
Identifying differential networks based on multi-platform gene expression data†
Le
Ou-Yang
ab,
Hong
Yan
b and
Xiao-Fei
Zhang
*c
aCollege of Information Engineering, Shenzhen University, Shenzhen, China
bDepartment of Electronic and Engineering, City University of Hong Kong, Hong Kong, China
cSchool of Mathematics and Statistics & Hubei Key Laboratory of Mathematical Sciences, Central China Normal University, Wuhan, China. E-mail: zhangxf@mail.ccnu.edu.cn
First published on 11th November 2016
Abstract
Exploring how the structure of a gene regulatory network differs between two different disease states is fundamental for understanding the biological mechanisms behind disease development and progression. Recently, with rapid advances in microarray technologies, gene expression profiles of the same patients can be collected from multiple microarray platforms. However, previous differential network analysis methods were usually developed based on a single type of platform, which could not utilize the common information shared across different platforms. In this study, we introduce a multi-view differential network analysis model to infer the differential network between two different patient groups based on gene expression profiles collected from multiple platforms. Unlike previous differential network analysis models that need to analyze each platform separately, our model can draw support from multiple data platforms to jointly estimate the differential networks and produce more accurate and reliable results. Our simulation studies demonstrate that our method consistently outperforms other available differential network analysis methods. We also applied our method to identify network rewiring associated with platinum resistance using TCGA ovarian cancer samples. The experimental results demonstrate that the hub genes in our identified differential networks on the PI3K/AKT/mTOR pathway play an important role in drug resistance.
1 Introduction
Understanding the molecular basis of disease requires characterization of complex regulatory interactions between genetic components such as mRNAs and proteins. Although computational approaches have been widely used to identify gene regulatory networks that are involved in certain biological processes or conditions,1–8 most of them are developed for a single static condition.9 Research studies have shown that the development and progression of cancer often involve the aberrations of interdependencies between genetic components.10–13 Therefore, exploring how the network changes with genetic and environmental alterations is of great interest for understanding the underlying biological mechanisms.In recent years, the accumulation of gene expression profiles has provided rich functional information on gene regulatory network analysis. Gaussian graphical models, which assume the available gene expression data are generated from a multivariate Gaussian distribution, have been widely used for estimating gene regulatory networks from gene expression data.14,15 Based on Gaussian graphical models, two genes are conditionally independent given the other genes if and only if the corresponding entry of the inverse covariance matrix, or precision matrix, is zero. Therefore, we can infer gene regulatory networks by estimating the corresponding precision matrices.16 Gaussian graphical models have the advantage of inferring direct dependencies between genes. However, most existing methods estimate a gene regulatory network for a specific condition, and do not consider the network rewiring between different conditions or disease states. Differential network analysis, which aims to gain insights into altered regulatory mechanisms between two different conditions or disease states, has recently emerged as an important complement to differential expression analysis.17 Several computational approaches have been proposed for differential network analysis.9,13,18–21 Based on Gaussian graphical models, the differential network between two conditions can be formulated as the difference between the two corresponding precision matrices. Thus, a straightforward strategy is to separately estimate the precision matrix for each group and then calculate their difference.13,18 To exploit the similarity between gene networks corresponding to distinct but related conditions, Danaher et al. introduced a joint graphical lasso model to estimate multiple graphical models that share certain characteristics, such as the locations or weights of non-zero edges.22 However, most of the above methods need to infer the network for individual conditions and assume that all the gene networks are sparse. But real gene networks often contain genes that interact with many other genes, which may violate the sparsity condition. Furthermore, there are many interactions that are not strong in a single static condition, but the changes in these interactions are large.9 Direct application of these sparse regularized network inference methods may not be able to detect these changes.
The direct estimation of differential networks has emerged as an alternative approach for differential network analysis. Zhao et al.20 proposed a differential network estimation method to directly estimate the difference between precision matrices. Their method does not require separate estimation of each precision matrix and does not require each precision matrix to be sparse. However, this estimator is computationally expensive, especially for large scale data sets.9,20 Recently, Yuan et al.9 proposed a new convex function to directly estimate the precision matrix difference based on a D-trace loss function.23 Their model can be solved efficiently using the alternating direction method of multiplier.9
Recently, advances in biotechnology have allowed biomedical researchers to collect a wide variety of gene expression data on a common set of patients from different platforms.24 For example, The Cancer Genome Atlas (TCGA) has provided gene expression data collected from multiple platforms (e.g., Agilent 244K Custom Gene Expression G450, Affymetrix HT Human Genome U133 Array Plate Set and Affymetrix Human Exon 1.0 ST Array), which enables the reconstruction of gene regulatory networks and differential networks between two patient groups from different data platforms.25,26 Gene expression data collected from different platforms may share some common characteristics; integrating multiple gene expression profiles could help to identify differential networks more accurately. Although some previous methods can be applied to infer gene regulatory networks based on gene expression profiles collected from multiple data platforms,22,25 few methods are designed for estimating differential networks from multiple data platforms. We need to develop new computational methods that can integrate information on multiple gene expression profiles to estimate the differential networks between two patient groups.
To address the above problems and make effective use of multi-platform gene expression data, in this study, we propose a novel multi-view differential network analysis model (MEDIA) to directly infer the differential network between two patient groups based on multiple gene expression data sets collected from different data platforms. Instead of assuming that each individual group-specific network is sparse, we only assume that the differential network between two patient groups is sparse. This assumption is reasonable since for different disease states or patient groups, the corresponding gene regulatory networks are very similar to each other.9,22 Based on the D-trace loss function,9,23 and the group lasso penalty,27 our model can exploit the characteristics shared by gene expression data collected from different types of platforms. We utilize the alternating direction method of multiplier (ADMM) to solve the optimization problem. To illustrate the effectiveness of MEDIA, we first perform simulation studies, and then apply MEDIA to TCGA ovarian cancer samples to identify network rewiring associated with platinum resistance. Through the simulation studies and real data analysis, MEDIA shows a superior performance compared to other state-of-the-art differential network inference methods, which demonstrates the effectiveness and power of our model. Moreover, we find that the hub genes in our identified differential networks on the PI3K/AKT/mTOR pathway play an important role in drug resistance.
2 Methods
In this section, we will formulate our problem and describe the details of the proposed multi-view differential network analysis model (MEDIA). The workflow of the MEDIA is shown in Fig. 1.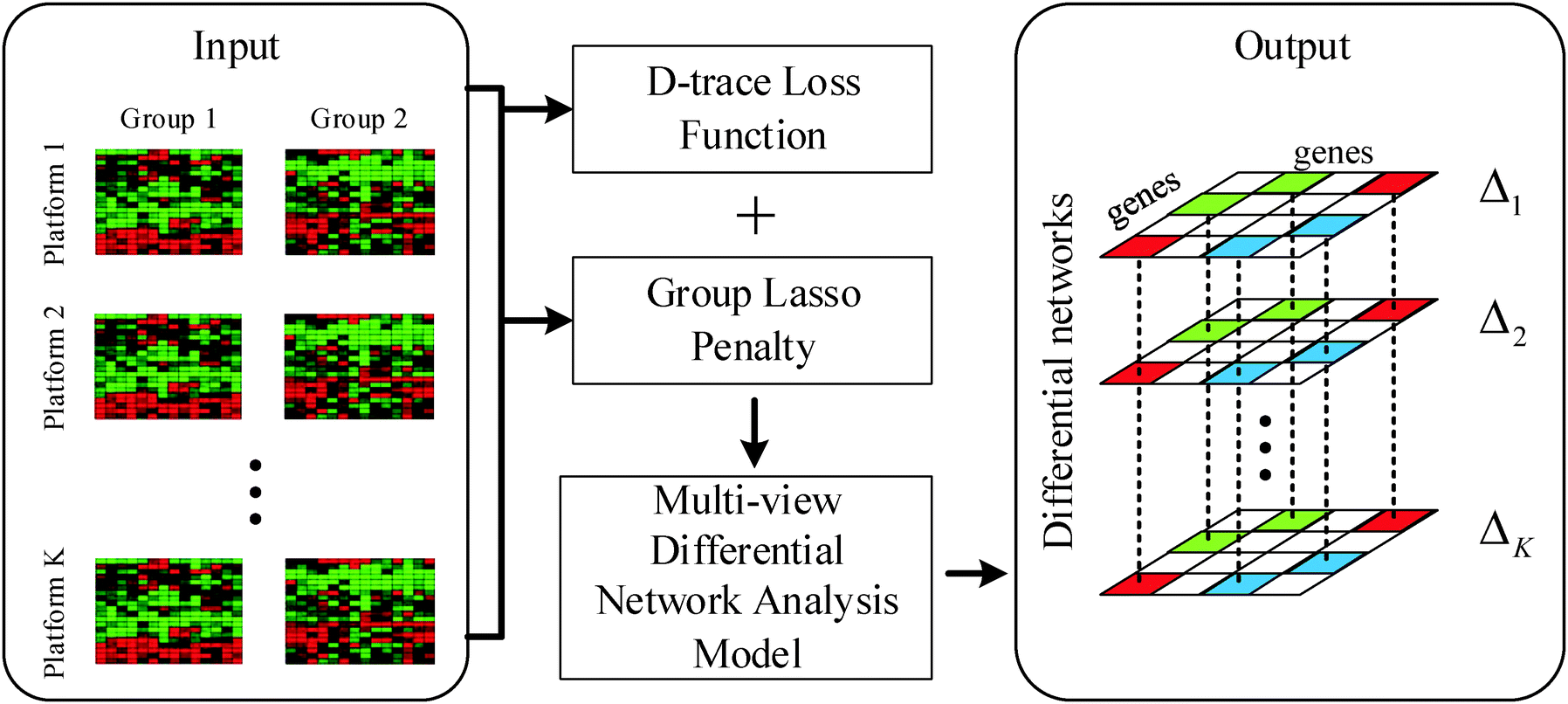 | ||
| Fig. 1 Illustration of the Multi-viEw DIfferential network Analysis model (MEDIA). The input of MEDIA is a set of gene expression profiles of two groups of patients collected from K microarray platforms. MEDIA utilizes D-trace loss function to directly estimate the differential network between two patient groups and uses group lasso penalty (i.e., l2,1-norm regularization) to enforce the differential networks Δk, k = 1,2,…,K for different platforms to have sparsity-consistent entries. The output of MEDIA is a set of differential networks that have same structures and different edge weights. | ||
2.1 Notations
Suppose that W = (wij) ∈![[Doublestruck R]](https://www.rsc.org/images/entities/char_e175.gif) p×p is a p × p matrix, and its Frobenius norm is
p×p is a p × p matrix, and its Frobenius norm is 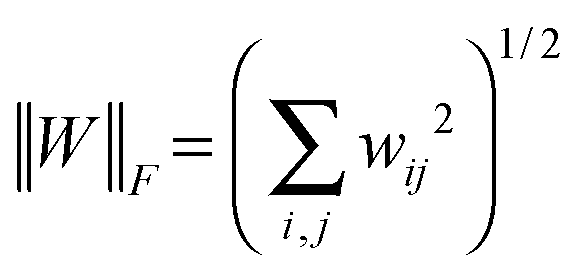 . The inner product of two same-sized matrix W and H is defined as 〈W,H〉 = tr(WHT) and we have 〈W,W〉 = ∥W∥F2.
