
The development of small-molecule modulators for ClpP protease activity
Fei
Ye
a,
Jiahui
Li
b and
Cai-Guang
Yang
*b
aCollege of Life Sciences, Zhejiang Sci-Tech University, Hangzhou, China
bLaboratory of Chemical Biology, State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China. E-mail: yangcg@simm.ac.cn
First published on 1st November 2016
Abstract
The global spread of antibiotic resistance among important human pathogens emphasizes the need to find new antibacterial drugs with a novel mode of action. The ClpP protease has been shown to demonstrate its pivotal importance to both the survival and the virulence of pathogenic bacteria during host infection. Deregulating ClpP activity either through overactivation or inhibition could lead to antibacterial activity, declaiming the dual molecular mechanism for small-molecule modulation. Recently, natural products acyldepsipeptides (ADEPs) have been identified as a new class of antibiotics that activate ClpP to a dysfunctional state in the absence of cognate ATPases. ADEPs in combination with rifampicin eradicate deep-seated mouse biofilm infections. In addition, several non-ADEP compounds have been identified as activators of the ClpP proteolytic core without the involvement of ATPases. These findings indicate a general principle for killing dormant cells, the activation and corruption of the ClpP protease, rather than through conventional inhibition. Deletion of the clpP gene reduced the virulence of Staphylococcus aureus, thus making it an ideal antivirulence target. Multiple inhibitors have been developed in order to attenuate the production of extracellular virulence factors of bacteria through covalent modifications on serine in the active site or disruption of oligomerization of ClpP. Interestingly, due to the unusual composition and activation mechanism of ClpP in Mycobacterium tuberculosis, mycobacteria are killed by ADEPs through inhibition of ClpP activity rather than overactivation. In this short review, we will summarize recent progress in the development of small molecules modulating ClpP protease activity for both antibiotics and antivirulence.
 Fei Ye | Fei Ye received a BS degree in Pharmacy from Zhejiang University in 2008 and earned a PhD in Drug Design from Shanghai Institute of Materia Medica, Chinese Academy of Sciences in 2013. In the same year, she was recruited to Zhejiang Sci-Tech University as an Assistant Professor. She is currently focusing on the discovery of novel lead compounds against various targets of diseases, such as bacterial infection and cancers. |
 Jiahui Li | Jiahui Li graduated from Henan Agricultural University in 2014 and received a BS degree in Biological Engineering. She is currently a PhD student in Yang's lab at Shanghai Institute of Materia Medica, Chinese Academy of Sciences. She is currently working on the development of novel anti-infective agents mainly targeting ClpP protease in Staphylococcus aureus. |
 Cai-Guang Yang | Cai-Guang Yang received a BS degree in Chemical Engineering from Huazhong University of Science and Technology and earned a PhD in Organic Chemistry from the Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences. He received his postdoctoral training in Structure Biology in the Chemistry Department at the University of Chicago. In 2008, he was recruited to Shanghai Institute of Materia Medica, Chinese Academy of Sciences, where he was appointed as a full Professor. Current research in his lab centres on the discovery of small-molecule modulators tackling undruggable proteins in order to probe biology and eventually develop new medicines. |
Introduction
In recent years, there has been an alarming increase in the number of bacterial strains resistant to most known antimicrobial agents, such as methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-resistant Enterococci (VRE), and penicillin-resistant Streptococcus pneumoniae (PRSP).1,2 To date, almost every class of antibiotics has encountered the problem of drug-resistance.3 As a result, many diseases that were thought to be controlled by currently available agents are re-emerging in hospitals, leading to a dramatic global health problem. Therefore, there is an urgent need for the discovery and development of novel antibiotics that can effectively treat multidrug-resistant bacteria.4 To avert cross-resistance with currently used antibacterial drugs, new classes of antibiotics with novel mechanisms of action are waiting to be discovered.In the past few decades, steady progress has been witnessed in the discovery of potential antibacterial drug targets.5 Caseinolytic protease P (ClpP), for example, has been the subject of numerous research studies. ClpP is a highly conserved serine protease present in bacteria and eukaryotes.6 The primary physiological role of the ClpP system is to maintain cellular homeostasis by controlled degradation of short-lived regulatory proteins as well as misfolded or damaged proteins, including those involved in regulating stress responses and virulence-factor production.7,8 Therefore, the ClpP protease plays an essential role in the virulence of pathogenic bacteria during host infection.9 Genetic clpP knockouts in S. aureus and Listeria monocytogenes resulted in strongly decreased extracellular virulence, leading to attenuated infections in murine abscess models.10,11 Taken together, the critical physiological roles of ClpP make it an attractive new target for the development of anti-infective agents.
