DOI:
10.1039/C6MB00691D
(Communication)
Mol. BioSyst., 2017,
13, 83-91
Competitive profiling of celastrol targets in human cervical cancer HeLa cells via quantitative chemical proteomics†
Received
5th October 2016
, Accepted 1st November 2016
First published on 7th November 2016
Abstract
Celastrol, isolated from the traditional Chinese medicinal herb Tripterygium wilfordii Hook. f. (Thunder God's Vine), has been used to treat cancer, chronic inflammatory, autoimmune and other human diseases. However, to date, the protein targets and the mechanism of action of celastrol have remained elusive. In this study, we find that celastrol can react with protein thiols in a unique covalent and reversible manner, while protein denaturing disrupts the interaction. Through a competitive chemoproteomics approach utilizing a cysteine-targeting activity-based probe, we report the proteome-wide quantitative profiling of cellular targets of celastrol in human cervical cancer HeLa cells. Representative targets are further validated via in vitro binding experiments and/or enzymatic activity assays. Bioinformatics analysis results suggest that celastrol exerts its numerous therapeutic effects through interaction with promiscuous proteins involved in various biological processes and cellular pathways.
Introduction
Natural products and their derivatives comprise a major source of small molecular drugs in the treatment of multiple human diseases.1 Celastrol, a quinone methide triterpene extracted from the medicinal plant Tripterygium wilfordii Hook. f., has been used as a traditional Chinese herb to treat cancer, chronic inflammatory, autoimmune and other human diseases for decades.2 Although celastrol has been proposed to form covalent Michael adducts with protein thiols (Fig. 1A),3 to date, only a limited number of protein targets have been reported, including Hsp90β, Cdc37, p23, IKKβ, proteasome, and Pdx1.4 Because the identification of the reported targets is almost all based on biochemical studies with recombinant proteins and special cellular fractions in vitro, they cannot fully address most of the aforementioned biological activities and therapeutic effects of celastrol. Recent advancement in proteomic technologies has greatly promoted the application of activity/affinity-based proteome profiling (ABPP) in proteome-wide target identification.5 In the case of celastrol, a biotinylated activity-based probe was designed to “fish” the cellular binding partners within Panc-1 cell lysates. Although multiple proteins have been enriched according to the silver staining gel, only three high abundant proteins, Annexin II, eEF1A, and β-tubulin, were reported.6 Despite being extremely powerful in its applications, the conventional ABPP strategy also has some shortcomings of its own.7 Recently, a competitive ABPP strategy combined with the quantitative proteomics technology has been developed and successfully applied in target profiling of cysteine-reactive small molecules in proteomes.8 Another similar strategy has been used in profiling oxidized cysteines in various biological systems.9 Compared with conventional ABPP, the competitive chemoproteomics approach affords a robust and facile approach for the profiling of potential targets of electrophilic compounds in native biological systems.10
