
Erythropoietin and co.: intrinsic structure and functional disorder†
Vladimir N.
Uversky
*abc and
Elrashdy M.
Redwan
*ad
aDepartment of Biological Sciences, Faculty of Sciences, King Abdulaziz University, P.O. Box 80203, Jeddah, Saudi Arabia. E-mail: redwan1961@yahoo.com
bLaboratory of Structural Dynamics, Stability and Folding of Proteins, Institute of Cytology, Russian Academy of Sciences, St. Petersburg, Russia
cDepartment of Molecular Medicine and USF Health Byrd Alzheimer's Research Institute, Morsani College of Medicine, University of South Florida, Tampa, FL, USA. E-mail: vuversky@health.usf.edu
dTherapeutic and Protective Proteins Laboratory, Protein Research Department, Genetic Engineering and Biotechnology Research Institute, City for Scientific Research and Technology Applications, New Borg EL-Arab 21934, Alexandria, Egypt
First published on 1st November 2016
Abstract
Erythropoietin (Epo) is a heavily glycosylated protein, with its main function being related to erythropoiesis, where it controls red blood cell production via interaction with the Epo receptor (EpoR). It also plays a number of important roles in various hormonal, growth factor, and cytokine pathways. These roles are defined by Epo partners, such as the homodimeric (EpoR)2 receptor, the heterodimeric EpoR/βCR receptor and hypoxia inducing factor (HIF). Although the main structural features of both Epo and EpoR are conserved in vertebrates, the secretion sites of Epo in mammals are different from those in other vertebrates. Both biosynthetic and synthetic analogues of this protein are available on the market. Several side effects, such as pure red cells aplaisa, increase the rate of cancer-related death in patients treated with recombinant Epo. The multifunctionality of Epo and the ability of this protein to serve as a hormone, a cytokine, and a growth factor suggest the presence of functional disorder, which is a typical “structural” feature of moonlighting proteins. The goal of this article is to evaluate the roles of intrinsic disorder in the functions of Epo and its primary interactors, EpoR, βCR, and HIF-1α.
Introduction
Erythropoietin (Epo) was the first cytokine whose existence was experimentally validated.1 Today, Epo rivals insulin in its importance, and millions of people benefit each year from this therapeutic protein. In fact, Epo has multibillion annual sales, thereby representing one of the largest biopharmaceuticals markets in the world.2,3A very peculiar feature of Epo is its multifunctionality since, depending on conditions, this protein can act as a cytokine, a hormone, and a growth factor. The cytokine activity of Epo is manifested via its interaction with specific receptors located on the surface of target cells, thereby opening a way to transduce signals from the exterior to interior of those target cells. Epo also acts as a classical hormone, since it is synthesized in one organ (interstitial cells in the peritubular capillary bed of the renal cortex of the mammalian kidney in adults or in perisinusoidal cells of the liver during the fetal and perinatal period), then secreted into the bloodstream to show its activity at another site, the blood-forming system (red blood cell progenitors and precursors found in the bone marrow in humans). Finally, in its role as a growth factor, Epo is absolutely required for the formation of mature red blood cells from multipotent progenitors. In adult humans, Epo regulates normal erythropoiesis, an important process leading to the production of ∼2.5 million red cells per second.
Similar to other Epos, human Epo (hEpo, UniProt ID: P01588) is synthesized in the form of a 193 residue-long precursor protein containing an N-terminally located signal peptide (residues 1–27). Although the theoretical mass of mature hEpo is 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 240 Da, the apparent molecular mass is 34 to 36 kDa according to SDS-PAGE analysis. This difference between the theoretical and experimentally determined masses was interpreted as an indication that the average carbohydrate content of hEpo is of ∼40%.4,5 However, one should keep in mind that SDS-PAGE cannot provide information on the actual molecular mass of a protein and cannot be directly used for estimation of the carbohydrate content in a protein since all glycosylated proteins have anomalous mobility on SDS-PAGE. Therefore, the abnormal electrophoretic mobility of hEpo can only be taken as a reflection of the fact that this protein is glycosylated. Glycosylation plays a fundamental role in the function of many biopharmaceutical proteins and it is important for the various biological activities of Epo, affecting the solubility, cellular processing, secretion, and in vivo metabolism of this protein.6,7 On the other hand, recombinant production of active and adequately glycosylated Epo is challenging. Owing to the complexity associated with the production of Epo, the therapeutic preparation of human recombinant Epo (hrEpo) is typically conducted in mammalian host cells, mainly in Chinese hamster ovary (CHO) cells. This approach ensures a complex glycan structure, which is required for the therapeutic efficacy of this molecule, including its in vivo activity and half-life. Epo was the first proteinaceous biopharmaceutical produced as a recombinant protein from mammalian cells (known as Epogen/Procrit, a drug approved in 1989 in the US, and in 1990 in the EU for anemia treatment), and was the first such drug to surpass an annual sales value of $1 billion.2,3
240 Da, the apparent molecular mass is 34 to 36 kDa according to SDS-PAGE analysis. This difference between the theoretical and experimentally determined masses was interpreted as an indication that the average carbohydrate content of hEpo is of ∼40%.4,5 However, one should keep in mind that SDS-PAGE cannot provide information on the actual molecular mass of a protein and cannot be directly used for estimation of the carbohydrate content in a protein since all glycosylated proteins have anomalous mobility on SDS-PAGE. Therefore, the abnormal electrophoretic mobility of hEpo can only be taken as a reflection of the fact that this protein is glycosylated. Glycosylation plays a fundamental role in the function of many biopharmaceutical proteins and it is important for the various biological activities of Epo, affecting the solubility, cellular processing, secretion, and in vivo metabolism of this protein.6,7 On the other hand, recombinant production of active and adequately glycosylated Epo is challenging. Owing to the complexity associated with the production of Epo, the therapeutic preparation of human recombinant Epo (hrEpo) is typically conducted in mammalian host cells, mainly in Chinese hamster ovary (CHO) cells. This approach ensures a complex glycan structure, which is required for the therapeutic efficacy of this molecule, including its in vivo activity and half-life. Epo was the first proteinaceous biopharmaceutical produced as a recombinant protein from mammalian cells (known as Epogen/Procrit, a drug approved in 1989 in the US, and in 1990 in the EU for anemia treatment), and was the first such drug to surpass an annual sales value of $1 billion.2,3
In its active form, the 165-amino-acid-long hEpo is glycosylated at a single O-glycosylation site (Ser126) and three N-glycosylation sites (Asn24, Asn38, and Asn83). Although these three N-glycosylation sites are conserved in Chordata from fish to primates, clearly indicating the functional importance of these posttranslational modifications, much less is known about the functional importance of O-glycosylation. These observations agree with the fact that the therapeutic activity of Epo depends on the three N-glycans (Epo with the removed N-glycans has a very short half-life and virtually no erythropoietic function in vivo, see ref. 5), whereas the O-glycosylation has no known function. Furthermore, it has been shown that variations in the glycosylation caused by different processing conditions can yield functionally different Epo samples, where different samples can have up to a five-fold difference in the erythropoietic function in vivo.8,9 Furthermore, recent work by Erbayraktar and colleagues revealed that a version of Epo molecule can be created that entirely lacks erythropoietic function, but shows strong neuroprotection.10
Because of the multifunctionality of Epo and its involvement in a wide range of biological processes, it is believed that this protein can be successfully used for the treatment of a variety of diseases. In fact, in addition to its applications for anemia treatment, Epo's abilities for improving cardiac function, reducing fatigue, improving cognition in patients with diabetes mellitus (DM), regulating cellular energy metabolism, controlling obesity, as well as regulating tissue repair and regeneration, apoptosis, and autophagy in experimental models of DM are under investigation.11–13 All this suggests that Epo can be considered as a great candidate for treatment of metabolism disorders, such as DM. This is a really important point since a significant portion of the global population is affected by DM and its complications, which result in disability and death, and which currently have rather limited therapeutic options.
