar fashion with a small initial clockwise (for the first ∼10 ns) rotation followed by a steady counterclockwise rotation varying from ∼−10° to −20°. These helix rotations were comparatively less for the ground state conformation (average rotation ∼−6.12°). In the signaling state, a small helical tilt (∼2°–3°) along the z-axis was revealed for both TM2 helices (see the ESI,† Fig. S2B). The prominent TM2 helix rotation and tilt in the intermediate state with light-activated retinal isomerization showed its important role in HAMP1 rotation and signal transduction and resembled other homologous proteins.17,43,44
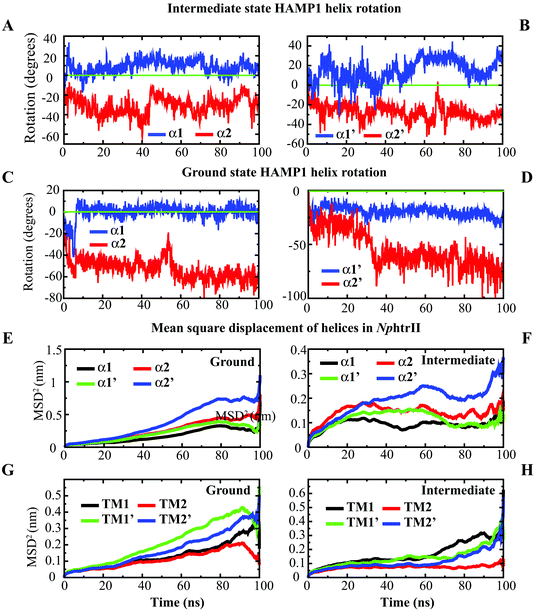 |
| | Fig. 3 Helix rotation and mean square displacement (MSD) analysis in the HAMP1 homodimer. (A) α1 and α2 rotations in the intermediate state, (B) α1′ and α2′ rotations in the intermediate state, (C) α1 and α2 rotations in the ground state, (D) α1′ and α2′ rotations in the ground state, (E) MSD of the HAMP1 helices in the ground state, (F) MSD of the HAMP1 helices in the intermediate state, (G) MSD of the NphtrII–TM helices in the ground state, and (H) MSD of the NphtrII–TM helices in the intermediate state. | |
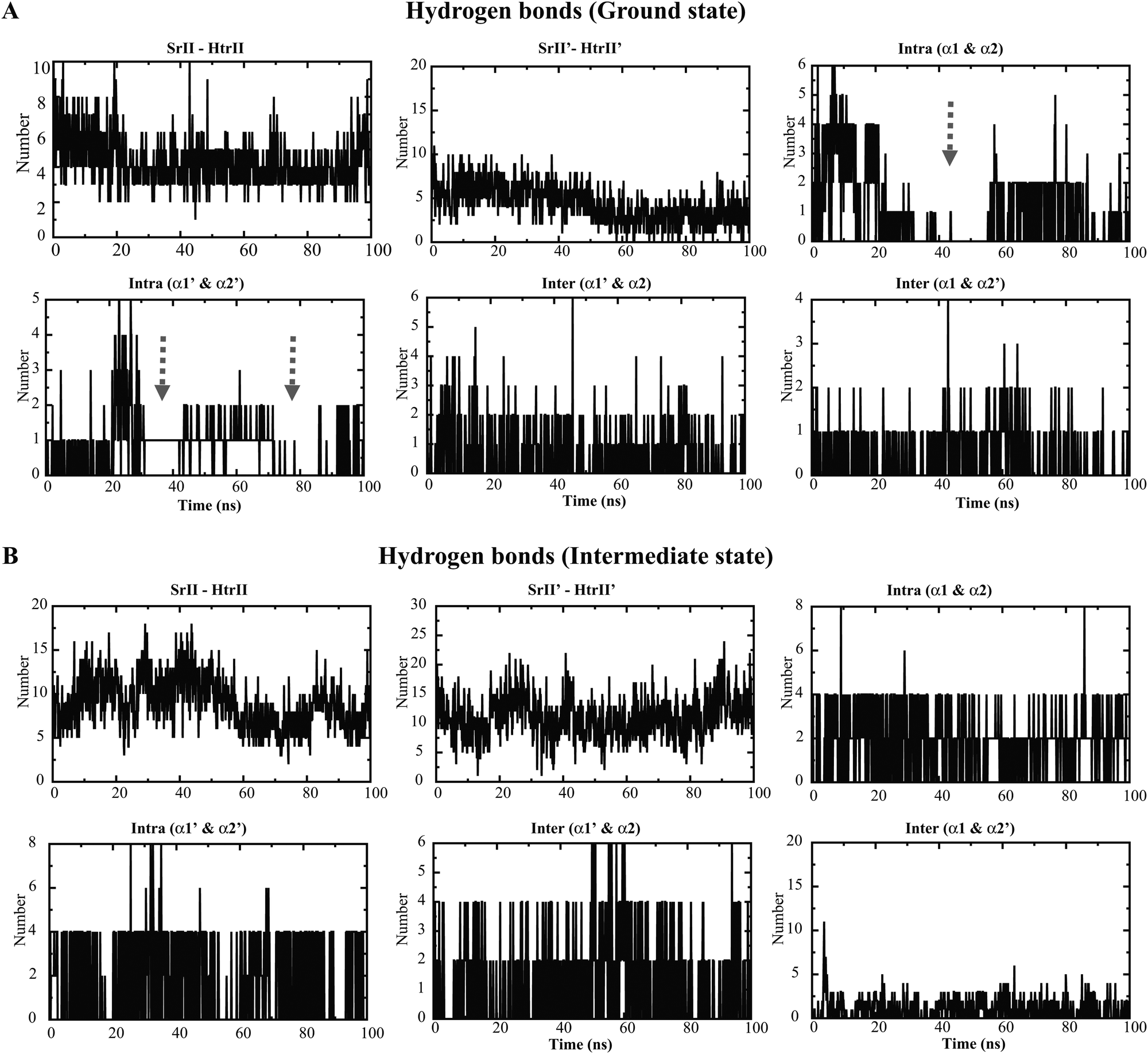
The mean square displacement (MSD) measurement of the TM helices presented an approximately equal helix displacement in both states. The tightly coupled TM2 helices depicted lower MSD values than the free TM1 helices. However, a relatively higher MSD was obtained for HAMP1 helices (>50 Å2) in the ground state conformation. In contrast, a smaller MSD (<30 Å2) was revealed in the intermediate state and suggested the existence of a comparatively rigid four-bundle helix conformation (Fig. 3E–H). The hydrogen bond (H-bond) network analysis between NpsrII and htrII yielded a high number of H-bonds for the intermediate state (∼5 to 10). On the other hand, a stable H-bond network was accounted for in the ground state conformation (Fig. 4). Intra and interdomain HAMP1 helix interaction analysis showed a relatively high and stable intermolecular H-bond network (between α and α′) in the intermediate state and suggested its active engagement in signal transduction. In contrast, the ground state conformation presented a discrete H-bond pattern in both the intra- and inter-HAMP helices (Fig. 4A and B). The C-terminal HAMP1-connecting helix (137–159) rendered a similar H-bond pattern (∼3 to 4 H-bonds) with a significant helical tilt (average tilt intermediate state: α3 = 9.85°, α3′ = 14.83°; average tilt ground state: α3 = 3.21°, α3′ = 3.63°) and rotation for the intermediate conformation.