The crystal structures from several different organisms revealed that ClpP monomers self-assemble into two heptameric rings, forming a cylindrical tetradecamer which enclose a large chamber containing 14 proteolytic active sites (Ser–His–Asp).12–15 ClpP switches dynamically between three states in order to perform its biological function: the active extended, the inactive compressed, and the intermediate compact state (Fig. 1a–c).15 Under normal physiological conditions, ClpP functions in conjunction with its AAA+ partners (ATPases associated with diverse cellular activities) such as ClpX and ClpA, which recognize, unfold, and translocate substrates into the proteolytic chamber of ClpP for degradation through axial pores lined by axial loops.16–21 Cryo-electron microscopy (EM)22 has revealed that binding of ATPases induces the enlargement of the axial pores of the tetradecameric ClpP chamber. Structural evidence12,23–28 and biochemical date29–33 have demonstrated that the major interactions of ATPases and ClpP proteases occur between the conserved IGF/L loops of ATPase and the hydrophobic pockets (H pocket) on the surface of ClpP (Fig. 1d). Only small peptides with six or fewer amino acids could be degraded without the involvement of ATPase chaperones.34
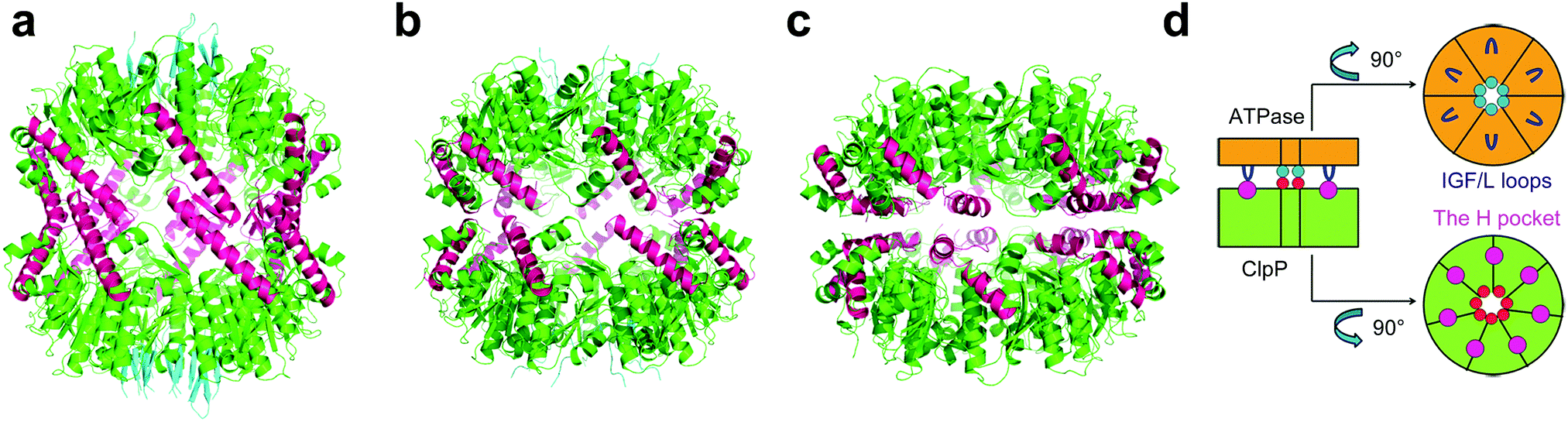 | ||
| Fig. 1 (a) Crystal structure of SaClpP (PDB ID: 3STA) showing the ClpP tetradecamer in the extended state. (b) Structure of SaClpP (PDB ID: 4EMM) showing ClpP in the compact state. (c) Structure of SaClpP (PDB ID: 3ST9) showing the enzyme in the compressed state. (d) Cartoons showing interactions of ClpX hexamer (orange) and ClpP heptamer (green) between ClpX IGF/L loops (blue) and hydrophobic ClpP pockets (magenta). | ||
Due to the essential role of ClpP in bacterial homeostasis and pathogenicity, both the overactivation and inhibition of this protease lead to bactericidal activity. Recently, several small molecules that perturb the physiological function of ClpP have been reported, which can be classified as either activators or inhibitors. Treatment of ClpP activators results in bacteria inhibition or death,35 whereas chemical knockout of clpP by ClpP inhibitors leads to a global decrease in virulence factor production,36–38 indicating the dual molecular mechanism for ClpP modulation. In this short review, we will summarize recent progress in the development of small-molecule modulators targeting ClpP.
ADEPs and derivatives mimic the docking interaction between ClpP and ATPases, leading to overactivation of ClpP
As we discussed above, in the absence of ATPase chaperones, ClpP can only degrade small peptides (<six amino acids in length).34 ADEPs, a new class of natural antibiotics,39,40 were reported to target ClpP protease.41 Remarkably, these compounds do not kill bacteria by inhibiting the essential protein in bacteria, but rather indiscriminately increase the activity of ClpP, converting it to an unregulated protease that is capable of degrading unfolded and nascent proteins without the involvement of the ATPase.41–43 Dysregulation of the ClpP protease can prevent physiological functions in bacteria, leading to inhibition of cell division and cell death.41,42,44 Collectively, ADEPs have been observed to exhibit potent activity against a broad range of Gram-positive bacterial pathogens (e.g., S. aureus, S. pneumoniae, Enterococci, and M. tuberculosis) and efficacy in animal models of bacterial infection.41,45–48The representative members of ADEP antibiotics are A54556A and B isolated from Streptococcus hawaiiensis (Fig. 2a).39 The correct structure of A54556A (also denoted as ADEP1) was determined by Brötz-Oesterhelt and colleagues, based on which they synthesized several improved congeners.41 Although the natural products have potent antibacterial activities