. The inner product of two same-sized matrix W and H is defined as 〈W,H〉 = tr(WHT) and we have 〈W,W〉 = ∥W∥F2.
2.2 Model formulation
The conditional dependencies among p genes are often modelled using a Gaussian graphical model, where the gene expressions of n observations are assumed to be independently drawn from a multivariate normal distribution N(0,Θ−1) (here Θ = Σ−1 denotes the precision matrix and Σ denotes the covariance matrix) and two genes are conditionally dependent if and only if the corresponding entry of the precision matrix is nonzero. In this study, we also model the gene network as the precision matrix. Suppose there are n = nx + ny subjects that can be divided into two groups (nx denotes the number of subjects in group 1 and ny denotes the number of subjects in group 2), and we have independent observations of p genes collected from K different data platforms for these two group of subjects, i.e., xki = (xki1,…,xkip)T ∈p for i = 1,…,nx, k = 1,…,K from group 1 and ykj = (ykj1,…,ykjp)T ∈ p for j = 1,…,ny, k = 1,…,K from group 2. The two groups can correspond to two different environmental conditions or two different genetic conditions. We assume that 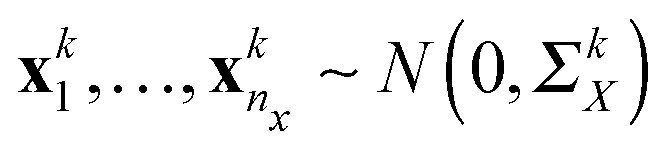 and
and 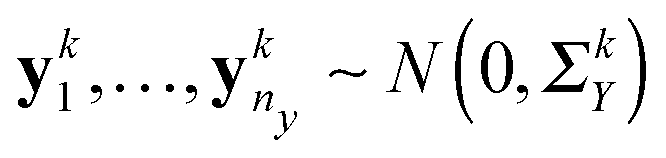 , where ΣkX and ΣkY are the covariance matrices for groups 1 and 2, corresponding to the k-th data platform for k = 1,…,K. Our goal is to simultaneously infer K differential networks that describe the change of conditional dependencies among p genes between two conditions (i.e., groups 1 and 2), based on gene expression data collected from K different data platforms. For each data platform, the differential network is defined as the difference between the two precision matrices, denoted by (ΣkY)−1 − (ΣkX)−1, k = 1,…,K.
, where ΣkX and ΣkY are the covariance matrices for groups 1 and 2, corresponding to the k-th data platform for k = 1,…,K. Our goal is to simultaneously infer K differential networks that describe the change of conditional dependencies among p genes between two conditions (i.e., groups 1 and 2), based on gene expression data collected from K different data platforms. For each data platform, the differential network is defined as the difference between the two precision matrices, denoted by (ΣkY)−1 − (ΣkX)−1, k = 1,…,K.
Following ref. 9, we use a symmetric matrix Δk to denote the precision matrix difference (ΣkY)−1 − (ΣkX)−1 and introduce a D-trace loss function LD(Δk,ΣkX,ΣkY), as follows:
| LD(Δk,ΣkX,ΣkY) = ½〈Δk2,(ΣkXΣkY + ΣkYΣkX)/2〉 − 〈Δk,ΣkX − ΣkY〉. | (1) |
![[capital Sigma, Greek, circumflex]](https://www.rsc.org/images/entities/i_char_e139.gif) kX,kY), where kX and kY are the sample covariance matrices for the k-th platform, to compute the estimator of Δk.
kX,kY), where kX and kY are the sample covariance matrices for the k-th platform, to compute the estimator of Δk.
Moreover, based on the assumption that the differential network between two patient groups is sparse, we need to introduce a lasso-type penalty on Δk for k = 1,…,K to encourage sparsity in each Δk. If we just apply l1 norm penalty on each Δk (i.e., ∥Δk∥1) in addition to the above D-trace loss function, it is equivalent to applying the lasso penalized D-trace loss method on each platform separately. However, the inference of the individual differential network Δk does not well utilize the cross-platform information, which is crucial to produce accurate and reliable results. Although different data platforms might reflect the dependencies between genes in different scales, the differential networks inferred from K different data platforms should have the same sparse structure with different edge weights. Therefore, to effectively fuse gene expression data collected from multiple platforms, we might wish to jointly estimate a collection of differential networks Δ1,Δ2,…,ΔK, where each Δk corresponds to the k-th platform. Here, to make effective use of the cross-platform information, the differential networks may be enforced to be sparsity-consistent. Based on this assumption, we formulated the following multi-view differential network analysis model (MEDIA). For the sake of convenience, we denote {kX}Kk=1, {kY}Kk=1 and {Δk}Kk=1 as {X}, {Y} and {Δ} respectively.
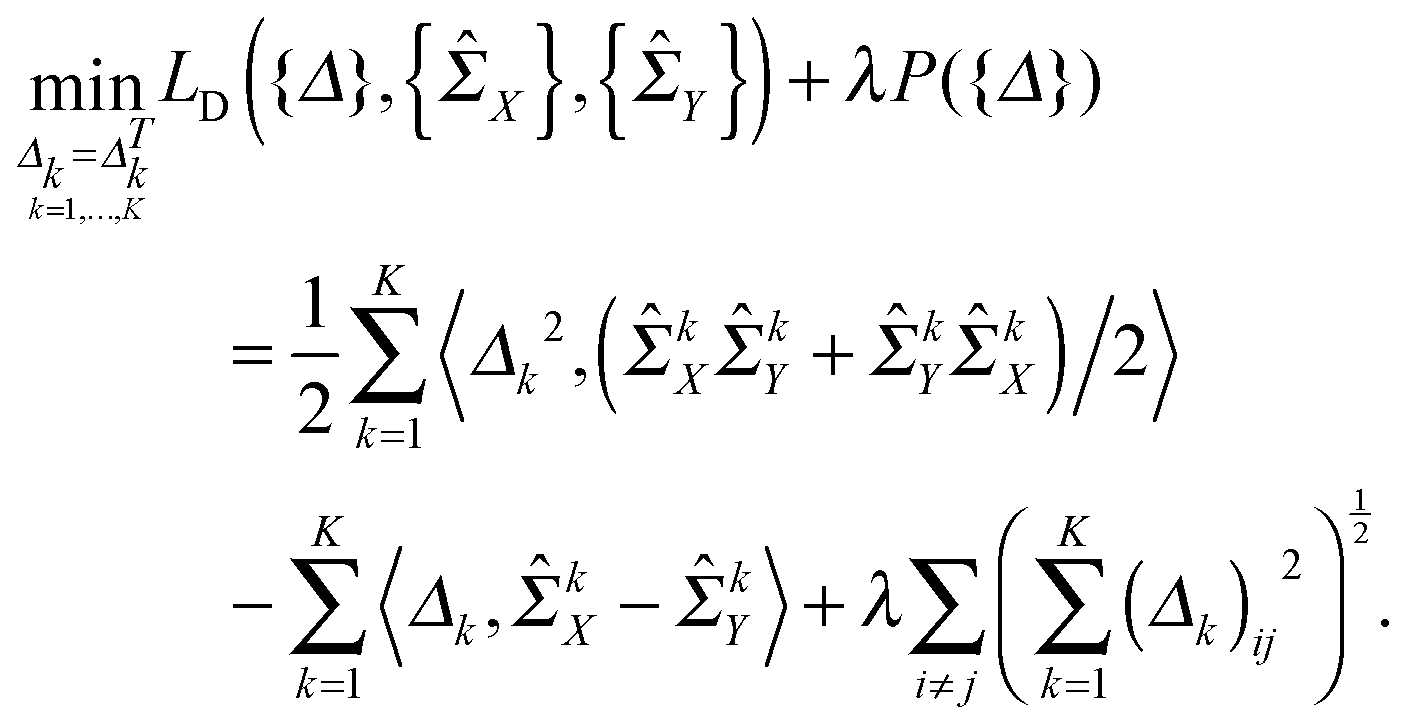 | (2) |
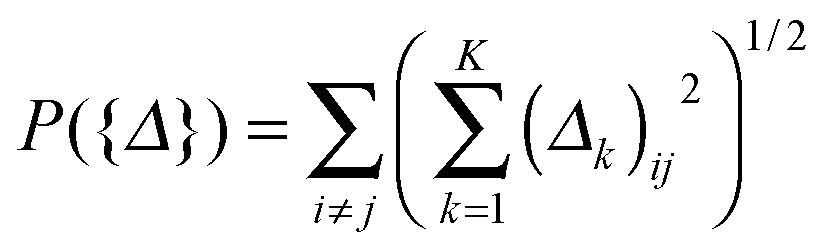 defined on {Δ} plays a key role in our MEDIA method: it is the minimization of P({Δ}) that enforces the entries (Δk)ij, k = 1, 2,…,K, to have consistent magnitudes, all either zeros or nonzeros, as shown in Fig. 1 (there will be a tendency for the 0s in the K estimated differential networks to occur in the same places). Without this regularization term, the formulation (2) will reduce to a “trivial” method that is equivalent to applying the D-trace loss function to infer each differential network Δk individually.
