
Uncoupling of an ammonia channel as a mechanism of allosteric inhibition in anthranilate synthase of Serratia marcescens: dynamic and graph theoretical analysis†
Ashutosh
Srivastava‡
and
Somdatta
Sinha
 *
*
Centre for Protein Science, Design, Engineering (CPSDE), Department of Biological Sciences, Indian Institute of Science Education Research Mohali, India. E-mail: ssinha@iisermohali.ac.in
First published on 1st November 2016
Abstract
Anthranilate synthase (AS) is the first branch node enzyme that catalyzes the conversion of chorismate to anthranilate in the high energy-consuming tryptophan biosynthetic pathway in Serratia marcescens. AS, with an allosterically-bound inhibitor (tryptophan), shows complete inhibition in its catalytic function, but the inhibitor-bound structure is very similar to that of the substrate-bound AS. Even though the reaction mechanisms of several chorismate-utilizing enzymes are known, the unusual structure–function relationship in catalysis and allosteric inhibition of AS by tryptophan, with an insignificant change in structure, remains elusive. In the absence of structural variation, we use an integrated computational approach of coarse-grained protein contact networks, Gaussian network model, and atomistic Molecular Dynamics simulations of the substrate-bound and inhibitor-bound AS structures, and show the role of small but critical allosteric changes that induce complete inhibition of AS activity. We predict, through dynamic correlation studies, perturbation in crucial inter-subunit interactions between the two substrate-binding sites (“ammonia channel”) and the allosteric inhibitor-binding site, and identify, through shortest path analysis, the non-active site residues participating in the communication pathways. We argue that such a regulatory mechanism (change in function without a significant change in the structure) for catalysis is useful for a branch point enzyme that has to undergo fast redistribution of fluxes according to different metabolic states of the organism. Being essential to the survival of microorganisms, including pathogenic ones, and absent in mammals, AS is a highly attractive drug target. Thus, the allosteric AS residues participating in catalysis identified in this study could be important for drugability.
Introduction
Anthranilate synthase (AS) catalyzes the first reaction in the tryptophan biosynthetic pathway present in bacteria, fungi and plants. AS being the branch point enzyme in the highly energy-consuming aromatic amino acid synthesis pathway, plays a key role in the overall regulation of the flux through this pathway and is essential for survival in several microorganisms. It undergoes allosteric inhibition by the end-product of the pathway, tryptophan, in many prokaryotes, thereby assuming a central role in regulating metabolism at the versatile metabolic node of chorismate.1 The absence of the tryptophan biosynthetic pathway in mammals makes anthranilate synthase a highly attractive potential drug target against pathogenic microbes.2–4 Serotonin, a product of tryptophan, is an important ingredient in medicines for humans, and the essential amino acid tryptophan is commercially used in animal feed. Thus, understanding the reaction mechanisms of chorismate-utilizing enzymes in general, and AS in particular, has been an active area of research,5–7 which has led to inhibition-resistant strains for overproduction8 and the structural characterization of the binding sites of chorismate (CA), and the allosteric inhibitor, tryptophan, of AS in some bacteria/archaea – Serratia marcescens,9Salmonella typhimurium,10Sulfolobus solfataricus11 and Mycobacterium tuberculosis.4AS is comprised of two subunits or components. The α subunit (TrpE in S. marcescens) catalyzes aromatization of CA, and the β subunit (TrpG in S. marcescens) transfers an amino group from glutamine to chorismate. Both the subunits are products of two different genes. The crystal structure of AS from S. marcescens, in complex with the substrate and the inhibitor, is a heterotetrameric complex with a TrpE:TrpE heterotetramer interface.9 (For details see the ESI†.)
In this work we have considered the AS in Serratia marcescens, which is a tetrameric TrpE2:TrpG2 complex.9 The TrpE subunit is a 520 amino acid long polypeptide with an anthranilate catalytic site and an allosteric site where tryptophan binds causing inhibition of the enzyme. The tryptophan binding site has been characterized and the residues involved in the feedback regulation have been identified.10,12 TrpG is a 193 amino acid long polypeptide, belonging to the glutamine amidotransferase family of enzymes that provides ammonia for anthranilate synthesis by breaking up glutamine into glutamate and ammonia. The ammonia formed at the active site of the TrpG subunit is transferred to the TrpE active site thus coupling the two reactions together. Both the activities – formation of anthranilate and formation of ammonia, are inhibited by a single molecule of tryptophan that binds at a site approximately 18 Å away from the TrpE active site. This suggests that the perturbation caused by the binding of an inhibitor is communicated through the protein to both the active sites leading to inhibition. More interestingly, AS from S. marcescens, in the substrate and inhibitor bound forms, showed little structural difference (1.02 Å on 471 aligned Cα atoms of TrpE and 0.713 Å on 181 aligned Cα atoms of TrpG).9Fig. 1(a) shows the superposed structures of the TrpE:TrpG heterodimer in the substrate bound and inhibitor bound forms. It may be noted that in the hetero-tetrameric form, the top part of helix α7, which is at the hetero-tetramer interface, shows a shift.9 Therefore, even though the superposition of the two structures shows very high similarity (low RMSD), there are small local variations as seen in Fig. 1(a).
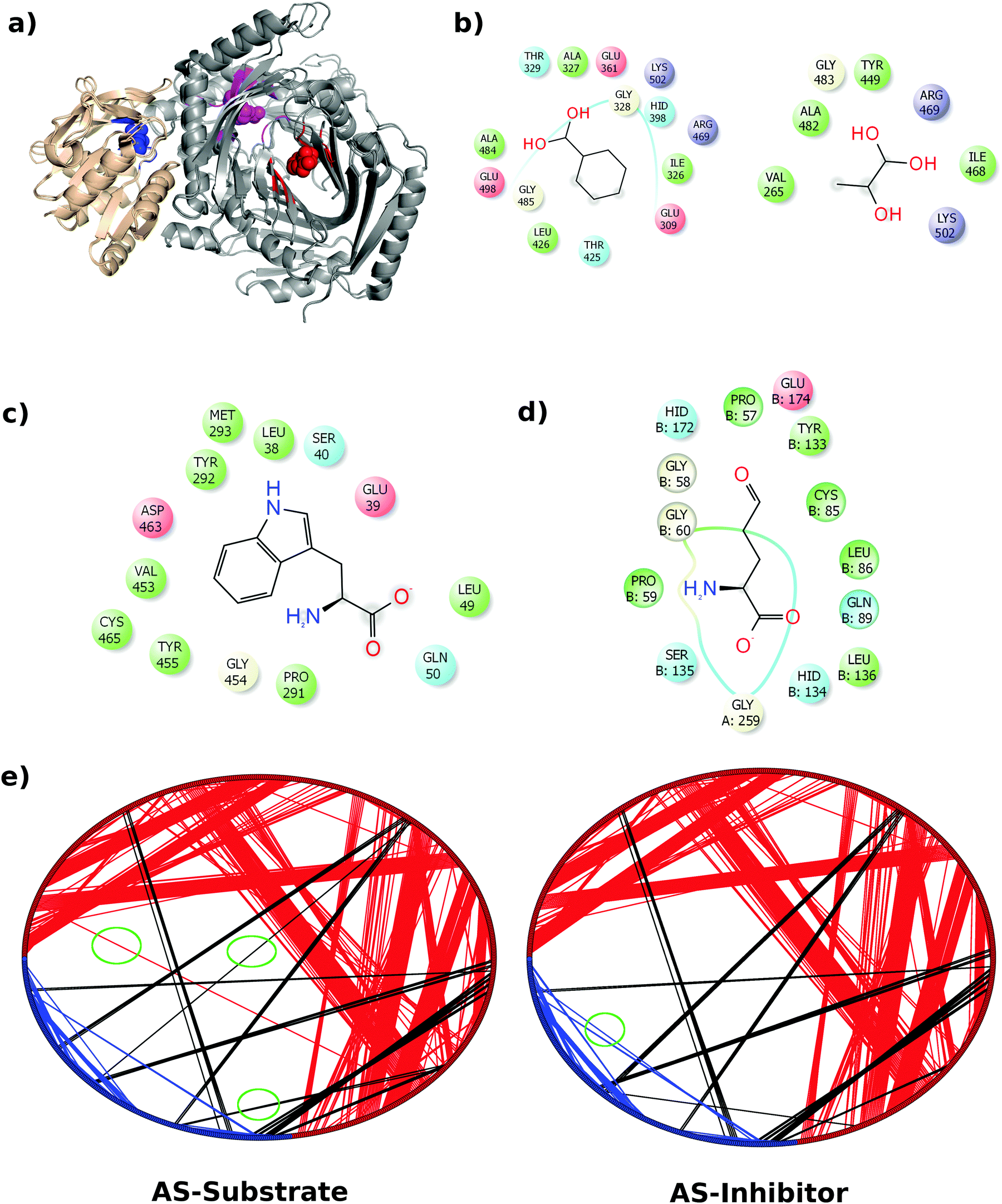 | ||
| Fig. 1 Network modelling of AS structures. (a) The superposed heterodimeric structure of the AS substrate and AS inhibitor shows little variation in the structure. TrpE and TrpG are shown in grey and wheat, respectively, the TrpG substrate (glutamine) binding site is shown in blue, the TrpE substrate (chorismate) binding site is shown in magenta and the inhibitor (tryptophan) binding site is shown in red. All the corresponding ligands are shown in the sphere representation in their respective colours. (b) Substrate (CA) binding residues in TrpE. (c) Inhibitor (trytophan) binding resides in TrpE. (d) Substrate (glutamine) binding residues in TrpG. (e) Ring graph representation of the AS-substrate and AS-inhibitor PCNs. Intra-chain edges (contacts) between the TrpE nodes are shown in red, whereas those between TrpG nodes are shown in blue. Inter-chain edges are shown in black. The differences between the two PCNs have been highlighted using green circles. | ||
In order to understand the mechanism of allosteric inhibition of the catalytic function of AS, we need to determine how the perturbation is transferred inside the AS molecule upon binding of the inhibitor, without inducing any significant overall structural change in the enzyme. Here we used the computational modeling methods of networks and molecular dynamics to analyze the small and local changes in the substrate-bound and inhibitor-bound structures of the AS and studied the important interactions along the communication pathways in the molecule. We further studied the dynamics of the TrpE:TrpG heterodimer in two states to understand the long range communication in AS that leads to the complete inhibition of enzyme activity. For the first time, we have identified the presence of an “Ammonia Channel” in S. marcescens to aid in the catalysis on substrate-binding, and have shown perturbation in it due to inhibitor (tryptophan) binding due to altered communication pathways through allosteric sites, which do not lead to any significant structural variation in this metabolically important branch point enzyme, but leads to complete inhibition in enzyme activity.