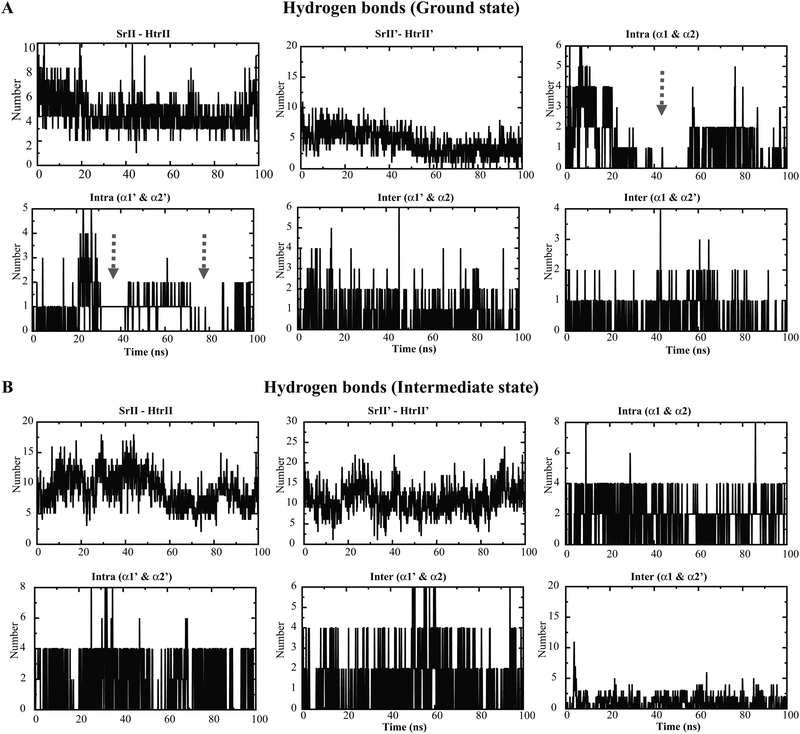 |
| | Fig. 4 Hydrogen bond analysis in the NpsrII–htrII complex during a 100 ns time period. The different molecules used for the hydrogen bond calculation are labeled on the top of each graph. Different domains in the 2:2 complex are denoted as srII/srII′, htrII/htrII′, α1/α′ and α2/α2′. Hydrogen bond analysis in the ground state (A) and in the intermediate state (B) complexes. The arrow marks indicate the discrete hydrogen bond pattern in the ground state HAMP1 dimer in the intra-helical domains. | |
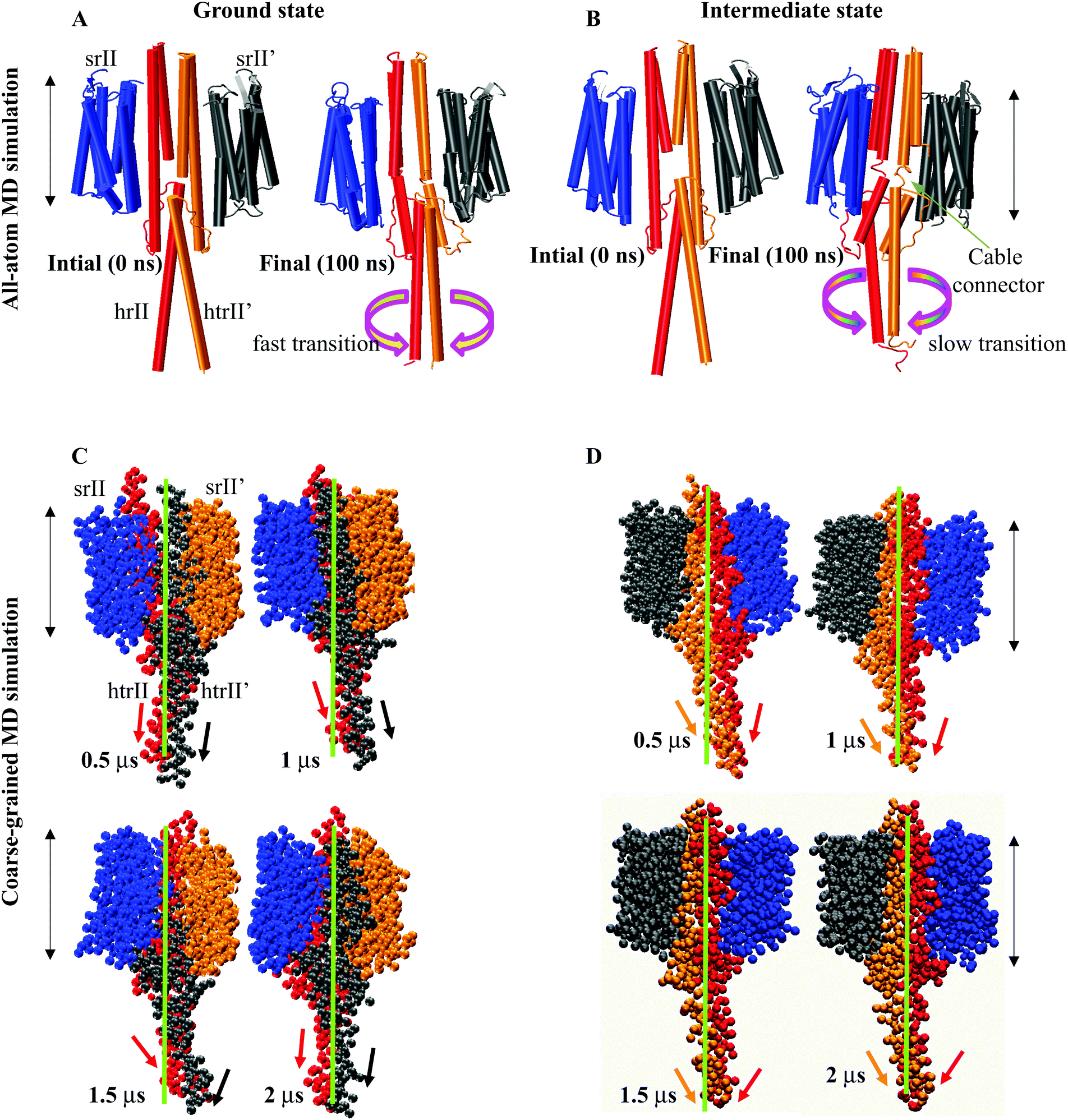
A close inspection of all-atom MD snapshots showed a unique small linker (control cable) between TM2 and the α1 helix in the intermediate state. As suggested earlier,17 in our initial model, we extended the TM2 helix with all helix conformations. The trajectory interpretation indicated a relatively fast and large transition at α3 regions in the ground state conformation (Fig. 5A). On the other hand, the HAMP1 helix arrangements controlled through small cables depicted a slow and small transition across the bilayer normal axis in the intermediate conformation (Fig. 5B). Our findings provided an insight into these small connectors that are important for the HAMP1 helix rotation and arrangement similar to other chemoreceptors.43,45 Mutation analysis in these linkers in some other HAMP containing proteins showed their crucial involvement in HAMP arrangement.43,45 The structural interpretation of the all-atom MD snapshots indicated the transient movement of the HAMP1-connecting helix along the XY plane. The intra and interhelical interactions in the four-bundle HAMP domain regulated the motion of the connecting helix (α3). The α3 helix traversed a path of 1 nm to 2 nm from the bilayer normal upon α1–α2, α1′–α2′, α1–α2′, and α1′–α2 helix packing. These rotations showed their crucial involvement in signal transmission from the HAMP1 to the HAMP2 domain. On the other hand, these rotations may also be misleading in the absence of HAMP2 for the truncated NphtrII. It may yield a large tilt over a longtime scale simulation (Fig. 5A and B). To investigate this condition, we analyzed the rotation of the HAMP-connecting helices by performing 2 μs long CG-MD simulations.