defined on {Δ} plays a key role in our MEDIA method: it is the minimization of P({Δ}) that enforces the entries (Δk)ij, k = 1, 2,…,K, to have consistent magnitudes, all either zeros or nonzeros, as shown in Fig. 1 (there will be a tendency for the 0s in the K estimated differential networks to occur in the same places). Without this regularization term, the formulation (2) will reduce to a “trivial” method that is equivalent to applying the D-trace loss function to infer each differential network Δk individually.
2.3 Algorithm for parameter estimation
In this section, we solve the optimization problem (2) by using an alternating direction method of multiplier (ADMM).28 We first introduce K auxiliary matrices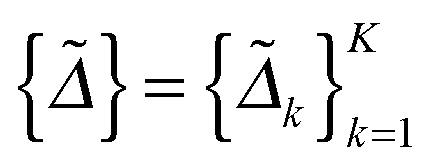 and rewrite (2) as
and rewrite (2) as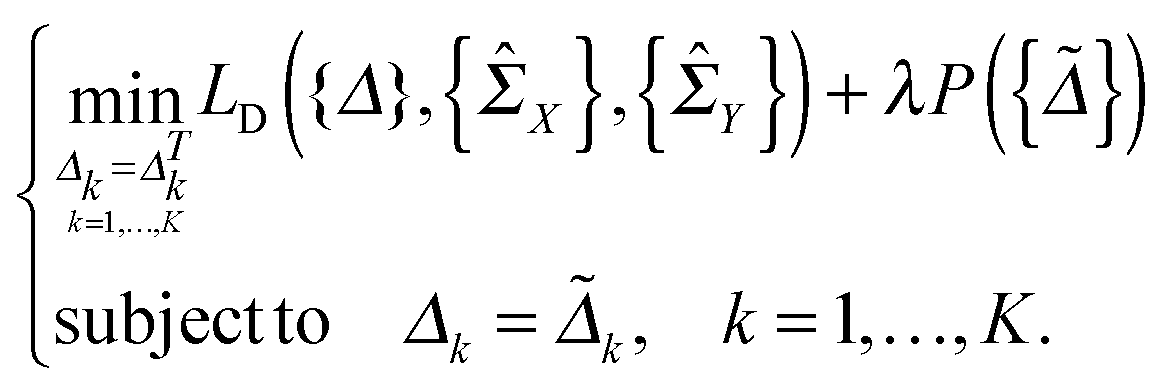 | (3) |
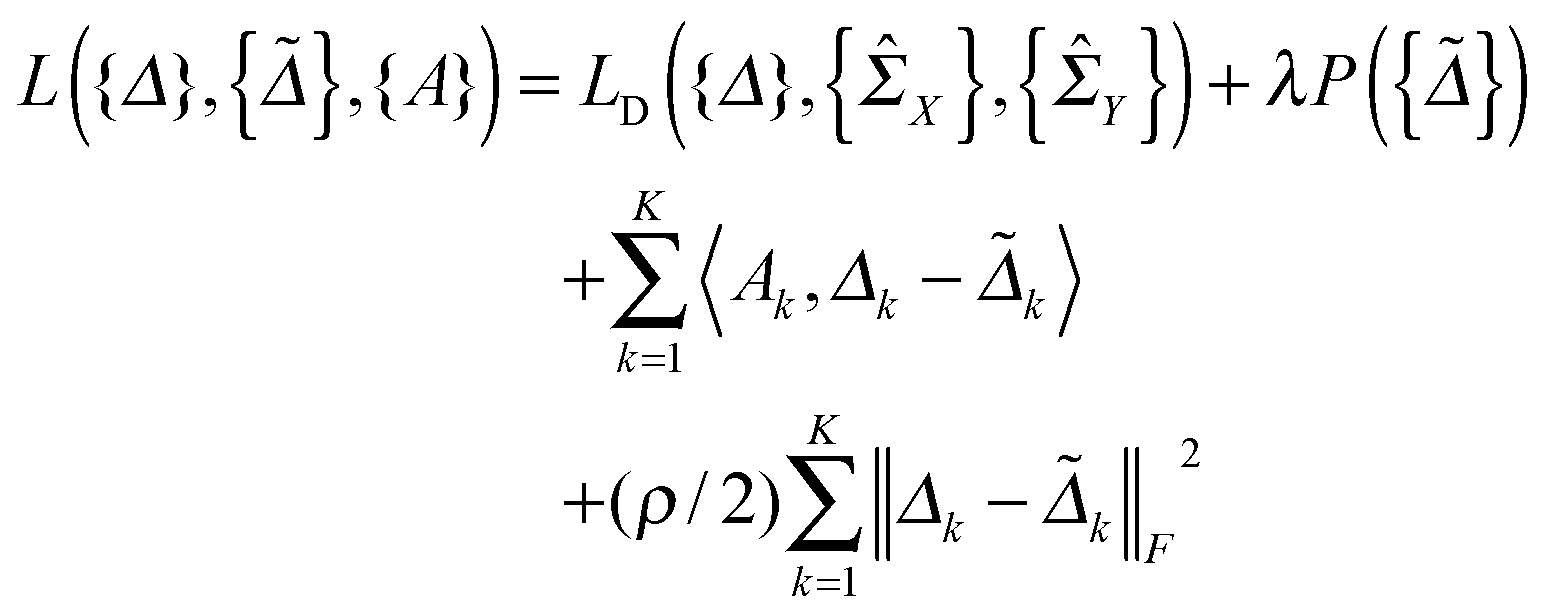 | (4) |
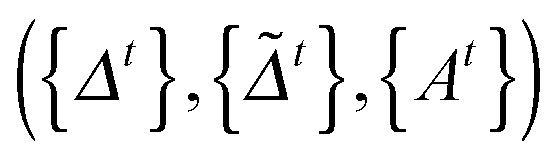 at the t-th step (t = 1, 2,…), we update
at the t-th step (t = 1, 2,…), we update  as follows:
as follows: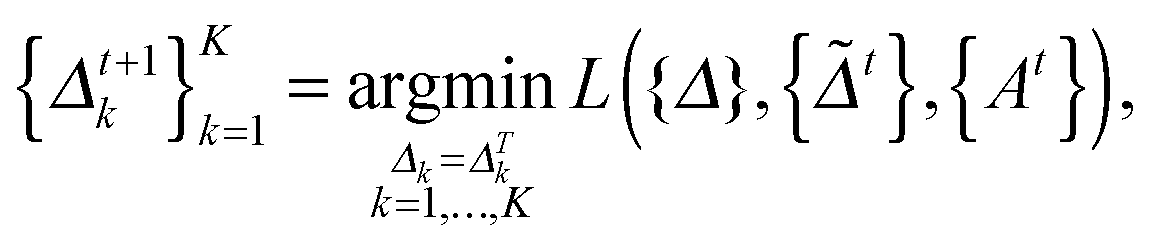 | (5) |
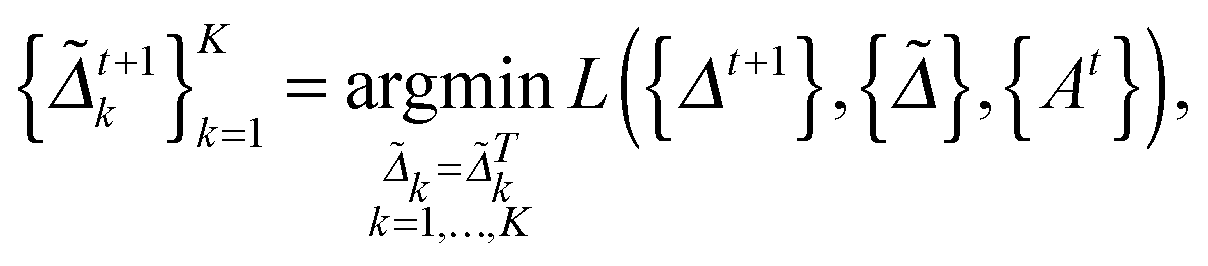 | (6) |
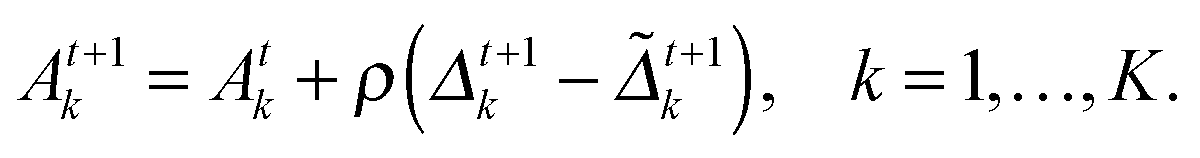 | (7) |
2.4 Selection of the tuning parameter
In this study, we use a model-free criterion called estimation stability with cross-validation (ESCV)9,29 to select the tuning parameter λ. In particular, we first randomly partition the samples into M subsets (Xk[m],Yk[m]) (m = 1,…,M), for k = 1,…,K (in this study, we set M = 5). For each data platform, we denote (Xk[−m],Yk[−m]) as the subsets by leaving out the m-th subset of samples (Xk[m],Yk[m]). For a given value of tuning parameter λ, we obtain the following solution: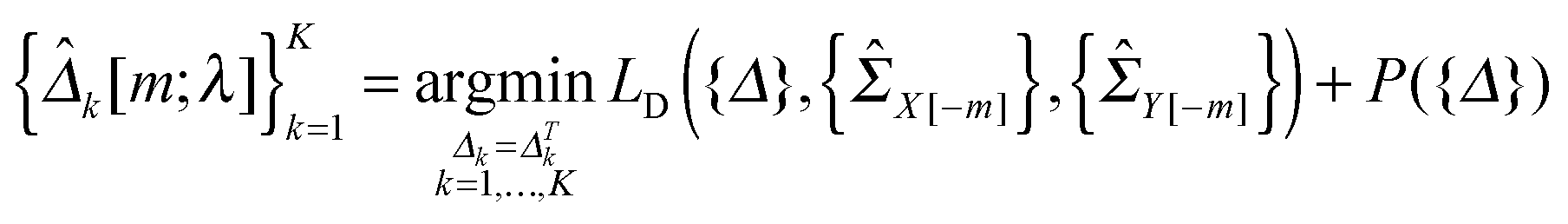 | (8) |
kX[−m] and kY[−m] are sample covariance matrices estimated from Xk[−m] and Yk[−m], respectively. Suppose that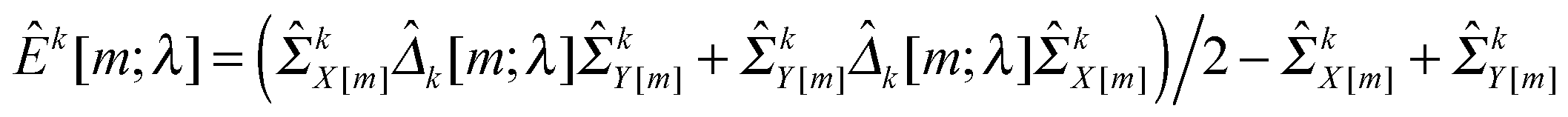 | (9) |
kX[m] and kY[m] are sample covariance matrices based on Xk[m] and Yk[m], respectively. Let 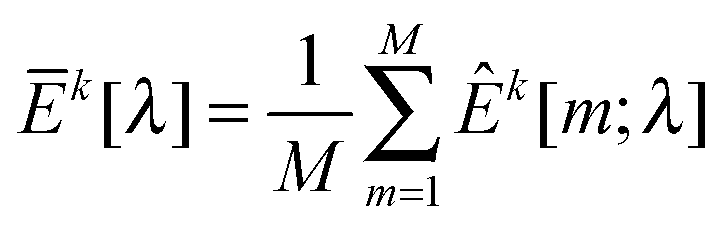 . We then define the sample variance of
. We then define the sample variance of 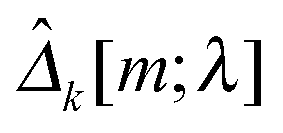 as
as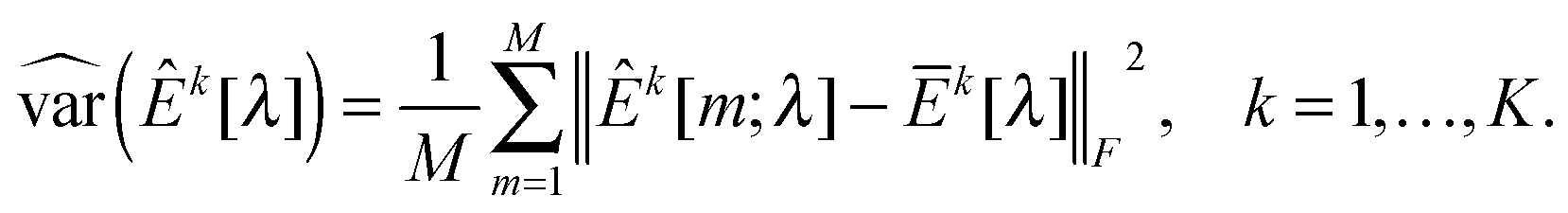 | (10) |
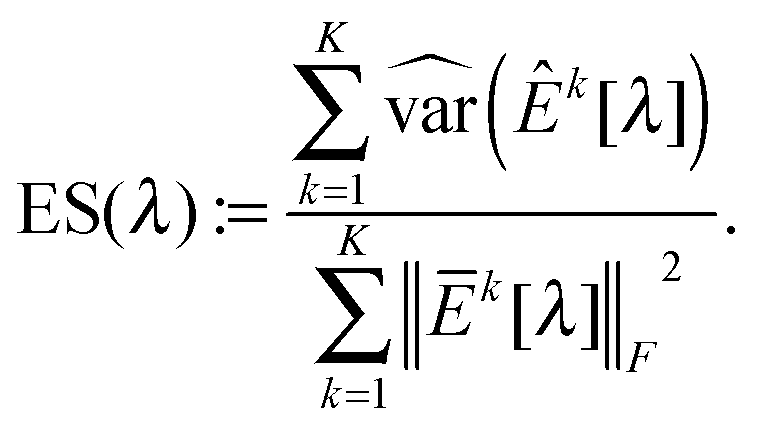 | (11) |