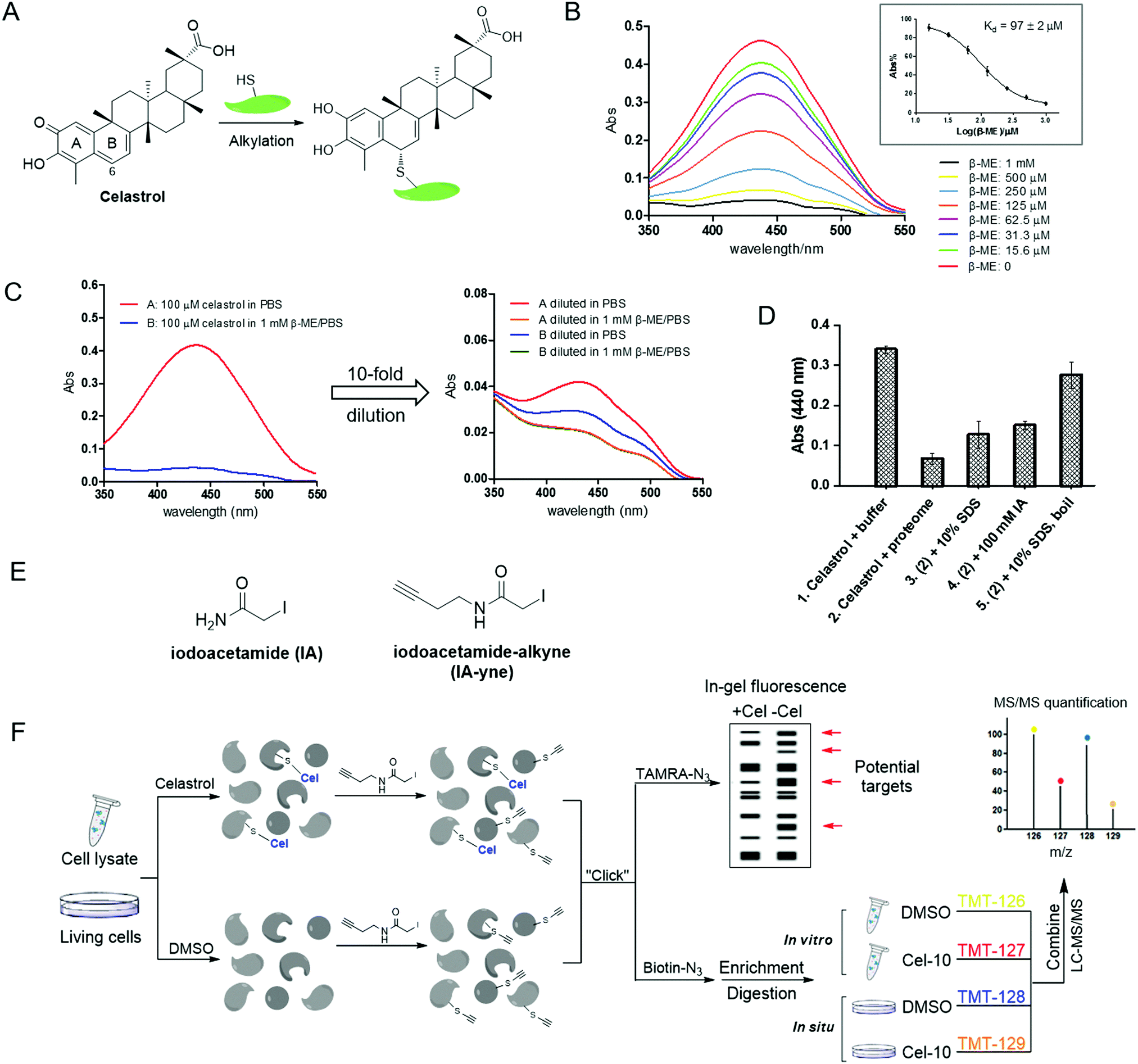
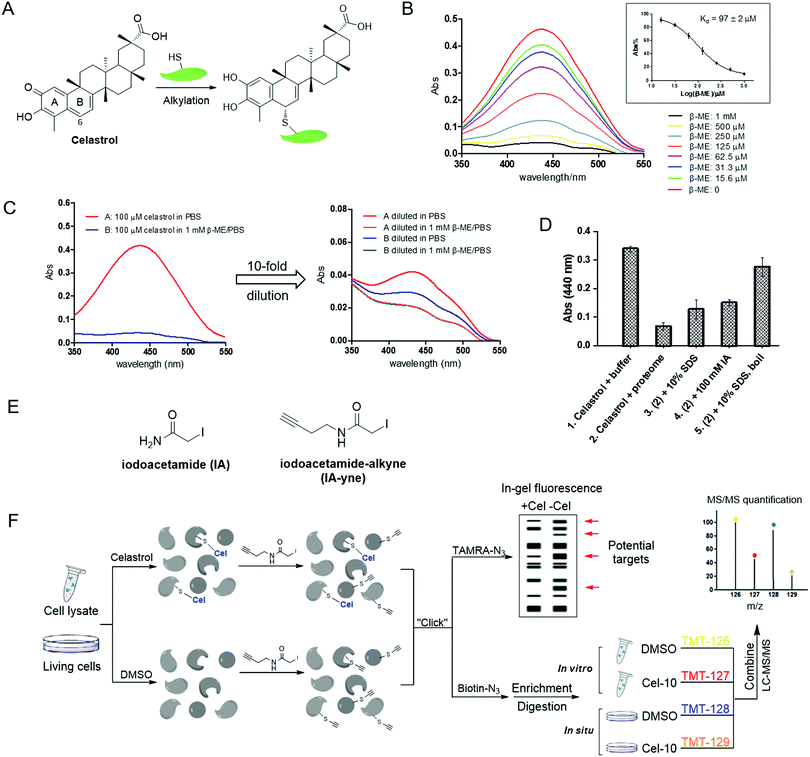 |
| | Fig. 1 Thiol reactivity of celastrol and the overall strategy of competitive proteomics-based celastrol target identification. (A) Alkylation of protein thiols by celastrol; (B) UV-visible absorption spectra of celastrol in the presence of increasing concentrations of β-ME in PBS (inset: equilibrium dissociation constants (Kd) of β-ME–celastrol adducts); (C) dilution experiments showing reversible formation of β-ME adducts with celastrol; (D) alkylation of HeLa cellular lysates with celastrol can be reversed by protein denaturing (SDS or/and boiling) and irreversible cysteine alkylation; (E) structures of iodoacetamide (IA) and its derived probe iodoacetamide–alkyne (IA-yne); and (F) overview of tandem mass tagging (TMT)-based quantitative competitive chemical proteomics strategy for celastrol target profiling. | |
In the present study, celastrol was found to alkylate model thiols and proteome samples through an unusual covalent and reversible paradigm. Therefore, we profiled its cellular targets via a competitive chemoproteomics approach by utilizing an iodoacetamide-derived cysteine-reactive probe. Using tandem mass tagging (TMT)-based quantitative mass spectrometry, we identified over 60 celastrol binding proteins in the human cervical cancer HeLa cell proteome. These proteins were extensively involved in various biological processes including metabolic processes, cellular processes, biological regulation, etc. Representative protein targets involved in the redox metabolic process were validated by in vitro labeling experiments and/or enzymatic activity assays. The promiscuous targeting activity of celastrol explained its diverse biological and therapeutic effects, while the unique reversible covalent alkylation mechanism could expand the medicinal application prospects of celastrol.
Results and discussion
Characterization of the reaction of celastrol with model thiols and cellular proteomes
Cysteine thiol is one of the most reactive nucleophilic residues due to the polarizability and electron-rich nature of the sulfur atom.11 Previous studies have revealed that quinone methide rapidly reacts with thiol groups under physiological conditions.12 Thus, we initiated our study by examining the reaction of celastrol with a model thiol, β-mercaptoethanol (β-ME). The incubation of celastrol with increasing concentrations of β-ME in phosphate-buffered saline (PBS, pH 7.4) caused a stepwise decrease of the UV-visible absorption of celastrol at around 440 nm (λmax), indicating the formation of covalent adducts; by fitting the absorption at 440 nm, the apparent binding affinity was calculated to be 96.7 μM (Fig. 1B). Further dilution experiments following literature procedures demonstrated that the formation of covalent adducts is reversible, because the UV absorption recovered after 10-fold dilution of the celastrol–β-ME adducts into PBS (Fig. 1C).13 Next, we investigated the reactivity of celastrol in freshly prepared HeLa cell proteomes. As predicted, the UV absorption at 440 nm reduced obviously in cellular