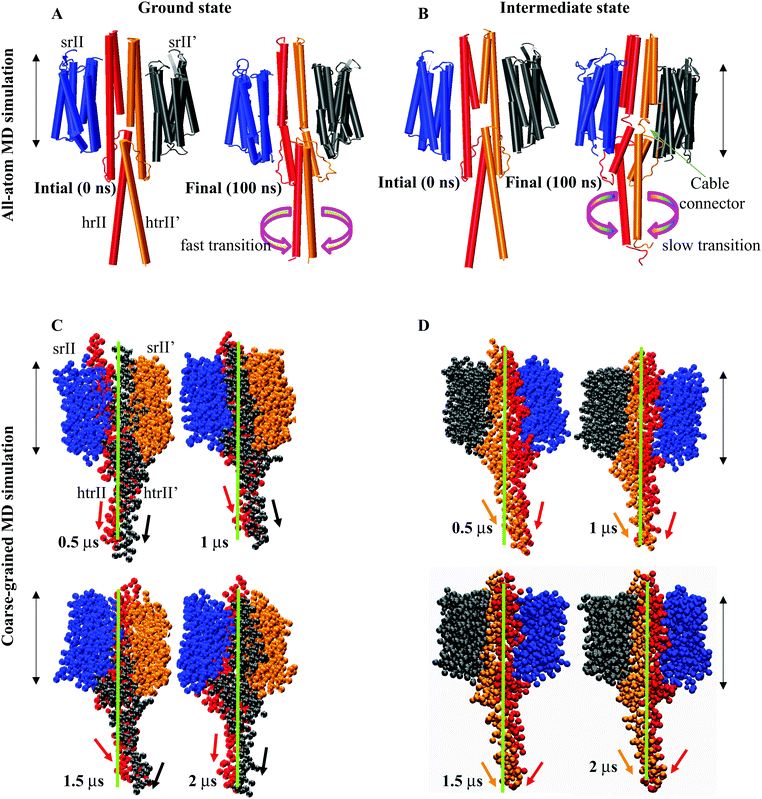 |
| | Fig. 5 Structural interpretation of the NpsrII–htrII complex (2:2) in the active and inactive states using all-atom MD and CG-MD simulations. The protein molecules are shown as a cartoon in VMD. The heterodimers are colored separately and are labelled. (A) All-atom MD in the ground state shows a fast and tightly placed C-terminal helix rotation; (B) in the intermediate state the C-terminal helix rotations are comparatively slow with distinct helical crossing angles. The distinguished control cables (small connectors) connecting TM2 and α1/α1′ are shown. The curve arrows show the helix translations and rotations at the C-terminus along the helix axis. Snapshots of different conformers derived from the 2 μs CG-MD simulations in the ground (C) and intermediate state (D) complexes. The protein molecules are drawn as “VDW” in VMD, and the helix movements and rotations are monitored using arrows with respect to the lipid bilayer normal. The membrane position is indicated by the double-sided black arrows. | |
CG-MD analysis
The HAMP-connecting helix (α3) rotation (137–159) in the XY plane was more prominent in the intermediate state (from 20° to 40°) and relatively smaller in the ground state (from 0° to −20°). The α3 movement for both states during 2 μs is illustrated in Fig. 5C and D. The rotations in the intermediate state are influenced by the intra and interhelical four-bundle helix packing. The transient motion of the connecting helix along the bilayer normal provided an insight into the downstream signaling in the NpsrII–htrII complex. These helix shifts and rotations were consistent in both the all-atom MD and CG-MD simulations and indicated distinct HAMP1 and α3 conformations for both states. The effect of helix rotation and tilt (α3 = 3.85°, α3′ = 4.81°) on structural drifts was quantified through their relative motions. A comparative analysis showed a well-oriented cross helix angle in the intermediate state. On the contrary, the ground state complex revealed a sharp kink (at T133 and S134) and unidirectional helix bending away from the bilayer normal along the horizontal axis. The distinguished crossing angle and rotation (average rotation: α3 = 23° and α3′ = 29°) of α3 in the intermediate state indicated the difference in the helical register in the multi-HAMP domain of NphtrII. In signaling states, such helical crossing and rotations may play an important role in downward signal propagation and HAMP2 four-bundle helix stabilization.
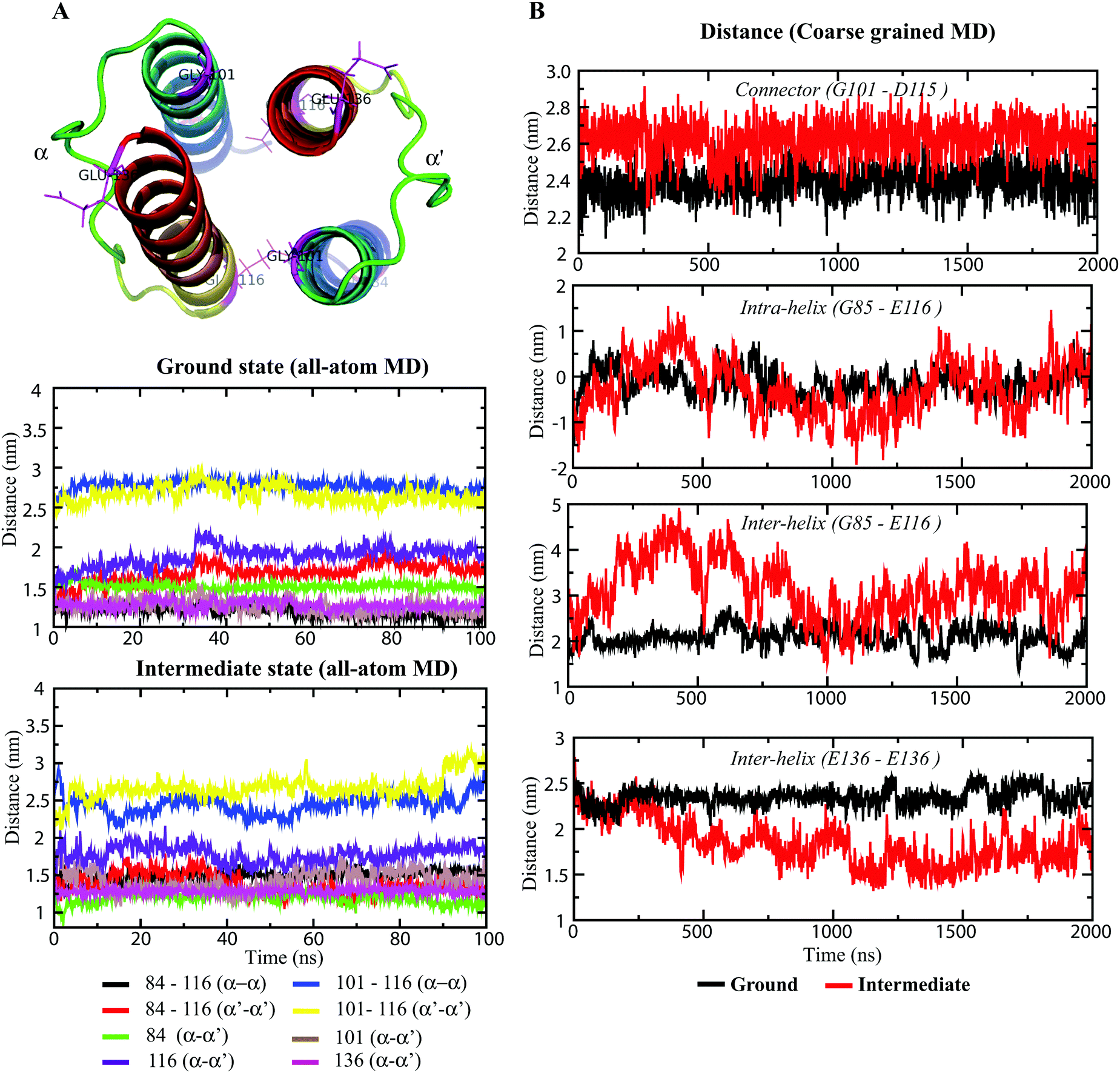
Longitudinal helix shift analysis
In recent years, several models have been proposed to describe the HAMP switching mechanism and downstream signal transduction, and these models include the gear-box model (based on knob-into-holes packing),17 dynamic-bundle model (based on the packing stability of four-bundle helices),44 piston-like model (based on HAMP-connected TM domain motion),46 and scissor-like motion (based on helix–helix crossing angles),16 among others. The differential states of HAMP1 domains13 and the dynamic behavior of HAMP1 helices suggest longitudinal and transverse displacement of the α1 and α2 helices. The distance measurement of the four-bundle helices from their respective terminus quantified these displacements for each helix and the connecting loop (connector). The distance measurement was performed between selective terminal residues (between their