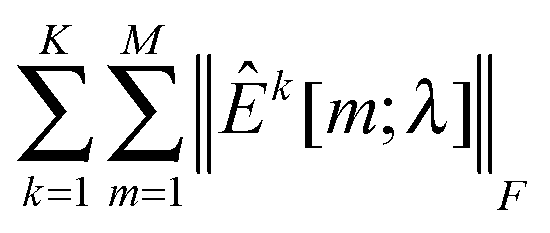 as λCV, and then choose λESCV < λCV that can minimize the estimation stability metric ES(λ).
as λCV, and then choose λESCV < λCV that can minimize the estimation stability metric ES(λ).
3 Simulation studies
In this section, we perform simulations to assess the performance of our multi-view differential network analysis model and compare it with other available algorithms. The algorithms we compare with include the joint graphical lasso with group lasso penalty (GGL) as proposed in ref. 22 and the D-trace loss estimator (D-trace loss) as proposed in ref. 9.The D-trace loss estimate method is designed to directly estimate the difference between two precision matrices, but this method can only analyze each data platform separately. The comparison between the MEDIA and D-trace loss estimate method will show the gain of performance when MEDIA utilizes group sparse penalty to borrow strength across multiple data platforms. GGL is designed to jointly estimate multiple precision matrices that share certain characteristics, while MEDIA directly estimates multiple differential networks from multiple data platforms. Comparing MEDIA with GGL would show the benefit of directly inferring multiple differential networks without estimating the individual precision matrices. We do not compare MEDIA with fused graphical lasso22 and the method of Zhao et al.20 as a previous study has shown that the D-trace loss estimate method outperforms these two approaches.9 In the following experiments, when applying the GGL, precision matrices corresponding to multiple platforms are fitted for each group separately, and then we estimate that the differential network corresponding to each platform by calculating the difference between the inferred group-specific precision matrices. When applying the D-trace loss estimate method, the differential network is inferred for each platform separately. For MEDIA, we can directly infer multiple differential networks from multiple data platforms simultaneously. For GGL, we use the JGL function with ‘penalty = group’ from the R package JGL. To ease interpretation, similar to ref. 22, we reparameterize the tuning parameters for GGL, where 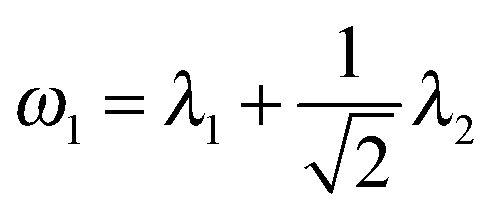 and
and 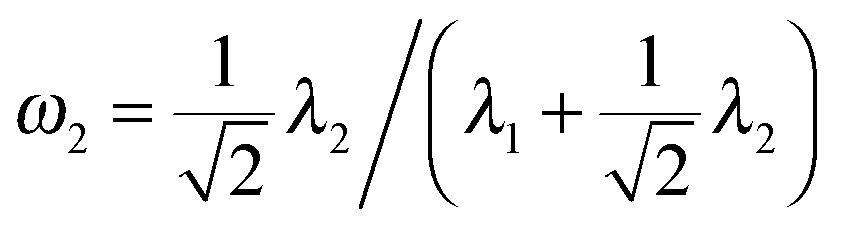 .
.