Materials and methods
The work has been done on the TrpE:TrpG dimer comprised of chains A and B (see the ESI† for details).Protein contact network (PCN) representation of AS structures
To understand the allosteric inhibition mechanism of the AS enzyme we have modelled the crystal structures of substrate bound (PDB ID: 1I7Q) and inhibitor bound AS (PDB ID: 1I7S) as a Protein Contact Network (PCN). PCN is a coarse-grained, graph representation of the three dimensional protein structure models as determined by various structure determination techniques (X-ray crystallography, NMR spectroscopy).13–15 Each Cα atom of the residues in the protein is considered as a node and the spatial proximity between them defines the link. Any two nodes are considered to be linked by an edge if the distance between them is less than 7 Å.In this way a protein structure consisting of N amino acid residues can be represented as an adjacency matrix (A) such that
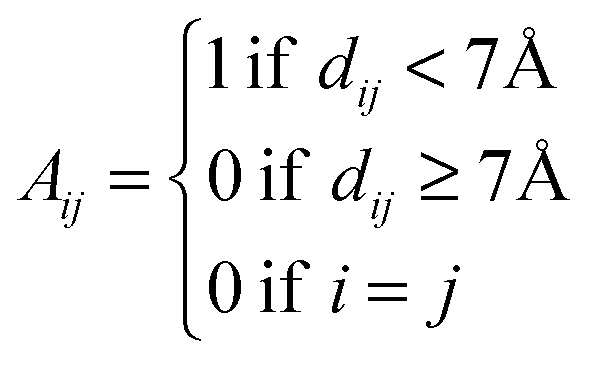
Network parameters
Network parameters can describe the different organizational properties of the whole network and of the individual nodes. The following parameters were used in this study.Degree, degree distribution and hubs
Degree of a node (k) is defined as the total number of nodes that it is directly connected to.Degree distribution represents the fraction of nodes P(k) having a particular degree k. It is an important parameter as it represents the topology and connection pattern of the network.
Hubs are defined as the nodes with high degree values. Here the nodes with a degree greater than or equal to 12 are considered as hubs. The cut-off for the definition of hub (i.e. 12) has been chosen by considering the degree distributions of the PCNs.
Betweenness centrality
Betweenness of a vertex can be roughly defined as the number of shortest paths going through the vertex.Thus, betweenness centrality (BC) of a node x is
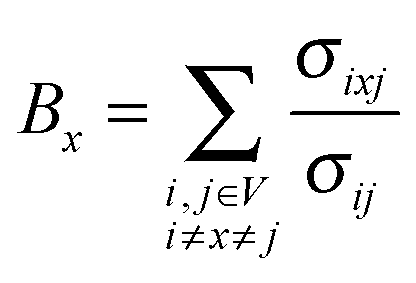
The residues with significantly high BC were determined by defining a z score
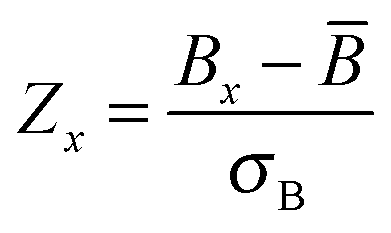
![[B with combining overline]](https://www.rsc.org/images/entities/i_char_0042_0305.gif) is the average BC and σB is the standard deviation. The residues with a Zx value greater than or equal to 2.5 are considered to have a high BC.
is the average BC and σB is the standard deviation. The residues with a Zx value greater than or equal to 2.5 are considered to have a high BC.
Shortest path and characteristic path length
The shortest path between a pair of nodes, for an unweighted graph, is defined as the smallest number of links that need to be traversed in order to reach from one node to another. However, for a weighted graph, with specific weights on the edges, the shortest path corresponds to the set of steps from one node to another that minimizes the sum of the weights on the corresponding edges. An average of the shortest paths between all the pairs of nodes in a network is defined as the Characteristic Path Length (CPL). CPL is a global property that determines the typical separation between two nodes in a network.16Gaussian network model of AS structures
Under the Gaussian network model, the protein in the folded state is assumed to be equivalent to a three dimensional elastic network.17 The adjacency matrix corresponding to each protein, formed as mentioned above, can be used to derive the Kirchoff’s Matrix as follows| Γ = D − A |
and A is the adjacency matrix.
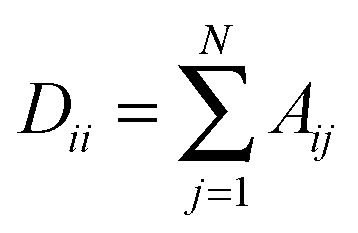
The inverse of this Kirchoff’s matrix provides the pair-wise residue cross correlations as per the following relation:18,19
| 〈ΔRi·ΔRj〉 = (3kBT/γ)[Γ−1]ij |
These cross correlations between pairs of residues are used to weigh the edges of the PCN in order to determine the unique shortest paths between nodes.
Communication pathways in protein
Communication pathways within a protein were found from the shortest paths between the inhibitor binding site and the active site and the pair-wise residue correlations. The shortest paths were calculated between inhibitor binding and catalytic residues using the weighted edges as calculated above. Thus a modified adjacency matrix is defined aswhere Cij is the correlation between residues i and j.
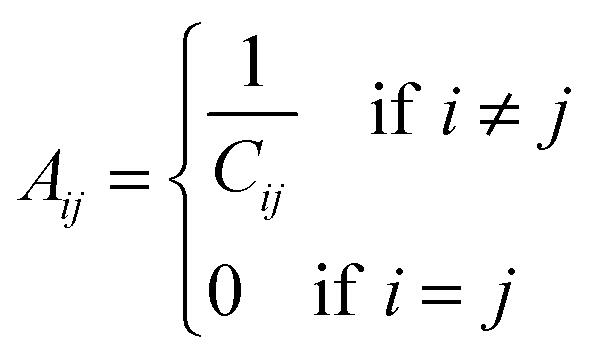
Community calculation
Communities in the PCNs of the AS-substrate and AS-inhibitor were calculated using the “igraph” package. The fast greedy algorithm proposed by Clauset et al.20 was used for calculating the best split of the communities.Tunnel determination using Mole 2
The tunnel between the substrate binding site of TrpE and the substrate binding site of TrpG was determined using Mole 2.0 software.21 The starting point was determined by the residues forming the substrate binding site in TrpG and the end point was given by the residues forming the substrate binding site in TrpE. The pymol plugin of Mole 2.0 was used for the visualization and to prepare the figure.Cavity determination
The cavities in the protein structures were determined by the CastP.22 The default values were used for the determination of cavities.Molecular dynamics simulation
An equilibrium molecular dynamics simulation was performed on the AS heterodimer (TrpE:TrpG) using GROMACS.23 The starting model for the simulation was obtained from the crystal structure of the AS in the presence of a substrate (PDB ID: 1I7Q) and an inhibitor (PDB ID: 1I7S). The dimers comprised of chains A and B of both the structures were used for the simulation. Six residues (43–48) in the TrpE chain of the inhibitor bound structure were not resolved in the crystal structure and hence were missing in the PDB file. These six residues were modeled based on the coordinates of the same residues in the substrate bound structure (1I7Q) in Pymol after superimposing the two structures. The final models consist of 517 residues in chain A (4–520) and 192 residues (2–193) in chain B making a total of 709 residues.The two starting models were put in a dodecahedron box and filled with water molecules giving the final box size of 929.98 nm3 and 121![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 645 atoms in the case of the 1I7Q system, and 891.05 nm3 and 116857 atoms in the case of the 1I7S system. The water model used was TIP4P and the force field used was OPLS-AA. The systems were then neutralized using counter ions (15 Na+ ions in the 1I7Q system and 16 Na+ ions in the 1I7S system) and energy minimization was performed until the maximum force acting on any atom was less than 1000 KJ mol−1 nm−1. The AS-substrate and AS-inhibitor systems were minimized in 1561 steps and 1248 steps respectively. The energy minimized systems were then equilibrated in an NVT ensemble for 500 ps. A Modified Berendsen Thermostat (V-rescale)24 was used for temperature coupling. The temperature of the system was maintained at 300 K. After this the systems were equilibrated in an NPT ensemble for 500 ps. The pressure was maintained at 1 bar using a Parrinello–Rahman barostat.25 After equilibration the production run was performed for 70 ns. The time step used for integration was 2 fs and the frames were saved after every 1000 steps, giving a total of 35001 frames in each trajectory.
645 atoms in the case of the 1I7Q system, and 891.05 nm3 and 116857 atoms in the case of the 1I7S system. The water model used was TIP4P and the force field used was OPLS-AA. The systems were then neutralized using counter ions (15 Na+ ions in the 1I7Q system and 16 Na+ ions in the 1I7S system) and energy minimization was performed until the maximum force acting on any atom was less than 1000 KJ mol−1 nm−1. The AS-substrate and AS-inhibitor systems were minimized in 1561 steps and 1248 steps respectively. The energy minimized systems were then equilibrated in an NVT ensemble for 500 ps. A Modified Berendsen Thermostat (V-rescale)24 was used for temperature coupling. The temperature of the system was maintained at 300 K. After this the systems were equilibrated in an NPT ensemble for 500 ps. The pressure was maintained at 1 bar using a Parrinello–Rahman barostat.25 After equilibration the production run was performed for 70 ns. The time step used for integration was 2 fs and the frames were saved after every 1000 steps, giving a total of 35001 frames in each trajectory.
Root mean square deviations (RMSD) were calculated for Cα atoms of the trajectory frames using the 0th frame of simulation as the reference. The RMSD was calculated using the g_rms function of GROMACS.
Root mean square fluctuations (RMSF) were also calculated for the Cα atoms of the protein using the g_rmsf function of GROMACS.
The dynamic cross correlation maps were calculated using the Bio3D package26 of R.
Results and discussion
Protein contact network analysis
Given the insignificant structural variation between the substrate-bound (PDB ID: 1I7Q) and inhibitor-bound (PDB ID: 1I7S) structures of AS in S. marcescens, the two proteins were modelled as networks, and the contacts were compared to map the small conformational changes throughout the structure. Fig. 1(e) shows the ring graph representation of the PCNs of the AS-substrate and AS-inhibitor. In this representation, all the nodes lie sequentially along the circumference of the ring and the edges are shown as lines. Using ring graph representation, the conformational changes can be visualized by comparing the contact patterns. The contacts lost and gained in the AS-inhibitor are represented on the structure of the heterodimer in Fig. S1 (ESI†).To study the changes in the contact pattern that may give clues for functional changes,27 we chose the residues participating in each of the three functional regions playing a crucial role in the over-all function of the enzyme – (a) inhibitor (tryptophan) binding region (Fig. 1(c)), (b) substrate (CA) binding region of TrpE (Fig. 1(b)) and (c) substrate (glutamine) binding region of TrpG (Fig. 1(d)). Fig. 1(a) shows these three regions on the heterodimer structure of AS in red, blue and magenta, respectively.