lysates but partially recovered due to protein denaturing (SDS, boiling) or the addition of iodoacetamide, an irreversible cysteine alkylation reagent (Fig. 1D). Taken together, celastrol can react with small molecule/protein thiols through covalent but reversible alkylation. Hence, when characterizing the target profile of celastrol via affinity purification, interacting proteins could be detached from the matrix during washing steps. We therefore intended to apply a competitive chemical proteomics approach to quantitatively profile potential cellular targets of celastrol across cellular proteomes by using an iodoacetamide (IA)-based cysteine-reactive probe (Fig. 1E and F).
Gel-based celastrol-competitive labeling of HeLa proteomes in vitro and in situ
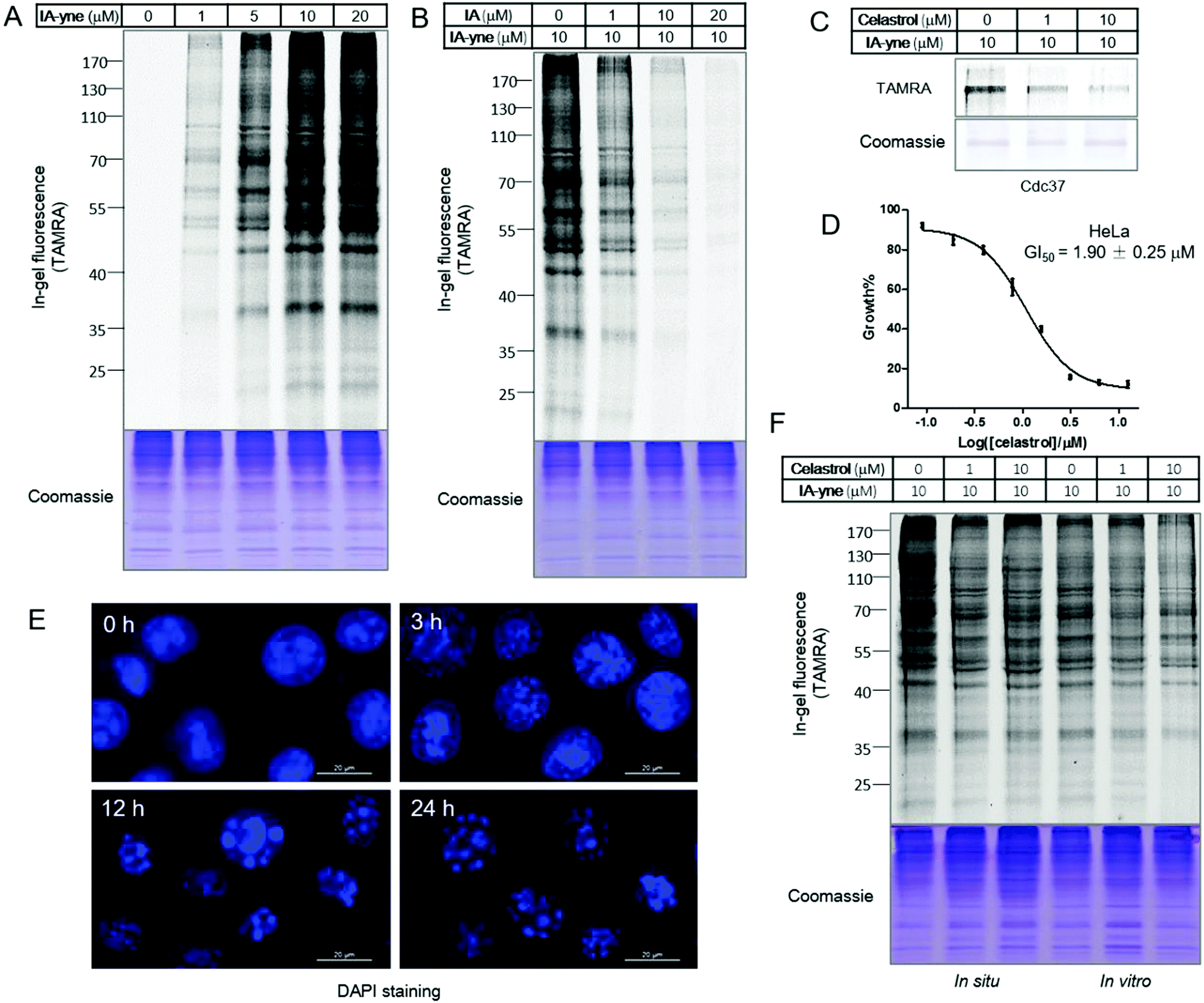
As a widely used cysteine alkylation reagent, the potency, selectivity and stability of iodoacetamide under physiological conditions have been well documented.14 The alkyne-tagged iodoacetamide probe IA-yne was synthesized according to literature procedures.14 Although 100 mM of iodoacetamide caused instantaneous release of celastrol in the proteome, iodoacetamide and IA-yne within the micro-molar range did not induce the disruption of the celastrol–protein complex (Fig. S1, ESI†). To demonstrate the feasibility and optimize the labeling conditions of this probe, the proteome labeling profile was assessed through gel-based experiments. We chose the well-known HeLa cell line as a model system as it represents a homogeneous and reproducible biological system, and was highly sensitive to celastrol in previous studies.4c,d,15IA-yne labeled HeLa lysates were subjected to the copper catalyzed alkyne–azide cycloaddition reaction (CuAAC) with TAMRA–azide, and proteome labeling was evaluated through in-gel fluorescence scanning following SDS-PAGE. 10 μM of IA-yne was sufficient for robust labeling and pretreatment of the lysates with IA decreased the overall signal (Fig. 2A and B). To prove the concept of competitive proteomics profiling, Cdc37, one of the known celastrol targets, was treated sequentially with celastrol (1.0 μM and 10 μM, 1 hour) and IA-yne (10 μM, 1 hour), followed by the click reaction with TAMRA–azide. As detected using in-gel fluorescence, the labeling was obviously blocked by celastrol (Fig. 2C). Next, we investigated the competition of the IA-yne profile by celastrol within whole proteomes of cellular lysates and living cells. To optimize the concentration of celastrol in living cell experiments, cell viability was determined after 24 hours of incubation and celastrol inhibited the HeLa cell growth with a GI50 of 1.9 μM (Fig. 2D); actually, celastrol showed potent inhibitory effects towards several normal mammalian cell lines, including human embryonic kidney HEK293T, human umbilical vein endothelial HUVEC and Madin-Darby Canine Kidney Epithelial MDCK cells (Fig. S2, ESI†). To optimize the incubation time, HeLa cells were treated with celastrol (10 μM) for various periods of time, followed by 4,6-diamidino-2-phenylindole (DAPI) staining. Microscopy results showed that the cell nuclei remained stable after 3 hours of treatment and appeared fragmented after 12 hours (Fig. 