Following previous studies,9 we use several metrics to evaluate the performance of various algorithms. If ![[small delta, Greek, circumflex]](https://www.rsc.org/images/entities/i_char_e0b1.gif) kij is the (i,j)th entry of an estimator
kij is the (i,j)th entry of an estimator 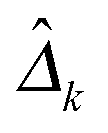 and δkij is the (i,j)th entry of the true Δk, for k = 1,…,K, the true positive rate (TPR), false positive rate (FPR), precision (Pre) and recall (Rec) are defined as
and δkij is the (i,j)th entry of the true Δk, for k = 1,…,K, the true positive rate (TPR), false positive rate (FPR), precision (Pre) and recall (Rec) are defined as
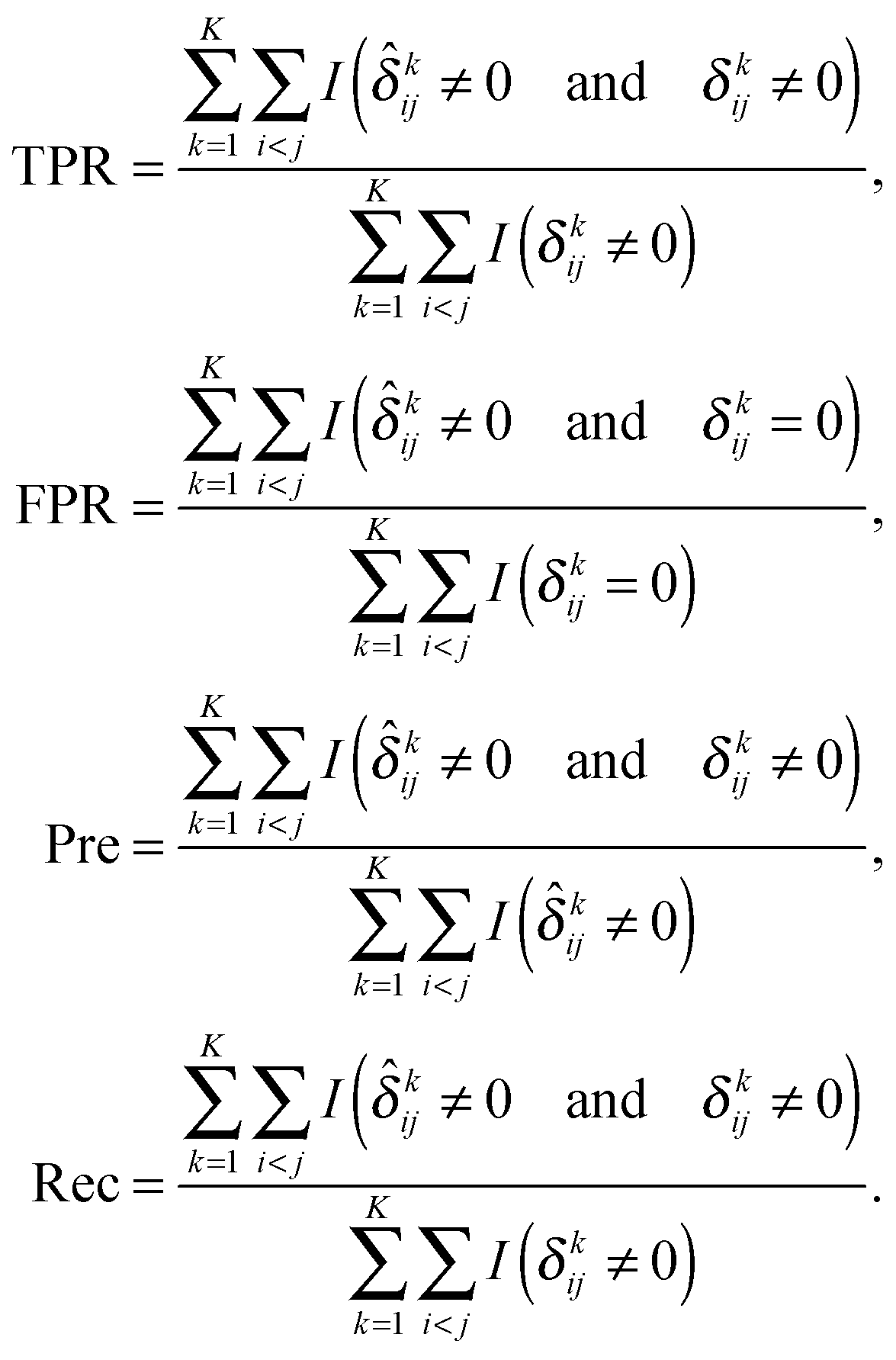
3.1 Simulation set-up
We consider two groups of subjects and their observations of p features collected from K = 3 data platforms. We generate 6 networks (i.e., scale-free, Erdos–Renyi and community) for the two groups of subjects and 3 data platforms, and all of them include the same set of p features. For each data platform, the networks corresponding to two groups have the same structure and only 10% of the edges have different values. The structure of networks and differential networks are preserved across the 3 data platforms. The scale-free, Erdos–Renyi and community are generated following the settings of Mohan et al.30 Across all simulation setups, we set p = 100 and nx = ny = 50, 100, 200.2. For k = 1,…,K, we repeat steps 3–5 to generate data sets for each data platform.
3. We generate a p × p symmetric matrix SkX to denote the adjacency matrix of the network corresponding to the first group. In particular, (SkX)ij = 0 if there is no edge between feature i and j, and a uniform distribution with support [−1, −0.5] ∪ [0.5,1] is used to generate the nonzero entry of SkX. We duplicate SkX into SkY. Then we set the entries of SkY that correspond to differential edges to be zeros or change their signs. Therefore, 10% of the entries in SkX and SkY have different values.
4. We let d = min(λmin(SkX),λmin(SkY)), where λmin(·) denotes the smallest eigenvalue of the matrix. To ensure positive definiteness, we set (ΣkX)−1 = SkX + (0.1 + |d|)I and (ΣkY)−1 = SkY + (0.1 + |d|)I, where I is a p × p identity matrix.
5. We generate nx = ny = n independent subjects for each group, i.e., xk1,…,xkn ∼ N(0,ΣkX) and yk1,…,xkn ∼ N(0,ΣkY).
3.2 Performance as a function of tuning parameters
The average performance of the compared methods on simulation data (Simulation 1) with p = 100 and n = 50, 100, 200 over 100 random generations of the data is shown in Fig. 2 (experimental results with respect to Simulations 2 and 3 are shown in Fig. S1 and S2, ESI†). The columns in Fig. 2 display the evaluation results with respect to different sample sizes (n = 50, 100, 200), while the rows in Fig. 2 display the evaluation results with respect to different evaluation metrics (the first row shows the ROC curves and the second row shows the precision–recall curves). In each plot, different colored curves correspond to the results of different methods, and different points on each curve correspond to the evaluation results with respect to different tuning parameter values. In particular, as MEDIA and D-trace loss have one tuning parameter, the curves for MEDIA and D-trace loss indicate the results obtained as the value of λ (for MEDIA) and λn (for D-trace loss) varied, respectively. Since GGL has two tuning parameters, each curve for GGL indicates the results obtained with a single value of the tuning parameter ω2, as the value of ω1 is varied. We observe that MEDIA outperforms GGL for a suitable range of parameters. Unlike GGL, which focuses on the joint estimation of the individual precision matrices and assumes individual precision matrices to be sparse, MEDIA does not need the sparsity assumption for individual precision matrices and is able to directly estimate multiple differential networks by integrating information across multiple data platforms. D-trace loss performs worse than MEDIA since it does not make use of the characteristics shared by different data platforms. Overall, our simulation results strongly suggest that MEDIA can be applied to a wide range of multi-view differential network inference scenarios.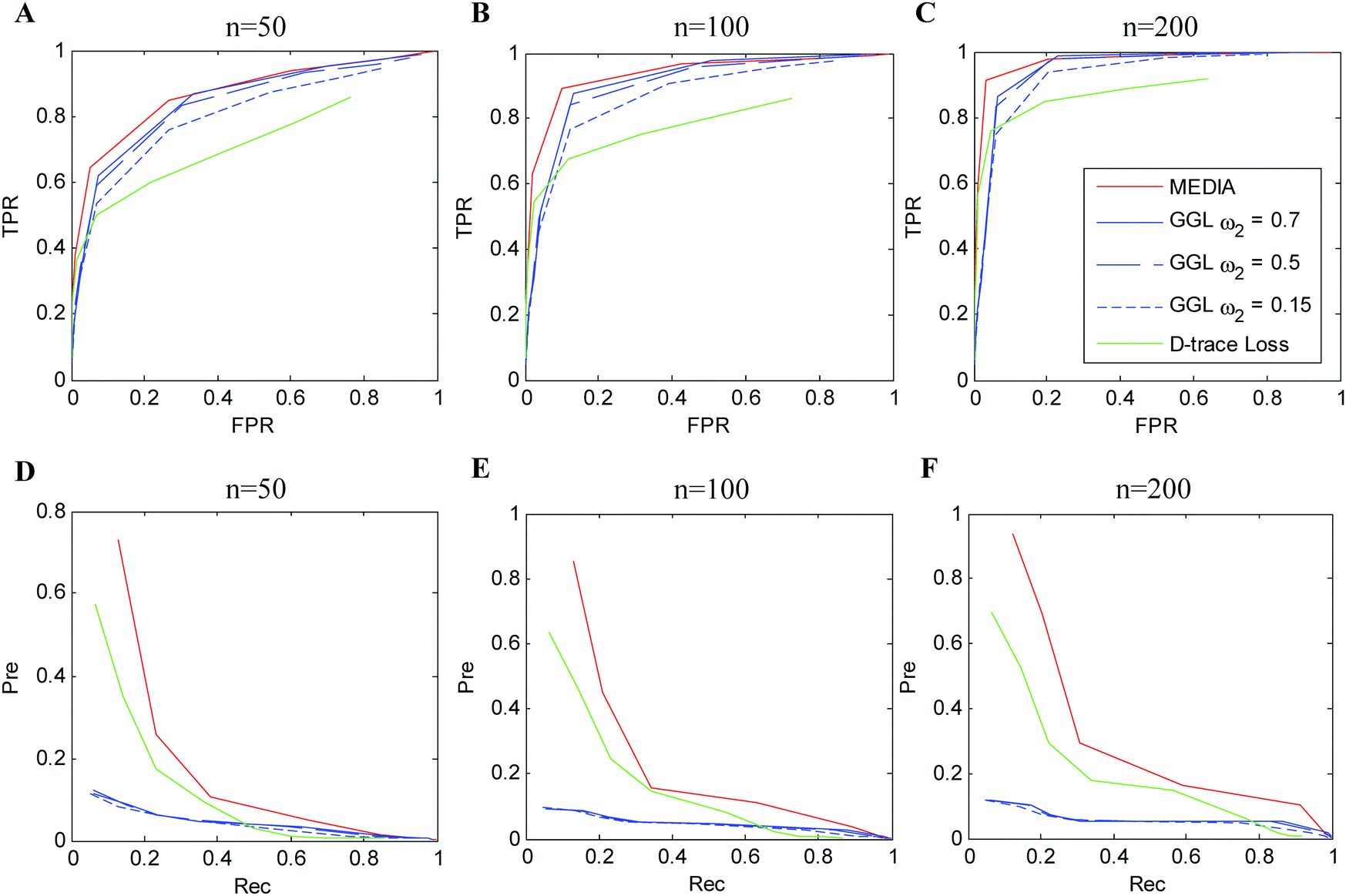 | ||
| Fig. 2 Performance of different methods on simulation data (Simulation 1) with p = 100, K = 3 and nx = ny = 50, 100, 200. (A–C) ROC curves. (D–F) Precision–recall curves. Red line: MEDIA; blue line: grouped graphical lasso (GGL); green line: D-trace loss estimator. For GGL, each colored curve corresponds to a fixed value of ω2, with the value of ω1 varied. Each curve is averaged over 100 random generations of the data. | ||
4 Gene expression study of ovarian cancer
4.1 Data sets
Ovarian cancer is the leading cause of cancer-related deaths among gynecologic tumors.31 As stated in ref. 32, the development of drug resistance is the predominant cause of treatment failure and death in ovarian cancer. Therefore, it is important to study the gene regulatory network difference between the drug sensitivity patient group and drug resistance patient group. The gene expression data sets for ovarian cancer patients were collected from The Cancer Genome Atlas (TCGA). In particular, three platforms, namely, Agilent 244K Custom Gene Expression G450, Affymetrix HT Human Genome U133 Array Plate Set, and Affymetrix Human Exon 1.0 ST Array, are considered in this study.26 For the sake of convenience, we refer to them as G450, U133 and HuEx, respectively. These gene expression profiles (level 3) were downloaded from the TCGA website (on February 2016), where the gene expression levels of 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 750 genes for 514 patients across all the three platforms are available. Logarithmic transformation is taken for each data set to make the data more normally distributed.