in vitro, they were found to be inactive in mouse models of lethal bacterial infection due to their poor solubility, chemical lability and rapid systemic clearance.47 To optimize the structures of ADEPs, preliminary structure–activity relationship (SAR) studies were carried out, which found that replacement of the N-methylalanine residue in the core macrocycle with a rigid pipecolate moiety lowers the entropic cost of ClpP binding. Besides, the chemical stability and bioavailability of the compounds could be further enhanced by substitution of the polyunsaturated side chain with a heptenoyl moiety and by replacement of phenylalanine in its side chain with 3,5-difluorophenylalanine. Consequently, the optimized derivative, ADEP4, exhibited more chemical stability and 160-fold enhanced activity (Fig. 2b).47 Importantly, ADEP4 has remarkable activity in vivo. Recently, it has been reported that combining ADEP4 with rifampicin kills bacterial persister cells of pathogenic S. aureus in vitro and in mouse models of chronic infection, suggesting a realistic path for developing therapies to treat chronic infections.49 Afterwards, Sello's group showed that the conformational flexibility of the core structure of ADEP peptidolactone strongly influences ClpP binding, replacing amino acids in the ADEP macrocycle with conformationally constrained residues, such as 4-methylpipecolate (Fig. 2c)45 and allo-threonine (ADEP B315, Fig. 2d),50 could further improve antibacterial activity. ADEP B315 is highly effective in mice that have lethal infections of methicillin-sensitive and resistant strains of S. aureus.51
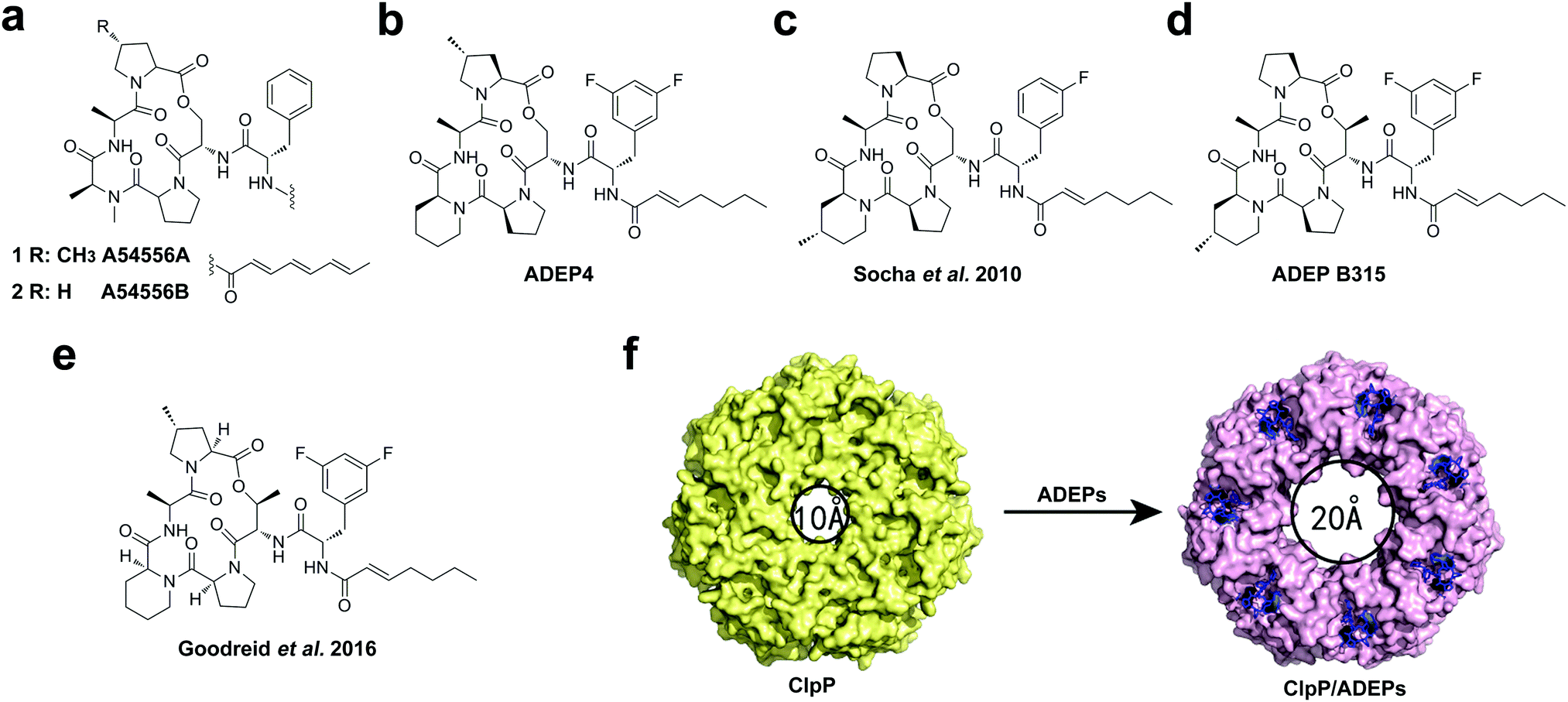 | ||
| Fig. 2 (a–e) Chemical structures of ADEP natural products and optimized synthetic analogues thereof. (f) ADEPs bind to the H pockets of ClpP, leading to the enlargement of the axial pores. ADEPs are shown as blue sticks. | ||
N-Acylphenylalanine moiety, a simple fragment of ADEPs is both necessary and sufficient for ClpP overactivation and antibacterial activity.52 This moiety is not simply a stand-in of the IGF/L loops of ATPase; it likely has enhanced physicochemical properties that are better suited for ClpP binding.53 Most of the ADEP antibiotics mentioned above are active against Gram-positive bacteria. Recently, optimization of the macrocyclic core residues and the N-acyl side chain led to an ADEP derivative (Fig. 2e), which shows potent activity against Gram-negative bacteria including Neisseria meningitidis and Neisseria gonorrheae, as well as increased activity against the Gram-positive species S. aureus and Enterococcus faecalis compared to the known derivatives.54
The structural changes of ClpP underlying activation by ADEPs have been determined by the co-crystal structures of Bacillus subtilis ClpP (BsClpP), Escherichia coli ClpP (EcClpP), M. tuberculosis ClpP (MtClpP1P2), and N. meningitides ClpP (NmClpP) bound to ADEPs.54–57 Analysis of these complex structures indicated that the activators bind in the H pockets located between adjacent subunits on the apical surface of ClpP, mimicking the docking interaction between the conserved IGF/L loops of ATPase and ClpP (Fig. 2f).55–57 A consequence of this competitive binding is reorientation of the ClpP subunits, leading to enlargement of the entrance pore, which allows substrates to access the degradation chamber in an uncontrolled way and results in cell death (Fig. 2f).42,44,58 Recently, we have found that there is a key component in the H pocket, Y63, that interacts with ATPases or ADEPs, the conformational twist of the side chain of Y63 that, when triggered by ATPase or ADEP binding, initiates the activation of ClpP.59 The understanding of the activation mechanism may aid in the development of small-molecule activators of ClpP through targeting the H pocket.