According to experimental and clinical studies, Epo shows significant efficiency in the treatment of several maladies, including those involving the developing brain.14 Administration of recombinant human Epo (rhEpo) to mice undergoing ischemic stroke induced by a platelet-rich thrombus formation was shown to liberate the mice from ischemic injury by reducing neuronal loss and blood–brain barrier (BBB) breakdown in an age-dependent manner, being more effective in young than aged animals.15 In the traumatic brain injury (TBI), administration of Epo was shown to help restore the axonal integrity, as well as promoting cellular proliferation, reducing brain edema, and preserving cellular homeostasis.16 In clinical studies of the neurodegenerative maladies associated with the loss of cognition, Epo was shown to prevent or reduce injury in the nervous system.16 It was also established that production of Epo is decreased in aging organisms, likely due to oxidative stress,17 and such a decline in the Epo expression can be related to some aging-related disorders, since this protein can control cell death pathways tied to apoptosis and autophagy as well as overseeing processes that affect cellular longevity and aging.16 On the other hand, supplements of exogenous Epo in aged rats partially rescued memory impairments, suppressed the oxidative stress and inflammatory response, and restored levels of brain-derived neurotrophic factor (BDNF), which is the critical neurotropic factor for synaptic plasticity and memory.18 Due to its importance for the adaptation of the body to habitation and exercise, Epo is illegal for use in sports, and this protein is listed by The International Olympic Committee (IOC/World Anti-Doping Agency, WADA) as one of the prohibited abuse substances under the class of “peptide hormones, mimics, and analogues”.19,20
Many features of Epo listed in this brief introduction (such as multifunctionality, ability to be engaged in numerous interactions with various partners, relation to various pathologies, and crucial dependence on posttranslational modifications) suggest that this protein might have functionally important intrinsically disordered regions. In fact, recent studies revealed that all proteomes analyzed so far contain numerous intrinsically disordered proteins (IDPs),21–25 which are biologically important proteins without unique 3D structures.24,26–35 These IDPs and hybrid proteins with ordered and intrinsically disordered domains/regions36 are known to be very common in nature21,23,24,37–39 and therefore represent an important part of the protein kingdom.26,31,34,40–44 IDPs have functions that complement activities of ordered proteins,45–47 with these highly dynamic proteins being commonly involved in signaling, regulation, and recognition.24,26–32,40,45–56 Functions of IDPs are modulated by various posttranslational modifications (PTMs), and phosphorylation sites,57 and furthermore the sites of many other PTMs are preferentially located within the intrinsically disordered regions.58 These flexible regulators and controllers of various biological processes are tightly controlled themselves,59,60 whereas deregulation of many IDPs is intimately associated with a variety of human diseases.35,61,62
Structural characterization of hEpo
Epo is a 34–36 kDa acidic glycoprotein hormone of 165 amino acids produced in the adult kidney and in the fetal liver.4 hEpo is synthesized as a precursor protein (UniProt ID: P01588) containing a signal peptide (residues 1–27) that is removed during protein maturation.63 Similar to other members of the cytokine family, structurally, human Epo (hEpo) is characterized by an up–up–down–down four-α-helical bundle architecture (helices αA (residues 8–26), αB (residues 55–83), αC (residues 90–112), and αD (residues 138–161)). In addition to this topology, in hEpo there are two mini-α-helices, αB′ (residues 47–52) located near the C-terminus of the AB loop and αC′ (residues 114–121), as well as two short antiparallel β-strands (residues 39–41 and 133–135).64 The structure of hEpo is stabilized by a disulfide bridge between Cys7 and Cys161 that links a pair of long antiparallel helices, αA and αD. The other pair of long antiparallel helices, αB and αC, is connected by a short loop. The longer AB-loop contains an additional mini-α-helix B′ and is involved in the interaction of Epo with its receptor, EpoR.64This loop is connected to the end of the αA by a second disulfide bond, Cys29–Cys33. The hydrophobic core of hEpo is formed by non-polar side-chains of the αA, αB, and αC helices packed against Phe138, Phe142, Tyr145, Phe148, Leu153, and Tyr156 located on the interior face of the αD-helix.64
Fig. 1A represents a crystal structure of the complex between the hEpo and its homodimeric (hEpoR)2 receptor (PDB ID: 1EER) and shows that hEpo occupies a specific position within the dimer of the extracellular ligand-binding domains of hEpoR. The interaction between hEpo and (hEpoR)2 relies on two binding interfaces defined as high- and low-affinity sites, with corresponding Kd of ∼1 nM and ∼1 μM, respectively.70 The high-affinity site of Epo includes residues from αA, αB′, and αD helices, and a part of the AB loop, whereas the low-affinity site of Epo contains residues located within helices αA and αC (Val11, Arg14, Tyr15, Ser100, Arg103, Ser104 and Leu108).64
 | ||
| Fig. 1 Structural characterization of human Epo. (A) The crystal structure of the complex between hEpo shown as a red ribbon and its homodimeric (hEpoR)2 receptor represented as blue and green surfaces (PDB ID: 1EER). (B) Analysis of the structural difference between the bound and unbound conformations of hEpo (PDB IDs 1EER and 1BUY, respectively) via structural alignment of these two forms. This structural alignment was conducted using the MultiProt web-based server.65 The structure of free hEpo is shown as a green ribbon (PDB ID: 1BUY), whereas the multicolored ribbon indicating different secondary structure elements corresponds to the (hEpoR)2-bound form of hEpo (PDB ID: 1EER). The shown protein structures were created using the Visual Molecular Dynamics (VMD) software.66 (C) Intrinsic disorder propensity of hEpo (UniProt ID: P01588) evaluated by the members of PONDR family of disorder predictors, PONDR® VLXT (black line),28 PONDR® VSL2 (yellow line),67 PONDR® VL3 (green line),68 and PONDR® FIT (red line).69 In these analyses, disorder scores above 0.5 are considered to correspond to the disordered residues/regions. The light red shadow around the PONDR® FIT curve reflects errors in disorder evaluation by this tool. Positions of α-helices and β-strands are shown correspondingly as blue and cyan bars in the middle of the plot. | ||
In its unbound form, hEpo is characterized by a simplified structure, consisting of four up–up–down–down long α-helices (helices αA (residues 9–26), αB (residues 56–83), αC (residues 92–111), and αD (residues 138–161)) connected by two long loops (AB and CD) and one short loop (BC), with the shortened αB′ helix (residues 49–51) being found within the long AB loop.71 Therefore, comparison of the solution structure of unbound hEpo with the (hEpoR)2 bound form of this protein clearly indicates the presence of noticeable conformational changes associated with the complex formation. In addition to shortening and reorientation of the αB′ helix, the region between the residues Leu112 and Thr132 that form the long CD loop has an extended conformation and is highly flexible.71 Furthermore, the unbound hEpo does not have the short antiparallel β-strands seen in the complexed form of this protein. The presence of noticeable structural differences between the bound and unbound hEpo conformations (PDB IDs 1EER and 1BUY, respectively) is further illustrated by Fig. 1B, which represents the structural alignment of these two forms. This structural alignment was conducted using the MultiProt web-based server.65
Fig. 1C presents the results of the evaluation of the intrinsic disorder predisposition of hEpo conducted by a series of disorder predictors from the PONDR family. These computational tools (PONDR® VLXT,28 PONDR® VSL2,67 PONDR® VL3,68 and PONDR® FIT69) were chosen based on their different angles of considering disorder propensity in a query protein, where PONDR® VLXT has high sensitivity to local sequence peculiarities associated with disorder-based interaction sites,28 PONDR® VL3 provides accurate evaluation of the presence of long disordered regions,68 PONDR® VSL2 is one of the more accurate stand-alone disorder predictors,67,72,73 and a meta-predictor PONDR-FIT is moderately more accurate than each of the component predictors,69 PONDR® VLXT,74 PONDR® VSL2,67 PONDR® VL3,68 FoldIndex,75 IUPred,76 and TopIDP.77 In this analysis, residues/regions are predicted to be disordered if they are characterized by predicted intrinsic disorder scores (PIDSs) above 0.5, and flexible regions are assumed to have PIDSs ranging from 0.2 to 0.5. Fig. 1C shows that the mature hEpo is predicted to have significant amounts of intrinsic disorder and to contain several flexible regions. Functionally important sites (i.e., most of the regions involved in the interaction with (hEpoR)2) are located either within or in close proximity to the disordered (PIDS > 0.5) or flexible (PIDS > 0.2) regions.
In fact, the average disorder propensities of the hEpo regions involved in the high-affinity site are 0.347 ± 0.021 and 0.245 ± 0.017 for the αA and αD helices, respectively. Similarly, some residues involved in the hEpo low-affinity site are characterized by an average disorder score of 0.397 ± 0.007 (a central part of the αC from the low-affinity site that includes Ser100, Arg103, Ser104, and Leu108). On the other hand, the αB′ helix from the high-affinity site and the N-terminal part of the αA helix that includes Val11, Arg14, Tyr15 forming the low-affinity site are located within the more ordered regions of hEpo, being characterized by average disorder scores of 0.120 ± 0.005 and 0.195 ± 0.020, respectively. Furthermore, long AB and CD loops are also predicted to be flexible/disordered, and a significant amount of conformational flexibility is expected for helices αA, αC, αC′, and αD and for both β-strands.
Evolution of erythropoietin
The 2144 bp-long human EPO gene found in chromosome 2 (7 pterq22) consists of 4 exons, which contain 582 bp and are connected by 4 introns, totalling 1562 bp. Although the Epo mRNA is translated into a polypeptide chain of 193 amino acids, the posttranslational removal of the N-terminal signal peptide (residues 1–27) and C-terminal arginine results in a circulating protein of 165 residues.78 Similar to human EPO, the erythropoietin gene from the pufferfish (Fugu rubripes) has 5 exons and 4 introns and encodes a protein that comprises 185 residues and has noticeable similarity to Epos from various mammals.79Although the primary site of hEpo production is the kidney, the primary fugu Epo producing organ is the heart. In addition to these primary Epo generators, this protein is also produced by the liver and brain of both fugu and human.
Fig. S1 (see ESI†) presents the results of multiple sequence alignment of Epo proteins from various organisms and show that these proteins are characterized by a relatively high level of overall conservation. To understand the prevalence of intrinsic disorder in these proteins, their sequences were subjected to computational analysis using a set of disorder predictors.
Fig. 2A and B present results of this analysis conducted for the precursors (i.e., natural products of the Epo genes that contain a signal peptide) of mammalian and non-mammalian Epo proteins, respectively. It is clear that substantial disorder is present in all these proteins. Furthermore, Epos possess great variability in their predisposition for intrinsic disorder, with some members of this protein family being predicted to be mostly structured, and with the overall levels of intrinsic disorder being noticeably increased in many other Epos. The idea that different Epos are characterized by different levels of overall intrinsic disorder predisposition is illustrated by Fig. 2C, which shows a phylogenic tree built for these proteins.
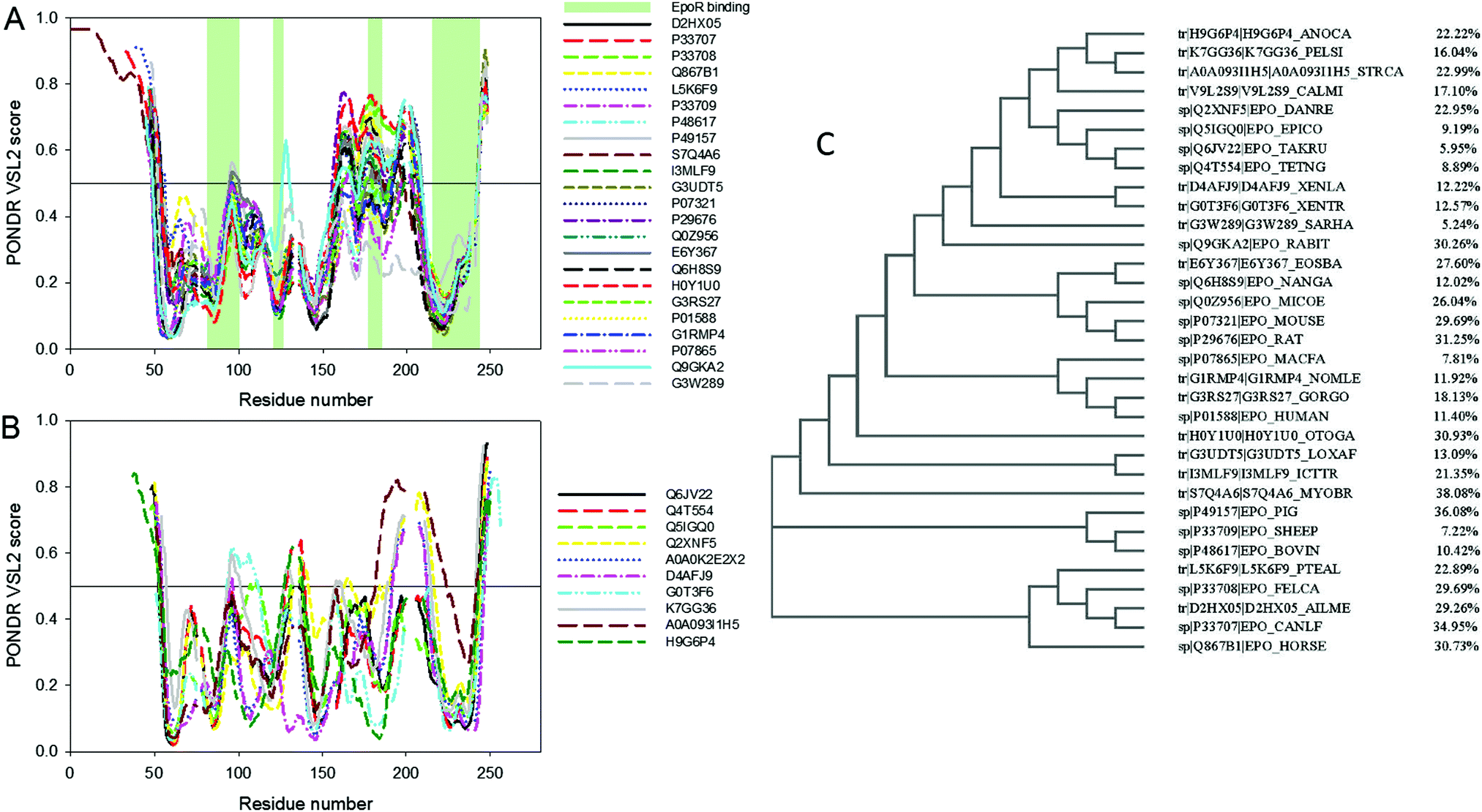 | ||
| Fig. 2 Aligned disorder PONDR® VSL2 profiles of various Epo proteins (A) mammalian and (B) non-mammalian precursors of Epo proteins. Sequences were first aligned using the ClustalW2 algorithm (http://www.ebi.ac.uk/Tools/msa/clustalw2/). The ClustalW2 alignment data are shown in Fig. S1 (see ESI†). The gaps in each line correspond to the gap in alignment. A disorder threshold is indicated as a thin line (at score of 0.5) to show a boundary between disorder (>0.5) and order (<0.5). Light-green-shaded areas in (A) show the locations of regions of human Epo involved in interaction with EpoR. (C) Phylogenic tree built for Epo proteins analyzed in this study. Corresponding percentages of predicted disordered residues for all Epo proteins are shown. | ||
Fig. 2C shows their corresponding percentages of predicted disordered residues, which range from as low as 5.2% to as high as 38.1% for Epos from Sarcophilus harrisii and Myotis brandtii, respectively. This variability in the overall levels of intrinsic disorder seems to contradict the hypothesis that the intrinsic disorder is of great importance for proper binding of Epo to its receptor. However, one should keep in mind that it is not the overall disorder levels in Epo that are crucial for the efficient interaction of this protein with its receptor, but the peculiarities of disorder distribution within its sequence. Fig. 2A shows that positions of disordered/flexible regions in mammalian Epos are generally conserved and these proteins are different from each other mostly by the intensities of the “peaks” in their disorder profiles. Furthermore, disorder patterns of regions potentially involved in interaction with EpoR are also conserved, providing further support to the idea of the functional importance of intrinsic disorder. A similar situation is observed for non-mammalian Epos, many of which also have similarly positioned regions of increased flexibility/disorder. In other words, the major difference between the proteins within one group (mammalian or non-mammalian) is the prediction “strength” of the similarly positioned disordered/flexible regions, whereas between these two groups, proteins differ in the positions of their disordered/flexible regions within the amino acid sequences.