Subgraphs of the functional regions show changes in the contact patterns primarily in the TrpG substrate (glutamine) binding region
The subgraphs of the functional regions mentioned above were extracted from the whole PCNs of the AS-substrate and AS-inhibitor (Fig. 2).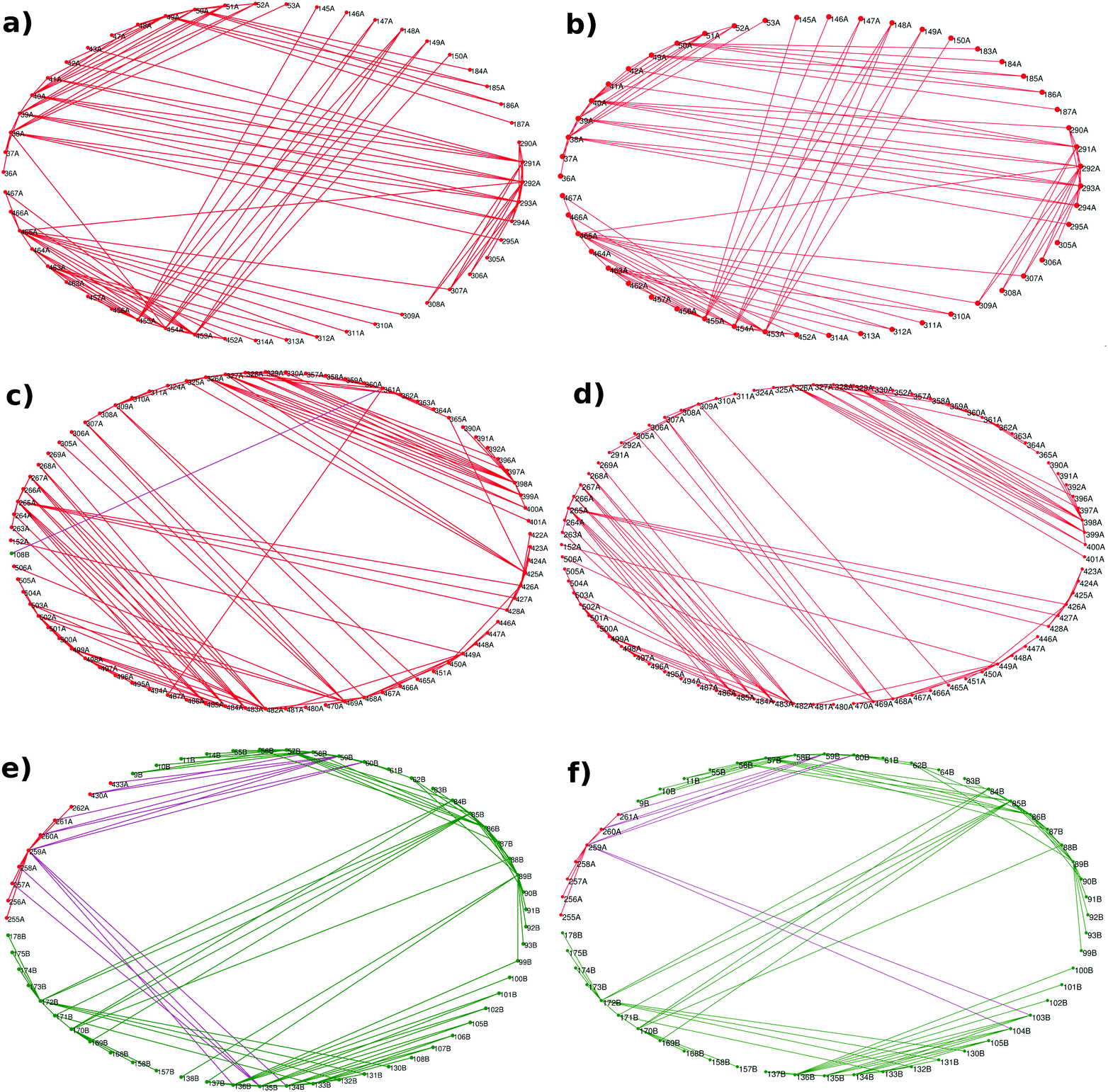 | ||
| Fig. 2 Subgraphs of the three functional regions show specific changes in contact patterns. Ring graph representation of the subgraphs of – inhibitor (tryptophan) binding region in the (a) AS-substrate and (b) AS-inhibitor; substrate (CA) binding region in the (c) AS-substrate and (d) AS-inhibitor; and substrate (glutamine) binding region in the (e) AS-substrate and (f) AS-inhibitor. Edges between the TrpE nodes have been shown in red, edges between the TrpG nodes in green, and edges between the TrpE and TrpG interface nodes in purple. | ||
The analysis of the subgraphs of the inhibitor and substrate binding regions reveals major rearrangement of the contacts caused by small conformational fluctuations occurring due to binding of the inhibitor. These rearrangements involve residues at the heterotetramer and heterodimer interfaces, thus playing an important role in transmission of perturbation between the two substrate binding subunits. The effects of the rearrangements in contact patterns of the inhibitor binding and substrate binding regions can be quantified by analyzing the network parameters.
Network parameters reveal residues showing changes due to inhibitor binding
The average network properties of the AS-substrate and AS-inhibitor showed no significant difference. The residue specific network parameters were calculated and compared for all the residues in the PCNs of the AS-substrate and AS-inhibitor.Degree
The residues showing a change in degree greater than or equal to 3 are listed in Table S1 (ESI†). Most residues that show any change lie between the substrate binding sites of TrpE and TrpG and the TrpE:TrpG interface. The effects of inhibitor binding in TrpE are communicated to the TrpG substrate binding site through the loss of crucial contacts in the β strands, β5 (98–115) and β6 (128–139) that are shifted in the AS-inhibitor. Note that the largest change in degree in this region (in 103, 104, 106, 108) belongs to the β sheet (103–109) that is largely disordered in the AS-inhibitor.9 In TrpE, residues showing a considerable decrease in the degree occur either at the TrpE substrate binding site (361 and 425) or in the residues surrounding it (262, 288, 422, 430, 505 and 306). These changes play an important role in the transfer of perturbation from the inhibitor binding region in TrpE to the substrate (CA) binding region of TrpE, and then to the substrate (Glutamine) binding region of TrpG.Hubs
High degree nodes in the PCN that connect to several residues are referred to as hubs. Hubs have been implicated to reveal the perturbations occurring in the protein structure networks due to ligand binding.28 Hubs were determined for the AS-substrate and AS-inhibitor as described in the Materials and methods section. All the hubs in the AS-substrate and AS-inhibitor are listed in Table S2 (ESI†). The total number of hub residues in the AS-substrate is forty-three, whereas in the AS-inhibitor there are thirty-seven hub residues. Several hub residues are common between the two structures – most of them being in the TrpG subunit. Invariant hubs have been previously shown to span the entire structure suggesting their role in maintaining structural stability.28,29 Few hubs are unique to each of the structures, suggesting their role specific to the inhibitor or substrate binding.Hub residues I256, G259 and P430 occur at the interface of TrpE and TrpG sharing contacts with both the subunits. This makes these residues crucial for communication between TrpE and TrpG. The absence of these residues as hubs in the AS-inhibitor suggests disruption in the inter subunit communication between the two substrate binding sites through them due to inhibitor binding. Among the above, P430 loses contacts with G58 and P59 of TrpG which interact directly with glutamate, and G259 loses contacts with residues H134, S135 and L136 which again interact directly with glutamate. These changes cause perturbation in the substrate binding region of TrpG. Residues R452 and I466 are hubs close to the inhibitor binding site (V453 and C465 respectively) in the substrate-bound PCN. Inhibitor binding causes them to lose contacts and thus affecting communication through these hubs. Residue 398, which is a hub in the AS-substrate but not in the AS-inhibitor, participates in substrate binding and its role has been confirmed by mutational studies.30
Betweenness centrality (BC)
Betweenness centrality is an important measure to determine the role of nodes in information flow in a network, and residues with a high BC have been found to play an important role in structural complexes.31 The BC for each residue in the AS-substrate and AS-inhibitor was calculated as described in the Materials and methods section, and high BC residues are listed in Table S3 (ESI†). Most of the residues with a high BC belong to the hetero-tetramer interface. The binding of an inhibitor causes small but important changes in the AS-inhibitor network, which lead to changes in the residue specific network parameters. Fig. 3(a) shows the difference in the BC between the AS-substrate and AS-inhibitor. As observed in the earlier sections, many interface residues show changes in BC, and therefore, for understanding the allosteric communication within AS upon binding of the inhibitor, the altered BCs of these residues become important.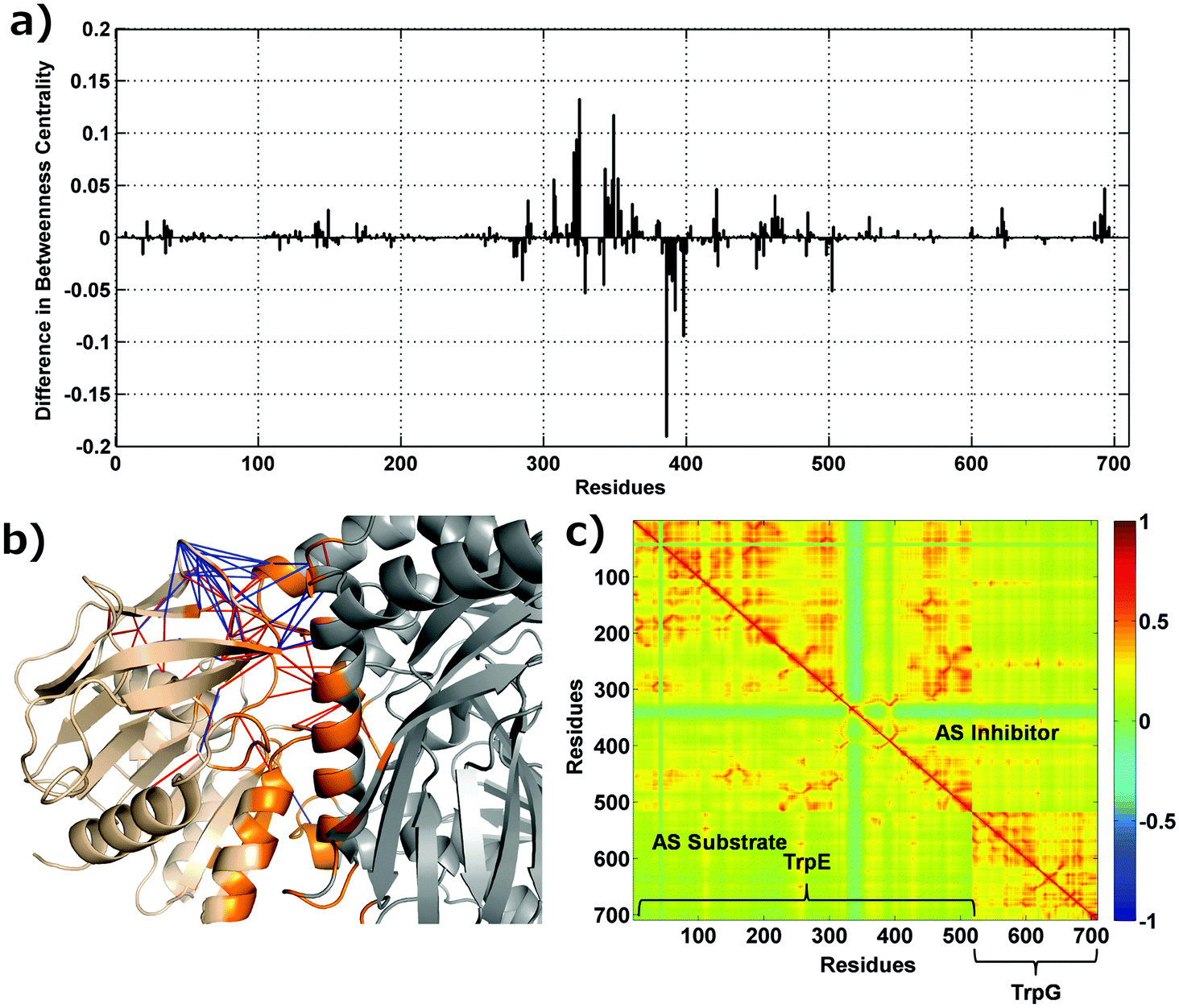 | ||