2E). Thus, the HeLa cells were incubated with DMSO and celastrol (10 μM) for 3 hours to allow the binding to cellular targets. The attached cells were harvested and equal lysates were labeled with IA-yne (10 μM). For the in vitro study, equal amounts of HeLa proteomes were incubated with DMSO and celastrol (10 μM), followed by IA-yne (10 μM) labeling and click chemistry. Celastrol exhibited a weaker competition effect than that of IA, indicating the higher selectivity of celastrol to protein thiols due to its structural complexity (Fig. 2F). Nevertheless, the celastrol-competed bands are potential target proteins of the natural product.
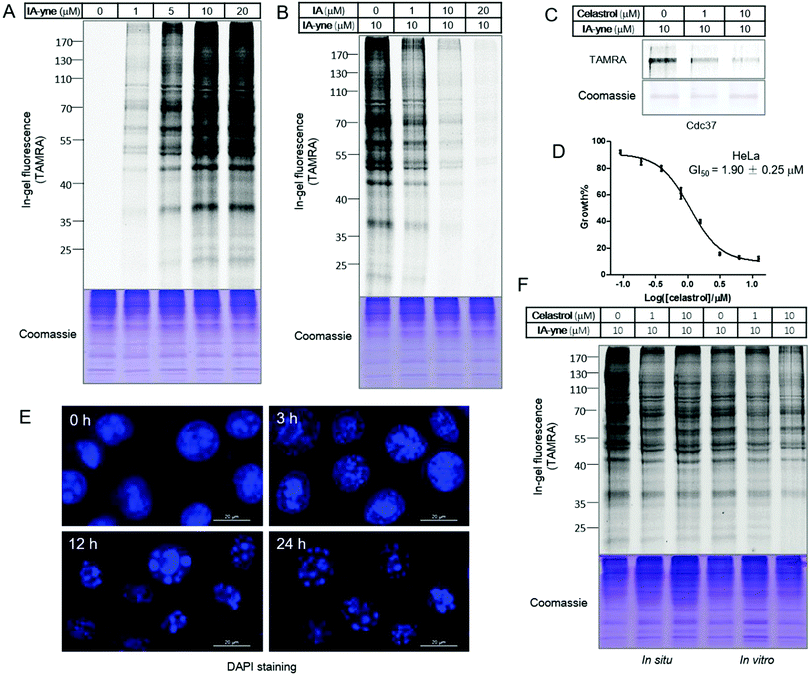 |
| | Fig. 2 Gel-based profiling of celastrol targets using the cysteine reactive probe IA-yne. (A) Labeling of HeLa proteomes with IA-yne; (B) IA-competitive labeling of HeLa proteomes; (C) celastrol-competitive labeling of recombinant human Cdc37 protein; (D) inhibition of HeLa cell proliferation by celastrol; (E) DAPI staining of celastrol treated HeLa cells with different time periods; and (F) celastrol-competitive labeling of living HeLa cells (in situ, lanes 1–3) and HeLa cell lysates (in vitro, lanes 4–6). | |
Identification of celastrol targets by competitive pull-down and quantitative chemical proteomics
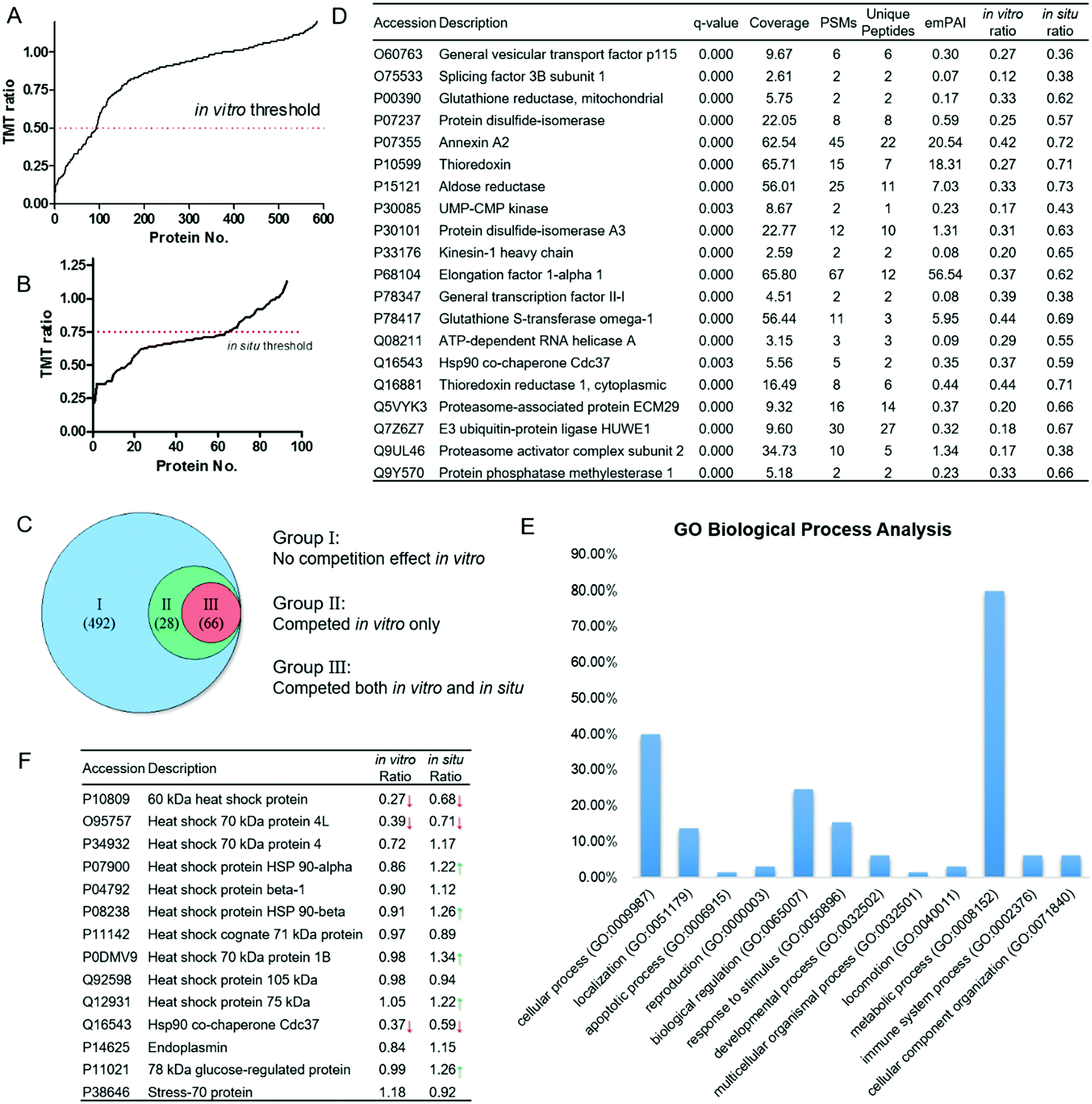
We then performed large-scale proteomics experiments using competitive pull-down combined with isobaric tandem mass tags (TMT)-based quantitative mass spectrometry. The proteomics experiment was carried out in duplicates. HeLa proteomes treated with celastrol/probe were subjected to click reactions with the biotin–azide affinity tag. After enrichment by pull-down with streptavidin beads, the bound proteins were subjected to on-bead tryptic digestion. The digested peptides were labeled with the respective TMT reagents, and combined for further identification using Thermo Q-Exactive LC-MS/MS mass spectrometry. The peptides, and hence proteins, were quantified by the intensities of the corresponding TMT reporter ions. Proteins competed by 10 μM celastrol with an average TMT ratio ≤ 0.5 were considered specific targets of celastrol. Overall, 586 proteins were identified in DMSO-treated lysates, 94 of which were significantly competed (Fig. 3A). The competition profile in living cells was quite different. The overall in situ TMT ratios were higher than in vitro ones (Fig. 3B), which could be explained by the reduced accessibility of celastrol to protein targets within cellular compartments, or protein overexpression caused by the celastrol treatment.16 Thus, we set the threshold of the in situ TMT ratio as 0.75, which was 50% higher than that of in vitro. As a result, the competitive proteomics study enables us to classify the 586 IA-yne enriched proteins into three groups: (1) 492 proteins did not show a significant competition effect in vitro; (2) 28 proteins were competed by celastrol in vitro but not in situ; (3) 66 proteins were competed by celastrol both in vitro and in situ (Fig. 3C), and were identified as potential targets of celastrol. Representative celastrol targets are listed in Fig. 3D and the full list of proteins are provided in Table S1 (ESI†).