Therefore, Fig. 2A and B can be used as illustrations of an important observation that there is a remarkable intergroup similarity in the overall appearance of the disorder profiles of mammalian and non-mammalian Epos. In fact, although the overall levels of intrinsic disorder in Epos are different, some of their parts are characterized by high flexibility manifested as high values of local intrinsic disorder propensity. Regions are considered as flexible or disordered if their disorder scores exceed the values of 0.25 or 0.5, respectively. Therefore, in the aligned profiles of mammalian Epos, N- and C-terminal tails and segments 60–70, 90–120, 130–140, and 160–210 are systematically predicted to have from one to three either flexible or disordered regions. Similarly, in the aligned profiles of non-mammalian Epos, flexibility/disorder is present in N- and C-tails, as well as in the segments including residues 70–80, 90–105, 125–140, 155–165, and 180–220.
Alternative splicing
A distal promoter is used for generation of the brain-specific fugu Epo and the corresponding transcript includes an alternatively spliced first coding exon.79 Despite a similar “mosaic” structure of EPO gene in mammals, with the exception being the PhD thesis by Christel Barbara Bonnas80 and a corresponding patent,81 there are no other reports on the presence of alternative splicing in mammalian Epos. The analysis of human and murine brain and kidney cDNA reported in ref. 80 and 81 revealed the presence of a major Epo product (600 bp) and several products of smaller sizes corresponding to incomplete Epo transcripts containing internal deletions.80,81 These studies also revealed that the human splice variant hS3 is generated by alternative splicing-induced removal of exon 3, whereas the second splice form, hS4, is characterized by the loss of the first 30 residues encoded by exon 4. The authors discussed seven isoforms for both human and mouse Epos and pointed out that these isoforms have different functional properties.80,81Fig. 3A shows the results of multiple sequence alignment of human Epo isoforms described in ref. 80 and 81 whereas Fig. 3B–H analyze the correlation between alternative splicing of hEPO mRNA and predisposition of the corresponding hEpo protein variants for intrinsic disorder. These data are consistent with important notions: (a) alternative splicing affects the intrinsic disorder propensity of hEpo; (b) alternative splicing causes an increase in the overall disorder of a protein; (c) alternative splicing preferentially occurs in disordered or flexible regions; and (d) various alternatively spliced variants of hEpo can be arranged in the following order according to their mean disorder scores evaluated as values averaged over the outputs of three disorder predictors:| hEpo canonical (0.279 ± 0.034) < hEpo h1-4 (0.286 ± 0.046) < hEpo hs4 (0.289 ± 0.049) < hEpo hs3 (0.296 ± 0.034) < hEpo h1-5 (0.290 ± 0.043) < hEpo h1-1 (0.331 ± 0.057) < hEpo h2-1 (0.363 ± 0.038). |
 | ||
| Fig. 3 Effect of alternative splicing on intrinsic disorder propensity of hEpo. (A) The results of multiple sequence alignment of human Epo isoforms described in ref. 80 and 81. Plots (B)–(H) show the results of the analysis of the effect of alternative splicing on intrinsic disorder predispositions of the corresponding hEpo protein variants evaluated by PONDR® VLXT (black lines), PONDR® VL3 (red lines), and PONDR® VSL2 (green lines). | ||
Hypoxia-dependent induction of the mammalian EPO gene: role of HIF-1α
The induction of the mammalian EPO gene can be hypoxia-dependent, being regulated by binding of a hypoxia-inducible factor 1 (HIF-1) to a hypoxic response element (HRE) located in the 3′ untranslated region (UTR) of the Epo and other hypoxia-dependent genes.86,87 The process starts with the hypoxia-induced increase in the stability of cytoplasmic HIF-1α, which then moves to the nucleus, dimerizes with HIF-1β to form active HIF-1 that binds to HRE and activates transcription of hypoxia-dependent genes.87 Although there are three HIF-α isoforms (HIF-1α, HIF-2α, HIF-3α), it is believed that HIF-1α plays the major role in activation of the Epo gene. Being expressed in virtually all organs of the body, this protein is known to control expression of about 200 genes, products of which are involved in angiogenesis, glycolysis, and erythropoiesis.88Fig. 4A shows that human HIF-1α (UniProt ID: Q16665) is predicted to be a highly disordered protein (in fact, more than 50% of its residues are predicted to be disordered), which is in line with the well-established fact that proteins involved in transcription regulation are highly disordered in general.89–92 Furthermore, long disordered regions found in this protein contain multiple predicted disorder-based protein–protein interaction sites (residues 399–413, 426–455, 465–495, 516–542, 555–583, 598–608, 627–647, 670–678, 687–698, 706–716, 726–738, and 771–782 identified by the ANCHOR algorithm45,46), suggesting that HIF-1α can be involved in one-to-many or polyvalent interactions, the mechanisms defining the ability of this protein to be engaged in a multitude of biologically important contacts with different binding partners (see Fig. 4B). Fig. 4C presents the results of the analysis of human HIF-1α by the D2P2 internet tool (http://d2p2.pro/),93 which provides the results of disorder evaluation in a query protein by PONDR® VLXT,74 IUPred,76 PONDR® VSL2B,68,94 PrDOS,95 ESpritz,96 and PV2,93 and shows the location of functional domains, curated cites of various PTMs and predicted disorder-based potential binding sites. Fig. 4C shows that this protein is predicted to have at least three long disordered regions, possess multiple functional domains, have potential disorder-based binding motifs, and various PTMs, such as acetylation, ubiquitylation, SUMOylation, nitrosylation and phosphorylation are spread throughout disordered regions of this protein.
 | ||
| Fig. 4 (A) Intrinsic disorder propensity of human HIF-1α (UniProt ID: Q16665) evaluated by the members of the PONDR family of disorder predictors, PONDR® VLXT (black line), PONDR® VSL2 (yellow line), PONDR® VL3 (green line), and PONDR® FIT (red line). (B) Analysis of the interactivity of human IF-1α (UniProt ID: Q16665) by the internet tool STRING.104 The output of STRING analyzer is the network of predicted interactions for a query protein, where nodes correspond to proteins, whereas the edges represent the predicted or known functional associations between these proteins. There are seven types of evidence used in predicting the associations, which are indicated in the resulting network by the differently colored lines, where a red line indicates the presence of fusion evidence; a green line – neighborhood evidence; a blue line – co-occurrence evidence; a purple line – experimental evidence; a yellow line – text mining evidence; a light blue line – database evidence; a black line – co-expression evidence.104 (C) Evaluation of the functional intrinsic disorder propensity of human HIF-1α (UniProt ID: Q16665) analyzed by the D2P2 database (http://d2p2.pro/).93 The top nine colored bars represent the location of disordered regions predicted by different disorder predictors (Espritz-D, Espritz-N, Espritz-X, IUPred-L, IUPred-S, PV2, PrDOS, PONDR® VSL2b, and PONDR® VLXT, see keys for the corresponding color codes). The green-and-white bar in the middle of the plot shows the predicted disorder agreement between these nine predictors, with green parts corresponding to disordered regions by consensus. Yellow bar shows the location of the predicted disorder-based binding site (MoRF region), whereas the differently colored circles at the bottom of the plots show the locations of various posttranslational modifications. | ||
Data on the HIF-1α PTMs are extracted from the D2P2 database which shows the positions of “curated PTM sites” within the sequence of a query protein. To the best of our knowledge, these PTMs are experimentally determined and not predicted. Furthermore, there are several studies discussing both the presence and functional outputs of various PTMs. For example, Ser257 was shown to be phosphorylated by casein kinase 1 and this modification represents an important mechanism for control of the HIF-1α activity during hypoxia, likely via regulation of the inter-subunit interactions,97 whereas the HIF-1α phosphorylation at Ser576 and Ser657 by Polo-like kinase 3 (Plk3) plays a role in regulation of the HIF-1α-based hypoxia signaling pathway,98 and GSK3β-driven phosphorylation of HIF-1α on a cluster of serine residues (Ser551, Ser555, and Ser589) within the oxygen-dependent degradation domain (ODDD) enhances HIF-1α degradation by the proteasomal pathway.99 Hydroxylation of Pro402 and Pro564 located within the HIF-α ODDD plays a role in controlling the ubiquitylation of this protein by VHLE3.100 Acetylation and SIRT2-mediated deacetylation of HIF-α at Lys532 and Lys709 regulate the stability of this protein in tumor cells.101S-Nitrosation of Cys800 activates HIF-α interaction with p300 and stimulates transcriptional activity of this protein.102 Finally, an asparaginyl hydroxylase enzyme, a factor inhibiting HIF-1 (FIH-1), controls hydroxylation of the key asparagine residue Asn803 located within the C-terminal transactivation domain (CAD) of HIF-α,and thereby regulates the transcriptional activity of this protein.103
The fact that the disordered regions of this protein are heavily decorated by various PTMs is in agreement with the well-known fact that sites of many enzymatically catalyzed PTMs are preferentially located within disordered regions, which makes them easily accessible to modifying enzymes.57,58 Furthermore, in addition to the full-length canonical form, HIF-1α is present in two alternatively spliced isoforms, with region 736–826 being missing in isoform-2, and with the N-terminal 1MEGAGGANDKKK12 region in isoform-3 being substituted to MSSQCRSLENKFVFLKEGLGNSKPEELEEIRIENGR.