| Fig. 3 Changes in betweenness centrality, interface contacts and cross-correlations. (a) Residue-wise difference in betweenness centrality between the AS-inhibitor and AS-substrate. (b) Contacts lost and gained at the interface of TrpE and TrpG. TrpE is shown in grey, TrpG in wheat and the interface region in orange. Contacts gained in the AS-inhibitor are shown in blue and contacts lost in red. (c) Cross correlation between all pairs of residues in the AS substrate (lower triangle) and AS-inhibitor (upper triangle) heterodimers. The residues specific to the two chains (TrpE and Trp G) are labelled. | ||
Contact changes upon inhibitor binding and Gaussian network modelling
The network analysis presented above points towards the perturbation of the contact patterns at the interface region, which has been corroborated by the changes in the network parameters. Fig. 3(b) shows the contact changes at the heterodimer interface – contacts lost (red) and gained (blue) at and near the TrpE:TrpG interface. As can be seen from the figure several contacts have been broken and made suggesting a major rearrangement of the contact pattern which may cause perturbation at the inter-chain communication due to inhibitor binding, where two substrate binding sites are present. In order to map the pathways of communication between the three regions defined above (Fig. 1), and to understand the perturbation in these pathways leading to inhibition of the enzyme activity, we used a combined PCN and Gaussian network model (GNM) approach, which gives the cross correlations between all the residues that can be used to weigh the edges of the PCN in order to determine the unique shortest paths between the nodes, through which perturbations may travel.The pair-wise cross correlations between the amino acids in two structures were determined as described in the Materials and methods section and are shown in Fig. 3(c) for the heterodimer. Fig. 3(c) shows the pair-wise cross correlations between residues in the AS-substrate (shown in the lower triangle) and the AS-inhibitor (shown in the upper triangle). The residues belonging to the same chain have more residues that are positively correlated as compared to the residues in the other chain (TrpE and TrpG). The correlations between residues in the AS inhibitor (upper triangle) have increased compared to those in the AS substrate (lower triangle). These correlations are utilised to assign weights to the edges of the protein contact network for calculation of the shortest paths.32
Communication between the functional regions is perturbed upon binding of the inhibitor – shortest path analysis
Allosteric communication within a protein molecule has been quite extensively studied by modelling proteins as information processing networks, where information transfer is assumed to occur along the shortest paths between different regions.33–35 Perturbations in the shortest paths can then be studied in order to understand the allosteric mechanisms leading to functional regulation. The shortest paths, among the functional regions, were calculated using the cross correlations (Fig. 3(c)) as edge weights in PCN from the Gaussian network model (see the Methods section for details).Perturbation due to the binding of an inhibitor may alter the inter- and intra-subunit communication, and the paths between a pair of nodes can change in one of the following ways – (1) increase in length, (2) decrease in length, and (3) change in paths with the same length. All the above possibilities were analysed for the set of nodes in the three functional regions (Fig. 1(a) and Fig. 4). Very few paths remain invariant upon inhibitor binding while the majority of the shortest paths have increased in length (yellow bars) in all three cases (58.7%, 64.3% and 70.7% respectively) – (i) from the tryptophan (inhibitor) binding site in TrpE to (a) the chorismate (substrate) binding site in TrpE, and (b) the glutamine (substrate) binding site in TrpG; and (ii) the chorismate (substrate) binding site in TrpE to the glutamine (substrate) binding site in TrpG. A small percentage of paths have decreased in length (red bars). Some new paths have arisen without any change in length (cyan bars). These alterations in the shortest path lengths between the sub-networks of functional regions indicate that the small conformational changes that occur due to the inhibitor binding to the enzyme (AS) caused alteration in the intra and inter-subunit communication, especially between the two substrate binding regions. These results match the residue-specific changes in the network parameters (degree, betweenness centrality), where most changes were observed between the two substrate-binding regions and at the heterodimer interface.
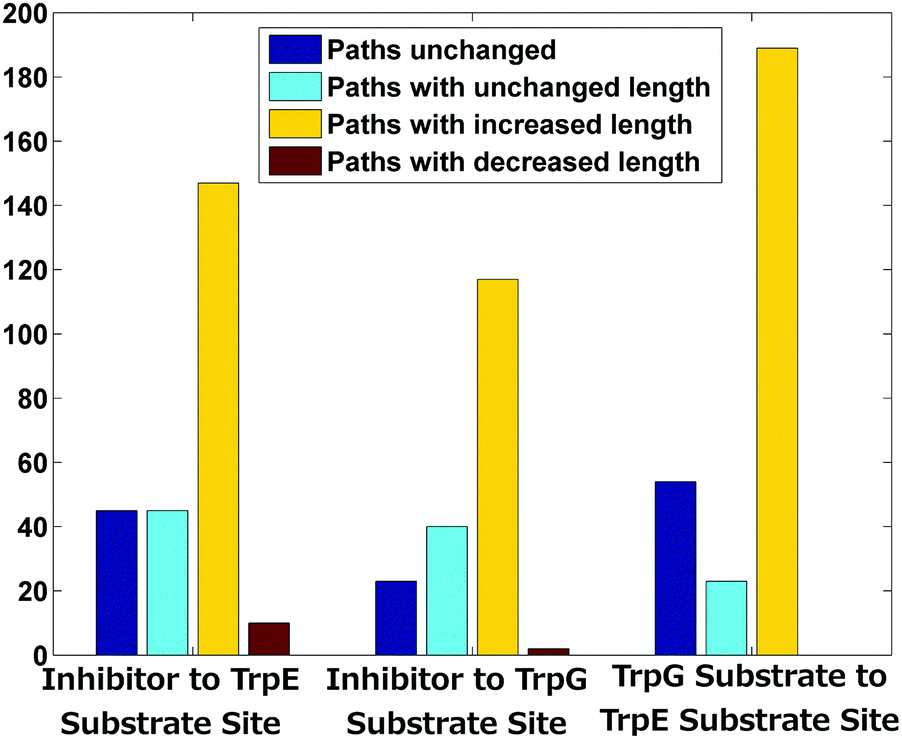 | ||
| Fig. 4 Summary of shortest path changes in the AS-inhibitor structure as compared to the AS-substrate. | ||
In order to gain more insight into the communication between the two active sites we analysed all the shortest paths between the two active sites in detail and mapped the residues participating in these paths (Fig. 5). Several perturbations in these paths can be observed in the AS-inhibitor. As mentioned earlier, the shortest path lengths between the two active sites have increased in the AS-inhibitor. The most prominent change that can be observed was the involvement of residues 103, 104 and 105 of TrpG on the paths between the TrpG substrate binding site and the TrpE substrate binding site. These three residues also showed the largest change in degree and are part of strand β5, which is disordered in the AS-inhibitor.