Small-molecule activators of ClpP may bind in the H pocket or allosteric site
Despite the potent bactericidal activity of ADEPs and related derivatives, the availability of this natural product family has been limited due to the lack of activity against Gram-negative pathogens and susceptibility to active efflux.41 In fact, the N-acylated phenylalanine pharmacophore of ADEPs is the recognized motif for drug efflux pumps.60 Hence, efforts have been made to discover non-ADEP activators in order to investigate the mechanism of ClpP activation and develop antibiotics with broad-spectrum activities.By using a high-throughput screening approach with a fluorescence-based readout against commercially available compound libraries, Leung and co-workers have identified several structurally diverse non-ADEP compounds that could activate the EcClpP protease in the absence of ATPases, called activators of self-compartmentalizing proteases 1 to 5 (ACP1 to 5, Fig. 3a).35,61 The chemical optimization of the most potent activator ACP1, which also displayed good drug-like characteristics, yielded compounds which had in vitro ClpP activation properties comparable to that of ADEPs (ACP1a-b, Fig. 3a). Notably, ACP1b also showed good bactericidal properties in cells, implying its potential application in therapeutics. Similar to ADEPs, the binding of ACP compounds stabilizes ClpP and promotes the formation of the double-ring tetradecamer.35,61 In the following work, the fluorescence-based protease assay was utilized to screen a library containing ∼450 structurally diverse fungal and bacterial secondary metabolites.62 Sclerotiamide, a paraherquamide-related indolinone, was identified as the first non-ADEP natural product activator of ClpP (Fig. 3b). The SAR study revealed that the C-10 α-hydroxy motif is a necessary group for ClpP activation. However, Sclerotiamide shows modest potency compared to ADEPs and ACPs, thus improvement of activity is required for further applications.62
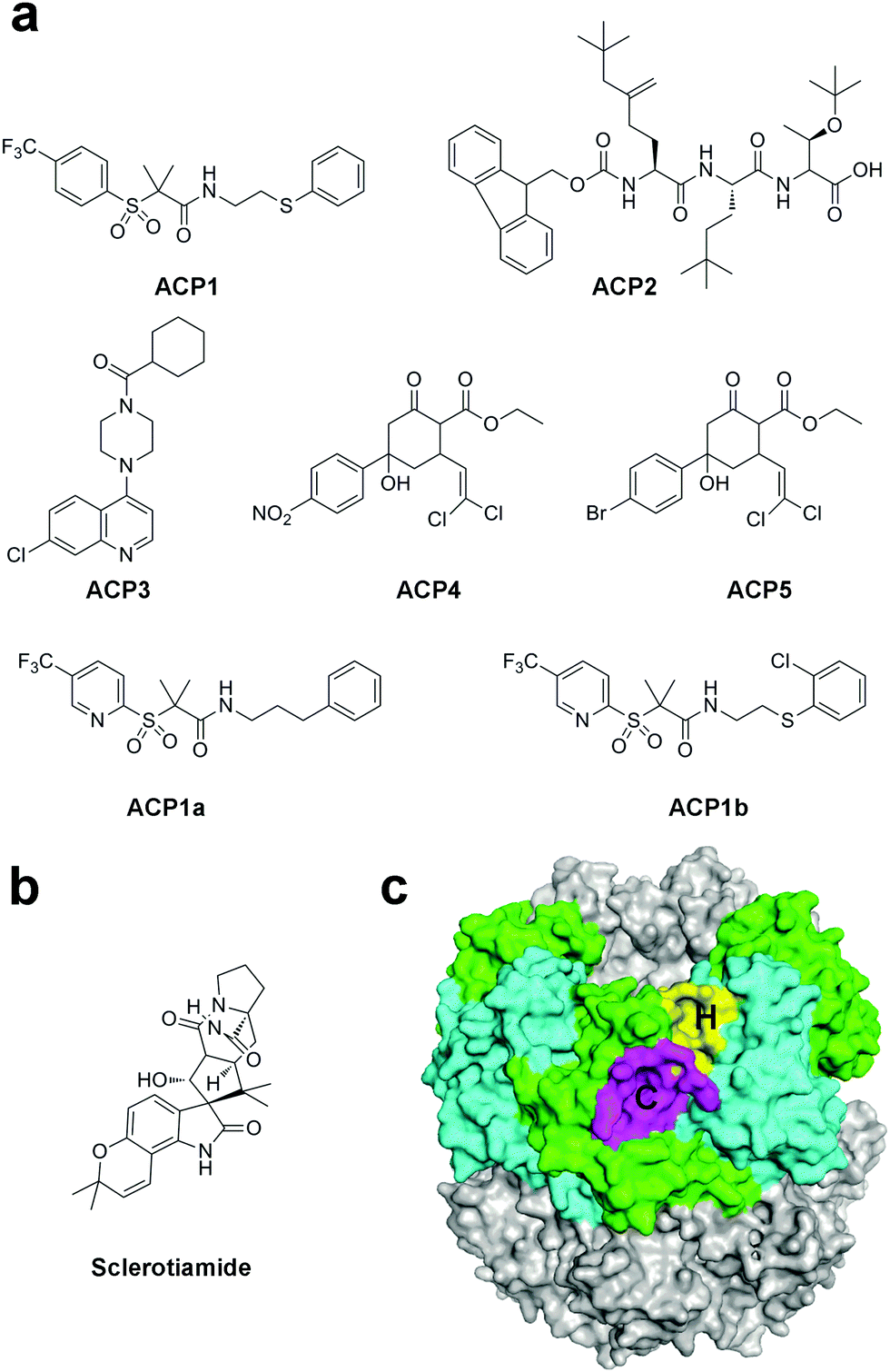 | ||
| Fig. 3 (a) Chemical structures of ACP1–ACP5 as well as of ACP1a and ACP1b. (b) Chemical structures of sclerotiamide. (c) Surface model of ClpP (PDB ID: 3MT6). The H pockets are colored in yellow, while the C pockets are colored in magenta. | ||
As we discussed above, the crystal structures of ClpP/ADEP complexes revealed that ADEPs occupy the H pocket on the apical surface of ClpP, therefore preventing the binding of the IGF/L motif of ATPase. The molecular docking simulation combined with mutational analysis suggested that the stronger ACPs bind to the H pocket, whereas the weaker ACPs appear to bind to both the H pocket and a second pocket called the C pocket, which comprises residues Y90, M94, Q95, D100, V101, and H170 of one subunit and residues H205 and N207 of the adjacent subunit (Fig. 3c). The two pockets are separated by residues at the very C terminus of ClpP.38 Recently, steady progress has been witnessed in developing allosteric drugs, which provide many benefits compared to normal orthosteric drugs.63 Hence, the C pocket could be used as an allosteric site to develop novel ClpP activators.