These alternative splicing events affect the disordered region and/or potentially modulate the interactivity of HIF-1α by removing a couple of predicted disorder-based interaction sites (residues 726–738 and 771–782). These observations are in line with the known association between alternative splicing and intrinsic disorder, where regions taken out of mRNA by alternative splicing preferentially encode for intrinsically disordered regions in proteins.82,84,85
Although the EPO gene from Fugu rubripes is homologous to the human gene, it lacks a flanking HRE, which is critical for Epo induction by hypoxia in mammals.79 Despite the fact that the fugu EPO gene has no functional HRE in the 5′ or 3′ flanking regions, there is some indication that hypoxia increases appropriate splicing of Epo transcripts. Much is known about hypoxia induction of Epo in the kidney and Epo stimulation of bone marrow erythropoiesis in mammals, especially humans and rodents. In contrast, fugu Epo is expressed mainly in the heart, with some expression in the brain and liver, but not in the kidney, and erythropoiesis takes place in the kidney rather than in the bone marrow. Nevertheless, it appears that Epo synthesis away from the site of erythropoiesis and its secretion into the circulation for transport to stimulate red cell production is conserved between mammals and fish.79
Of note is the similarity of Epo expression in the brain and liver. In mammalian model systems, the effects of Epo are extended beyond erythropoiesis and provide protection for the embryo and selected adult organs against ischemia or stress. In addition to Epo requirement for erythropoiesis, developmental defects in brain neuroepithelium and in heart endocardium and myocardium as well as increased neuron sensitivity to hypoxia were described in mice that lack the erythropoietin receptor.87,105 Neuroprotection by Epo has been demonstrated in several adult animal models for brain hypoxia and mechanical trauma. This includes Epo protection against brain ischemia, which reduces hippocampal neuron damage and memory loss.87,106
A single dose of Epo following myocardial infarction in rats reduced infarct size and functional decline 8 weeks after insult.87,107 Fugu EPO expression in the heart and brain raises the possibility that its neuroprotective and cardioprotective activities require local Epo production, may be evolutionarily conserved, and perhaps were among the original functions of this molecule.79
Non-erythropoietic functions of Epo: role of Epo receptor
Being expressed in several non-hematopoietic tissues, Epo plays a role in protection from apoptosis and inflammation due to hypoxia, toxicity or injury and has protective and proliferative activities on non-hematopoietic cells, as well as on the erythroid progenitor cells in bone marrow. In particular, interest has increased in the anti-apoptotic effects of Epo in the nervous system and in the possible use of this protein as a neuroprotective agent.108 In recent clinical trials, researchers successfully used Epo to treat tissue injury induced by a stroke.109,110 Medical practitioners, however, should consider some important side effects of using Epo, such as hypertension, thrombosis, and tumor growth, for this clinical indication.109Furthermore, Epo has a number of crucial immunoregulatory effects. Although such immunoregulatory activities of Epo are realized via different mechanisms, an important role is attributed to the Epo–EpoR signaling initiated by interaction of Epo with its receptor EpoR (either in its homodimeric form, (EpoR)2, or in the heterodimeric form EpoR/βCR resulting from the interaction of EpoR with the β common receptor (βCR)), which is part of the granulocyte macrophage colony stimulating factor (GM-CSF) and the IL-3 and IL-5 receptors and is found in a wide array of cells, such as microglia, cardiomyocytes, astrocytes, renal tubular and collecting duct cells, neurons, smooth muscle cells, myoblasts, retina and vascular cells. Related protective effects of Epo were shown to play an important role in neuroprotection and cardioprotection, and were also reported to be applicable in pancreatic-related diseases, auditory injury, and retina degeneration.111 This wide tissue distribution of EpoR opens the possibility for endogenous Epo to be engaged in and control signaling in various non-hematopoietic tissues, and it also provides means for the exogenous Epo to be responsible for modulation of the function of various organs and to control cellular responses to diverse types of injury.111 Furthermore, analysis of the experimental autoimmune encephalomyelitis (EAE) multiple sclerosis model revealed that the pro-inflammatory responses of antigen-specific T-cells can be inhibited by Epo, thereby inducing immune tolerance.112
Therefore, these observations clearly indicate that the non-erythropoietic biological effects of Epo based on the activation of Epo–EpoR signaling in non-hematopoietic organs and cell types deserve close attention of researchers, since these mechanisms are directly related to the clinical applications of Epo and its derivatives for treatment of chemotherapy-induced anemia in cancer patients or anemia in chronic kidney disease.111 It is believed that the induction of non-erythropoietic functions of Epo is related to the interaction of this protein with the heterodimeric EpoR/βCR receptor, which requires high local concentrations of Epo.112 This is in a contrast to the initiation of the erythropoietic functions of Epo, which relies on the binding of picomolar concentrations of this cytokine to the canonical (EpoR)2 homodimers.112 This principle difference in initiation of the non-erythropoietic and erythropoietic functions of Epo defines the need to search for low affinity forms of this hormone/cytokine as well as development of its low affinity analogues.112–117
One should keep in mind that although in many preclinical studies Epo and its various derivatives and/or analogues were shown to serve as efficient cytoprotective agents,118 there is remarkable controversy among the results generated by small and large clinical studies.10,119,120 Therefore, despite large expectations related to the discovered potential anti-apoptotic and cytoprotective effects of recombinant human erythropoietin (hrEpo) and hopes that high-dose, short-term hrEpo therapy can be used for tissue protection in myocardial infarction and stroke, a systematic analysis of the related clinical trials revealed a lack of reproducible benefits from this treatment combined with the increased risk of mortality and some adverse side effects.121 This could be associated with the use of excessive Epo concentrations, which can initiate side effects via the cross-talk between the protective routes and the hematopoietic activity. On the other hand, clinical trials on the effects of the Epo analogues known as erythropoiesis-stimulating agents (ESAs) showed that these agents can substantially reduce the mortality of critically ill trauma patients.114 In addition, despite the fact that Epo is believed to initiate its non-erythropoietic effects via interaction with the heterodimeric EpoR/βCR complex,88,109 the presence of βCR is not always needed for the transduction of the Epo signal toward protection from apoptosis.88,122 All these observations clearly show that better understanding is needed of the cellular receptors used for mediation of the Epo pleiotropic effects in various non-hematopoietic cells.88
It was established that functional variability of Epo can be ascribed to variability of its modifications, with some non-erythropoietic functions of this important cytokine being initiated by variously modified Epo. For example, although the carbamylated Epo (cEpo) failed to interact with the (EpoR)2 homodimers, this Epo form served as an efficient and specific neuronal protector and a general cytoprotector.122 The signal transduction route of cEpo was shown to be associated with the initiation of the JAK2-STAT5-mediated cascade known to play a neuroprotective role itself.122 The cardioprotective effects of Epo were also retained by cEpo.88,123 Another Epo variant, isaloerythropoietin (asialo-Epo), which is a short half-life Epo form generated by stripping of sialic acid from this protein, was not able to promote erythropoiesis. However, in experimental models of cerebral ischaemia, spinal compression, and sciatic nerve crush, asialo-Epo granted neuron protection, being able to cross the blood–brain barrier and bind to neurons within the cortex and hippocampus.10,119 Similarly, other non-erythropoietic Epo derivatives, such as glutaraldehyde Epo (gEpo) and ARA290, despite their lack of ability to bind to the classic homodimeric (EpoR)2 complex possess various cytoprotective effects.124
Clinical side effects of Epo treatment
According to the recent US Food and Drug Administration (FDA) review, treatment with erythropoietin analogues led to earlier progression or sooner death of cancer patients in comparison with patients that did not receive such treatment. This statement was based on the analysis of eight studies presented to the FDA in which it was shown that patient survival is decreased or tumor progression is accelerated by administration of erythropoietin analogues.125 In fact, several clinical studies have shown increased rates of cancer-related deaths in patients treated with recombinant Epo.108,126–128 Some explanations for this adverse effect of recombinant Epo in anemic cancer patients include stimulation of tumor cells via Epo receptors, EpoRs,86 a higher risk of thromboembolic events,105 and failure to respond to recombinant Epo.4,108Mechanistically, the overall danger of using erythropoietin analogues as drugs is supported by the fact that tumor cells commonly have functional EpoRs. As a result, various tumor-promoting functions can be promoted via signaling involving Epo and its receptors. In agreement with this hypothesis, several preclinical studies revealed the existence of Epo-induced proliferation of cancer cells, stimulation of chemo-taxis, migration and invasion of cancer cells, inhibition of apoptosis, development of drug resistance, and protection of cancerous tissues from ischemic injury. In fact, the presence of EPOR mRNA and/or EpoR protein has been reported in breast carcinoma,129–131 renal carcinoma,132 lung carcinoma,133 endometrial carcinoma,134 tumors of the cervix and of other organs of the female reproductive tract,135–137 thyroid cancer,138 and various pediatric tumors.139,140 Although these observations raised significant concerns, it has been also emphasized that patients with tumors that do not have EpoRs might not suffer from the harmful effects of the Epo analogues.141
Almost all biopharmaceuticals induce an immune response, although its incidence varies from extremely rare to more than 50% of the patients. Antibodies to therapeutic proteins can not only compromise efficacy, but can also cross-react with endogenous factors to cause serious toxicity, if these factors have an essential biological function. This was illustrated by reports of an upsurge of pure red blood cell aplasia, a severe form of anemia, in patients receiving recombinant erythropoietin α. The aplasia appeared to be mediated by the antibodies to the recombinant Epo. This incident has obviously inspired the regulatory authorities to pay special attention to Epo's immunogenicity.142,143
Erythropoietin analogues
Currently, clinics use three types of recombinant Epo: (1) Epoetin alfa (which is hEpo produced in cell culture using recombinant DNA technology),(2) Epoetin beta (which is a synthetic, recombinant form of Epo), and
(3) darbEpoetin alfa (which is also a synthetic form of erythropoietin).
Epoetin alfa and beta show differences in carbohydrate moieties but no differences in clinical efficacy.144 DarbEpoetin alfa contains five N-linked glycosylation sites, compared to three for Epoetin, and shows a prolonged half-life relative to Epoetin preparations.145
In fact, hEpo and Epoetin are known to contain three N-linked and one O-linked carbohydrate chains attached to residues Asn24, Asn38, and Asn83, and Ser126, respectively,63,146–148 whereas the two new sites of N-linked glycosylation in DarbEpoetin alfa were introduced by changing five amino acid residues in hEpo by site-directed mutagenesis.145
In comparison with hEpo, DarbEpoetin has more sialic acid-containing oligosaccharides, and this increased content of the sialic acid-containing oligosaccharides has a profound effect on receptor binding, clearance, and in vivo biological activity.145 Recently, researchers have developed other EpoR activating molecules that have a long-acting effect, permit oral administration, and are useful in patients with pure red cell aplasia (PRCA).149,150 Qureshi et al. developed a small molecule for oral administration that activates EpoR homodimers.151
This small compound served as a competitor for Ep, and, at the same time, was able to switch on the Epo receptor.151 Although this small molecule was a competitive inhibitor of Epo–EpoR binding, it was not, however, effective enough to be used as a therapeutic agent.150 Despite the fact that the FDA has not approved any natural erythropoietic agents, a long list of traditional medicines has been tried to correct anemia.108 Chemical synthesis of native Epo and/or new Epo analogous is a very promising area of research152–154 waiting for discovery of safe and effective analogues that would support tissues undergoing neurodegeneration.