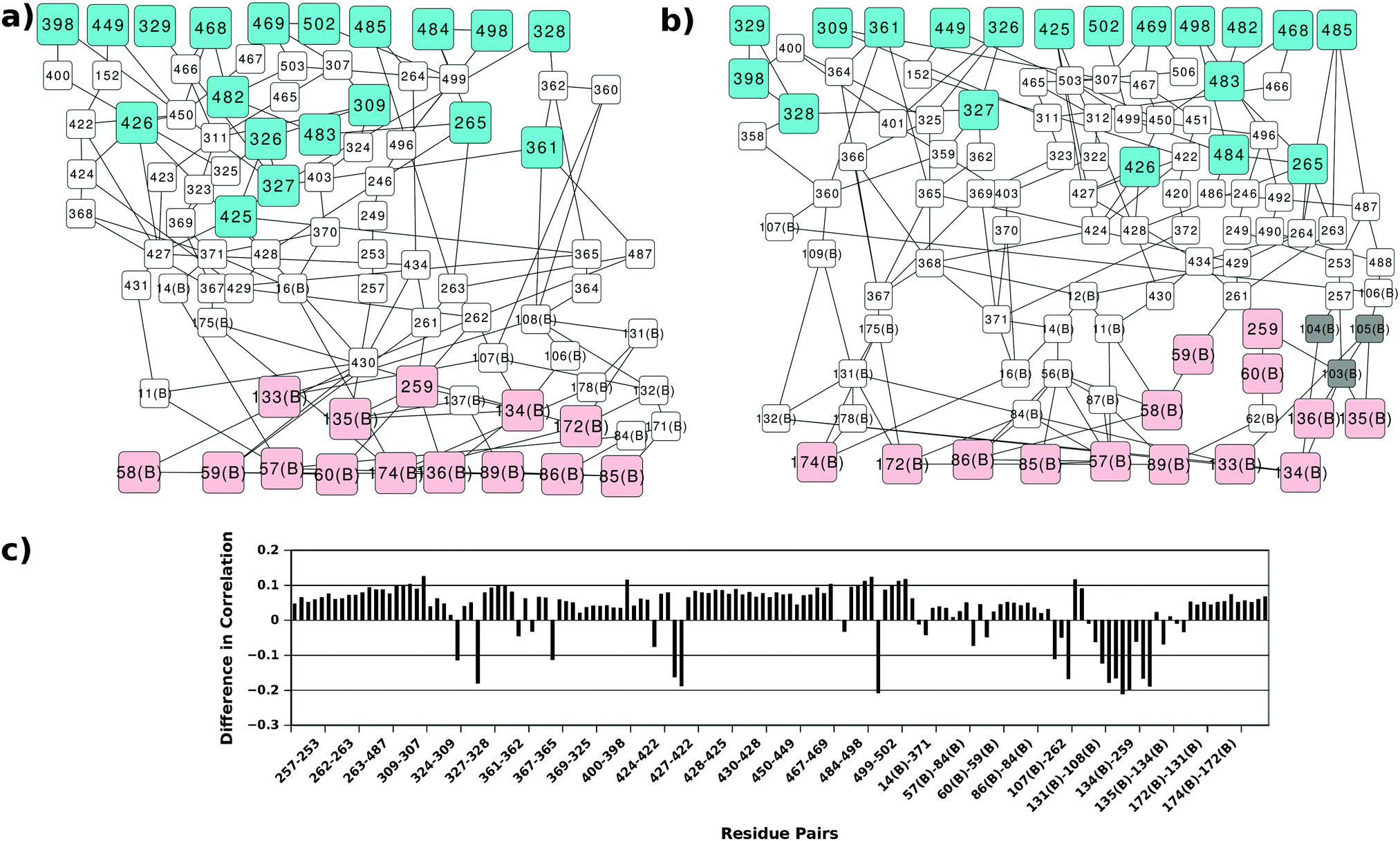 | ||
| Fig. 5 Perturbations in the shortest paths and cross-correlations. Shortest paths between the substrate binding sites of Trp G (pink) and TrpE (cyan) in the (a) AS-substrate and (b) AS-inhibitor. Grey nodes (103, 105 and 105 of TrpG) show the TrpG residues present in the AS-inhibitor shortest paths and not in the AS-substrate. (c) Specific residue pairs in the shortest paths between two substrate-binding regions show a considerable decrease in the cross-correlation in the AS inhibitor. | ||
Changes in the paths can be caused by alterations in the contact pattern and/or correlations between the residues. Contact pattern changes at the functional sites and the interface region have been described earlier. Changes in correlations between the residue pairs participating in the shortest paths between the two active sites were calculated (Fig. 5(c)). Even though most residue pairs exhibit a small increase in cross correlations, few residue pairs show a considerable decrease. Most of these residue pairs play important roles in the communication between the two active sites and occur at the interface of TrpE and TrpG.
Community analysis of AS-substrate and AS-inhibitor PCNs: rearrangement in the membership of residues in the shortest paths connecting the two substrate-binding regions
The network parameter, contact and shortest path analyses of the AS-substrate and AS-inhibitor PCNs described above clearly point towards perturbation of communication between the two substrate binding sites in the two chains (TrpE and TrpG) of AS upon inhibitor binding at TrpG, and thereby regulating its catalytic activity. Communication between the two chains of AS (TrpE and TrpG) is crucial for the function of the enzyme, as during catalysis the ammonia formed at the TrpG active site is transferred to the TrpE active site for the formation of anthranilate from chorismate, thus coupling the two reactions. The two active sites, therefore, need to communicate through the network of amino acids between them, for the successful reaction to take place. Most real world networks possess modules or communities that correspond to functional clustering of the nodes.36 In the case of PCNs, the modules have been found to correspond to the functional regions of the proteins.34 In order to study the effect of inhibitor binding on the overall network architecture, the community structure of the PCNs of the AS-substrate and AS-inhibitor were determined and compared. The AS-substrate heterodimer divides into five communities whereas the AS-inhibitor heterodimer divides into six communities. All the communities for both the PCNs have been listed in Table S4 (ESI†) along with the node membership. The communities in the two PCNs were mapped on to the protein structures.In the case of the AS-substrate structure, the three functional regions (the substrate binding region of TrpE, the substrate binding region of TrpG, and the inhibitor binding region in TrpE) were part of two large communities CS1 and CS2 (Fig. 6(a) in green and blue respectively). Almost all the residues of TrpG and some residues from TrpE form the community CS2 (Blue) in the AS-substrate. In the case of the AS-inhibitor (Fig. 6(b)) all three functional regions are also part of two large communities CI1 and CI2 (Fig. 6(b) red and yellow respectively). However, there is some rearrangement of membership of the residues participating in these communities. Residues 422–443 that include the inhibitor-binding region and the substrate-binding region of TrpE belong to CS1 in the AS-substrate. Interestingly, in the case of the AS-inhibitor, these residues are part of CI2, which also contains residues belonging to the substrate-binding region of TrpG. These residues also participate in the shortest paths between the two substrate-binding active sites belonging to two different chains. Since most of the rearrangement is observed in the region between the two active sites this also indicates the perturbation in the connectivity pattern between them. These results suggest that inhibitor binding can induce perturbation in the network connectivity at the global level in terms of the community structure, even though there may not be significant conformational changes observed in the three-dimensional structure of the two proteins.
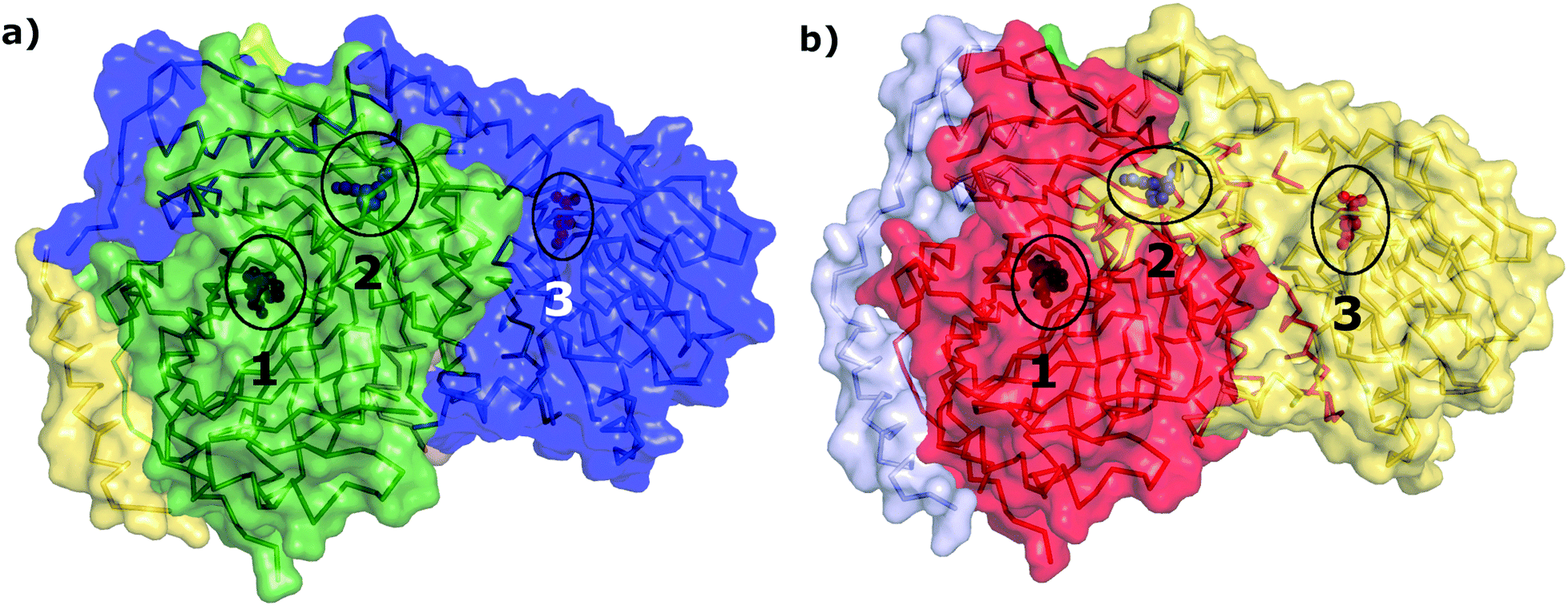 | ||
| Fig. 6 Community membership of different residues. The structures of (a) AS-substrate and (b) AS-inhibitor with different communities shown in different colors. The ligands present in the substrate binding sites of TrpE (numbered 2) and TrpG (numbered 3) and the inhibitor binding site in TrpE (numbered 1) have been shown as blue, red and black sphere representations respectively (encircled in black). | ||
Physical path between the two substrate-binding regions is perturbed upon inhibitor binding
All the coarse-grained network analyses of the two protein structures mentioned above indicate that the perturbation in the paths between the two substrate-binding regions is the most important change happening upon inhibitor binding in AS. It is known that during the catalytic reaction of AS, aromatization of the substrate chorismate takes place in the TrpE in S. marcescens, and the ammonia, obtained from the glutamine to glutamate reaction in the TrpG active site, is transferred to the product anthranilate. No path has been observed in the crystal structure of AS in Serratia marcescens9 for the ammonia to be directly transferred from the TrpG active site to the TrpE active site through cavities present between them, even though, based on the crystal structure of AS from Salmonella typhimurium10 such a path has been shown to be present through cavities present between them.In order to relate the perturbation in the shortest path results from PCN analyses to the possibility of the existence of a physical path (possibly transient), we used Mole 2 to determine the physical path between the substrate-binding active sites of TrpG and TrpE21,37 (Fig. 7). The tunnels determined by Mole 2 for the AS-inhibitor are remarkably different from that of the AS-substrate: (a) the path is much shorter (33.82 Å) in the AS-substrate, compared to that for the AS-inhibitor (52.24 Å); and (b) the path between the two active sites in the case of the AS-substrate has low average solvent accessible surface area (SASA) per residue (15.4 Å2), whereas the path in the AS-inhibitor is solvent exposed with a high SASA per residue (44.1 Å2). These indicate that, upon inhibitor binding, the path to transfer ammonia from its site of synthesis in the TrpG to the anthranilate synthesis site in TrpE during catalysis is not only extended in length, but also becomes more solvent-exposed. Both these indicate disruption in catalytic activity due to inhibitor binding. Table S5 (ESI†) shows the SASA for all residues lining the paths from the TrpG active site to the TrpE active site in the AS-substrate and AS-inhibitor.