Cα atoms), as shown in Fig. 6. The ground state complex showed very little fluctuations in distances between intra and interhelical HAMP1 domains. Moreover, the flexible 14-residue (102–115) long loop presented a very small conformational rearrangement within the defined length of ∼24 Å (Fig. 6B). The distance between the N-terminal residues (E116) in α2 and α2′ exhibited an initial rise (∼30 ns) followed by a constant plateau (∼3 nm) for the ground state conformations (Fig. 6A). On the other hand, in the intermediate state, this distance showed a periodic terminal switching (Fig. 6A). Interestingly, the C- and N-terminus of α1 (G101) and α2 (E116), respectively, rendered an opposite helical register (Fig. 6A). When the distance at one terminus (α1 or α2) increases, the corresponding opposite terminus (α1′ or α2′) decreases. The switching persists for several nanoseconds (∼20 ns), and the graph appears like a sine curve with uniform repeats. The CG-MD simulation also rendered a curve-shaped distance graph for the intermediate conformation as compared with the flattened ground state distance graph (Fig. 6B). The changes in distances in the intermediate state conformation indicated the relatively large inter and intrahelical motions in the four-bundled HAMP1 dimer. The 14-residue connector also depicted a significant distance alternation (Fig. 6A) in the signaling state conformation during the simulation in comparison with the resting state complex. The uniform alternative helical shifts in the HAMP1 helices were significantly influenced by the NphtrII TM2 rotation connecting HAMP1 through well-defined control cables, and the conformational changes were channeled to the HAMP1 domain through piston-like motions.43,45
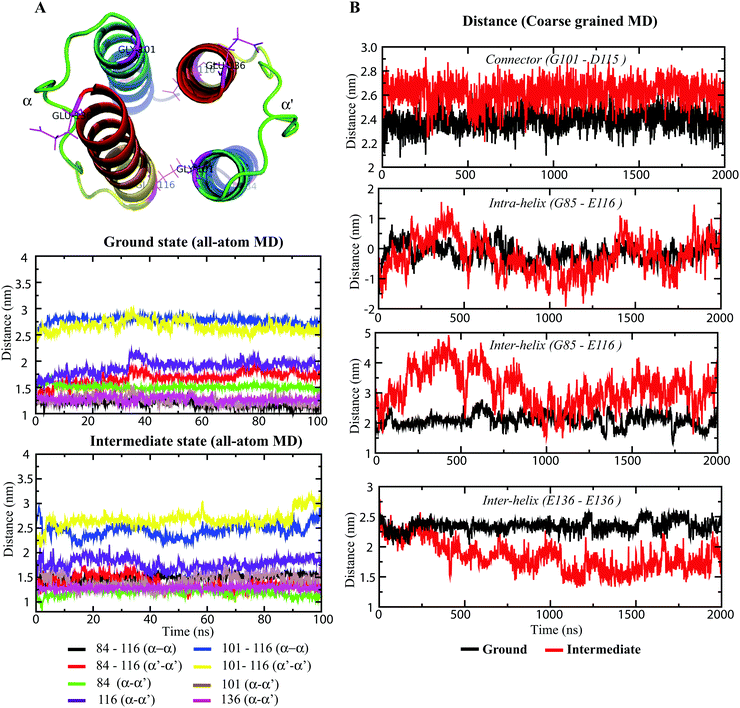 |
| | Fig. 6 Longitudinal distance measurement between selected HAMP residues using Cα-atoms. (A) The residues utilized for distance measurement are labeled in the protein cartoon representation. The distance graphs during 100 ns in the all-atom MD simulations are shown in both states using different colors. The color legends are given below the graph. (B) Graphs showing inter and intra helical distance changes during a 2 μs CG-MD simulation. Black and red represent distances for the inactive and active states, respectively. The distances are calculated between the Cα-atoms of the labeled residues. | |
Four-bundle HAMP helix packing
In multi-HAMP proteins, the HAMP conformations were significantly influenced by the neighboring HAMP domains and varied in terms of helix orientation and crossing angles. In P. aeruginosa (Aer2), the clockwise and counterclockwise rotations of α1 and α2 were associated with other HAMPs and modulated the inward and outward motions forming a trapezoidal shape for HAMP2.18 In NphtrII, previous studies reported a distinguished HAMP1 conformation (rectangular/rhomboid) in the absence of HAMP2.13 The results of our study provide structural insights into these two differential HAMP1 shapes in the ground (square) and intermediate (rhomboid) states. A sharp tilt in the α-helices in the active state resulted in a more similar rhomboid conformation with alternative helix movements (see the ESI,† Fig. S3). The crossing angle in α3 depicted substantial rotation and indicated its role in HAMP2 helical rearrangement. Both the all-atom and CG-MD simulations showed two distinct HAMP1 conformations, as shown in Fig. S3 (see the ESI†).