Epo receptor and βCR
Because of the important roles of EpoR and βCR in erythropoietic and various non-erythropoietic activities of Epo, some information should be provided about the functional and structural properties of these receptors. Epo receptors (EpoRs) mediate the activities of Epo. EpoRs belong to the type I cytokine receptor superfamily, members of which are characterized by a single transmembrane domain, a WSXWS motif, and contain four cysteine residues.110 The Epo binding to the homodimeric (EpoR)2 interferes with the Fas Ligand (FasL)–Fas signaling, thereby inducing erythropoiesis.105 Here, terminal erythroid differentiation is enabled by shutting down the apoptosis of erythroid progenitor cells.There is also a contact-dependent negative feedback mechanism, where the production of immature progenitors is suppressed by mature erythroid cells via up-regulation of the apoptosis-inducing ligands, such as tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) and FasL. As a result, immature erythroid precursors are eliminated by the regulated apoptosis caused by the caspase-mediated cleavage of one of the essential transcription factors in erythroid development, GATA1. Therefore, Epo-induced down-regulation of FasL and Fas expression increases the production of red blood cells (RBCs) and oxygen transport capacity in the circulation.112 Epo binding induces dimerization and conformational changes in EpoR, which activate Janus tyrosine kinase-2 (JAK-2) leading to its auto-phosphorylation, as well as promoting phosphorylation of the cytoplasmic domain of EpoR at eight conserved tyrosine residues, and phosphorylation of several other proteins involved in signal transduction. Among targets of this phosphorylation events is the signal transducer and activator of transcription-5 (STAT-5),155–157 an important protein that interacts with a multitude of signaling proteins containing SRC homology 2 (SH2) domains. Activated transcription factors, such as STAT-5, move to the nucleus and activate genes for anti-apoptotic molecules.158
The structure of the crystallizable part of human EpoR (residues 25–249 that corresponds to the extracellular domain of the mature protein with the removed signaling peptide, residues 1–24) contains a short N-terminal α-helix (∼15 residues) followed by two β-sandwich domains, D1 and D2, each consisting of ∼100 residues organized in eight (4 + 4, D1 domain) or seven (3 + 4, D2 domain) β-strands.64 In the Epo-(EpoR)2 structure, two EpoR monomers interact with each other preferentially through their D2 domains. Interaction of EpoR with Epo mostly relies on the loops between the β-strands in both domains of the receptor, such as three loops of domain D1, two loops of the domain D2 and a loop connecting domains D1 and D2.64
In addition to rather extensive structural characterization of the extracellular domain, solution NMR analysis of the 237–284 fragment of human EpoR corresponding to the transmembrane domain (TMD, residues 251–272) and juxtamembrane region (JM, residues 273–284) revealed that in the presence of the dodecylphosphocholine (DPC) micelles, TMD and JM were folded in a long α-helix, whereas N-terminally located residues 237–272 were not structured.159 There is no structural information on the C-terminal half of the protein. Fig. 5 provides a simple explanation for this lack of structural information by showing that the cytoplasmic domain of human EpoR (residues 274–508 in UniProt ID: P19235) is predicted to be mostly disordered. Fig. 5 also shows that this disordered cytoplasmic domain is crucial for the functionality of EpoR since it contains several predicted disorder-based interaction sites (residues 365–376, 391–400, 404–409, 422–432, 434–441, 452–463, and 503–508) and multiple phosphorylation sites.
 | ||
| Fig. 5 (A) Intrinsic disorder propensity of human EpoR (UniProt ID: P19235) evaluated by PONDR® VLXT (black line), PONDR® VSL2 (yellow line), PONDR® VL3 (green line), and PONDR® FIT (red line). (B) Analysis of the interactivity of human EpoR (UniProt ID: P19235) by STRING. (C) D2P2 output for human EpoR (UniProt ID: P19235). | ||
Furthermore, EpoR exists as three proteoforms generated by alternative splicing, EpoR-F (full-length protein), EpoR-S (soluble protein), and EpoR-T (truncated protein). The EpoR-F is a canonical polypeptide, which, in its mature form, consists of 484 residues and has all three topological domains, extracellular (residues 25–250), transmembrane (residues 251–273) and cytoplasmic (residues 274–508). The EpoR-S form has 217 residues after maturation and removal of a signal peptide (residues 1–24) and is different from the EpoR-F form by lacking both transmembrane and cytoplasmic domains (residues 242–508), and having region 196VEILEGRTECVLSNLRGRTRYTFAVRARMAE PSFGGFWSAWSEPVS241 changed to 196GTVFLSPDWLSSTRARPH VIYFCLLRVPRPDSAPRWRSWRAAPSVC241. In the EpoR-T (which has 304 residues in the mature form), a significant part of the cytoplasmic domain (residues 329–508) is removed by alternative splicing, and the preceding 306LWLYQNDGCLWWS PCTPFTEDPP328 region is changed to 306VGGLVVPSVPGLPCF LQPNCRPL328.
Importantly, both EpoR-S and EpoR-T might be of functional importance. For example, it was pointed out the serum of end stage kidney disease patients contained detectable amounts of EpoR-S, and the higher serum levels of this form were shown to correlate with increased Epo requirements, suggesting that enhanced production of EpoR-S may contribute to Epo resistance in end stage renal disease.160 It was also shown that some human tumor cells can express and secrete EpoR-S.161 As far as EpoR-T is concerned, it has been established that the dopaminergic neurons of the Substantia nigra can express this form, which operates as a decoy, being co-expressed with the EpoR-F.162 In addition, EpoR-T was shown to act as a dominant negative regulator of Epo signaling, leading to hypertension.163 Finally, based on the analysis of the transfection of human leukemic cells that can differentiate into mature erythroid cells with Epo treatment with EpoR-T, it has been concluded that the dysregulated expression of EpoR-T may cause apoptosis and blockage of erythroid differentiation, resulting in ineffective erythropoiesis.164 As was already discussed for Epo and HIF-1α, alternative spicing dramatically affects the intrinsic disorder predisposition of the protein and modifies its interactability.
The β common receptor (βCR) is a member of the type I cytokine receptor superfamily. It is a common part of several hetero-oligomeric cytokine receptors, and serves as a high affinity receptor for granulocyte macrophage colony stimulating factor (GM-CSF) and interleukin-3 (IL-3) and IL-5.
Brines et al. proposed that activation and phosphorylation of JAK-2 can be induced via binding of Epo to the intracellular domain of the heterotrimeric receptor consisting of an EpoR and a βCR homodimer, which is associated with the heterotrimer.165 Following JAK-2 activation, signaling cascades similar to hematopoiesis might occur and result in tissue protection.109 Furthermore, researchers have reported the presence of Epo/EpoR-based signaling events leading to the modulation of the intracellular concentrations of nitric oxide and cyclic guanosine monophosphate (cGMP).166
As with many other cytokine receptors, human βCR has an extracellular domain (residues 17–443), a transmembrane domain (residues 444–460) and a cytoplasmic domain (residues 461–897). Although βCR is almost two-fold larger than EpoR, these two receptors have some similarity, where structural information is available for their extracellular domains, whereas cytoplasmic domains are not characterized structurally.
Fig. 6 shows that these two proteins have a similar predisposition for intrinsic disorder, since the cytoplasmic C-terminal domain of human βCR is predicted to be mostly disordered. This disordered cytoplasmic domain of βCR has multiple predicted disorder-based protein–protein interaction sites (residues 518–547, 55–563, 579–610, 622–751, 759–772, 810–835, and 870–883) and several phosphorylation sites. Furthermore, another disorder-based interaction site (residues 272–278) is located in the middle of the extracellular domain of βCR, in close proximity to the region of missing electron density (residues 280–294 in PDB ID: 1GH7).
 | ||
| Fig. 6 (A) Intrinsic disorder propensity of human βCR (UniProt ID: P32927) evaluated by PONDR® VLXT (black line), PONDR® VSL2 (yellow line), PONDR® VL3 (green line), and PONDR® FIT (red line). (B) Analysis of the interactivity of human βCR (UniProt ID: P32927) by STRING. (C) D2P2 output for human βCR (UniProt ID: P32927). | ||
Fig. 7A shows that the extracellular domain of human βCR has a complex multidomain structure with four mostly β-structural fibronectin domains connected by long linkers.167 This protein exists as a highly intertwined, head-to-tail dimer with the potential ligand-binding sites being located between domain 1 of one monomer and domain 4 of another monomer. Domain 4 (which in vivo is tethered to the membrane via the transmembrane domain) is characterized by the highest mobility with respect to the rest of the structure.167 To understand which part of the human βCR might be involved in interaction with Epo, structural alignment of the βCR and EpoR structures has been conducted using the MultiProt algorithm,65 which is a fully automated technique to detect multiple alignments within the compared protein structures (http://bioinfo3d.cs.tau.ac.il/MultiProt/). The results of this structural alignment of the full-length extracellular domains of human EpoR (PDB ID: 1EER, 213 residues) and human βCR (PDB ID: 2GYS, 397 residues) are shown in Fig. 7B and illustrate that remarkably good alignment (RMSD of 1.56 Å) can be achieved for one domain consisting of 93 residues (see Fig. 7). This structurally alignable region corresponds to domain 2 of βCR and domain D2 of EpoR, and is therefore located away from the βCR ligand binding site and includes only a part of the functional loops that EpoR uses for interaction with Epo. This reduction in the number of potential Epo binding loops might explain the reduced affinity of βCR to Epo.
 | ||
| Fig. 7 (A) Structural characterization of the extracellular domain of human βCR (PDB ID: 2GYS). The protein exists as a highly intertwined dimer and the corresponding monomers are shown as green and blue ribbons. (B) Results of multiple structural alignment of the full-length extracellular domains of human EpoR (PDB ID: 1EER, 213 residues; red ribbon) and human βCR (PDB ID: 2GYS, 397 residues; blue ribbon). This structural alignment was conducted using the MultiProt web-based server.65 The shown protein structures were created using the Visual Molecular Dynamics (VMD) software.66 | ||
Disease-related mutations in hEpo and its major interactors
There are no known mutations in human Epo that would be directly associated with diseases. The only known pathological correlation is related to the increased levels of Epo in patients with proliferative diabetic retinopathy (PDR) and end-stage renal disease (ESRD), which are two of the most common and severe microvascular complications of diabetes. It is believed that these increased levels of Epo are caused by the polymorphism in the promoter region of the EPO gene.168 Similarly, no pathological mutations have been reported for human HIF-1α.On the contrary, two mutations in EpoR (Asn487Ser and Pro488Ser) are known to be associated with familial erythrocytosis 1 (ECYT1), which is an autosomal dominant, relatively benign disease that does not progress to leukemia (OMIM entry # 133100). Some major characteristics of this malady include low serum levels of Epo, increased mass of serum red blood cell combined with no increase in platelets nor leukocytes, elevated levels of hemoglobin and hematocrit, and hypersensitivity of erythroid progenitors to erythropoietin.169,170 Both of the ECYT1-related mutations found in EpoR are located within the disordered C-terminal tail of this protein in close proximity to two predicted disorder-based binding sites (residues 452–463 and 503–508) and two phosphorylation sites (Tyr485 and Tyr489), suggesting that at least some of the pathological consequences of these mutations can be related to their potential effects on the disorder-based functions of EpoR.