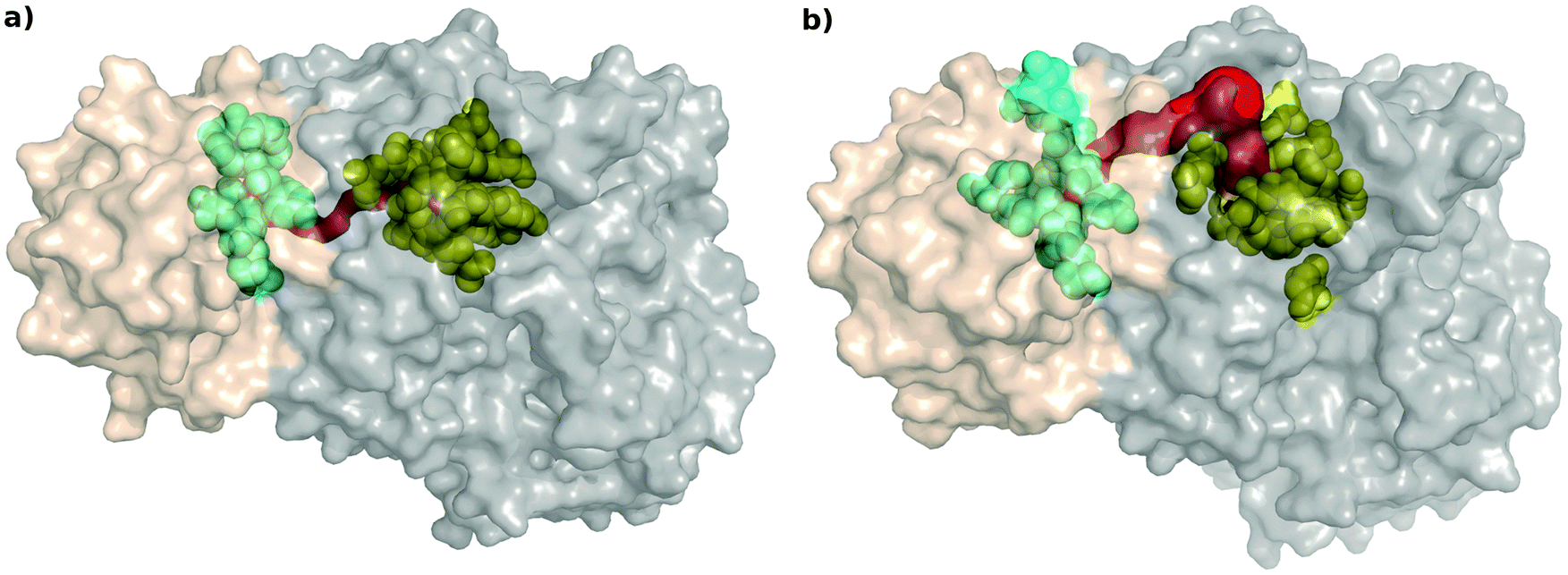 | ||
| Fig. 7 Channel between two active sites. Path determined by Mole 2 (Red) is more solvent exposed in the AS-inhibitor structure (b) as compared to the AS-substrate structure (a). Substrate (glutamine) binding site of TrpG is shown in cyan and the substrate (chorismate) binding site of TrpE in yellow. | ||
The typical radius of the path required for the transfer of an ammonia molecule is ∼3.2 Å,38 however the path determined by Mole 2 was narrow. We determined the cavities present between the two active sites using CASTp22 to determine if the path obtained by Mole 2 coincided with the cavities calculated for both the proteins. Fig. S2 (ESI†) shows the cavities for the AS-substrate and AS-inhibitor between the two active sites. The details of the pockets showed in the figure have been mentioned in Table S6 (ESI†). It was found that there are solvent protected cavities present between the two active sites in the AS-substrate, whereas, the cavity containing the TrpG substrate-binding site is solvent exposed in the AS-inhibitor. Several residues lining the cavities between the two active sites obtained by CASTp also line the path determined by Mole 2, thus suggesting that, at the time of catalysis, the conformational changes lead to transfer of ammonia through the cavities between the two substrate binding sites, but binding of the inhibitor causes conformational changes leading to the loss of this path thus blocking the catalysis. The binding of the inhibitor causes conformational changes in the path between the two active sites, thus perturbing the communication between the two sites. The residues participating in the shortest paths between the two active sites determined in earlier analysis were also found to line the path determined by Mole 2.
Residues in the shortest paths show specific changes in network parameters and are conserved
The analysis of the AS-substrate and AS-inhibitor discussed above reveals residues that play an important part in maintaining the communication pathways in the functional protein, and perturbation of which causes the inhibition of the enzyme. The properties of these residues with respect to their role in the network and structure were studied in detail to further illustrate their importance in the inhibition mechanism. Changes in the degree of all the residues participating in the shortest paths between the two active sites upon binding of the inhibitor were observed (Fig. 8). Of the 87 residues, 42 show a decrease in degree whereas just seven show an increase (of which the degree of six residues increase by one) suggesting a loss of interactions in the communication paths. The most prominent decrease is observed in the case of residues 106, 107, 108, 135, 136, 138 of Trp G, and 262, 306, 361, 422, 430 of TrpE. Loss of degree indicates loss of crucial contacts that may lead to decreased communication between the two active sites.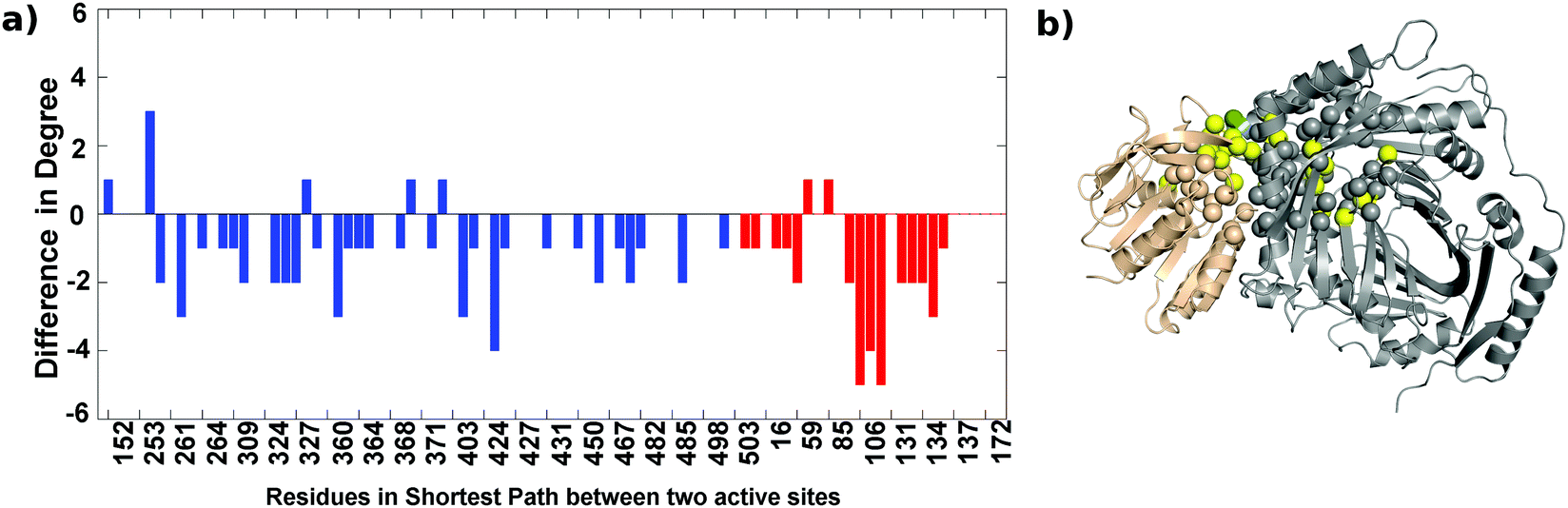 | ||
| Fig. 8 Majority of the shortest path residues between two active sites show a reduced degree upon inhibitor binding. (a) Difference in degree – blue bars for residues of TrpE, and red bars for residues in the TrpG active sites. (b) Residues showing a high increase (>3) (green spheres) and a high decrease (yellow spheres) (>3). The TrpE subunit is shown in grey and the TrpG subunit in a wheat color. | ||
As the communication between different sites on the bienzyme complex plays a crucial role in the overall function of the protein, it is expected that the residues participating in such communication should be evolutionarily conserved. In order to test this, conservation scores of the residues involved in the shortest paths between the three regions defined above were obtained from the Consurf server39 (Fig. S3, ESI†). As can be seen from the figure, most of the residues participating in the shortest paths are conserved. This suggests that the information transfer between the two functional regions of AS occurs through a network of evolutionarily-conserved residues.35 In the case of the AS-inhibitor, however, there are a few residues (G103, E104, I105 of TrpG and Q488 of TrpE) occurring in these shortest paths that show very low conservation suggesting non-optimal paths existing in the AS-inhibitor complex, which might be due to conformational changes leading to inhibition of the enzyme.
These results, using a combination of network analysis and computational probing of the structures, give new insights into the mechanism of allosteric inhibition through perturbation in the communication between the two substrate-binding regions of the enzyme anthranilate synthase in S. marcescens. However, in vivo these perturbations due to ligand-binding are transferred by the continuous breathing motion of the molecule. In order to understand changes in the dynamics of the enzyme, molecular dynamics simulations were performed using the AS-substrate and AS-inhibitor heterodimers as the starting models.
Molecular dynamics simulations highlight dynamical changes upon inhibitor binding
Molecular Dynamics (MD) simulations of the TrpE:TrpG heterodimer, in the two states AS-substrate and AS-inhibitor, were performed for a period of 70 ns (see the Materials and methods section for details). The RMSD values for the AS-inhibitor MD trajectory are slightly higher than the AS-substrate MD trajectory (Fig. S4, ESI†). Both the trajectories stabilised after 10 ns and the average RMSD from 10 ns to 70 ns was 0.22 ± 0.01 nm and 0.24 ± 0.01 nm for the AS-substrate and AS-inhibitor respectively. All subsequent analyses were performed on the trajectory from 10 ns to 70 ns (30001 frames).
Root mean square fluctuations increase for certain residues in inhibitor bound proteins
Root mean square fluctuation (RMSF) is a measure of the flexibility of atoms during dynamic simulations. The RMSF in both the proteins do not show much difference, but a few residues exhibit a marked difference in flexibility as shown in Fig. S5(a) (ESI†) and represented on the crystal structure in Fig. S5(b) (see the ESI† for details).The change in the flexibility of the residues participating in the shortest paths between the TrpG active site and the TrpE active site are studied and the difference in RMSF for these residues in the AS-inhibitor and AS-substrate was calculated. For most of these residues, the RMSF has increased (Fig. 9). The residues that show a remarkable increase in the flexibility are A360 of TrpE and L106-G108 of TrpG.