750 genes for 514 patients across all the three platforms are available. Logarithmic transformation is taken for each data set to make the data more normally distributed.
Following the criterion used in ref. 33, we can define platinum-based chemotherapy response groups. In particular, patients are defined as platinum sensitive if there is no evidence of disease progression within 6 months from the end of the last primary treatment, and the follow-up interval is at least 6 months from the date of last primary treatment. Patients with evidence of disease progression within 6 months from the end of primary treatment are defined as platinum resistant. Among the 514 patients, 340 patients have explicit platinum status, and 242 are platinum sensitive and 98 are platinum resistant. For each platform, the gene expression data within each sub-group of patients are standardized to have a mean of zero and a standard deviation of one.
In this study, we take a pathway-based analysis. We focus our attention on a specific pathway, namely the PI3K/AKT/mTOR pathway, which is frequently mutated or altered in ovarian cancer26 and is frequently implicated in resistance to anticancer therapies,34 to investigate the change in gene interactions between platinum sensitive and platinum resistant ovarian cancer patients. We downloaded the PI3K/AKT signaling pathway and the mTOR signaling pathway from the Kyoto Encyclopedia of Genes and Genomes database.35 Out of the 362 genes in the PI3K/AKT/mTOR pathway, 301 genes are included in our considered gene expression data sets. The standardized gene expression data for each sample can be downloaded from http://https://github.com/Zhangxf-ccnu/MEDIA.
4.2 Identifying differential networks associated with drug resistance in ovarian cancer
We applied MEDIA to the gene expression data from the above three platforms with respect to platinum sensitive patients and platinum resistant patients. The tuning parameter λ of MEDIA is tuned by ESCV. According to ESCV, we set λ = 0.418. Owing to the fact that MEDIA can encourage a shared pattern of sparse structures across different data platforms, the differential networks identified using our MEDIA from K data platforms have the same structures, which involves 430 differential edges among 267 genes.Within biological networks, hub genes always play important roles in biological processes. Thus, we are interested in the biological significance of hub genes in our identified differential networks. The list of hub genes that have degrees greater than 5 in the differential networks is presented in Table 1. As the estimated differential networks describe the change of conditional dependencies among 301 genes between the platinum sensitive patient group and the platinum resistant patient group, we first analyze whether the hub genes in the identified differential networks are associated with drug resistance and cancer development. We collected 161 genes related to cisplatin resistance and 758 genes related to drug resistance from the database of Genomic Elements Associated with drug Resistance (GEAR), and collected 572 genes for which mutations have been causally implicated in cancer from the Cancer Gene Census (CGC) database.36 Among the 301 genes in the PI3K/AKT signaling pathway, there are 26 genes, 74 genes and 60 genes associated with cisplatin resistance, drug resistance and cancer development respectively. We observe that out of the 36 hub genes in our identified differential networks, 7 of them are related to cisplatin resistance, 14 of them are related to drug resistance and 12 of them are related to cancer (the corresponding results are shown in Table 1). Based on Fisher's exact test, we find that our identified hub genes are significantly enriched with all three types of cancer-related genes (the p-values are 0.0234, 0.0401 and 0.0439 respectively).
| Evidence | Genes (degree of connectivity) |
|---|---|
| Here “GEARcisplatin” denotes the hubs detected by MEDIA that are associated with resistance to cisplatin, “GEARdrug” denotes the hubs detected by MEDIA that are associated with resistance to drugs and “CGC” denotes the hubs detected by MEDIA that are causally implicated in cancer according to the Cancer Gene Census (CGC) database. | |
| Hub genes detected by MEDIA | CDKN1A (9), TP53 (9), IRS1 (8), KIT (8), MAP2K1 (8), MYC (8), PRLR (8), RPS6KB1 (8), THBS4 (8), CCND2 (7), CCNE1 (7), COL4A6 (7), FGF13 (7), GNG12 (7), ITGA9 (7), LAMC3 (7), MET (7), PDGFA (7), PPP2R2A (7), BCL2L1 (6), CCND3 (6), COL4A2 (6), FGF20 (6), FGFR3 (6), GNB1 (6), IL6 (6), IL6R (6), ITGA6 (6), KITLG (6), PDPK1 (6), PPP2R5B (6), PTEN (6), RAC1 (6), RXRA (6), TSC2 (6), VEGFA (6) |
| GEARcisplatin | CDKN1A (9), TP53 (9), MYC (8), BCL2L1 (6), CCND3 (6), IL6 (6), PTEN (6) |
| GEARdrug | CDKN1A (9), TP53 (9), KIT (8), MYC (8), RPS6KB1 (8), CCND2 (7), FGF13 (7), BCL2L1 (6), CCND3 (6), FGFR3 (6), IL6 (6), PTEN (6), RAC1 (6), VEGFA (6) |
| CGC | TP53 (9), KIT (8), MAP2K1 (8), MYC (8), CCND2 (7), CCNE1 (7), MET (7), CCND3 (6), FGFR3 (6), PTEN (6), RAC1 (6), TSC2 (6) |
To give an example of the differential networks inferred by MEDIA on the PI3K/AKT/mTOR pathway, we show the top three hub genes (CDKN1A, TP53, IRS1) and their neighbors in Fig. 3. Note that if these three hub genes connect with other hub genes, we also show the neighbors of those hub genes. Therefore, there are six hub genes shown in Fig. 3, where CCNE1, FGF13 and COL4A6 are hub genes that connect with CDKN1A, TP53 and IRS1 respectively. Among the six hub genes shown in Fig. 3, CDKN1A and TP53 are known to be associated with cisplatin drug resistance, while FGF13 is known to be associated with drug resistance (Table 1). The other three hub genes (i.e., IRS1, CCNE1 and COL4A6) may be potential genes that are related to platinum resistance. IRS1 plays a key role in transmitting signals from the IGF1 receptors to the PI3K-Akt signalling pathway and the Erk MAP kinase pathway.37 As a signalling adapter protein, IRS1 can integrate different signalling cascades and has a functional role in cancer progression and platinum resistance.38 Ravikumar et al. have reported that IRS1 is an important mediator of ovarian cancer cell growth suppression and could be a potential effective target for chemotherapeutic intervention.31 Amplification of CCNE1 (cyclin E1) is the most prominent structural variant associated with primary treatment failure in high-grade serous ovarian cancer.39 Etemadmoghadam et al. have revealed that amplification of CCNE1 is a preadaptation that confers primary platinum resistance.40 We find that the associations between BCL2 and other two hub genes (FGF13 and CCNE1) undergo changes between the two patient groups (Fig. 3). Researchers have found that overexpression of BCL2 plays a role in the development of drug resistance.41,42 Eliopoulos et al. have revealed that BCL2 is frequently expressed in fresh biopsies of primary ovarian carcinoma and that it is an important determinant of drug-induced apoptosis thereby modulating resistance to chemotherapy.43 Okada et al. have reported that FGF13 plays a pivotal role in platinum resistance and in reducing intracellular platinum concentrations in cancer cells.44 Thus, the change in associations between BCL2 and two hub genes (CCNE1, FGF13) may be related to drug resistance. COL4A6 is involved in ECM-receptor interaction pathways. ECM can affect drug resistance by preventing drug penetration into the cancer tissue and the expression of COL4A6 is downregulated in drug-resistant cell lines.45 Among the seven neighbors of COL4A6 in the differential network, five of them (CCND3, FGF13, SPP1, IRS1 and CCNE1) are associated with ovarian cancer and drug resistance (e.g., CCND3 and FGF13 have been reported to be associated with drug resistance,44,46 SPP1 has been reported to be associated with ovarian cancer and cancer metastasis,47 and both IRS1 and CCNE1 are closely related to ovarian cancer and drug resistance38,40). Therefore, COL4A6 and its neighbors in the differential network may potentially be used as a molecular target to improve the treatment of platinum-resistant tumors.