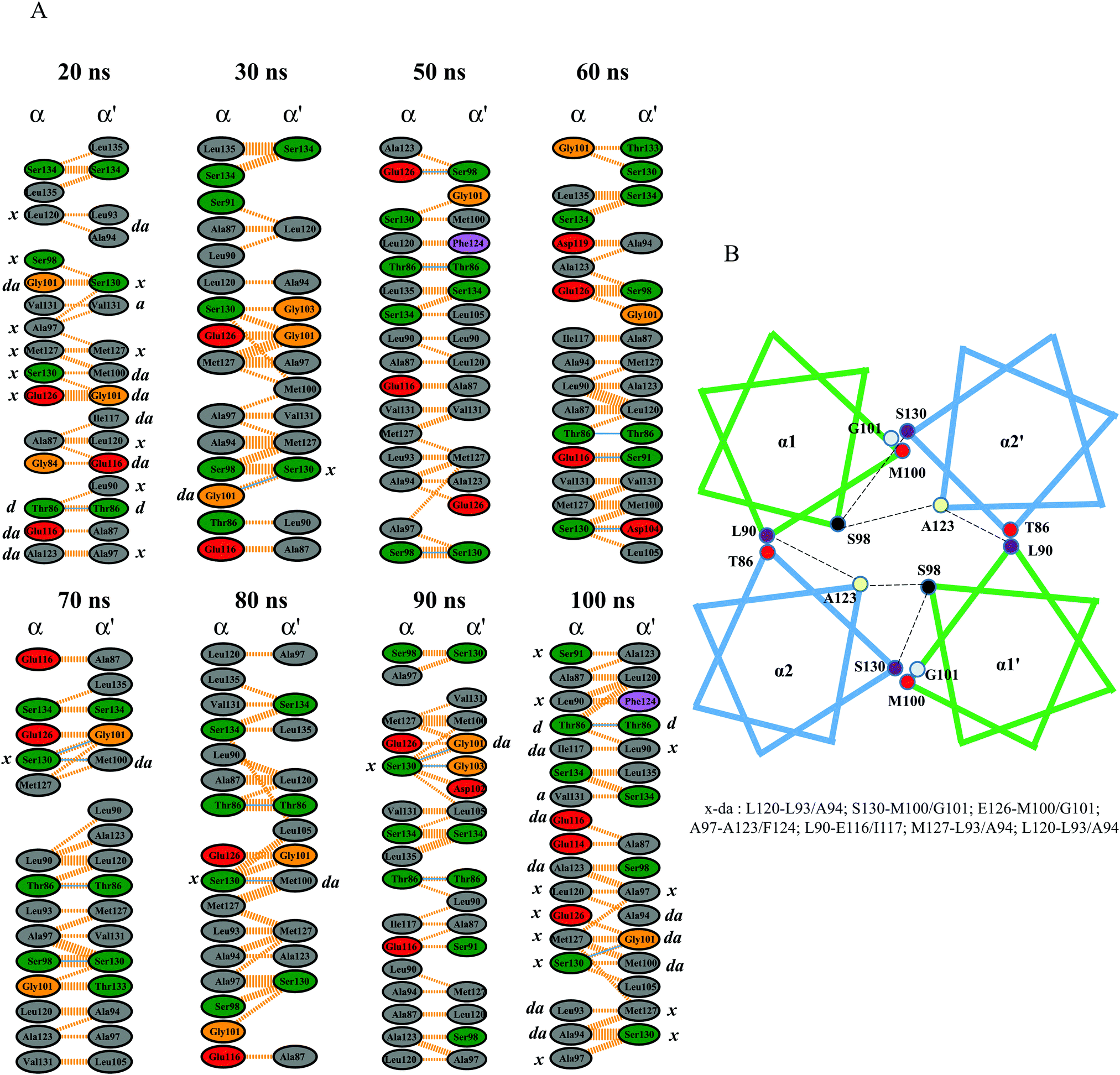
The helix packing analysis was performed by extracting a few snapshots from the MD trajectory. The Gly101 formed potential hydrogen and hydrophobic interactions with the “a” position of the heptad repeat (Ser130) at the end of α2′. This residue was also previously reported to be crucial in providing a sharp turn at the α1 helix,17 and its flexibility indicates its essential role in HAMP packing. These crossing angles and bindings are very important for complex stabilization, as seen in Aer2.18 The interaction map showed other potential hydrogen and hydrophobic interactions that mainly include leucine, threonine, and serine residues. The importance of leucine, serine, and threonine residues in intra/interhelical packing as well as homo or hetero-oligomerization has been widely investigated.47,48 The results of our study show conserved serine and threonine packing in the NpHAMP1 four-bundle helices (Fig. 7A). In addition, hydrophobic heptad packing analysis revealed a discrete helix packing map for our chosen MD snapshots. The different heptad packing along with its corresponding heptad position is illustrated in Fig. 7B. Serine and threonine also show a small helical kink at the N- and C-terminus (includes T86, S98, S130, and S134) of α1′ and α2′, respectively. These helical kinks were caused by irregular helix movements that destabilized the helix packing and thus decreased the phi and increased the psi angles.49 The “x–da” layer packing was switched and displaced along with the inward/outward and upward/downward helical shifts in the α-helices to stabilize the helix packing. These findings resemble Af1503 HAMP packing.17 The acidic residues (D104, E116, D119, and E126) in both α-helices were engaged in forming hydrogen and hydrophobic interactions. Residues such as T86, S98, M100, G101, and S130 were also actively engaged in the interaction network model throughout the simulation. Overall, the “x–da” packing pattern and opposite helix rotation signified the knob-into-holes packing and highlighted the proposed gear-box model mechanism in NphtrII for downstream signal transduction.
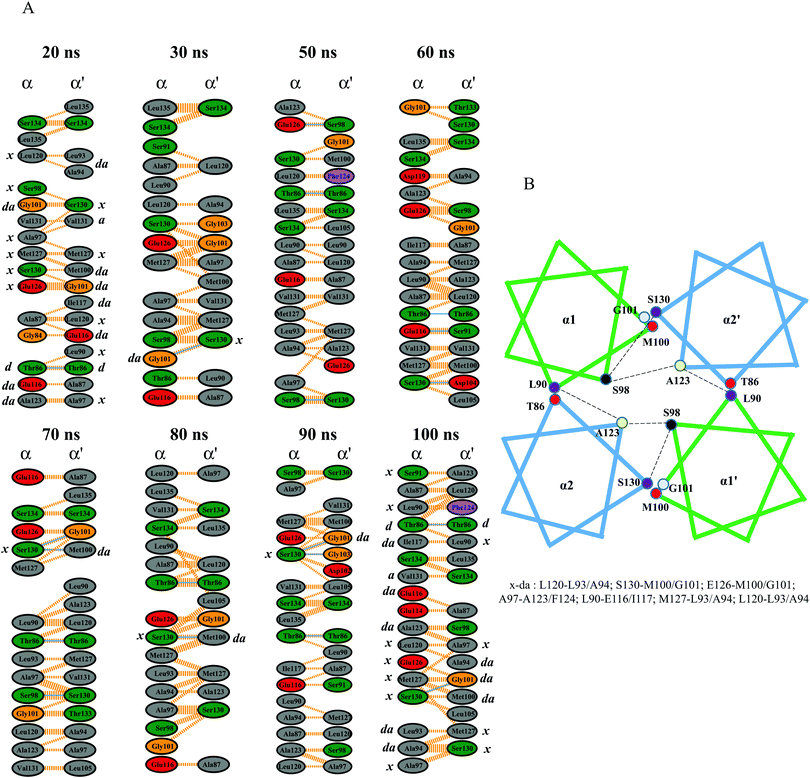 |
| | Fig. 7 Inter-residual interaction map in the HAMP1 homodimer generated using LigPlot+ (academic license). (A) Different snapshots and the interaction pattern of variable time scales are shown. H-bonds and hydrophobic interactions are shown as solid and dotted lines, respectively. Different amino acids are labeled inside the circle and are colored according to their hydrophobic properties. The “x–da’ packing pattern is shown at the bottom and the corresponding “x/da” residues are labeled for the H-bonded residues, 20 ns and 100 ns snapshots. (B) Schematic representation of the four-bundle HAMP helix packing representing a gear-box model HAMP signal propagation. | |
Effect of salt and temperature on HAMP1
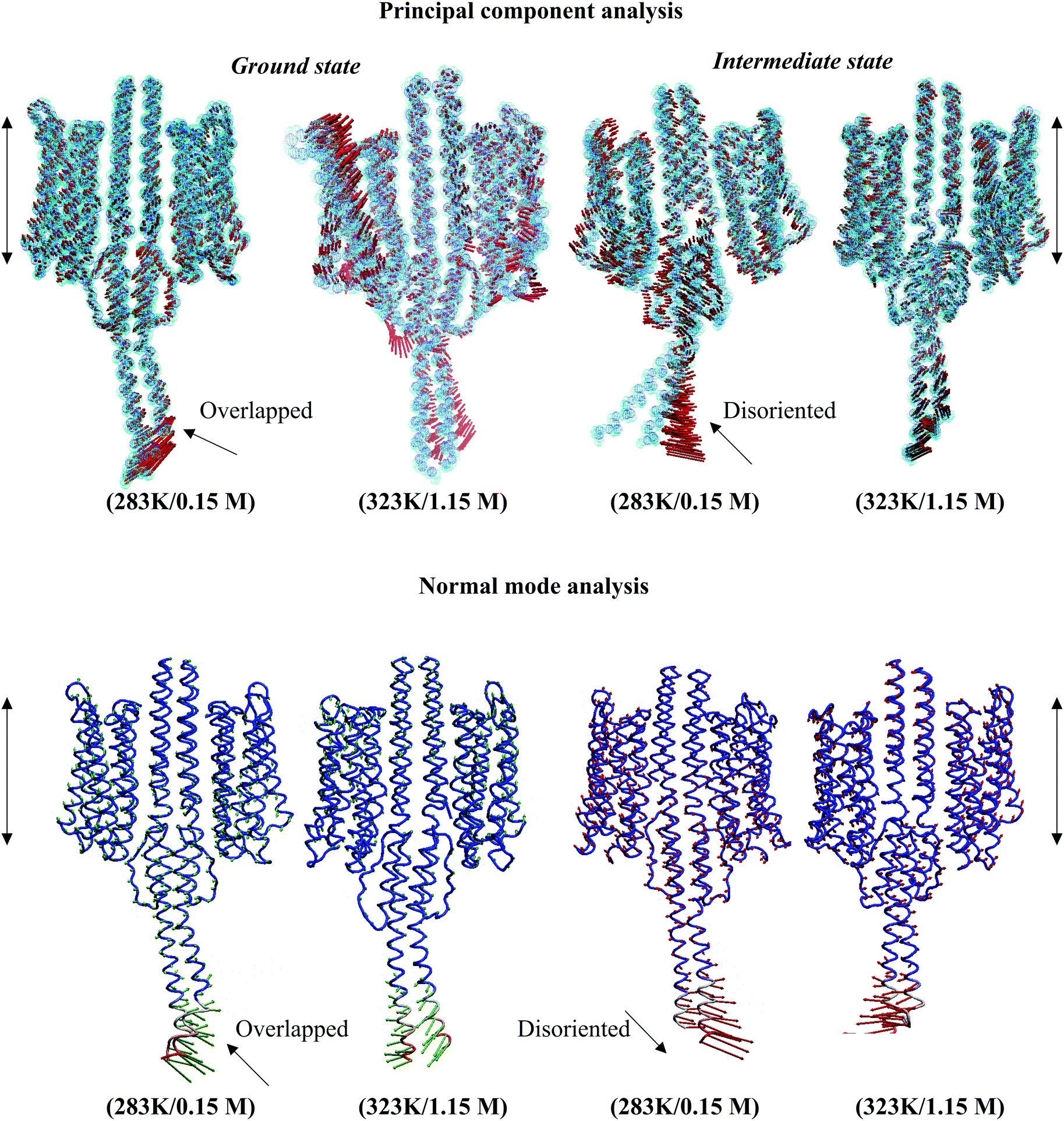