Alterations in the CSF2RB gene encoding human βCR are associated with pulmonary surfactant metabolism dysfunction-5 (SMDP5; OMIM: 614370), which is an autosomal recessive lung disease manifested as pulmonary alveolar proteinosis (PAP) characterized by the ineffective clearance of surfactant by alveolar macrophages.171,172 The PAP-causing Ser271Leu mutation is located in the middle of the extracellular domain, within one of the regions with heterogeneous predictions of disorder propensity. Being positioned in close proximity to the predicted disorder-based binding site 272–278, this mutation might affect the interactivity of βCR. In addition to the single point mutation Ser271Leu,171 PAP can be caused by a truncating mutation of the CSF2RB gene leading to the degradation of the mRNA and complete lack of the βCR in the affected patient.172
Concluding remarks
This work represents an “unstructural biology” view of Epo and three major players related to the multifaceted activity of this protein in organisms, HIF-1α, EpoR, and βCR. We show that these four Epo and co. proteins are predicted to have long intrinsically disordered regions that are directly related to the functionality and regulation of these important proteins. These disordered regions contain sites of protein–protein interactions and also include various PTMs likely needed for their regulation.Notes and references
- A. J. Erslev, ASAIO J., 1993, 39, 89–92 Search PubMed.
- R. M. Redwan el, Hum. Antibodies, 2007, 16, 137–158 Search PubMed.
- G. Walsh, Nat. Biotechnol., 2014, 32, 992–1000 Search PubMed.
- J. L. Spivak, P. Gascon and H. Ludwig, Oncologist, 2009, 14(suppl 1), 43–56 Search PubMed.
- M. Takeuchi, N. Inoue, T. W. Strickland, M. Kubota, M. Wada, R. Shimizu, S. Hoshi, H. Kozutsumi, S. Takasaki and A. Kobata, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 7819–7822 Search PubMed.
- J. C. Egrie and J. K. Browne, Nephrol., Dial., Transplant., 2001, 16(suppl 3), 3–13 Search PubMed.
- J. C. Egrie and J. K. Browne, Br. J. Cancer, 2001, 84(suppl 1), 3–10 Search PubMed.
- C. T. Yuen, P. L. Storring, R. J. Tiplady, M. Izquierdo, R. Wait, C. K. Gee, P. Gerson, P. Lloyd and J. A. Cremata, Br. J. Haematol., 2003, 121, 511–526 Search PubMed.
- P. L. Storring and C. T. Yuen, Blood, 2003, 101, 1204 Search PubMed ; author reply 1204–1205.
- S. Erbayraktar, G. Grasso, A. Sfacteria, Q. W. Xie, T. Coleman, M. Kreilgaard, L. Torup, T. Sager, Z. Erbayraktar, N. Gokmen, O. Yilmaz, P. Ghezzi, P. Villa, M. Fratelli, S. Casagrande, M. Leist, L. Helboe, J. Gerwein, S. Christensen, M. A. Geist, L. O. Pedersen, C. Cerami-Hand, J. P. Wuerth, A. Cerami and M. Brines, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 6741–6746 Search PubMed.
- K. Maiese, World J. Diabetes, 2015, 6, 1259–1273 Search PubMed.
- K. Maiese, Biomed. Pharmacother., 2008, 62, 218–232 Search PubMed.
- K. Maiese, Front. Biosci., Landmark Ed., 2016, 21, 561–596 Search PubMed.
- K. Maiese, Curr. Neurovasc. Res., 2016, 13, 329–340 Search PubMed.
- P. Theriault, A. Le Behot, A. ElAli and S. Rivest, Oncotarget, 2016, 7, 35552–35561 Search PubMed.
- K. Maiese, J. Transl. Sci., 2016, 2, 140–144 Search PubMed.
- X. Li, Y. Chen, S. Shao, Q. Tang, W. Chen, Y. Chen and X. Xu, Exp. Gerontol., 2016, 83, 89–93 Search PubMed.
- Z. Jia, R. Xue, S. Ma, J. Xu, S. Guo, S. Li, E. Zhang, J. Wang and J. Yang, Mol. Neurobiol., 2016, 53, 5664–5670 Search PubMed.
- J. A. Pascual, V. Belalcazar, C. de Bolos, R. Gutierrez, E. Llop and J. Segura, Ther. Drug Monit., 2004, 26, 175–179 Search PubMed.
- E. Brzezianska, D. Domanska and A. Jegier, Biol. Sport, 2014, 31, 251–259 Search PubMed.
- A. K. Dunker, Z. Obradovic, P. Romero, E. C. Garner and C. J. Brown, Genome Inform. Ser. Workshop Genome Inform., 2000, 11, 161–171 Search PubMed.
- C. J. Oldfield, Y. Cheng, M. S. Cortese, C. J. Brown, V. N. Uversky and A. K. Dunker, Biochemistry, 2005, 44, 1989–2000 Search PubMed.
- J. J. Ward, J. S. Sodhi, L. J. McGuffin, B. F. Buxton and D. T. Jones, J. Mol. Biol., 2004, 337, 635–645 Search PubMed.
- V. N. Uversky, J. Biomed. Biotechnol., 2010, 2010, 568068 Search PubMed.
- B. Xue, A. K. Dunker and V. N. Uversky, J. Biomol. Struct. Dyn., 2012, 30, 137–149 Search PubMed.
- P. E. Wright and H. J. Dyson, J. Mol. Biol., 1999, 293, 321–331 Search PubMed.
- V. N. Uversky, J. R. Gillespie and A. L. Fink, Proteins, 2000, 41, 415–427 Search PubMed.
- A. K. Dunker, J. D. Lawson, C. J. Brown, R. M. Williams, P. Romero, J. S. Oh, C. J. Oldfield, A. M. Campen, C. M. Ratliff, K. W. Hipps, J. Ausio, M. S. Nissen, R. Reeves, C. Kang, C. R. Kissinger, R. W. Bailey, M. D. Griswold, W. Chiu, E. C. Garner and Z. Obradovic, J. Mol. Graphics Modell., 2001, 19, 26–59 Search PubMed.
- P. Tompa, Trends Biochem. Sci., 2002, 27, 527–533 Search PubMed.
- V. N. Uversky, Eur. J. Biochem., 2002, 269, 2–12 Search PubMed.
- V. N. Uversky, Protein Sci., 2002, 11, 739–756 Search PubMed.
- H. J. Dyson and P. E. Wright, Nat. Rev. Mol. Cell Biol., 2005, 6, 197–208 Search PubMed.
- V. N. Uversky and A. K. Dunker, Biochim. Biophys. Acta, 2010, 1804, 1231–1264 CrossRef CAS PubMed.
- A. K. Dunker, C. J. Oldfield, J. Meng, P. Romero, J. Y. Yang, J. W. Chen, V. Vacic, Z. Obradovic and V. N. Uversky, BMC Genomics, 2008, 9(suppl 2), S1 Search PubMed.
- V. N. Uversky, C. J. Oldfield and A. K. Dunker, Annu. Rev. Biophys., 2008, 37, 215–246 Search PubMed.
- A. K. Dunker, M. Babu, E. Barbar, M. Blackledge, S. E. Bondos, Z. Dosztányi, H. J. Dyson, J. Forman-Kay, M. Fuxreiter, J. Gsponer, K.-H. Han, D. T. Jones, S. Longhi, S. J. Metallo, K. Nishikawa, R. Nussinov, Z. Obradovic, R. Pappu, B. Rost, P. Selenko, V. Subramaniam, J. L. Sussman, P. Tompa and V. N. Uversky, Intrinsically Disord. Proteins, 2013, 1, e24157 Search PubMed.
- N. Tokuriki, C. J. Oldfield, V. N. Uversky, I. N. Berezovsky and D. S. Tawfik, Trends Biochem. Sci., 2009, 34, 53–59 Search PubMed.
- B. Xue, R. W. Williams, C. J. Oldfield, A. K. Dunker and V. N. Uversky, BMC Syst. Biol., 2010, 4(suppl 1), S1 Search PubMed.
- B. Xue, A. K. Dunker and V. N. Uversky, J. Biomol. Struct. Dyn., 2012, 30, 137–149 Search PubMed.
- V. N. Uversky, Cell. Mol. Life Sci., 2003, 60, 1852–1871 Search PubMed.
- K. K. Turoverov, I. M. Kuznetsova and V. N. Uversky, Prog. Biophys. Mol. Biol., 2010, 102, 73–84 Search PubMed.
- H. J. Dyson, Q. Rev. Biophys., 2011, 44, 467–518 Search PubMed.
- P. Tompa, Trends Biochem. Sci., 2012, 37, 509–516 Search PubMed.
- V. N. Uversky, Protein Sci., 2013, 22, 693–724 Search PubMed.
- H. Xie, S. Vucetic, L. M. Iakoucheva, C. J. Oldfield, A. K. Dunker, V. N. Uversky and Z. Obradovic, J. Proteome Res., 2007, 6, 1882–1898 Search PubMed.
- S. Vucetic, H. Xie, L. M. Iakoucheva, C. J. Oldfield, A. K. Dunker, Z. Obradovic and V. N. Uversky, J. Proteome Res., 2007, 6, 1899–1916 Search PubMed.
- H. Xie, S. Vucetic, L. M. Iakoucheva, C. J. Oldfield, A. K. Dunker, Z. Obradovic and V. N. Uversky, J. Proteome Res., 2007, 6, 1917–1932 Search PubMed.
- G. W. Daughdrill, G. J. Pielak, V. N. Uversky, M. S. Cortese and A. K. Dunker, in Handbook of Protein Folding, ed. J. Buchner and T. Kiefhaber, Wiley-VCH, Verlag GmbH & Co., Weinheim, Germany, 2005, pp. 271–353 Search PubMed.
- A. K. Dunker, C. J. Brown and Z. Obradovic, Adv. Protein Chem., 2002, 62, 25–49 Search PubMed.
- A. K. Dunker, C. J. Brown, J. D. Lawson, L. M. Iakoucheva and Z. Obradovic, Biochemistry, 2002, 41, 6573–6582 Search PubMed.
- A. K. Dunker, M. S. Cortese, P. Romero, L. M. Iakoucheva and V. N. Uversky, FEBS J., 2005, 272, 5129–5148 Search PubMed.
- A. K. Dunker, E. Garner, S. Guilliot, P. Romero, K. Albrecht, J. Hart, Z. Obradovic, C. Kissinger and J. E. Villafranca, Pac. Symp. Biocomput., 1998, 473–484 Search PubMed.
- P. Tompa, FEBS Lett., 2005, 579, 3346–3354 Search PubMed.
- P. Tompa and P. Csermely, FASEB J., 2004, 18, 1169–1175 Search PubMed.