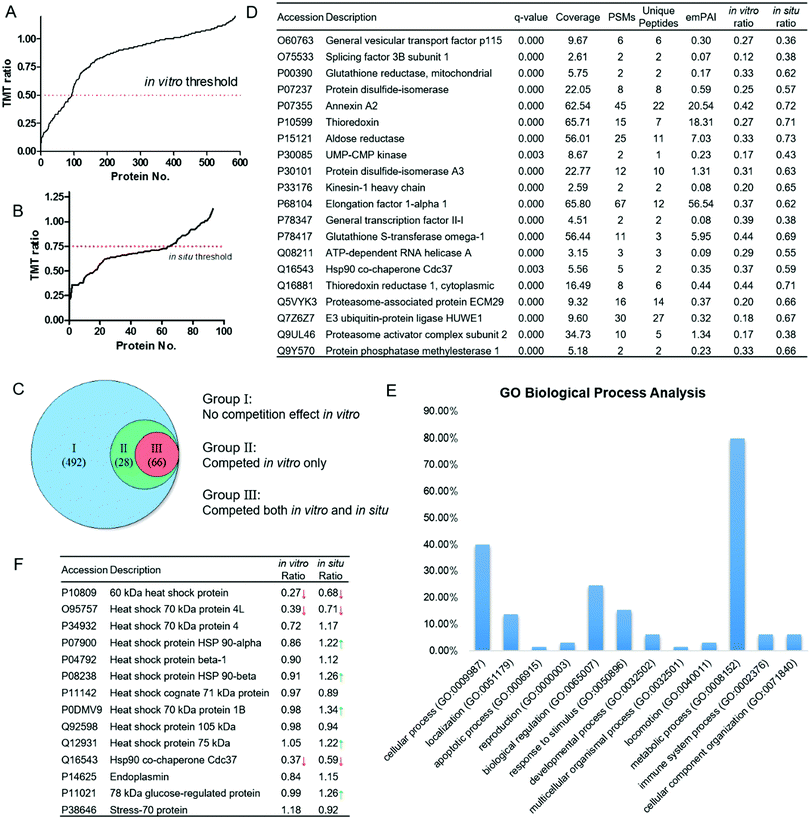 |
| | Fig. 3 Competitive proteomics profiling of celastrol targets and functional analysis. (A) 586 proteins were enriched and identified by IA-ynein vitro, 94 of which were competed in vitro; (B) 66 of the 94 proteins were competed in situ; (C) classification of IA-yne enriched proteins through the in vitro and in situ competition effect of celastrol; (D) representative celastrol-competed targets from both the in vitro and in situ experiments; (E) bioinformatic analysis of the potential biological processes targeted by celastrol using Gene Ontology (GO) tools; and (F) competition effect of celastrol on the enrichment of heat shock (related) proteins by IA-ynein vitro and in situ. Red/green arrows indicate the significant decrease/increase of the enrichment effect due to celastrol. | |
Bioinformatics analysis of targets of celastrol
Next, to reveal the key biological processes and pathways in HeLa cell lines, the 66 high-confidence proteins were classified with the Gene Ontology (GO) PANTHER software. These targets were extensively involved in multiple vital biological processes, among which the metabolic process showed the highest percentage (Fig. 3E). A number of proteins related to the thiol redox metabolism were classified into this category, including thioredoxins (TRs), thioredoxin reductase 1 (TRX1), glutathione reductase (GR), protein disulfide isomerases (PDIs) and glutathione S-transferase omega 1 (GSTO1). The physiological roles of these proteins are directly controlled by the oxidation status of the redox-sensitive cysteines within their active sites.17 Like many other natural products possessing extremely strong electrophilicity, celastrol is also likely to alkylate the hyper-reactive functional cysteines in cells, leading to multiple therapeutic anti-cancer and anti-inflammatory effects.18,19 Other bioinformatics analysis results, including cellular localizations, molecular functions, and protein classes, are provided in Fig. S2 (ESI†). Importantly, the PANTHER pathway analysis showed that the ubiquitin proteasome pathway was the most significantly engaged pathway of celastrol (Fig. S2D, ESI†). The 26S proteasome, comprised of two 19S regulatory particles and one 20S core particle, is now one of the attractive targets in the treatment of cancer.20 Celastrol has been reported to promote cancer cell apoptosis through potently inhibiting the chymotrypsin-like activity of the 20S proteasome.4e Our data suggest that the alkylation of the regulatory subunits of the 26S proteasome might also confer to its inhibitory effect on the ubiquitin-proteasome system. It was also of interest to find that multiple celastrol-related heat shock family proteins reported previously were enriched by the probe in our study (Fig. 3F).4a,b,21 Although it has been suggested that celastrol could disrupt Hsp90–Cdc37 binding through interaction with the C-terminal domain of Hsp90,4a our data clearly demonstrate that celastrol is more likely to bind Cdc37 both in vitro and in situ (TMT ratios = 0.37 and 0.59, respectively), instead of Hsp90α (TMT ratios = 0.86 and 1.22) and Hsp90β (TMT ratios = 0.91 and 1.26). Notably, the in situ TMT ratios of Hsp90α, Hsp90β and several other members even increased after the celastrol treatment, which is possibly caused by the comprehensive effect of proteasome inhibition and heat shock response induced by the natural product.
Target validation of representative celastrol-competed proteins
To validate the identified targets of celastrol, we performed competitive labeling of three reported functional cysteine-dependent proteins, GSTO1, PDI, and GR, in the list. As expected, all proteins were labeled nicely by IA-yne, and the labeling was obviously abolished by celastrol pretreatment (Fig. 4A). As a member of the GST superfamily account for cellular detoxification, GSTO1 is involved in exporting a wide spectrum of endogenous and xenobiotic electrophiles by conjugating glutathione to the compounds.22 GSTs have emerged as a promising therapeutic target because they are overexpressed in cancer cells and may play a significant role in other human diseases.23 The UV-visible spectra demonstrate the decrease in absorbance of celastrol at 440 nm after the addition of various concentrations of GSTO1, indicating the occurrence of covalent alkylation (Fig. 4B). Further enzymatic activity assays suggested that GSTO1 was potently inhibited by celastrol with an IC50 of 93 nM (Fig. 4C). Unfortunately, when trying to investigate the modification site of GSTO1 by tryptic digestion and mass spectrometry, we could not identify any celastrol-modified peptide (data not shown). We speculated that the reversible Michael adduct could be disrupted after proteolysis due to the disintegration of the protein secondary structure, or the peptide–celastrol adducts might not be stable under mass spectrometry conditions. Finally, we carried out pull-down/immunoblotting experiments on both cellular lysates and live cells with antibodies of GSTO1 and PDI. The results further confirmed GSTO1 and PDI as celastrol targets (Fig. 4D).
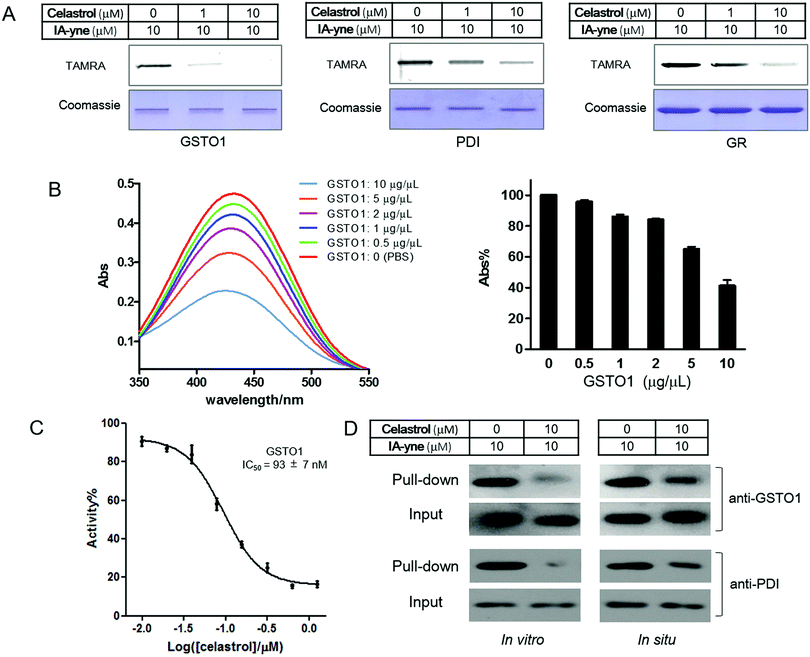 |
| | Fig. 4 Validation of selected targets of celastrol identified by competitive proteomics. (A) Celastrol-competitive labeling of recombinant proteins by IA-ynein vitro; (B) UV-visible absorption spectra of 100 μM of celastrol in the presence of increasing concentrations of GSTO1; (C) inhibitory effect of celastrol against recombinant GSTO1 protein; and (D) western blotting (by using anti-GSTO1 and anti-PDI antibodies) of the pull-down samples upon labeling with IA-yne. | |
Experimental section
Chemicals
Celastrol was obtained from MedChemExpress. TAMRA–azide and biotin–azide were ordered from Lumiprobe and Biomatrik, respectively. β-Mercaptoethanol (β-ME), iodoacetamide (IA), tris(3-hydroxypropyltriazolylmethyl)amine (THPTA), tris(2-carboxyethyl)phosphine (TCEP), sodium ascorbate (NaVc), and other chemicals were ordered from Sigma-Aldrich.