The development of active-site-directed inhibitors targeting ClpP
By utilizing a chemical proteomic strategy called activity-based protein profiling (ABPP),64,65 Sieber and co-workers identified a selection of trans-β-lactone probes, which are cell-permeable and can sensitively label and inhibit ClpP of nonpathogenic bacterial strains.66 Inspired by these results, a small library of reactive β-lactone probes was applied to S. aureus cells and their corresponding MRSA strains.36 After cell lysis, an azid-rhodamine tag was appended to the labeled proteins of the cytosolic proteome fraction via click chemistry. Labeled proteomes were separated via SDS-PAGE analysis and detected using fluorescence scanning.67–69 Three lactone probes, D3, G2 and E2, were identified as novel inhibitors for specific and selective targeting of ClpP in S. aureus and MRSA strains (Fig. 4a). Among them, the most potent compound D3 is able to completely abolish or significantly decrease the expression of major extracellular virulence factors including hemolysins, proteases, lipases, and DNases, which are key players in the elimination of the host immune response, tissue necrosis, and inflammation.10,36 Different from traditional antibiotic chemotherapy, targeting virulence factors would not necessarily kill bacteria, but neutralize the harmful effects of bacterial pathogens. Thereby it could lead to the elimination of disarmed bacteria and less selective pressure on bacteria and resistance development.3,70,71 The phenotypic properties of the chemical clpP knockout by ClpP inhibitors exactly correspond to those reported for the ΔclpP mutant.10 Subsequent structural refinement yielded optimized inhibitors of ClpP, lactone U1 and U1P with higher potency (Fig. 4a).37,72 U1 shows 3 to 5-fold increased potency than lactone D3 in the inhibition of crucial virulence factors in S. aureus and MRSA strains.37 Furthermore, inhibitor U1 is able to abolish the production of the devastating pyrogenic toxin superantigens (PTSAs) including enterotoxins and toxic shock syndrome toxin 1 (TSST-1),37 which is directly related to severe diseases such as toxic shock syndrome.73,74 In addition, the lactone U1 down-regulates expression of crucial virulence factors for L. monocytogenes pathogenesis including listeriolysin O (LLO) and phospholipases C (PLC),75 which are important for the invasion of Listeria from the phagosome to the eukaryotic host cell.76,77 Consequently, inhibitor U1 decreases the intracellular replication of L. monocytogenes in mouse macrophages by about 24%.75 Lactone U1 is also able to block the activity of the ClpP protease in Plasmodium falciparum, thereby inhibiting the development of the parasite apicoplast as well as the progression of the asexual stages of parasites.78 Moreover, β-lactones were found to be toxic to M. tuberculosis through inhibition of ClpP1P2 peptidase.38 Overall, these data emphasized the utility of β-lactone-mediated virulence inhibition for an intracellular pathogen. Follow-up in vivo skin infection studies showed that lactone U1 is effective for the treatment of subcutaneous S. aureus abscesses in mice.79 A single dose of U1 resulted in a significant decrease in abscess size even when applied 6 h post-infection.79 Besides, A2-32-01, a synthesized β-lactone, could inhibit the enzymatic activity of recombinant human mitochondrial ClpP, thereby being cytotoxic to acute myeloid leukemia (AML) cells.80,81 Taken together, these data underscore the fact that the chemical inhibition of ClpP sheds light on the development of potential drugs for anti-virulence, anti-infective and anti-AML therapies.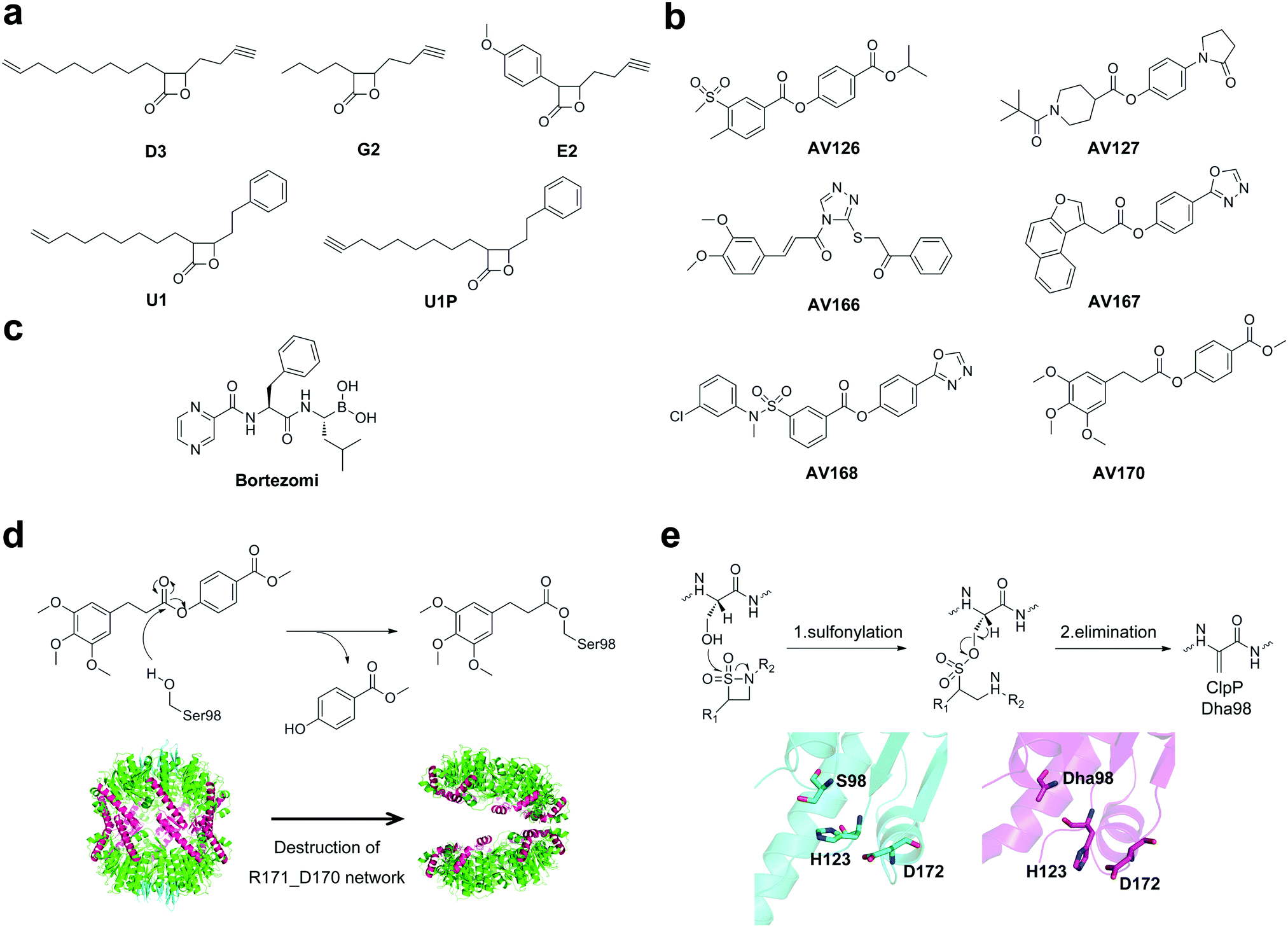 | ||
| Fig. 4 (a) Chemical structures of β-lactone inhibitors of ClpP. (b) Chemical structures of phenyl ester inhibitors of ClpP. (c) Chemical structures of bortezomib. (d) Upper panel, suggested mode of action for modification on S98 in the active site of ClpP, illustrated for AV170; bottom panel, modification of the active site leads to ClpP deoligomerization. (e) Upper panel, mechanism for the formation of Dha involving sulfonylation of the active site serine and subsequent elimination; bottom panel, crystal structure of wild-type SaClpP (cyan, PDB ID: 3STA) and Dha–SaClpP (magenta, PDB ID: 4MXI). | ||
Although β-lactones have been applied as chemical tools for the investigation of ClpP in several organisms,37,75,82 the limitation of potency, selectivity and stability restricted their applications. Since β-lactones are labile electrophiles which quickly hydrolyze in human plasma within minutes,79 identification of ClpP inhibitors with more stable moieties is necessary. A novel class of phenyl ester and triazole amide compounds was discovered based on an unbiased high-throughput screen (HTS) against more than 137![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 compounds (Fig. 4b).83 Compared to β-lactones, phenyl ester inhibitors exhibit superior potency, inhibition kinetics, plasma half-life, and target selectivity.83 Besides, phenyl ester compounds were selective for bacterial, but not for human ClpP, thus the potency of phenyl esters largely exceeded that of β-lactones. In addition, based on a target mechanism-based whole-cell screening method, bortezomi, a 26S proteasome drug, was identified as a potent inhibitor of MtClpP1P2 and bacterial growth (Fig. 4c).84