- P. Tompa, C. Szasz and L. Buday, Trends Biochem. Sci., 2005, 30, 484–489 Search PubMed.
- V. N. Uversky, C. J. Oldfield and A. K. Dunker, J. Mol. Recognit., 2005, 18, 343–384 Search PubMed.
- L. M. Iakoucheva, P. Radivojac, C. J. Brown, T. R. O'Connor, J. G. Sikes, Z. Obradovic and A. K. Dunker, Nucleic Acids Res., 2004, 32, 1037–1049 Search PubMed.
- V. Pejaver, W. L. Hsu, F. Xin, A. K. Dunker, V. N. Uversky and P. Radivojac, Protein Sci., 2014, 23, 1077–1093 Search PubMed.
- J. Gsponer, M. E. Futschik, S. A. Teichmann and M. M. Babu, Science, 2008, 322, 1365–1368 Search PubMed.
- V. N. Uversky and A. K. Dunker, Science, 2008, 322, 1340–1341 Search PubMed.
- V. N. Uversky, V. Dave, L. M. Iakoucheva, P. Malaney, S. J. Metallo, R. R. Pathak and A. C. Joerger, Chem. Rev., 2014, 114, 6844–6879 Search PubMed.
- V. N. Uversky, Front. Mol. Biosci., 2014, 1, 6 Search PubMed.
- P. H. Lai, R. Everett, F. F. Wang, T. Arakawa and E. Goldwasser, J. Biol. Chem., 1986, 261, 3116–3121 Search PubMed.
- R. S. Syed, S. W. Reid, C. Li, J. C. Cheetham, K. H. Aoki, B. Liu, H. Zhan, T. D. Osslund, A. J. Chirino, J. Zhang, J. Finer-Moore, S. Elliott, K. Sitney, B. A. Katz, D. J. Matthews, J. J. Wendoloski, J. Egrie and R. M. Stroud, Nature, 1998, 395, 511–516 Search PubMed.
- M. Shatsky, R. Nussinov and H. J. Wolfson, Proteins, 2004, 56, 143–156 Search PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14(33–38), 27–38 Search PubMed.
- K. Peng, S. Vucetic, P. Radivojac, C. J. Brown, A. K. Dunker and Z. Obradovic, J. Bioinf. Comput. Biol., 2005, 3, 35–60 Search PubMed.
- K. Peng, P. Radivojac, S. Vucetic, A. K. Dunker and Z. Obradovic, BMC Bioinf., 2006, 7, 208 Search PubMed.
- B. Xue, R. L. Dunbrack, R. W. Williams, A. K. Dunker and V. N. Uversky, Biochim. Biophys. Acta, 2010, 1804, 996–1010 Search PubMed.
- J. S. Philo, K. H. Aoki, T. Arakawa, L. O. Narhi and J. Wen, Biochemistry, 1996, 35, 1681–1691 Search PubMed.
- J. C. Cheetham, D. M. Smith, K. H. Aoki, J. L. Stevenson, T. J. Hoeffel, R. S. Syed, J. Egrie and T. S. Harvey, Nat. Struct. Biol., 1998, 5, 861–866 Search PubMed.
- Z. L. Peng and L. Kurgan, Curr. Protein Pept. Sci., 2012, 13, 6–18 Search PubMed.
- X. Fan and L. Kurgan, J. Biomol. Struct. Dyn., 2014, 32, 448–464 Search PubMed.
- P. Romero, Z. Obradovic, X. Li, E. C. Garner, C. J. Brown and A. K. Dunker, Proteins, 2001, 42, 38–48 Search PubMed.
- J. Prilusky, C. E. Felder, T. Zeev-Ben-Mordehai, E. H. Rydberg, O. Man, J. S. Beckmann, I. Silman and J. L. Sussman, Bioinformatics, 2005, 21, 3435–3438 Search PubMed.
- Z. Dosztanyi, V. Csizmok, P. Tompa and I. Simon, Bioinformatics, 2005, 21, 3433–3434 Search PubMed.
- A. Campen, R. M. Williams, C. J. Brown, J. Meng, V. N. Uversky and A. K. Dunker, Protein Pept. Lett., 2008, 15, 956–963 Search PubMed.
- S. C. Chang, D. Sikkema and E. Goldwasser, Biochem. Biophys. Res. Commun., 1974, 57, 399–405 Search PubMed.
- C. F. Chou, S. Tohari, S. Brenner and B. Venkatesh, Blood, 2004, 104, 1498–1503 Search PubMed.
- C. B. Bonnas, PhD thesis, Charité – Universitätsmedizin, 2009.
- A. Meisel, J. Priller, C. Bonnas and U. Dirnagl, Google Pat., US20110008363, 2011 Search PubMed.
- P. R. Romero, S. Zaidi, Y. Y. Fang, V. N. Uversky, P. Radivojac, C. J. Oldfield, M. S. Cortese, M. Sickmeier, T. LeGall, Z. Obradovic and A. K. Dunker, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8390–8395 Search PubMed.
- V. N. Uversky, Curr. Pharm. Des., 2013, 19, 4191–4213 Search PubMed.
- M. Buljan, G. Chalancon, S. Eustermann, G. P. Wagner, M. Fuxreiter, A. Bateman and M. M. Babu, Mol. Cell, 2012, 46, 871–883 Search PubMed.
- M. Buljan, G. Chalancon, A. K. Dunker, A. Bateman, S. Balaji, M. Fuxreiter and M. M. Babu, Curr. Opin. Struct. Biol., 2013, 23, 443–450 Search PubMed.
- L. E. Huang and H. F. Bunn, J. Biol. Chem., 2003, 278, 19575–19578 Search PubMed.
- N. T. Noguchi, Blood, 2004, 104, 1238 Search PubMed.
- T. Tanaka and M. Nangaku, Exp. Cell Res., 2012, 318, 1068–1073 Search PubMed.
- J. Liu, N. B. Perumal, C. J. Oldfield, E. W. Su, V. N. Uversky and A. K. Dunker, Biochemistry, 2006, 45, 6873–6888 Search PubMed.
- Y. Minezaki, K. Homma, A. R. Kinjo and K. Nishikawa, J. Mol. Biol., 2006, 359, 1137–1149 Search PubMed.
- H. V. Erkizan, V. N. Uversky and J. A. Toretsky, Clin. Cancer Res., 2010, 16, 4077–4083 Search PubMed.
- S. D. Westerheide, R. Raynes, C. Powell, B. Xue and V. N. Uversky, Curr. Protein Pept. Sci., 2012, 13, 86–103 Search PubMed.
- M. E. Oates, P. Romero, T. Ishida, M. Ghalwash, M. J. Mizianty, B. Xue, Z. Dosztanyi, V. N. Uversky, Z. Obradovic, L. Kurgan, A. K. Dunker and J. Gough, Nucleic Acids Res., 2013, 41, D508–D516 Search PubMed.
- Z. Obradovic, K. Peng, S. Vucetic, P. Radivojac and A. K. Dunker, Proteins, 2005, 61(suppl 7), 176–182 Search PubMed.
- T. Ishida and K. Kinoshita, Nucleic Acids Res., 2007, 35, W460–W464 Search PubMed.
- I. Walsh, A. J. Martin, T. Di Domenico and S. C. Tosatto, Bioinformatics, 2012, 28, 503–509 Search PubMed.
- A. Kalousi, I. Mylonis, A. S. Politou, G. Chachami, E. Paraskeva and G. Simos, J. Cell Sci., 2010, 123, 2976–2986 Search PubMed.
- D. Xu, Y. Yao, L. Lu, M. Costa and W. Dai, J. Biol. Chem., 2010, 285, 38944–38950 Search PubMed.
- D. Flugel, A. Gorlach, C. Michiels and T. Kietzmann, Mol. Cell. Biol., 2007, 27, 3253–3265 Search PubMed.
- N. Masson, C. Willam, P. H. Maxwell, C. W. Pugh and P. J. Ratcliffe, EMBO J., 2001, 20, 5197–5206 Search PubMed.
- K. S. Seo, J. H. Park, J. Y. Heo, K. Jing, J. Han, K. N. Min, C. Kim, G. Y. Koh, K. Lim, G. Y. Kang, J. Uee Lee, Y. H. Yim, M. Shong, T. H. Kwak and G. R. Kweon, Oncogene, 2015, 34, 1354–1362 Search PubMed.
- I. M. Yasinska and V. V. Sumbayev, FEBS Lett., 2003, 549, 105–109 Search PubMed.
- D. Lando, D. J. Peet, J. J. Gorman, D. A. Whelan, M. L. Whitelaw and R. K. Bruick, Genes Dev., 2002, 16, 1466–1471 Search PubMed.
- D. Szklarczyk, A. Franceschini, M. Kuhn, M. Simonovic, A. Roth, P. Minguez, T. Doerks, M. Stark, J. Muller, P. Bork, L. J. Jensen and C. von Mering, Nucleic Acids Res., 2011, 39, D561–D568 Search PubMed.
- X. Yu, J. J. Shacka, J. B. Eells, C. Suarez-Quian, R. M. Przygodzki, B. Beleslin-Cokic, C. S. Lin, V. M. Nikodem, B. Hempstead, K. C. Flanders, F. Costantini and C. T. Noguchi, Development, 2002, 129, 505–516 Search PubMed.
- M. Sakanaka, T. C. Wen, S. Matsuda, S. Masuda, E. Morishita, M. Nagao and R. Sasaki, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4635–4640 Search PubMed.
- C. Moon, M. Krawczyk, D. Ahn, I. Ahmet, D. Paik, E. G. Lakatta and M. I. Talan, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 11612–11617 Search PubMed.
- S. J. Kim, Y. W. Chin and T. H. Heo, Altern. Ther. Health Med., 2013, 19, 54–60 Search PubMed.
- M. Brines and A. Cerami, Nat. Rev. Neurosci., 2005, 6, 484–494 Search PubMed.
- H. Youssoufian, G. Longmore, D. Neumann, A. Yoshimura and H. F. Lodish, Blood, 1993, 81, 2223–2236 Search PubMed.
- M. O. Arcasoy, Br. J. Haematol., 2008, 141, 14–31 Search PubMed.
- M. Nairz, T. Sonnweber, A. Schroll, I. Theurl and G. Weiss, Microbes Infect., 2012, 14, 238–246 Search PubMed.
- M. Alnaeeli, L. Wang, B. Piknova, H. Rogers, X. Li and C. T. Noguchi, Anatomy Research International, 2012, 2012, 953264 Search PubMed.
- C. J. French, N. J. Glassford, D. Gantner, A. M. Higgins, D. J. Cooper, A. Nichol, M. B. Skrifvars, G. Imberger, J. Presneill, M. Bailey and R. Bellomo, Ann. Surg., 2016 DOI:10.1097/SLA.0000000000001746.
- A. I. Khan, S. M. Coldewey, N. S. Patel, M. Rogazzo, M. Collino, M. M. Yaqoob, P. Radermacher, A. Kapoor and C. Thiemermann, Dis. Models & Mech., 2013, 6, 1021–1030 Search PubMed.
- H. E. Broxmeyer, Immunity, 2011, 34, 6–7 Search PubMed.