Equilibrium dissociation constants for β-ME/celastrol adducts
Celastrol (100 μM in PBS, pH 7.4) was incubated with β-ME (0–1.0 mM in PBS, pH 7.4) for 30 minutes at room temperature. The absorption spectra (350–550 nm) were recorded using an Agilent Cary 60 UV-visible spectrometer. The formation of adducts was quantified by monitoring the maximum absorbance (λmax) at 440 nm and the data were fitted using GraphPad Prism 5.0 to obtain the equilibrium dissociation constant (KD) value.
Dilution experiment with β-ME/celastrol adducts
Celastrol (100 μM in PBS, pH 7.4) was incubated with PBS and 1.0 mM β-ME in PBS for 30 minutes at room temperature. The solutions were 10-fold diluted in PBS containing 1.0 mM of β-ME and PBS alone, respectively. The final solutions were incubated at room temperature for 30 minutes and the disassociation of adducts was demonstrated by the absorption spectra (350–550 nm).
Determination of the reaction of celastrol with HeLa proteomes
100 μM of celastrol (10 mM stock in DMSO) was incubated with HeLa cell lysates (1.0 mg mL−1) suspended in lysis buffer (50 mM HEPES pH 7.4, 150 mM NaCl, 0.1% Triton X-100, 3.0 mM MgCl2, 1.0 mM EDTA and complete protease inhibitor (Roche)) and an equal volume of lysis buffer for 30 minutes at room temperature in triplicate. For protein denaturing experiments, the celastrol-treated proteomes were (1) added with 10% SDS (w/v) and incubated for 10 minutes at room temperature; (2) boiled for 10 minutes; (3) added with 10% SDS (w/v) and boiled for 10 minutes; and (4) added with 100 mM iodoacetamide and incubated for 1 hour at room temperature. The amount of free celastrol was determined by monitoring the absorption at 440 nm.
Cell culture
A human cervical carcinoma HeLa cell line (ATCC) was incubated in Dulbecco's Modified Eagle Medium (DMEM, Gibco) supplemented with 1% L-glutamine, 1% penicillin–streptomycin and 10% fetal bovine serum (FBS, Gibco). The culture incubator was set at 37 °C with 5% CO2.
Gel-based IA-yne profiling of HeLa cell lysates
HeLa cell lysates (1.0 mg mL−1) were incubated with DMSO or IA-yne for 30 minutes as indicated. 1% SDS (w/v) was added to each sample and the click reaction was performed as follows: for each reaction, 20 μL of the protein samples were added to freshly prepared 0.25 μL each of TAMRA–azide (10 mM stock in DMSO), CuSO4 (100 mM stock in H2O), THPTA (10 mM stock in H2O) and NaVc (100 mM stock in H2O). The samples were incubated at room temperature for 1 hour, added with SDS sample loading buffer, applied to SDS-PAGE and imaged using a FUJIFILM FLA 9000 plus DAGE fluorescence scanner.
Competition of IA-yne labeling profiles with iodoacetamide
HeLa cell lysates (1.0 mg mL−1) suspended in lysis buffer were incubated with iodoacetamide (0.1 μM, 10 μM, 20 μM) for 30 minutes. IA-yne (10 μM) was added and the protein samples were incubated at room temperature for another 30 minutes. The following click reaction and fluorescence scan were performed as above.
Competition of IA-yne labeling of recombinant proteins with celastrol
Recombinant human GSTO1 was ordered from GeneCopoeia™. Recombinant human CDC37 protein was ordered from Sino Biological Inc. Recombinant human glutathione reductase and protein disulfide isomerase were ordered from Sangon Biotech. The recombinant proteins reconstituted in PBS (final concentration 0.1 mg mL−1) were incubated with celastrol (1.0 μM, 10 μM) at room temperature for 3 hours. IA-yne (10 μM) was added and the protein samples were incubated at room temperature for another 30 minutes. The following click reaction and fluorescence scan were performed as above.
Cell proliferation assay
HeLa, HEK293T, HUVEC and MDCK cells were cultured in DMEM (Gibco) for three passages and diluted in culture medium to 8000 cells per mL. 100 μL of the cell suspension was seeded in each well of a 96-well plate and incubated at 37 °C overnight. Various concentrations of celastrol were dissolved in culture medium containing 0.5% DMSO. Cells in the 96-well plate were treated with 100 μL of various concentrations of celastrol and DMSO (negative control) for 24 hours in a 37 °C incubator. Cell viability was assessed using a CellTiter-Glo® Luminescent Kit (Promega).
Fluorescence microscopy study
HeLa cells were treated with 10 μM of celastrol in DMEM for 0–24 hours in 6-well plates with glass cover slips. The cells were washed twice with PBS and treated with 10 μg mL−1 of 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI, Invitrogen) dissolved in PBS for 15 minutes at room temperature, followed by imaging using a ZEISS LSM510 META fluorescence microscope.
Gel- and mass-based competitive labeling and pull-down of HeLa proteomes in vitro and in situ
For the in vitro study, each of the 1.0 mL HeLa cell lysates (1.0 mg mL−1) suspended in lysis buffer [50 mM HEPES pH 7.4, 150 mM NaCl, 0.1% Triton X-100, 3.0 mM MgCl2, 1.0 mM EDTA and complete protease inhibitor (Roche)] were incubated with DMSO (vehicle) and celastrol (10 μM) for 3 hours at room temperature. For the in situ study, HeLa cells were incubated with celastrol (10 μM) or DMSO for 3 hours. The cells were harvested and lysed in lysis buffer. Proteome samples of the in vitro and in situ experiments were incubated with 10 μM IA-yne at room temperature for 30 minutes. The samples were collected into new tubes and quickly precipitated via the sequential addition of CH3OH (600 μL), CHCl3 (150 μL) and H2O (400 μL). The precipitates were re-suspended in 520 μL of click buffer (50 mM HEPES pH 8.0, 1% SDS (w/v)). 20 μL of the protein samples were subjected to a click reaction with TAMRA–N3 as mentioned above, and the remaining 500 μL of the samples were subjected to a click reaction with Biotin–N3 (500 μM, Biomatrik Inc.), CuSO4 (1.0 mM), THTPA (100 μM) and NaVc (1.0 mM). The samples were precipitated again with CH3OH/CHCl3, washed with CH3OH and re-dissolved in 200 μL of click buffer via boiling and sonication. 30 μL of streptavidin–sepharose (GE Healthcare) beads were added to each sample and incubated at room temperature with continuous rotation for 1 hour. The beads were washed with PBS with 1% SDS (w/v) three times, PBS with 0.5 M NaCl three times, 4.0 M urea in 50 mM triethylammoniumbicarbonate (TEAB) twice, and 100 mM TEAB five times. Each wash was performed on a rotator for 15 minutes. The beads after wash were subjected to on-bead reductive alkylation with 200 μL of 10 mM of TCEP at 50 °C for 30 minutes and 200 μL of 55 mM iodoacetamide at 37 °C in the dark for another 30 minutes, followed by washing with 100 mM TEAB three times. The bound proteins on the beads were digested with 1.0 μg of sequencing grade modified trypsin (Promega) reconstituted in 50 μL of 100 mM TEAB for 16 hours at 37 °C. The digests were labeled with the respective TMTsixplex™ Isobaric Label Reagent Set (Thermo Scientific) according to the manufacturer's instructions. The labeled peptides were desalted using C18 columns and evaporated to dryness on a SpeedVac instrument. The dried peptides were resuspended in 30 μL of formic acid/acetonitrile/H2O (v![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :v:v = 0.5%/2%/97.5%) with sonication. The proteomics experiment was carried out in biological duplicates.