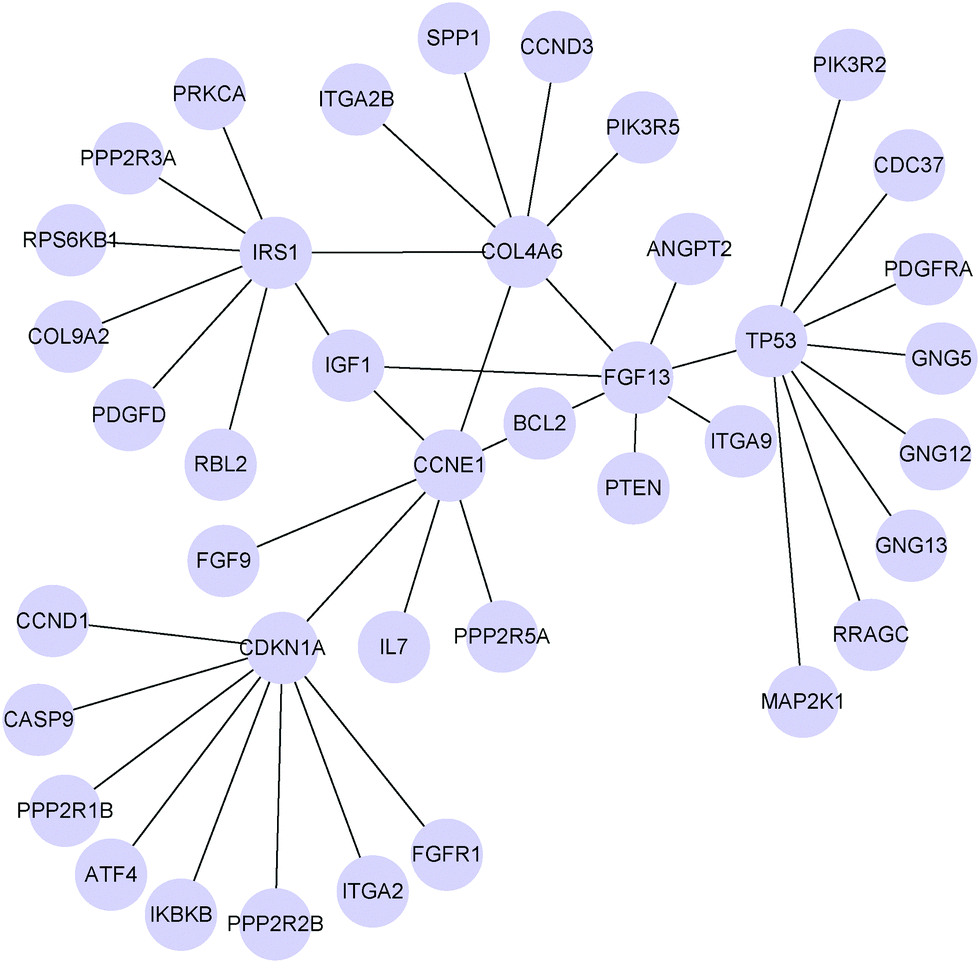 | ||
| Fig. 3 Six hub genes (CDKN1A, TP53, IRS1, CCNE1, FGF13 and COL4A6) and their neighbors in the differential network between platinum sensitive and platinum resistant ovarian cancers (PI3K/AKT/mTOR pathway) inferred by MEDIA based on gene expression data collected from G450, U133 and HuEx platforms with λ = 0.418. | ||
Besides these hub genes in the differential network, genes whose associations with hub genes result in changes between two patient groups could also be possible therapeutic targets in ovarian serous carcinoma. For example, FGFR1, a neighbor of CDKN1A in the differential network, has been found to associate with resistance to endocrine therapy in ER+/FGFR1-amplified breast cancer.48 CCND1, another neighbor of CDKN1A in the differential network, has been previously reported in ovarian cancer for its role in cisplatin resistance.49 Therefore, it is of interest to study how the associations between hub genes and their neighbors in the inferred differential networks correlate with platinum response in ovarian cancer.
We compare MEDIA with D-trace loss and GGL on the ovarian cancer data. For GGL, we run it separately for each patient group and each time it is applied across all the three platforms, then we estimate the differential network corresponding to each platform by calculating the difference between the inferred group-specific precision matrices. For D-trace loss, we run it separately for each platform and each time it is applied across the two patient groups. As suggested by Yuan et al.,9 the tuning parameter of D-trace loss is tuned by ESCV. Following Danaher et al.,22 the tuning parameters of GGL are tuned using the Akaike information criterion (AIC).
A common challenge in evaluating the differential network inference method comprehensively using real data is the lack of a gold standard. As we cannot obtain the true gene networks in the platinum-sensitive and platinum-resistant tumors, it is difficult to compare different methods according to the accuracy of the identified differential networks. In this study, we adopt an alternative strategy to evaluate the performance of various differential network analysis methods. In particular, for each method, we count the number of hub nodes in its reconstructed differential networks that match with known drug resistance-related or cancer-related genes. Therefore, a method that could well capture known functionally important genes may have a better performance in differential network identification. For D-trace loss and GGL, we consider the first 36 genes that have the highest degrees of connectivity as hub genes. From Table 2, we find that by considering the information shared by different data platforms and estimating the differential network between two patient groups directly, MEDIA can detect more cisplatin resistance-related, drug resistance-related and cancer-related genes than D-trace loss and GGL.
| Methods | GEARcisplatin | GEARdrug | CGC |
|---|---|---|---|
| MEDIA | 7 | 14 | 12 |
| D-trace loss | 6 | 11 | 7 |
| GGL | 2 | 6 | 8 |
5 Conclusions
The fast development of high-throughput technologies makes it possible to collect gene expression profiles of the same patients from multiple platforms. During the past few years, a number of computational algorithms have been proposed for differential network analysis. However, until now, most of these algorithms have focused on estimating the differential network from a single data platform without considering the common information shared across multiple platforms. In this paper, we propose a novel multi-view differential network analysis model, called MEDIA, to simultaneously infer multiple differential networks from gene expression data collected from different platforms. Unlike previous differential network analysis methods that need to infer each group-specific network separately and estimate the differential network for individual data platform, our method can directly estimate the differential network between two groups and borrow strength across multiple data platforms to achieve reliable differential network estimation. The analysis on simulated data sets shows that our method consistently outperforms existing state-of-the-art differential network analysis algorithms. We applied our method to TCGA ovarian gene expression data collected from three platforms in order to study network rewiring associated with drug resistance. The experimental results demonstrate the potential of our method in detecting edges that could provide insights into the molecular mechanisms of drug resistance.Besides graphical models, correlation-based approaches have also been developed to identify gene regulatory networks and differential networks. Although these approaches have addressed some biological problems, each group-specific network is inferred by computing the pairwise correlation (e.g., Pearson correlation coefficient or mutual information) between each pair of genes, which includes both direct and indirect dependencies.50 To address this problem, conditional mutual information (CMI) has been proposed to infer the nonlinear direct dependencies among genes.51 Recently, Zhang et al.52 and Zhao et al.53 proposed new concepts with new measures (conditional mutual inclusive information and part mutual information) of causal strength to overcome the underestimation problem of CMI. Compared with graphical models, correlation-based approaches are much easier to understand. To infer differential networks, a typical method uses some correlation-based approaches to compute the group-specific gene regulatory networks and estimates the differential networks by edge-wise subtraction of the group-specific gene networks.19,54,55 However, estimating each group-specific gene regulatory network separately will totally ignore the similarity between the two group-specific networks. On the other hand, based on graphical models, the differential network estimation problem can be formulated as a principle statistical learning problem. The similarity between the two groups and the consistency across multiple data platforms can be exploited by imposing a group sparsity penalty on the difference between group-specific networks. Moreover, other structures of interest can be easily imposed on the resulting differential networks by utilizing suitable penalty functions. Therefore, in this study, we focus on developing a graphical model that can directly estimate the differential network between two groups of samples and borrow strength across multiple data platforms to achieve more reliable results. As our model is based on the D-trace loss function and group lasso penalty, the comparison with D-trace loss and GGL is a compelling illustration of how graphical models improve their performance by imposing reasonable penalty functions.
Recently, Liu et al.21 developed a statistical method to construct sample-specific networks which reflect the variation between each single sample and the reference network. Personalized characterization of diseases is the key to achieving personalized medicine. However, similar to traditional differential network estimation methods and graphical models, our model can only infer an aggregated network for multiple samples. In the future, we will investigate how to utilize data collected from multiple platforms to identify sample-specific differential networks.
Competing interests
The authors declare that they have no competing interests.Authors contributions
LOY, XFZ and HY conceived and designed the experiments. LOY and XFZ performed the experiments. LOY and XFZ analyzed the data. LOY and XFZ drafted the manuscript under the guidance and supervision of HY. All co-authors have seen a draft copy of the manuscript and agree with its publication.Acknowledgements
This work is supported by the National Science Foundation of China (61402190, 61532008, 61602309), Self-determined Research Funds of CCNU from the colleges' basic research and operation of MOE (CCNU15A05039 and CCNU15ZD011), and Hong Kong Research Grants Council (Project C1007-15G).References
- A. A. Margolin, I. Nemenman, K. Basso, C. Wiggins, G. Stolovitzky, R. D. Favera and A. Califano, BMC Bioinf., 2006, 7, S7 Search PubMed.