000 compounds (Fig. 4b).83 Compared to β-lactones, phenyl ester inhibitors exhibit superior potency, inhibition kinetics, plasma half-life, and target selectivity.83 Besides, phenyl ester compounds were selective for bacterial, but not for human ClpP, thus the potency of phenyl esters largely exceeded that of β-lactones. In addition, based on a target mechanism-based whole-cell screening method, bortezomi, a 26S proteasome drug, was identified as a potent inhibitor of MtClpP1P2 and bacterial growth (Fig. 4c).84
Analytical studies revealed that β-lactone, phenyl esters and bortezomi covalently modify the active site of ClpP,36,83 causing either dissociation of the proteolytic complex into inactive heptamers or retention of the tetradecameric state (Fig. 4d and e). For example, β-lactones E2 and phenyl esters, which mimic peptide binding, undergo nucleophilic attack by S98 in the catalytic site, resulting in the acyl–enzyme intermediate (Fig. 4d). The steric bulk at the active site might move the catalytic H123, thereby shifting the catalytic D172. As a result, the R171–D170 network located at the equator of the tetradecameric cylinder, which is important for the stability of tetradecamers,15 was disrupted, leading to ClpP deoligomerization (Fig. 4d).52,83 A different inhibition mechanism for ClpP is conversion of the active site serine into a dehydroalanine (Dha) by β-sultams through sulfonylation and subsequent elimination (Fig. 4e), which was confirmed using mass spectrometry and crystallography data.52 The crystal structure of Dha–SaClpP revealed that no other structural changes were observed by comparison of the modified enzyme with the wild-type protein.52 These findings extend the view on protease inhibition and provide probes to investigate the activity and structure of ClpP.
Reversible inhibitors arrest ClpP in a defined conformation that can be revoked by ClpX binding
Both β-lactones and phenyl esters covalently modify the serine residue in the active site of ClpP, however, the application of covalent, irreversible inhibitors are limited because of the low stability of their electrophilic motifs.80 Compound AV145 is the first non-covalent inhibitor of ClpP protease (Fig. 5a).85 The crystal structure of SaClpP in complex with AV145 revealed that AV145 binds to the pocket close to the active site between α-helix E and strand β9 (Fig. 5b). AV145 binding leads to a significant rotation of the conserved residue P125 and distortion of the catalytic triad, thereby ClpP is arrested in an unprecedented inactive conformational state and lost the peptidase activity (Fig. 5b). The novel binding mode provides key information regarding structural optimization, resulting in several derivatives with increased inhibitor potency. Among them, compound AV286 with an IC50 value of <1 μM is the most potent non-covalent inhibitor (Fig. 5a). However, ClpX overrides the inhibitor-induced conformational lock. Thus, regulation of inhibitor binding by ClpX chaperones represents an important strategy for future drug development through targeting ClpP.85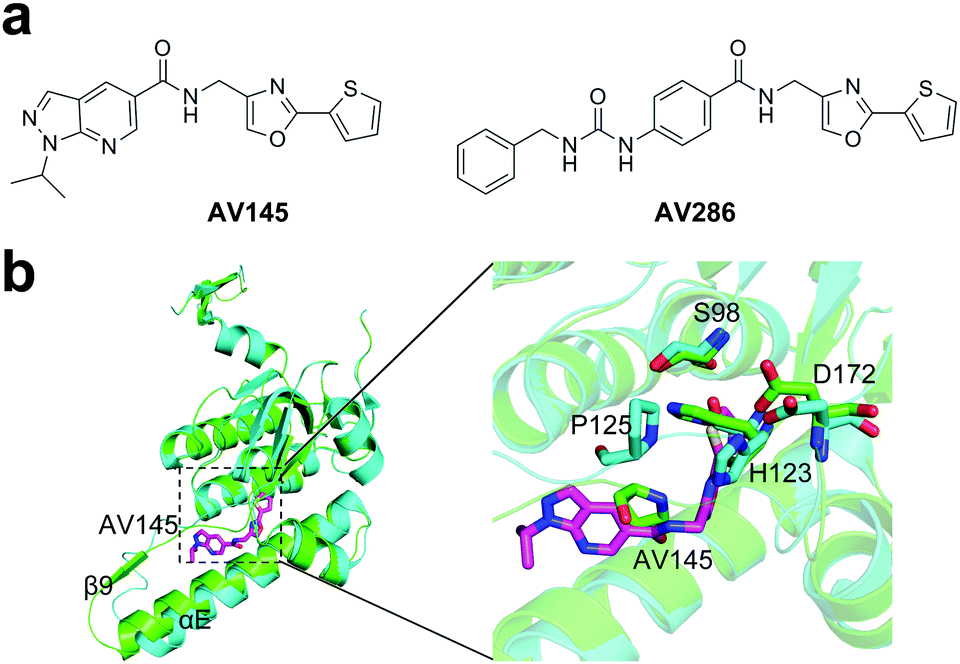 | ||
| Fig. 5 (a) Chemical structures of AV145 and AV286. (b) Left panel, binding of AV145 to SaClpP (PDB ID: 5DL1). Right panel, detailed exploration of the structural changes of the active site and P125 compared to the extended state. | ||
ADEPs kill mycobacteria through inhibition of ClpP function, by occupying the ATPase-binding site rather than conformational activation
ADEPs inhibit the growth of Gram-positive bacterial pathogens by activating ClpP and causing dysregulated protein degradation. However, a recent study revealed that mycobacteria are killed by ADEPs through inhibition of ClpP function.48 Different from most bacterial species possessing a single ClpP protein, M. tuberculosis contains two ClpPs, ClpP1 and ClpP2, which together form the ClpP1P2 tetradecamer. Additional activating factors such as the dipeptide benzyloxycarbonyl-leucyl-leucine (Z-LL) are needed in the organization of the active tetradecamer.86 The co-crystal structure of MtClpP1P2/ADEPs indicates that ADEPs lie in the H pocket on the surface of the ClpP2 ring.57 Although ADEPs were shown to possess antibacterial activities against M. tuberculosis by stimulating MtClpP1P2 to degrade unstructured proteins and larger peptides,46,57 the effect is weaker than that for ClpP from other bacteria and requires the involvement of an additional activating factor.48 The cell division protein FtsZ, which is dysfunctionally degraded by AEDP-activated ClpP in other bacteria, is not degraded in mycobacteria in the presence of ADEPs. Hence, it is demonstrated that ADEPs kill mycobacteria by preventing the interaction between ClpP1P2 and its cognate Clp-ATPases.86 The different mode of action of ADEPs reveals that the H pocket is a potential target for the development of both activators and inhibitors based on its dual molecular mechanism.Concluding remarks