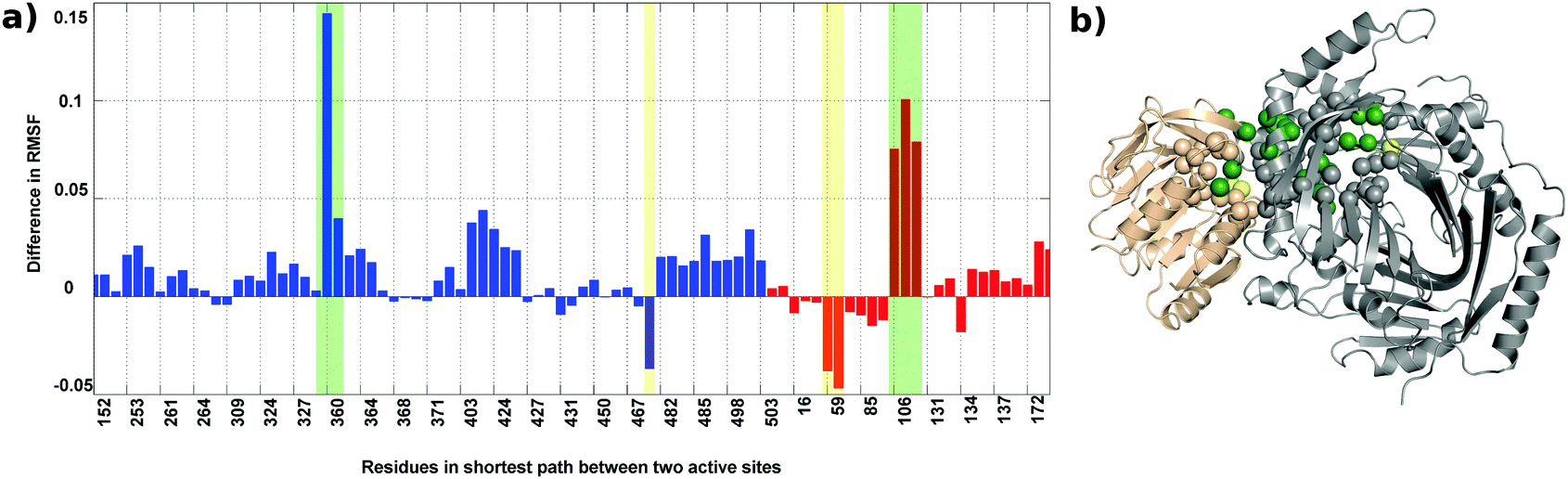 | ||
| Fig. 9 Most residues participating in the shortest paths show an increase in RMSF. (a) Difference in RMSF for the shortest path residues in TrpG (red) and TrpE (blue) active sites. (b) Residues showing a high increase (green spheres) and high decrease (yellow spheres) in RMSF in the inhibitor bound complex. The TrpE subunit is shown in grey and the TrpG subunit in a wheat color. | ||
Decrease in dynamic cross correlations between the residues at the heterodimer interface suggests a loss of communication between the functional regions upon inhibitor binding
Dynamic Cross Correlation Maps (DCCMs) give the pairwise correlations in the motions of the amino acid residues in the protein during the MD simulations and are calculated from the simulation trajectories. Fig. 10(a) shows the DCCM for the AS-substrate (upper triangle) and AS-inhibitor (lower triangle) from the MD simulations. The regions that show variation among the two have been highlighted in boxes. There is predominance of negative cross correlations in the AS-inhibitor part. Several residues belonging to the heterodimer interface and the substrate-binding region of the TrpE subunit show a decrease in correlation with the residues of the TrpG subunit, further emphasizing the loss of communication between the two chains upon binding of the inhibitor.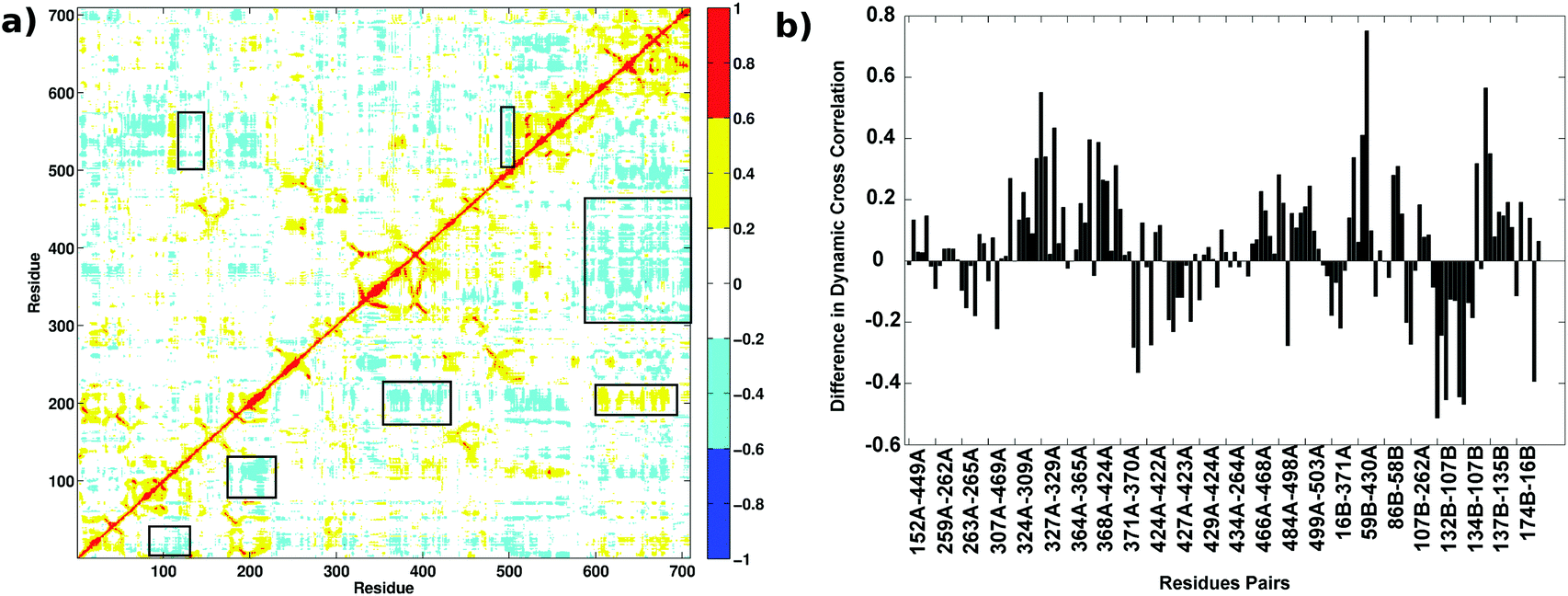 | ||
| Fig. 10 Dynamic cross correlations in the AS-substrate and AS-inhibitor. (a) Dynamic cross correlation map shows variation in the AS-inhibitor (lower triangular part) as compared to the AS-substrate (upper triangular part). The X and Y axes represent the residues in the primary sequence of the enzyme. The variations in the correlations are highlighted in boxes. (b) The correlations between the residues at the interface belonging to two different subunits decrease upon inhibitor binding. The difference in dynamic correlations between the residue pairs in the shortest paths between two active sites. | ||
Correlations between the residues participating in the shortest paths between the two substrate-binding active sites were found to increase, as well as decrease, in several residue pairs (Fig. 10(b)). These residue pairs and their connections were mapped on the crystal structure of the AS (Fig. S6, ESI†). Most of the residue pairs showing a decrease in the correlations (blue) belong to the heterodimer interface, whereas the residue pairs showing an increase in correlation (orange) mostly belong to the intra-subunit regions. Thus, even though the RMSD for the two structures in MD simulations do not show much difference, these observations on RMSF, along with previous analysis showing the loss of key connections at the interface and an increase in the flexibility of residues at the interface, suggest that binding of the inhibitor perturbs the communication between the two active sites leading to the loss of function in the enzyme.
A major paradigm shift in the understanding of allostery in recent years has indicated that allostery may not necessarily be associated with large scale conformational changes between the effector bound and unbound states,40 as was implicit in earlier structure-based models of allostery.41,42 Instead, emphasis is being laid on the dynamics and small conformational changes in the protein that might be causing the allosteric effects.43–45 Allosteric proteins have been studied computationally as information processing systems for finding the communication pathways between the allosteric site and the active sites, through which the perturbations might be transferred within various proteins.32,34,46 Computational methods have played an important role in mapping the allosteric pathways within the protein.47 In this study, we chose the substrate-bound and inhibitor-bound AS from S. marcescens, which have very similar conformations,9 and used computational approaches at the coarse grained level as well detailed atomistic simulations, to understand the details of the allosteric inhibition mechanism.
The AS structure from Serratia marcescens was modeled as a protein contact network (PCN) using Cα atoms as nodes and the distance between them as a measure of connectivity. We specifically studied the contact subgraphs of the functional regions – substrate (chorismate and glutamine) binding sites in TrpE and TrpG, and the inhibitor (tryptophan) binding sites in TrpE subunits. This coarse-grained representation of the structures gave important insight into the contacts lost and gained upon binding of the inhibitor (Fig. 2). A comparison of a few residue-specific network parameters, such as degree, hubs and betweenness centrality, indicated that the heterodimer interface residues might play an important role in the transfer of perturbation from the inhibitor-binding site in TrpE to the two active sites residing in two different subunits, TrpE and TrpG (Fig. 3). In order to obtain a detailed map of the path of communication between the important sites in the protein, weighted shortest paths were determined using Gaussian network analysis, and the perturbation in communication between the two substrate-binding sites was found by studying changes in the shortest paths (Fig. 4 and 5). These were correlated with conformational changes in the inhibitor-bound complex and hence to the inhibition of the catalytic activity of AS. All these results point towards perturbation in the paths connecting the two substrate-binding sites in the two subunits as being primarily affected due to allosteric binding of the inhibitor. The community analysis of the two PCNs, formed by highly connected groups of residues, showed rearrangements in the residues in the connecting region between the two active sites, indicating global level changes in network architecture due to inhibitor binding at a distant site (Fig. 6).