- R. Rong and X. Xijun, Exp. Ther. Med., 2015, 10, 413–418 Search PubMed.
- S. K. Barton, A. R. McDougall, J. M. Melville, T. J. Moss, V. A. Zahra, T. Lim, K. J. Crossley, G. R. Polglase and M. Tolcos, J. Physiol., 2016, 594, 1437–1449 Search PubMed.
- W. Jelkmann, Eur. J. Haematol., 2007, 78, 183–205 Search PubMed.
- R. G. Pearl, Crit. Care, 2014, 18, 526 Search PubMed.
- A. Lund, C. Lundby and N. V. Olsen, Eur. J. Clin. Invest., 2014, 44, 1230–1238 Search PubMed.
- M. Um, A. W. Gross and H. F. Lodish, Cell. Signalling, 2007, 19, 634–645 Search PubMed.
- F. Fiordaliso, S. Chimenti, L. Staszewsky, A. Bai, E. Carlo, I. Cuccovillo, M. Doni, M. Mengozzi, R. Tonelli, P. Ghezzi, T. Coleman, M. Brines, A. Cerami and R. Latini, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 2046–2051 Search PubMed.
- W. G. van Rijt, H. van Goor, R. J. Ploeg and H. G. Leuvenink, Transplant Int., 2014, 27, 241–248 Search PubMed.
- The Lancet Oncology, Leading Edge – Erythropoietin analogues: an unnecessary class of drugs, Lancet Oncol., 2008, 9, 81 Search PubMed.
- M. Henke, R. Laszig, C. Rube, U. Schafer, K. D. Haase, B. Schilcher, S. Mose, K. T. Beer, U. Burger, C. Dougherty and H. Frommhold, Lancet, 2003, 362, 1255–1260 Search PubMed.
- B. Leyland-Jones, B. Investigators and G. Study, Lancet Oncol., 2003, 4, 459–460 Search PubMed.
- J. R. Wright, Y. C. Ung, J. A. Julian, K. I. Pritchard, T. J. Whelan, C. Smith, B. Szechtman, W. Roa, L. Mulroy, L. Rudinskas, B. Gagnon, G. S. Okawara and M. N. Levine, J. Clin. Oncol., 2007, 25, 1027–1032 Search PubMed.
- G. Acs, P. Acs, S. M. Beckwith, R. L. Pitts, E. Clements, K. Wong and A. Verma, Cancer Res., 2001, 61, 3561–3565 Search PubMed.
- G. Acs, P. J. Zhang, T. R. Rebbeck, P. Acs and A. Verma, Cancer, 2002, 95, 969–981 Search PubMed.
- M. O. Arcasoy, K. Amin, A. F. Karayal, S. C. Chou, J. A. Raleigh, M. A. Varia and Z. A. Haroon, Lab. Invest., 2002, 82, 911–918 CrossRef CAS PubMed.
- C. Westenfelder and R. L. Baranowski, Kidney Int., 2000, 58, 647–657 Search PubMed.
- K. Kayser and H. J. Gabius, Zentralbl. Pathol., 1992, 138, 266–270 Search PubMed.
- G. Acs, X. Xu, C. Chu, P. Acs and A. Verma, Cancer, 2004, 100, 2376–2386 Search PubMed.
- G. Acs, P. J. Zhang, C. M. McGrath, P. Acs, J. McBroom, A. Mohyeldin, S. Liu, H. Lu and A. Verma, Am. J. Pathol., 2003, 162, 1789–1806 Search PubMed.
- Y. Yasuda, Y. Fujita, T. Musha, H. Tanaka, S. Shiokawa, K. Nakamatsu, S. Mori, T. Matsuo and Y. Nakamura, Ital. J. Anat. Embryol., 2001, 106, 215–222 Search PubMed.
- Y. Yasuda, T. Musha, H. Tanaka, Y. Fujita, H. Fujita, H. Utsumi, T. Matsuo, S. Masuda, M. Nagao, R. Sasaki and Y. Nakamura, Br. J. Cancer, 2001, 84, 836–843 Search PubMed.
- C. M. Yates, A. Patel, K. Oakley, A. Helms, R. M. Tuttle and G. L. Francis, J. Endocrinol. Invest., 2006, 29, 320–329 Search PubMed.
- B. M. Fine, M. Stanulla, M. Schrappe, M. Ho, S. Viehmann, J. Harbott and L. M. Boxer, Blood, 2004, 103, 1043–1049 Search PubMed.
- S. Batra, N. Perelman, L. R. Luck, H. Shimada and P. Malik, Lab. Invest., 2003, 83, 1477–1487 Search PubMed.
- D. P. Steensma, Lancet Oncol., 2008, 9, 316 Search PubMed.
- S. Louet, Nat. Biotechnol., 2003, 21, 956–957 Search PubMed.
- H. Schellekens, Nat. Biotechnol., 2004, 22, 1357–1359 Search PubMed.
- P. L. Storring, R. J. Tiplady, R. E. Gaines Das, B. E. Stenning, A. Lamikanra, B. Rafferty and J. Lee, Br. J. Haematol., 1998, 100, 79–89 Search PubMed.
- J. C. Egrie, E. Dwyer, J. K. Browne, A. Hitz and M. A. Lykos, Exp. Hematol., 2003, 31, 290–299 Search PubMed.
- J. K. Browne, A. M. Cohen, J. C. Egrie, P. H. Lai, F. K. Lin, T. Strickland, E. Watson and N. Stebbing, Cold Spring Harbor Symp. Quant. Biol., 1986, 51(Pt 1), 693–702 Search PubMed.
- M. A. Recny, H. A. Scoble and Y. Kim, J. Biol. Chem., 1987, 262, 17156–17163 Search PubMed.
- J. C. Egrie, T. W. Strickland, J. Lane, K. Aoki, A. M. Cohen, R. Smalling, G. Trail, F. K. Lin, J. K. Browne and D. K. Hines, Immunobiology, 1986, 172, 213–224 Search PubMed.
- D. L. Johnson and L. K. Jolliffe, Nephrol., Dial., Transplant., 2000, 15, 1274–1277 Search PubMed.
- A. Mikhail, A. Covic and D. Goldsmith, Kidney Blood Pressure Res., 2008, 31, 234–246 Search PubMed.
- S. A. Qureshi, R. M. Kim, Z. Konteatis, D. E. Biazzo, H. Motamedi, R. Rodrigues, J. A. Boice, J. R. Calaycay, M. A. Bednarek, P. Griffin, Y. D. Gao, K. Chapman and D. F. Mark, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 12156–12161 Search PubMed.
- M. Murakami, T. Kiuchi, M. Nishihara, K. Tezuka, R. Okamoto, M. Izumi and Y. Kajihara, Sci. Adv., 2016, 2, e1500678 Search PubMed.
- P. Wang, S. Dong, J. H. Shieh, E. Peguero, R. Hendrickson, M. A. Moore and S. J. Danishefsky, Science, 2013, 342, 1357–1360 Search PubMed.
- R. M. Wilson, S. Dong, P. Wang and S. J. Danishefsky, Angew. Chem., Int. Ed., 2013, 52, 7646–7665 Search PubMed.
- P. A. Tilbrook and S. P. Klinken, Int. J. Biochem. Cell Biol., 1999, 31, 1001–1005 Search PubMed.
- P. A. Tilbrook and S. P. Klinken, Growth Factors, 1999, 17, 25–35 Search PubMed.
- S. N. Constantinescu, T. Keren, M. Socolovsky, H. Nam, Y. I. Henis and H. F. Lodish, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 4379–4384 Search PubMed.
- F. Merchionne and F. Dammacco, Br. J. Haematol., 2009, 146, 127–141 Search PubMed.
- Q. Li, Y. L. Wong, Q. Huang and C. Kang, Biophys. J., 2014, 107, 2325–2336 Search PubMed.
- E. V. Khankin, W. P. Mutter, H. Tamez, H. T. Yuan, S. A. Karumanchi and R. Thadhani, PLoS One, 2010, 5, e9246 Search PubMed.
- G. Westphal, K. Braun and J. Debus, Clin. Exp. Med., 2002, 2, 45–52 Search PubMed.
- F. Marcuzzi, S. Zucchelli, M. Bertuzzi, C. Santoro, G. Tell, P. Carninci and S. Gustincich, J. Neurochem., 2016 DOI:10.1111/jnc.13757.
- T. Ioka, S. Tsuruoka, C. Ito, H. Iwaguro, T. Asahara, A. Fujimura and E. Kusano, Clin. Pharmacol. Ther., 2009, 86, 154–159 Search PubMed.
- R. Shimizu, N. Komatsu and Y. Miura, Exp. Hematol., 1999, 27, 229–233 Search PubMed.
- M. Brines, G. Grasso, F. Fiordaliso, A. Sfacteria, P. Ghezzi, M. Fratelli, R. Latini, Q. W. Xie, J. Smart, C. J. Su-Rick, E. Pobre, D. Diaz, D. Gomez, C. Hand, T. Coleman and A. Cerami, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 14907–14912 Search PubMed.
- B. B. Beleslin-Cokic, V. P. Cokic, X. Yu, B. B. Weksler, A. N. Schechter and C. T. Noguchi, Blood, 2004, 104, 2073–2080 Search PubMed.
- P. D. Carr, F. Conlan, S. Ford, D. L. Ollis and I. G. Young, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2006, 62, 509–513 Search PubMed.
- Z. Tong, Z. Yang, S. Patel, H. Chen, D. Gibbs, X. Yang, V. S. Hau, Y. Kaminoh, J. Harmon, E. Pearson, J. Buehler, Y. Chen, B. Yu, N. H. Tinkham, N. A. Zabriskie, J. Zeng, L. Luo, J. K. Sun, M. Prakash, R. N. Hamam, S. Tonna, R. Constantine, C. C. Ronquillo, S. Sadda, R. L. Avery, J. M. Brand, N. London, A. L. Anduze, G. L. King, P. S. Bernstein, S. Watkins, L. B. Jorde, D. Y. Li, L. P. Aiello, M. R. Pollak and K. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6998–7003 Search PubMed.
- J. P. Le Couedic, M. T. Mitjavila, J. L. Villeval, F. Feger, S. Gobert, P. Mayeux, N. Casadevall and W. Vainchenker, Blood, 1996, 87, 1502–1511 Search PubMed.
- L. Sokol, J. F. Prchal, A. D'Andrea, T. A. Rado and J. T. Prchal, Exp. Hematol., 1994, 22, 447–453 Search PubMed.
- T. Suzuki, B. Maranda, T. Sakagami, P. Catellier, C. Y. Couture, B. C. Carey, C. Chalk and B. C. Trapnell, Eur. Respir. J., 2011, 37, 201–204 Search PubMed.
- T. Tanaka, N. Motoi, Y. Tsuchihashi, R. Tazawa, C. Kaneko, T. Nei, T. Yamamoto, T. Hayashi, T. Tagawa, T. Nagayasu, F. Kuribayashi, K. Ariyoshi, K. Nakata and K. Morimoto, J. Med. Genet., 2011, 48, 205–209 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Multiple sequence alignment of the erythropoietin from different organisms. See DOI: 10.1039/c6mb00657d |
| This journal is © The Royal Society of Chemistry 2017 |