Collectively, this short review reinforces the potential of ClpP as a new antibacterial drug target and provides a number of novel structural scaffolds and diverse mechanisms from which novel modulators of ClpP activity could be further developed. Of note, the hydrophobic pocket, which is the binding site of ATPase, ADEPs activators, is a significant site for the development of small-molecule activators of ClpP protease. The binding of the modulators to these sites allows for differential regulation of protease activity. This defines a fundamental principle for antibiotics development through dysfunctional activation of ClpP by small-molecule activators. It is clear now that such activators need to be able to efficiently twist the conformation of the side chain of Tyr63.59 On the other hand, the hydrophobic pocket is also a potential target for the development of ClpP inhibitors by disruption of the binding of ATPases to ClpP. In addition to the antibacterial potential, inhibition of ClpP in human mitochondria may be used as a therapeutic strategy for the treatment of AML.80,81 Thus, it is important to note that the selectivity of inhibitors for bacterial ClpP over the mitochondrial ClpP must be adequately demonstrated. Developing potent, stable, and specific ClpP inhibitors should be based on the sequence homology and structural diversity of ClpP in different organisms. In addition, cellular target validations were performed by ABPP for most ClpP inhibitors.36,66,72 However, the direct interaction between ClpP and most activators in cells was not investigated, which should be considered in future studies.In a more general sense, this review highlights the fact that antibiotics may perform their functions via multiple pathways; the highly conserved proteolytic systems in bacteria are excellent targets for the development of novel anti-infective agents as either inhibiting or overactivating these systems.
Acknowledgements
We gratefully acknowledge financial support from the Public Projects of Zhejiang Province (2015C33159 to F. Y.), the Zhejiang Province Natural Science Foundation (LQ14H300003 to F. Y.), the National Natural Science Foundation of China (81402849 to F. Y., and 21172234 to C. Y.), and the Institute for Drug Discovery and Development, Chinese Academy of Sciences (CASIMM0120154016 to C. Y.).References
- C. Walsh and G. Wright, Chem. Rev., 2005, 105, 391–394 CrossRef CAS PubMed.
- K. J. Williams and R. P. Bax, Curr. Opin. Invest. Drugs, 2009, 10, 157–163 CAS.
- A. E. Clatworthy, E. Pierson and D. T. Hung, Nat. Chem. Biol., 2007, 3, 541–548 CrossRef CAS PubMed.
- M. A. Fischbach and C. T. Walsh, Science, 2009, 325, 1089–1093 CrossRef CAS PubMed.
- R. M. Raju, A. L. Goldberg and E. J. Rubin, Nat. Rev. Drug Discovery, 2012, 11, 777–789 CrossRef CAS PubMed.
- P. Wong and W. A. Houry, J. Struct. Biol., 2004, 146, 79–89 CrossRef CAS PubMed.
- R. T. Sauer, D. N. Bolon, B. M. Burton, R. E. Burton, J. M. Flynn, R. A. Grant, G. L. Hersch, S. A. Joshi, J. A. Kenniston, I. Levchenko, S. B. Neher, E. S. Oakes, S. M. Siddiqui, D. A. Wah and T. A. Baker, Cell, 2004, 119, 9–18 CrossRef CAS PubMed.
- T. A. Baker and R. T. Sauer, Trends Biochem. Sci., 2006, 31, 647–653 CrossRef CAS PubMed.
- D. Frees, A. Chastanet, S. Qazi, K. Sorensen, P. Hill, T. Msadek and H. Ingmer, Mol. Microbiol., 2004, 54, 1445–1462 CrossRef CAS PubMed.
- D. Frees, S. N. Qazi, P. J. Hill and H. Ingmer, Mol. Microbiol., 2003, 48, 1565–1578 CrossRef CAS PubMed.
- O. Gaillot, S. Bregenholt, F. Jaubert, J. P. Di Santo and P. Berche, Infect. Immun., 2001, 69, 4938–4943 CrossRef CAS PubMed.
- J. Wang, J. A. Hartling and J. M. Flanagan, Cell, 1997, 91, 447–456 CrossRef CAS PubMed.
- A. Y. Yu and W. A. Houry, FEBS Lett., 2007, 581, 3749–3757 CrossRef CAS PubMed.
- J. Zhang, F. Ye, L. Lan, H. Jiang, C. Luo and C. G. Yang, J. Biol. Chem., 2011, 286, 37590–37601 CrossRef CAS PubMed.
- F. Ye, J. Zhang, H. Liu, R. Hilgenfeld, R. Zhang, X. Kong, L. Li, J. Lu, X. Zhang, D. Li, H. Jiang, C. G. Yang and C. Luo, J. Biol. Chem., 2013, 288, 17643–17653 CrossRef CAS PubMed.
- F. Beuron, M. R. Maurizi, D. M. Belnap, E. Kocsis, F. P. Booy, M. Kessel and A. C. Steven, J. Struct. Biol., 1998, 123, 248–259 CrossRef CAS PubMed.
- T. Ishikawa, F. Beuron, M. Kessel, S. Wickner, M. R. Maurizi and A. C. Steven, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 4328–4333 CrossRef CAS PubMed.
- A. Martin, T. A. Baker and R. T. Sauer, Nat. Struct. Mol. Biol., 2008, 15, 139–145 CAS.
- J. R. Hoskins, M. Pak, M. R. Maurizi and S. Wickner, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 12135–12140 CrossRef CAS.
- S. E. Glynn, A. R. Nager, T. A. Baker and R. T. Sauer, Nat. Struct. Mol. Biol., 2012, 19, 616–622 CAS.