It is known that chorismate and glutamine bind to the two subunits and that catalysis can take place when ammonia is transferred from one site to the other. Such substrate channelling is known as a form of intra-molecular communication, where the substrates and/or intermediates are transferred from one active site to the other active site of the enzyme or in multi-enzyme complexes involved in the process.48 But such a channel was not shown to be present in the S. marcescens crystal structure.9 Our network analysis provided a residue-wise map of the communication path between the two active sites, and a tentative network of amino acids that might be involved in the substrate channeling within the bienzyme complex (Fig. 5). The path obtained by the network analysis was further corroborated by the physical presence of cavities in the substrate bound structure that might be combining to form a transient tunnel for the transfer of ammonia from the TrpG active site to the TrpE active site. These paths were found to be disturbed in the inhibitor bound structure thus confirming that binding of the inhibitor causes structural changes that block the communication between the two active sites. The physical path obtained using Mole 2 in the case of the AS-inhibitor was found to be solvent exposed (Fig. 7). In the case of the AS-substrate, the tunnel coincided with the solvent protected cavities determined by CastP. The cross correlations, between the residues at the interface and the residues participating in the shortest paths in the substrate-bound complex, were also found to be decreased in the case of the inhibitor bound structure. Analysis of the Molecular Dynamics simulations of the substrate-bound enzyme and the inhibitor-bound AS did not show much variation in RMSD as both the structures and sequences are quite similar, but they showed an increase in the flexibility of the residues at the interface and the residues participating in the shortest paths (Fig. 9 and Fig. S6, ESI†). The dynamic cross correlations obtained from the equilibrium MD simulations also decrease in the residues at the heterodimer interface suggesting a loss of communication between the active sites. It is not clear if this effect seen in the MD simulations of AS in Serratia marcescens is common in other bacteria. The recent application of co-evolution analysis to study interacting interfaces might be useful to address such evolutionary questions.49
Based on the results obtained from multiple computational analyses – both from the coarse-grained static network approach and the detailed atomistic MD simulations – we conclude that the perturbation in the communication pathways between two active sites plays a major role in the allosteric inhibition of anthranilate synthase. The conformational changes due to allosteric binding of the inhibitor about 18 Å away from the substrate-binding site in TrpE leads to these perturbations in residues and makes the path of ammonia transfer solvent-accessible, thereby uncoupling the two reactions in the two active sites in the TrpE and TrpG subunits.
Conclusions
Our study leads to two important issues. First, the information extracted about specific residues making up the cavities for ammonia tunneling can be harnessed to predict mutations or discover novel inhibitors against anthranilate synthase. Such predictions can have far-reaching and substantial specific applications in drug development against bacterial pathogens, as the aromatic amino acid biosynthetic pathway is absent in mammals and many other organisms.The second one relates to the efficacy of this mode of regulation. Chorismate is a versatile central branch-point metabolic node in bacterial, plant, and fungal metabolism. Several of the chorismate-utilizing enzymes catalyzing the first committed steps in the formation of several different compounds show structural and mechanistic homology.1 Thus, the flux through this important metabolic node is distributed across a number of reactions, and the regulation of distribution of flux is crucial for proper functioning of the biochemical reactions around this node. The mode of regulation of AS is such that even one molecule of tryptophan can stop its catalytic activity in this particular reaction pathway by reorganizing the local positions and dynamics of the channel residues, without involving large scale structural changes in the AS structure that perform catalytic activities in other reaction pathways. This design of regulation of enzyme activity, with allosterically controlled small local perturbations in substrate channeling, can easily turn the enzyme activity on or off, thereby being able to regulate metabolic flux across multiple reactions from a single substrate efficiently. Thus our theoretical study elucidates a novel regulatory mechanism (change in function without significant change in the structure) for catalysis in such multi-functional enzymes that has to undergo fast redistribution of fluxes according to different metabolic states of the organism. The evolutionary implications of this mechanism needs to be further explored by analysing the other connected pathways, as was shown in a recent study27 on Class A G-protein-coupled receptors that mediate a wide variety of physiological functions initiated by diverse ligands, through structural identification of a region of convergence of different activation pathways near the G-protein-coupling region.
Acknowledgements
Part of this work is included in the PhD thesis submitted at the CSIR-Centre for Cellular and Molecular Biology, India, by AS. SS acknowledges support from the Department of Science & Technology as a J C Bose Fellowship.References
- F. Dosselaere and J. Vanderleyden, Crit. Rev. Microbiol., 2001, 27, 75–131 Search PubMed.
- X. Lin, S. Xu, Y. Yang, J. Wu, H. Wang, H. Shen and H. Wang, Protein Expression Purif., 2009, 64, 8–15 Search PubMed.
- Y. J. Zhang, M. C. Reddy, T. R. Ioerger, A. C. Rothchild, V. Dartois, B. M. Schuster, A. Trauner, D. Wallis, S. Galaviz, C. Huttenhower, J. C. Sacchettini, S. M. Behar and E. J. Rubin, Cell, 2013, 155, 1296–1308 Search PubMed.
- G. Bashiri, J. M. Johnston, G. L. Evans, E. M. M. Bulloch, D. C. Goldstone, E. N. M. Jirgis, S. Kleinboelting, A. Castell, R. J. Ramsay, A. Manos-Turvey, R. J. Payne, J. S. Lott and E. N. Baker, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2015, 71, 2297–2308 Search PubMed.
- C. T. Walsh, J. Liu, F. Rusnak and M. Sakaitani, Chem. Rev., 1990, 90, 1105–1129 Search PubMed.
- K. T. Ziebart, S. M. Dixon, B. Avila, M. H. El-Badri, K. G. Guggenheim, M. J. Kurth and M. D. Toney, J. Med. Chem., 2010, 53, 3718–3729 Search PubMed.
- J. E. Culbertson, D. Hee Chung, K. T. Ziebart, E. Espiritu and M. D. Toney, Biochemistry, 2015, 54, 2372–2384 Search PubMed.
- S. Sinha, Biotechnol. Bioeng., 1988, 31, 117–124 Search PubMed.
- G. Spraggon, C. Kim, X. Nguyen-Huu, M. C. Yee, C. Yanofsky and S. E. Mills, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 6021–6026 Search PubMed.
- A. A. Morollo and M. J. Eck, Nat. Struct. Biol., 2001, 8, 243–247 Search PubMed.
- T. Knöchel, A. Ivens, G. Hester, A. Gonzalez, R. Bauerle, M. Wilmanns, K. Kirschner and J. N. Jansonius, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9479–9484 Search PubMed.
- M. G. Caligiuris and R. Bauerles, J. Biol. Chem., 1991, 266, 8328–8335 Search PubMed.
- G. Bagler and S. Sinha, Phys. A, 2008, 346, 27–33 Search PubMed.
- G. Bagler and S. Sinha, Bioinformatics, 2007, 23, 1760–1767 Search PubMed.
- A. Srivastava and S. Sinha, PLoS One, 2014, 9, e102856 Search PubMed.
- D. J. Watts and S. H. Strogatz, Nature, 1998, 393, 440–442 Search PubMed.
- I. Bahar, A. R. Atilgan and B. Erman, Folding Des., 1997, 2, 173–181 Search PubMed.
- C. Chennubhotla and I. Bahar, PLoS Comput. Biol., 2007, 3, e172 Search PubMed.
- B. Isin, P. Doruker and I. Bahar, Biophys. J., 2002, 82, 569–581 Search PubMed.
- A. Clauset, M. E. J. Newman and C. Moore, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2004, 70, 66111 Search PubMed.
- D. Sehnal, R. Svobodová Vařeková, K. Berka, L. Pravda, V. Navrátilová, P. Banáš, C.-M. Ionescu, M. Otyepka and J. Koča, J. Cheminf., 2013, 5, 39 Search PubMed.
- T. A. Binkowski, S. Naghibzadeh and J. Liang, Nucleic Acids Res., 2003, 31, 3352–3355 Search PubMed.
- S. Pronk, S. Pall, R. Schulz, P. Larsson, P. Bjelkmar, R. Apostolov, M. R. Shirts, J. C. Smith, P. M. Kasson, D. Spoel, B. Hess and E. Lindahl, Bioinformatics, 2013, 29, 845–854 Search PubMed.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 14101 Search PubMed.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182 CrossRef CAS.
- B. J. Grant, A. P. C. Rodrigues, K. M. ElSawy, J. A. McCammon and L. S. D. Caves, Bioinformatics, 2006, 22, 2695–2696 Search PubMed.
- A. J. Venkatakrishnan, X. Deupi, G. Lebon, F. M. Heydenreich, T. Flock, T. Miljus, S. Balaji, M. Bouvier, D. B. Veprintsev, C. G. Tate, G. F. X. Schertler and M. M. Babu, Nature, 2016, 536, 484–487 Search PubMed.
- R. Sathyapriya and S. Vishveshwara, Proteins, 2007, 68, 541–550 Search PubMed.
- K. V Brinda and S. Vishveshwara, Biophys. J., 2005, 89, 4159–4170 Search PubMed.
- R. Bauerle, J. Hess and S. French, Methods Enzymol., 1987, 142, 366–386 Search PubMed.
- A. del Sol and P. O’Meara, Proteins, 2005, 58, 672–682 Search PubMed.
- A. Ghosh and S. Vishveshwara, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 15711–15716 Search PubMed.
- A. Ghosh, R. Sakaguchi, C. Liu, S. Vishveshwara and Y.-M. Hou, J. Biol. Chem., 2011, 286, 37721–37731 Search PubMed.
- A. del Sol, M. J. Araúzo-Bravo, D. Amoros and R. Nussinov, Genome Biol., 2007, 8, R92 Search PubMed.
- G. M. Süel, S. W. Lockless, M. A. Wall and R. Ranganathan, Nat. Struct. Biol., 2003, 10, 59–69 Search PubMed.
- M. Newman, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8577–8582 Search PubMed.
- M. G. Plach, P. Löffler, R. Merkl and R. Sterner, Angew. Chem., 2015, 54, 11270–11274 Search PubMed.
- J. B. Thoden, H. M. Holden, G. Wesenberg, F. M. Raushel and I. Rayment, Biochemistry, 1997, 36, 6305–6316 Search PubMed.
- H. Ashkenazy, E. Erez, E. Martz, T. Pupko and N. Ben-Tal, Nucleic Acids Res., 2010, 38, W529–W533 Search PubMed.
- C. Tsai, A. del Sol and R. Nussinov, J. Mol. Biol., 2009, 378, 1–11 Search PubMed.
- J. Monod, J. Wyman and J.-P. Changeux, J. Mol. Biol., 1965, 12, 88–118 Search PubMed.
- D. E. Koshland, G. Némethy and D. Filmer, Biochemistry, 1966, 5, 365–385 Search PubMed.
- A. del Sol, C.-J. Tsai, B. Ma and R. Nussinov, Structure, 2009, 17, 1042–1050 Search PubMed.
- I. Bahar, C. Chennubhotla and D. Tobi, Curr. Opin. Struct. Biol., 2007, 17, 633–640 Search PubMed.
- L. Vuillon and C. Lesieur, Curr. Opin. Struct. Biol., 2015, 31, 1–8 Search PubMed.
- K. Park and D. Kim, BMC Bioinf., 2011, 12, S23 Search PubMed.
- V. a. Feher, J. D. Durrant, A. T. Van Wart and R. E. Amaro, Curr. Opin. Struct. Biol., 2014, 25, 98–103 Search PubMed.
- X. Huang, H. M. Holden and F. M. Raushel, Annu. Rev. Biochem., 2001, 70, 149–180 Search PubMed.
- T. Gueudré, C. Baldassi, M. Zamparo, M. Weigt and A. Pagnani, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 12186–12191 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6mb00646a |
| ‡ Present address: Institute of Transformative Bio-molecules, Nagoya University, Nagoya, Japan. |
| This journal is © The Royal Society of Chemistry 2017 |