Structural analysis of the NpsrII–htrII complex under nonphysiological conditions presented an irregular helix kink (at T86, T92, Q129, S130, T132, Q147, and Q149) and a relatively high tilt for α1–α3 during the 100 ns period. The tilt angles of the α-helices in the ground state conformation were α1 = 3.77°, α2 = 6.02°, α3 = 16.83°, α1′ = 8.09°, α2′ = 5.66°, and α3′ = 7.60°, and for the intermediate states, α1 = 1.98°, α2 = 7.83°, α3 = 5.09°, α1′ = 3.21°, α2′ = 2.35°, and α3′ = 8.32°. The backbone RMSD comparison depicted an irregular graph for both ground and intermediate state NpsrII–htrII complexes in nonphysiological MD systems (see the ESI,† Fig. S4). CG-MD studies for 2 μs also presented an irregular helix bending in α3 (see the ESI,† Fig. S5). The PCA and ANM analyses showed disordered helix movements at the C-terminal end of NphtrII (Fig. 8). In addition, the irregular helix tilt arising from the α2/α2′ end restricted uniform helix rotation in the XY plane. Under physiological conditions, the helix movements were uniform, and the displacements and crossing angles at the C-terminal ends were nearly identical. Similarly, in the intermediate state, the low temperature and salt concentration significantly affected the helical tilt and helix packing (Fig. 8). The PCA and ANM analyses suggested that NpsrII–htrII complex formation and signaling were well achieved under physiological conditions and were significantly affected by the onset of unfavorable conditions.
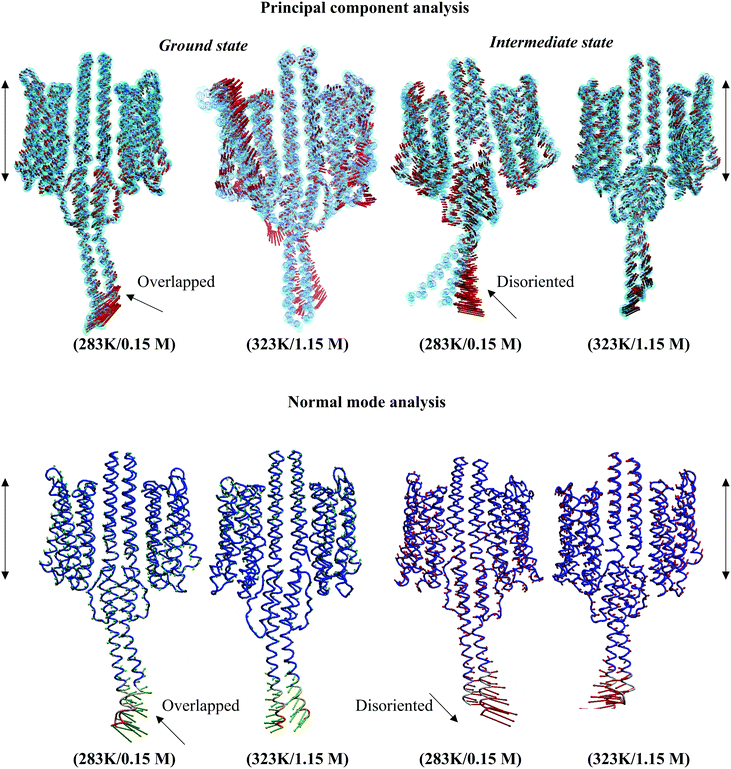 |
| | Fig. 8 Molecular dynamics trajectory analysis in active and inactive NpsrII–htrII complexes. The illustrations were generated using the ProDy program. The displacement and elasticity of the helices in low and physiological environments are shown using arrows. The backbone atoms are shown as dotted spheres and sticks in the PCA and ANM analyses, respectively. The membrane position is indicated by the double-sided black arrows. | |
Free binding energy analysis
Binding kinetics analysis in favoring bimolecular complex formation is very useful in exploring the biochemical pathways and biological functions. In addition to experimental techniques, computational methods, such as MM/PBSA and MM/GBSA, are very useful and have high accuracy. In this study, the execution of MM/PBSA in the truncated NphtrII homodimers yielded a ΔG that correlated with the experimental values described earlier for the srII–htrII complex in a detergent system. Here, in the heteromer system (2:2), the ΔG between srII and htrII was estimated to be −436.53 and −331.11 kJ mol−1 for the ground and intermediate states, respectively. Experimental reports depict a dissociation value (Kd) of ∼200 nM in detergent. However, the binding affinity in the lipid bilayer/membrane for these heterodimers is suggested to be at least one order of magnitude stronger.42 Thus, the computed ΔG value in the 2:2 complex closely follows the experimental values at 323 K in the ground state conformation as per their Kd in detergent is concerned.