:v:v = 0.5%/2%/97.5%) with sonication. The proteomics experiment was carried out in biological duplicates.
Mass spectrometry and proteomics data analysis
After filtration through a 22 μm membrane, the clear solution was subjected to nano LC-MS/MS separation. A volume of 3.0 μL of each sample was desalted by loading onto a Thermo C18 PepMap100 precolumn (300 μm × 5 mm) and eluted on a Thermo Acclaim PepMap RSLC analytical column (75 μm × 15 cm). Mobile phase A (0.1% formic acid in H2O) and mobile phase B (0.1% formic acid in acetonitrile) were used to establish the 120 minute gradient comprising 85 minutes of 4–30% B, 15 minutes of 30–50% B, and 5 minutes of 90% B, followed by re-equilibration at 4% B for 15 min. The flow rate was 0.3 μL min−1. Peptides were than analyzed on a Q-Exactive proteomic mass spectrometer (Thermo Scientific) in a data-dependent manner, with automatic switching between MS and MS/MS scans using a top-20 method. The MS spectra were recorded at a resolution of 70000 with a target value of 3 × 106 ions for a maximum integration time of 50 ms. The scan range was limited from 380 to 1800 m/z. Peptide fragmentation was performed via higher-energy collision dissociation (HCD) with the energy set at 32 normalized collision energy (NCE). The MS/MS spectra were recorded at a resolution of 17500 with a target value of 1 × 105 ions or a maximum integration time of 60 ms. The fixed first m/z value was 100, and the isolation window was 1.6 m/z. Protein identification and TMT quantification were performed using the Proteome Discoverer 2.1 software (Thermo Scientific). Peptide sequences (and hence protein identity) were determined by matching protein databases (Uniprot) with the acquired fragmentation pattern using the SEQUEST HT algorithm. The precursor mass tolerance was set to 10 ppm and the fragment ion mass tolerance to 0.02 Da. One missed cleavage site of trypsin was allowed. Carbamidomethyl (C) and TMT-sixplex (K and N-terminal) were used as a fixed modification. Oxidation (M) and deamination (N, Q) were used as variable modifications. All spectra were searched against a protein database using a target false discovery rate (FDR) of 1%. The proteins identified in the DMSO-treated samples were additionally filtered by taking a minimum protein score of 10 with two spectral counts and at least one unique peptide in each experimental replicate. Protein ratios were calculated as the median of all peptide hits belonging to a protein.
GSTO1 enzymatic assay
Recombinant GSTO1 (5.0 μg mL−1) dissolved in GST assay buffer (50 mM sodium phosphate buffer, pH 6.5, 0.1% Triton X-100) was incubated with various concentrations of celastrol on ice for 30 minutes followed by the addition of 2.0 mM GSH (Sigma) and 1.0 mM CDNB (Sigma). The reaction was monitored at 340 nm every 30 seconds for 5 minutes. Assays were repeated at least three times independently, and IC50 values were calculated and plotted using GraphPad Prism 5.0 software.
Determination of the reaction of celastrol with recombinant GSTO1
Celastrol (100 μM in PBS, pH 7.4) was incubated with recombinant GSTO1 proteins (0–10 μg μL−1 in PBS, pH 7.4) for 30 minutes at room temperature. The absorption spectra (350–550 nm) were recorded using an Agilent Cary 60 UV-visible spectrometer.
Target validation by pull-down/immunoblotting experiments
IA-yne affinity pull-down experiments upon DMSO- and celastrol-treated proteomes were carried out as described above. Bound proteins were eluted by boiling in SDS-PAGE sample loading buffer for 15 minutes and separated using SDS-PAGE together with the whole proteomes (input). Then the proteins were transferred onto nitrocellulose membranes (Millipore) and the membrane was blocked with 3% (w/v) BSA in tris-buffered saline (TBS) for 2 hours at room temperature. After blocking, the membranes were incubated with the primary antibodies of GSTO1 (Sangon Biotech) and PDI (Sino Biological Inc.) for 1 hour each, respectively. After washing with TBST (TBS containing 0.1% Tween-20) three times, the blots were further incubated with the HRP-conjugated anti-rabbit (Santa Cruz) secondary antibody for 1 hour at room temperature. After incubation, the blot was washed again with TBST three times and developed using an enhanced ECL chemiluminescent substrate kit (Pierce).
Conclusions
We have demonstrated here the successful target profiling of celastrol, a cysteine-reactive natural product, in human cervical cancer HeLa cell lines via a competitive proteomics approach utilizing an iodoacetamide-derived probe IA-yne. The multi-target activity of celastrol might explain its therapeutic effect in a broad range of human diseases. Besides, we compared our discovery with previously reported celastrol binding proteins (Table S2, ESI†). Three reported targets including Cdc37, Annexin A2, and eEF1A were reconfirmed in our study; however, celastrol showed less or no effect on the other proteins in Table S2 (ESI†), which could be explained by the relatively low alkylation efficiency of celastrol to the proteins, even though the absolute amount of the proteins alkylated by celastrol was detectable. Moreover, although cysteine-targeting irreversible inhibitors have attracted more attention due to their high selectivity and prolonged duration of action, covalent and reversible inhibitors maintain high ligand efficiencies for target proteins without the accumulation of permanent modified proteins and peptides.13 Thus, celastrol and other quinone methide-containing natural products can be applied to the development of covalent and reversible drugs of reactive cysteine-containing proteins.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grants 21572243 and 21502206), the “Thousand Talents Program” young investigator award (to Y. X.), and the Science and Technology Commission of Shanghai Municipality (grant 15JC1400402). We thank Ms Yunxiao He and Dr Yuanhong Shan of the Core Facility Centre of the Institute of Plant Physiology and Ecology for assistance in fluorescence microscopy and mass spectrometry.
Notes and references
-
(a) E. Carlson, ACS Chem. Biol., 2010, 5, 639 CrossRef CAS PubMed;
(b) A. Harvey, R. Edrada-Ebel and R. Quinn, Nat. Rev. Drug Discovery, 2015, 14, 111 CrossRef CAS PubMed.
-
(a) G. Sethi, K. Ahn, M. Pandey and B. Aggarwal, Blood, 2007, 109, 2727 CAS;
(b) R. Kannaiyan, M. Shanmugam and G. Sethi, Cancer Lett., 2011, 303, 9 CrossRef CAS PubMed.
- A. Salminen, M. Lehtonen, T. Paimela and K. Kaarniranta, Biochem. Biophys. Res. Commun., 2010, 394, 439 CrossRef CAS PubMed.