- R. De Smet and K. Marchal, Nat. Rev. Microbiol., 2010, 8, 717–729 Search PubMed.
- D. Marbach, J. C. Costello, R. Küffner, N. M. Vega, R. J. Prill, D. M. Camacho, K. R. Allison, M. Kellis, J. J. Collins and G. Stolovitzky, et al. , Nat. Methods, 2012, 9, 796–804 Search PubMed.
- R. Küffner, T. Petri, P. Tavakkolkhah, L. Windhager and R. Zimmer, Bioinformatics, 2012, 28, 1376–1382 Search PubMed.
- L. Ou-Yang, D.-Q. Dai, X.-L. Li, M. Wu, X.-F. Zhang and P. Yang, BMC Bioinf., 2014, 15, 335 Search PubMed.
- X. F. Zhang, L. Ou-Yang, Y. Zhu, M. Y. Wu and D. Q. Dai, BMC Bioinf., 2015, 16, 146 Search PubMed.
- X.-F. Zhang, L. Ou-Yang, X. Hu and D.-Q. Dai, BMC Genomics, 2015, 16, 745 Search PubMed.
- W. Kolch, M. Halasz, M. Granovskaya and B. N. Kholodenko, Nat. Rev. Cancer, 2015, 15, 515–527 Search PubMed.
- H. Yuan, R. Xi and M. Deng, 2015, arXiv preprint arXiv:http://1511.09188.
- E. E. Schadt, Nature, 2009, 461, 218–223 Search PubMed.
- Y. Cao, R. A. DePinho, M. Ernst and K. Vousden, Nat. Rev. Cancer, 2011, 11, 749–754 Search PubMed.
- A. V. Biankin, N. Waddell, K. S. Kassahn, M.-C. Gingras, L. B. Muthuswamy, A. L. Johns, D. K. Miller, P. J. Wilson, A.-M. Patch and J. Wu, et al. , Nature, 2012, 491, 399–405 Search PubMed.
- M. J. Ha, V. Baladandayuthapani and K.-A. Do, Bioinformatics, 2015, 31, 3413–3420 Search PubMed.
- A. Dobra, C. Hans, B. Jones, J. R. Nevins, G. Yao and M. West, Journal of Multivariate Analysis, 2004, 90, 196–212 Search PubMed.
- J. Schäfer and K. Strimmer, et al. , Stat. Appl. Genet. Mol. Biol., 2005, 4, 32 Search PubMed.
- M. Yuan and Y. Lin, Biometrika, 2007, 94, 19–35 Search PubMed.
- A. de la Fuente, Trends Genet., 2010, 26, 326–333 Search PubMed.
- J. Guo, E. Levina, G. Michailidis and J. Zhu, Biometrika, 2011, 98, 1–15 Search PubMed.
- C.-H. Zheng, L. Yuan, W. Sha and Z.-L. Sun, BMC Bioinf., 2014, 15, S3 Search PubMed.
- S. D. Zhao, T. T. Cai and H. Li, Biometrika, 2014, 101, 253–268 Search PubMed.
- X. Liu, Y. Wang, H. Ji, K. Aihara and L. Chen, Nucleic Acids Res., 2016 DOI:10.1093/nar/gkw772.
- P. Danaher, P. Wang and D. M. Witten, Journal of the Royal Statistical Society: Series B (Statistical Methodology), 2014, 76, 373–397 Search PubMed.
- T. Zhang and H. Zou, Biometrika, 2014, 101, 103–120 Search PubMed.
- A. G. Deshwar and Q. Morris, Bioinformatics, 2014, 30, 956–961 Search PubMed.
- Y. Wang, T. Joshi, X.-S. Zhang, D. Xu and L. Chen, Bioinformatics, 2006, 22, 2413–2420 Search PubMed.
- C. G. A. R. Network, et al. , Nature, 2011, 474, 609–615 Search PubMed.
- J. Friedman, T. Hastie and R. Tibshirani, 2010, arXiv preprint arXiv:http://1001.0736.
- S. Boyd, N. Parikh, E. Chu, B. Peleato and J. Eckstein, Foundations and Trends in Machine Learning, 2011, 3, 1–122 Search PubMed.
- C. Lim and B. Yu, Journal of Computational and Graphical Statistics, 2016, 25, 464–492 Search PubMed.
- K. Mohan, P. London, M. Fazel, D. M. Witten and S.-I. Lee, Journal of Machine Learning Research, 2014, 15, 445–488 Search PubMed.
- S. Ravikumar, G. Perez-Liz, L. Del Vale, D. R. Soprano and K. J. Soprano, Cancer Res., 2007, 67, 9266–9275 Search PubMed.
- D. D. Bowtell, S. Böhm, A. A. Ahmed, P.-J. Aspuria, R. C. Bast Jr, V. Beral, J. S. Berek, M. J. Birrer, S. Blagden and M. A. Bookman, et al. , Nat. Rev. Cancer, 2015, 15, 668–679 Search PubMed.
- S. Nabavi, D. Schmolze, M. Maitituoheti, S. Malladi and A. H. Beck, Bioinformatics, 2015, btv634 Search PubMed.
- H. A. Burris III, Cancer Chemother. Pharmacol., 2013, 71, 829–842 Search PubMed.
- M. Kanehisa and S. Goto, Nucleic Acids Res., 2000, 28, 27–30 Search PubMed.
- P. A. Futreal, L. Coin, M. Marshall, T. Down, T. Hubbard, R. Wooster, N. Rahman and M. R. Stratton, Nat. Rev. Cancer, 2004, 4, 177–183 Search PubMed.
- R. K. Dearth, X. Cui, H.-J. Kim, D. L. Hadsell and A. V. Lee, Cell Cycle, 2007, 6, 705–713 CrossRef CAS PubMed.
- N. Eckstein, J. Exp. Clin. Cancer Res., 2011, 30, 1 Search PubMed.
- D. Etemadmoghadam, G. Au-Yeung, M. Wall, C. Mitchell, M. Kansara, E. Loehrer, C. Batzios, J. George, S. Ftouni and B. A. Weir, et al. , Clin. Cancer Res., 2013, 19, 5960–5971 Search PubMed.
- D. Etemadmoghadam, R. Beroukhim, C. Mermel, J. George, G. Getz, R. Tothill, A. Okamoto, M. B. Raeder, P. Harnett and S. Lade, et al. , Clin. Cancer Res., 2009, 15, 1417–1427 Search PubMed.
- W. H. Wilson, J. Teruya-Feldstein, T. Fest, C. Harris, S. M. Steinberg, E. S. Jaffe and M. Raffeld, Blood, 1997, 89, 601–609 CAS.
- J. M. Davis, P. M. Navolanic, C. R. Weinstein-Oppenheimer, L. S. Steelman, W. Hu, M. Konopleva, M. V. Blagosklonny and J. A. McCubrey, Clin. Cancer Res., 2003, 9, 1161–1170 Search PubMed.
- A. G. Eliopoulos, D. J. Kerr, J. Herod, L. Hodgkins, S. Krajewski, J. C. Reed and L. S. Young, Oncogene, 1995, 11, 1217–1228 Search PubMed.
- T. Okada, K. Murata, R. Hirose, C. Matsuda, T. Komatsu, M. Ikekita, M. Nakawatari, F. Nakayama, M. Wakatsuki and T. Ohno, et al. , Sci. Rep., 2013, 3, 2899 Search PubMed.
- R. Januchowski, P. Zawierucha, M. Ruciński and M. Zabel, Oncol. Rep., 2014, 32, 1981–1990 Search PubMed.
- P. Zhang, Z. Zhang, X. Zhou, W. Qiu, F. Chen and W. Chen, BMC Cancer, 2006, 6, 1 Search PubMed.
- L. Steinman, Nat. Med., 2007, 13, 139–145 Search PubMed.
- C. Cole, S. Lau, A. Backen, A. Clamp, G. Rushton, C. Dive, C. Hodgkinson, R. McVey, H. Kitchener and G. C. Jayson, Cancer Biol. Ther., 2010, 10, 495–504 Search PubMed.
- E. E. Noel, M. Yeste-Velasco, X. Mao, J. Perry, S. C. Kudahetti, N. F. Li, S. Sharp, T. Chaplin, L. Xue and A. McIntyre, et al. , Am. J. Pathol., 2010, 176, 2607–2615 Search PubMed.
- S. Feizi, D. Marbach, M. Médard and M. Kellis, Nat. Biotechnol., 2013, 31, 726–733 CrossRef CAS PubMed.
- X. Zhang, X.-M. Zhao, K. He, L. Lu, Y. Cao, J. Liu, J.-K. Hao, Z.-P. Liu and L. Chen, Bioinformatics, 2012, 28, 98–104 Search PubMed.
- X. Zhang, J. Zhao, J.-K. Hao, X.-M. Zhao and L. Chen, Nucleic Acids Res., 2015, 43, e31 Search PubMed.
- J. Zhao, Y. Zhou, X. Zhang and L. Chen, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5130–5135 Search PubMed.
- H. Yu, B.-H. Liu, Z.-Q. Ye, C. Li, Y.-X. Li and Y.-Y. Li, BMC Bioinf., 2011, 12, 315 CrossRef PubMed.
- Y. Rahmatallah, F. Emmert-Streib and G. Glazko, Bioinformatics, 2014, 30, 360–368 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6mb00619a |
| This journal is © The Royal Society of Chemistry 2017 |