In the absence of experimental binding energy information for the intermediate state, we first calculated the ΔG value between srII and htrII in the experimental structure (PDB ID: 2F95), which is −211.85 kJ mol−1. A higher binding energy may be expected for the intermediate complex in the presence of HAMP1 and the C-terminal helix (α3/α3′). MM/PBSA estimated a ΔG value of −331.11 kJ mol−1 at 323 K for the intermediate state. Among various energetic parameters, the coulombic, van der Waals, and nonpolar solvation energies favor heteromer formation. In contrast, the polar solvation energy opposes the binding. The coulombic and polar solvation energies in the intermediate complex are much smaller than those in the ground state conformations. The relatively smaller ΔG value suggests loose packing between srII and htrII, that facilitate signal transduction. In the NphtrII dimer and truncated HAMP1 complexes, the polar solvation energy favored homodimerization and the coulombic terms restricted complex binding. This condition indicates that homodimerization in NphtrII is preferably contributed by the hydrophobic interactions, whereas heterodimerization is favored by the coulombic forces. The different binding energy parameters and the overall ΔG values for all systems are listed in Table 2. The binding energy calculated between the only HAMP1 domains presented a very small ΔG value (−21.03 kJ mol−1) for the ground state conformation as compared with the intermediate (−54.68 kJ mol−1) conformation in the 2:2 complex (Table S2, ESI†). In contrast, a comparatively high binding energy between the HAMP1 domains was estimated in the NphtrII homodimer (1:1) suggesting a more compact conformation in the absence of the sensory rhodopsin molecule.
Mutagenesis study of the thermodynamic stability of the srII–htrII complex
The contribution of individual interacting residues is highlighted by performing in silico virtual alanine scanning, which is widely used to predict hot spots in protein receptors.50 The effects of point mutations on the thermodynamic stability in NpsrII–htrII are presented in Table S3 (ESI†). The ΔG calculation showed a significant difference in the coulombic and polar solvation energy scores. The consequence of the mutation of the key hydrogen-bonded residues3,37 depicted significant ΔG alteration in the srII–htrII complex. The mutation of residues Y199srII, T189srII, and E43htrII resulted in substantial ΔG alteration compared with other mutations. As reported,37 the alanine mutation of Y199srII and F28htrII was shown to abolish heterodimer formation by destabilizing the aromatic side-chain interactions. However, our study showed a decrease in ΔG upon Y199srII mutations because of the loss of one potential hydrogen bond (BE altered ∼1.73 kJ mol−1).37 We speculated that the MM/PBSA computation on post-MD mutated snapshots may be the reason for a net ΔG (Table S3, ESI†). To further investigate, a 10 μs long CG-MD simulation was performed on the mutated (Y199srII and F289srII to alanine) ground state conformation (PDB: 1H2S). The results revealed no srII/htrII segregation during the 10 μs simulation period. This indicates that either (1) the influence of other bonding residues stabilizes complex formation or (2) a very longtime scale MD simulation is required to achieve such complex disintegration. Mutation at S189srII (bonded to E43htrII and S62htrII) and E43htrII rendered a significant ΔG alteration (Table S3, ESI†). Altogether, the mutational analysis of the intermediate state complex highlights the crucial role of Y199srII, T189srII, and E43htrII in complex stabilization and heterodimerization during signal propagation similar to the ground state conformation.
Conclusion
The different states of the HAMP1 domain are influenced by different environmental conditions as well as the NpsrII–htrII complex state. The enforcement of geometrical and conformational states in HAMP1 was largely regulated by the homodimer and heterodimer arrangements. Structural analysis showed that a physiological cell environment is necessary for the phototaxis in the haloalkaliphilic archaeon. The photo-activated conformational changes in NpsrII–htrII transferred to the HAMP1 domain through a piston-like helical shift and TM2 rotary motion. The “x–da” arrangements in HAMP1 suggested the knob-into-holes packing with a gear-box-like model for HAMP activation. The MM/PBSA calculations indicated that substantial ΔG is required for homo and heterodimerization in both the ground and intermediate states. The convergence of the present set of binding energy calculations was carefully monitored and could also be considered in the experimental and theoretical free-energy estimations. The key binding residues (i.e., Y199srII, T189srII, E43htrII, T86htrII, M100htrII, E116htrII, E126htrII, and S130htrII) predicted in srII and htrII may assist in modulating the phototaxis in N. pharaonis. Moreover, this analysis provides structural insights into the longitudinal helical shift in the four-bundle HAMP1 helices (α/α′). The conformational arrangement mediated by the NphtrII rotation through control cables and potential hydrophobic (x–da) packing for downstream signal transduction in different complex states could be useful in exploring the mechanism of phototaxis.
Acknowledgements
This work was supported by the Ministry of Education, Science, Sports and Culture of Japan.
References
- S. Kitanovic, P. Ames and J. S. Parkinson, J. Bacteriol., 2011, 193, 5062–5072 CrossRef CAS PubMed.
- A. A. Wegener, J. P. Klare, M. Engelhard and H. J. Steinhoff, EMBO J., 2001, 20, 5312–5319 CrossRef CAS PubMed.
- V. I. Gordeliy, J. Labahn, R. Moukhametzianov, R. Efremov, J. Granzin, R. Schlesinger, G. Büldt, T. Savopol, A. J. Scheidig, J. P. Klare and M. Engelhard, Nature, 2002, 419, 484–487 CrossRef CAS PubMed.
- J. Spudich and H. Luecke, Curr. Opin. Struct. Biol., 2002, 12, 540–546 CrossRef CAS PubMed.
- J. P. Klare, V. I. Gordeliy, J. Labahn, G. Büldt, H. J. Steinhoff and M. Engelhard, FEBS Lett., 2004, 564, 219–224 CrossRef CAS PubMed.
- I. Gushchin, A. Reshetnyak, V. Borshchevskiy, A. Ishchenko, E. Round, S. Grudinin, M. Engelhard, G. Büldt and V. Gordeliy, J. Mol. Biol., 2011, 412, 591–600 CrossRef CAS PubMed.
- A. Ishchenko, E. Round, V. Borshchevskiy, S. Grudinin, I. Gushchin, J. P. Klare, T. Balandin, a. Remeeva, M. Engelhard, G. Büldt and V. Gordeliy, J. Photochem. Photobiol., B, 2013, 123, 55–58 CrossRef CAS PubMed.