-
(a) T. Zhang, A. Hamza, X. Cao, B. Wang, S. Yu, C. Zhan and D. Sun, Mol. Cancer Ther., 2008, 7, 162 CrossRef CAS PubMed;
(b) S. Sreeramulu, S. Gande, M. Göbel and H. Schwalbe, Angew. Chem., Int. Ed., 2009, 48, 5853 CrossRef CAS PubMed;
(c) A. Chadli, S. Felts, Q. Wang, W. Sullivan, M. Botuyan, A. Fauq, M. Ramirez-Alvarado and G. Mer, J. Biol. Chem., 2010, 285, 4224 CrossRef CAS PubMed;
(d) J. Lee, T. Koo, H. Yoon, H. Jung, H. Jin and K. Lee, Biochem. Pharmacol., 2006, 72, 1311 CrossRef CAS PubMed;
(e) H. Yang, D. Chen, Q. Cui, X. Yuan and Q. Dou, Cancer Res., 2006, 66, 4758 CrossRef CAS PubMed;
(f) Q. Zhao, Y. Ding, Z. Deng, O. Lee, P. Gao, P. Chen, R. Rose, H. Zhao, Z. Zhang, X. Tao, A. Heck, R. Kao and D. Yang, Chem. Sci., 2015, 6, 4124 RSC.
-
(a) Y. Su, J. Ge, B. Zhu, Y. Zheng, Q. Zhu and S. Yao, Curr. Opin. Chem. Biol., 2013, 17, 768 CrossRef CAS PubMed;
(b) A. Speers and B. Cravatt, Chem. Biol., 2004, 11, 535 CrossRef CAS PubMed.
- L. Klaić, R. Morimoto and R. Silverman, ACS Chem. Biol., 2012, 7, 928 CrossRef PubMed.
-
(a) D. Titov and J. Liu, Bioorg. Med. Chem., 2012, 20, 1902 CrossRef CAS PubMed;
(b) S. Ziegler, V. Pries, C. Hedberg and H. Waldmann, Angew. Chem., Int. Ed., 2013, 52, 2744 CrossRef CAS PubMed;
(c) U. Rix and G. Superti-Furga, Nat. Chem. Biol., 2009, 5, 616 CrossRef CAS PubMed.
-
(a) D. Abegg, R. Frei, L. Cerato, D. Prasad Hari, C. Wang, J. Waser and A. Adibekian, Angew. Chem., Int. Ed., 2015, 54, 10852 CrossRef CAS PubMed;
(b) D. Medina-Cleghorn, L. Bateman, B. Ford, A. Heslin, K. Fisher, E. Dalvie and D. Nomura, Chem. Biol., 2015, 22, 1394 CrossRef CAS PubMed.
-
(a) J. Yang and D. Liebler, Mol. Cell. Proteomics, 2015, 15, 1 CrossRef PubMed;
(b) S. Leonard and K. Carrol, Curr. Opin. Chem. Biol., 2011, 15, 88 CrossRef CAS PubMed;
(c) J. Paige, G. Xu, B. Stancevic and S. Jaffrey, Chem. Biol., 2008, 15, 1307 CrossRef CAS PubMed.
- K. Backus, B. Correia, K. Lum, S. Forli, B. Horning, G. González-Páez, S. Chatterjee, B. Lanning, J. Teijaro, A. Olson, D. Wolan and B. Cravatt, Nature, 2016, 534, 570 CrossRef CAS PubMed.
- K. Reddie and K. Carroll, Curr. Opin. Chem. Biol., 2008, 12, 746 CrossRef CAS PubMed.
- J. Bolton, S. Turnipseed and J. Thompson, Chem.-Biol. Interact., 1997, 107, 185 CrossRef CAS PubMed.
- I. Serafimova, M. Pufall, S. Krishnan, K. Duda, M. Cohen, R. Maglathlin, J. McFarland, R. Miller, M. Frödin and J. Taunton, Nat. Chem. Biol., 2012, 8, 471 CrossRef CAS PubMed.
- E. Weerapana, C. Wang, G. Simon, F. Richter, S. Khare, M. Dillon, D. Bachovchin, K. Mowen, D. Baker and B. Cravatt, Nature, 2010, 468, 790 CrossRef CAS PubMed.
- Y. Hu, Y. Qi, H. Liu, G. Fan and Y. Chai, Biochim. Biophys. Acta, 2013, 1830, 2779 CrossRef CAS PubMed.
- J. Hansen, J. Palmfeldt, S. Vang, T. Corydon, N. Gregersen and P. Bross, PLoS One, 2011, 6, e26634 CAS.
- C. Klomsiri, P. Karplus and L. Poole, Antioxid. Redox Signaling, 2011, 14, 1065 CrossRef CAS PubMed.
-
(a) S. Tao, J. Tillotson, E. Wijeratne, Y. Xu, M. Kang, T. Wu, E. Lau, C. Mesa, D. Mason, R. Brown, J. La Clair, A. Gunatilaka, D. Zhang and E. Chapman, ACS Chem. Biol., 2015, 10, 1916 CrossRef CAS PubMed;
(b) J. Yang, C. Li, L. Ding, Q. Guo, Q. You and S. Jin, J. Nat. Prod., 2012, 75, 1108 CrossRef CAS PubMed.
- A. Trott, J. West, L. Klaić, S. Westerheide, R. Silverman, R. Morimoto and K. Morano, Mol. Biol. Cell, 2008, 19, 1104 CrossRef CAS PubMed.
- J. Almond and G. Cohen, Leukemia, 2002, 16, 433 CrossRef CAS PubMed.
-
(a) S. Westerheide, J. Bosman, B. Mbadugha, T. Kawahara, G. Matsumoto, S. Kim, W. Gu, J. Devlin, R. Silverman and R. Morimoto, J. Biol. Chem., 2004, 279, 56053 CrossRef CAS PubMed;
(b) W. Wang, L. Feng, Q. Yue, W. Wu, S. Guan, B. Jiang, M. Yang, X. Liu and D. Guo, J. Cell. Physiol., 2012, 227, 2196 CrossRef CAS PubMed;
(c) T. Paimela, J. Hyttinen, J. Viiri, T. Ryhänen, R. Karjalainen, A. Salminen and K. Kaarniranta, Pharmacol. Res., 2011, 64, 501 CrossRef CAS PubMed.
- A. Salinas and M. Wong, Curr. Med. Chem., 1999, 6, 279 CAS.
-
(a) P. Ruzza, A. Rosato, C. Rossi, M. Floreani and L. Quintieri, Anti-Cancer Agents Med. Chem., 2009, 9, 763 CrossRef CAS;
(b) D. Townsend and K. Tew, Oncogene, 2003, 22, 7369 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Quantitative proteomics information and classification of 586 proteins enriched in this study, and bioinformatics analysis of celastrol target proteins. See DOI: 10.1039/c6mb00691d |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Weichao
Li
ab,
Mingli
Wang
ac,
Xixi
Zhang
bd,
Haibing
Zhang
d,
Xiaofeng
Tong
c and
Youli
Xiao
*ab
a,
Weichao
Li
ab,
Mingli
Wang
ac,
Xixi
Zhang
bd,
Haibing
Zhang
d,
Xiaofeng
Tong
c and
Youli
Xiao
*ab