- R. Moukhametzianov, J. P. Klare, R. Efremov, C. Baeken, A. Göppner, J. Labahn, M. Engelhard, G. Büldt and V. I. Gordeliy, Nature, 2006, 440, 115–119 CrossRef CAS PubMed.
- E. Bordignon, J. P. Klare, M. Doebber, A. a. Wegener, S. Martell, M. Engelhard and H. J. Steinhoff, J. Biol. Chem., 2005, 280, 38767–38775 CrossRef CAS PubMed.
- J. Wang, J. Sasaki, A. L. Tsai and J. L. Spudich, J. Biol. Chem., 2012, 287, 21316–21325 CrossRef CAS PubMed.
- M. V. Airola, N. Sukomon, D. Samanta, P. P. Borbat, J. H. Freed, K. J. Watts and B. R. Crane, PLoS Biol., 2013, 11, e1001479 CAS.
- L. Zhu, P. G. Bolhuis and J. Vreede, PLoS Comput. Biol., 2013, 9, e1002913 CAS.
- I. Gushchin, V. Gordeliy and S. Grudinin, PLoS One, 2013, 8, e66917 CAS.
- K. Hayashi, Y. Sudo, J. Jee, M. Mishima, H. Hara, N. Kamo and C. Kojima, Biochemistry, 2007, 46, 14380–14390 CrossRef CAS PubMed.
- J. S. Parkinson, Annu. Rev. Microbiol., 2010, 64, 101–122 CrossRef CAS PubMed.
- K. E. Swain and J. J. Falke, Biochemistry, 2007, 46, 13684–13695 CrossRef CAS PubMed.
- M. Hulko, F. Berndt, M. Gruber, J. U. Linder, V. Truffault, A. Schultz, J. Martin, J. E. Schultz, A. N. Lupas and M. Coles, Cell, 2006, 126, 929–940 CrossRef CAS PubMed.
- M. V. Airola, K. J. Watts, A. M. Bilwes and B. R. Crane, Structure, 2010, 18, 436–448 CrossRef CAS PubMed.
- M. Doebber, E. Bordignon, J. P. Klare, J. Holterhues, S. Martell, N. Mennes, L. Li, M. Engelhard and H. J. Steinhoff, J. Biol. Chem., 2008, 283, 28691–28701 CrossRef CAS PubMed.
- D. Spiliotopoulos, A. Spitaleri and G. Musco, PLoS One, 2012, 7, e46902 CAS.
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed.
- N. A. Baker, D. Sept, S. Joseph, M. J. Holst and J. a McCammon, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10037–10041 CrossRef CAS PubMed.
- A. Sali and T. L. Blundell, J. Mol. Biol., 1993, 234, 779–815 CrossRef CAS PubMed.
- K. Nishikata, M. Ikeguchi and A. Kidera, Biochemistry, 2012, 51, 5958–5966 CrossRef CAS PubMed.
- B. R. Sahoo, P. K. Dubey, S. Goyal, G. K. Bhoi, S. K. Lenka, J. Maharana, S. K. Pradhan and R. S. Kataria, Chem.-Biol. Interact., 2014, 220, 255–268 CrossRef CAS PubMed.
- M. G. Wolf, M. Hoefling, C. Aponte-Santamaría, H. Grubmüller and G. Groenhof, J. Comput. Chem., 2010, 31, 2169–2174 CrossRef CAS PubMed.
- W. D. Cornell, P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell and P. A. Kollman, J. Am. Chem. Soc., 1995, 117, 5179–5197 CrossRef CAS.
- K. Lindorff-Larsen, S. Piana, K. Palmo, P. Maragakis, J. L. Klepeis, R. O. Dror and D. E. Shaw, Proteins, 2010, 78, 1950–1958 CAS.
- S. Hayashi, E. Tajkhorshid and K. Schulten, Biophys. J., 2002, 83, 1281–1297 CrossRef CAS PubMed.
- B. R. Sahoo, J. Maharana, G. K. Bhoi, S. K. Lenka, M. C. Patra, M. R. Dikhit, P. K. Dubey, S. K. Pradhan and B. K. Behera, Mol. BioSyst., 2014, 10, 1104–1116 RSC.
- B. R. Sahoo, M. R. Dikhit, G. K. Bhoi, J. Maharana, S. K. Lenka, P. K. Dubey and D. K. Tiwari, Amino Acids, 2015, 47, 381–400 CrossRef CAS PubMed.
- C. Oostenbrink, A. Villa, A. E. Mark and W. F. van Gunsteren, J. Comput. Chem., 2004, 25, 1656–1676 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- S. J. Marrink, H. J. Risselada, S. Yefimov, D. P. Tieleman and A. H. de Vries, J. Phys. Chem. B, 2007, 111, 7812–7824 CrossRef CAS PubMed.
- B. A. Hall, A. P. Chetwynd and M. S. P. Sansom, Biophys. J., 2011, 100, 1940–1948 CrossRef CAS PubMed.
- B. A. Hall, J. P. Armitage and M. S. P. Sansom, PLoS Comput. Biol., 2011, 7, e1002204 CAS.
- S. Hippler-Mreyen, J. P. Klare, A. A. Wegener, R. Seidel, C. Herrmann, G. Schmies, G. Nagel, E. Bamberg and M. Engelhard, J. Mol. Biol., 2003, 330, 1203–1213 CrossRef CAS PubMed.
- A. Bakan, L. M. Meireles and I. Bahar, Bioinformatics, 2011, 27, 1575–1577 CrossRef CAS PubMed.
- J. M. Swanson, R. H. Henchman and J. A. McCammon, Biophys. J., 2004, 86, 67–74 CrossRef CAS PubMed.
- B. R. Sahoo, J. Maharana, M. C. Patra, G. K. Bhoi, S. K. Lenka, P. K. Dubey, S. Goyal, B. Dehury and S. K. Pradhan, Colloids Surf., B, 2014, 121, 307–318 CrossRef CAS PubMed.
- J. Kyte and R. F. Doolittle, J. Mol. Biol., 1982, 157, 105–132 CrossRef CAS PubMed.
- J. Kriegsmann, M. Brehs, J. P. Klare, M. Engelhard and J. Fitter, Biochim. Biophys. Acta, 2009, 1788, 522–531 CrossRef CAS PubMed.
- G. A. Wright, R. L. Crowder, R. R. Draheim and M. D. Manson, J. Bacteriol., 2011, 193, 82–90 CrossRef CAS PubMed.
- Q. Zhou, P. Ames and J. S. Parkinson, Mol. Microbiol., 2009, 73, 801–814 CrossRef CAS PubMed.
- S. Kitanovic, P. Ames and J. S. Parkinson, J. Bacteriol., 2011, 193, 5062–5072 CrossRef CAS PubMed.
- J. J. Falke and G. L. Hazelbauer, Trends Biochem. Sci., 2001, 26, 257–265 CrossRef CAS PubMed.
- T. M. Gray and B. W. Matthews, J. Mol. Biol., 1984, 175, 75–81 CrossRef CAS PubMed.
-
D. Krylov and C. R. Vinson, Encyclopedia of Life Sci., 2001, pp. 1–7 Search PubMed.
- J. A. Ballesteros, X. Deupi, M. Olivella, E. E. Haaksma and L. Pardo, Biophys. J., 2000, 79, 2754–2760 CrossRef CAS PubMed.
- S. A. Martins, M. A. S. Perez, I. S. Moreira, S. F. Sousa, M. J. Ramos and P. A. Fernandes, J. Chem. Theory Comput., 2013, 9, 1311–1319 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6mb00730a |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here.