- J. Ortega, S. K. Singh, T. Ishikawa, M. R. Maurizi and A. C. Steven, Mol. Cell, 2000, 6, 1515–1521 CrossRef CAS PubMed.
- G. Effantin, T. Ishikawa, G. M. De Donatis, M. R. Maurizi and A. C. Steven, Structure, 2010, 18, 553–562 CrossRef CAS PubMed.
- M. C. Bewley, V. Graziano, K. Griffin and J. M. Flanagan, J. Struct. Biol., 2006, 153, 113–128 CrossRef CAS PubMed.
- S. E. Glynn, A. Martin, A. R. Nager, T. A. Baker and R. T. Sauer, Cell, 2009, 139, 744–756 CrossRef CAS PubMed.
- F. Guo, M. R. Maurizi, L. Esser and D. Xia, J. Biol. Chem., 2002, 277, 46743–46752 CrossRef CAS PubMed.
- D. Y. Kim and K. K. Kim, J. Biol. Chem., 2003, 278, 50664–50670 CrossRef CAS PubMed.
- A. Szyk and M. R. Maurizi, J. Struct. Biol., 2006, 156, 165–174 CrossRef CAS PubMed.
- B. M. Stinson, A. R. Nager, S. E. Glynn, K. R. Schmitz, T. A. Baker and R. T. Sauer, Cell, 2013, 153, 628–639 CrossRef CAS PubMed.
- Y. I. Kim, I. Levchenko, K. Fraczkowska, R. V. Woodruff, R. T. Sauer and T. A. Baker, Nat. Struct. Biol., 2001, 8, 230–233 CrossRef CAS PubMed.
- A. Martin, T. A. Baker and R. T. Sauer, Mol. Cell, 2007, 27, 41–52 CrossRef CAS PubMed.
- S. K. Singh, J. Rozycki, J. Ortega, T. Ishikawa, J. Lo, A. C. Steven and M. R. Maurizi, J. Biol. Chem., 2001, 276, 29420–29429 CrossRef CAS PubMed.
- S. A. Joshi, G. L. Hersch, T. A. Baker and R. T. Sauer, Nat. Struct. Mol. Biol., 2004, 11, 404–411 CAS.
- A. Gribun, M. S. Kimber, R. Ching, R. Sprangers, K. M. Fiebig and W. A. Houry, J. Biol. Chem., 2005, 280, 16185–16196 CrossRef CAS PubMed.
- S. Gottesman, M. R. Maurizi and S. Wickner, Cell, 1997, 91, 435–438 CrossRef CAS PubMed.
- E. Leung, A. Datti, M. Cossette, J. Goodreid, S. E. McCaw, M. Mah, A. Nakhamchik, K. Ogata, M. El Bakkouri, Y. Q. Cheng, S. J. Wodak, B. T. Eger, E. F. Pai, J. Liu, S. Gray-Owen, R. A. Batey and W. A. Houry, Chem. Biol., 2011, 18, 1167–1178 CrossRef CAS PubMed.
- T. Bottcher and S. A. Sieber, J. Am. Chem. Soc., 2008, 130, 14400–14401 CrossRef PubMed.
- T. Bottcher and S. A. Sieber, ChemBioChem, 2009, 10, 663–666 CrossRef PubMed.
- C. L. Compton, K. R. Schmitz, R. T. Sauer and J. K. Sello, ACS Chem. Biol., 2013, 8, 2669–2677 CrossRef CAS PubMed.
- R. E. K. K. H. Michel, US Pat., 4492650, 1985 Search PubMed.
- H. Osada, T. Yano, H. Koshino and K. Isono, J. Antibiot., 1991, 44, 1463–1466 CrossRef CAS PubMed.
- H. Brötz-Oesterhelt, D. Beyer, H. P. Kroll, R. Endermann, C. Ladel, W. Schroeder, B. Hinzen, S. Raddatz, H. Paulsen, K. Henninger, J. E. Bandow, H. G. Sahl and H. Labischinski, Nat. Med., 2005, 11, 1082–1087 CrossRef PubMed.
- J. Kirstein, A. Hoffmann, H. Lilie, R. Schmidt, H. Rubsamen-Waigmann, H. Brötz-Oesterhelt, A. Mogk and K. Turgay, EMBO Mol. Med., 2009, 1, 37–49 CrossRef CAS PubMed.
- M. Gersch, M. Stahl, M. Poreba, M. Dahmen, A. Dziedzic, M. Drag and S. A. Sieber, ACS Chem. Biol., 2016, 11, 389–399 CrossRef CAS PubMed.
- P. Sass, M. Josten, K. Famulla, G. Schiffer, H. G. Sahl, L. Hamoen and H. Brötz-Oesterhelt, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 17474–17479 CrossRef CAS PubMed.
- A. M. Socha, N. Y. Tan, K. L. LaPlante and J. K. Sello, Bioorg. Med. Chem., 2010, 18, 7193–7202 CrossRef CAS PubMed.
- J. Ollinger, T. O'Malley, E. A. Kesicki, J. Odingo and T. Parish, J. Bacteriol., 2012, 194, 663–668 CrossRef CAS PubMed.
- B. Hinzen, S. Raddatz, H. Paulsen, T. Lampe, A. Schumacher, D. Habich, V. Hellwig, J. Benet-Buchholz, R. Endermann, H. Labischinski and H. Brötz-Oesterhelt, ChemMedChem, 2006, 1, 689–693 CrossRef CAS PubMed.
- K. Famulla, P. Sass, I. Malik, T. Akopian, O. Kandror, M. Alber, B. Hinzen, H. Ruebsamen-Schaeff, R. Kalscheuer, A. L. Goldberg and H. Brötz-Oesterhelt, Mol. Microbiol., 2016, 101, 194–209 CrossRef CAS PubMed.
- B. P. Conlon, E. S. Nakayasu, L. E. Fleck, M. D. LaFleur, V. M. Isabella, K. Coleman, S. N. Leonard, R. D. Smith, J. N. Adkins and K. Lewis, Nature, 2013, 503, 365–370 CrossRef CAS PubMed.
- D. W. Carney, K. R. Schmitz, J. V. Truong, R. T. Sauer and J. K. Sello, J. Am. Chem. Soc., 2014, 136, 1922–1929 CrossRef CAS PubMed.
- M. Arvanitis, G. Li, D. D. Li, D. Cotnoir, L. Ganley-Leal, D. W. Carney, J. K. Sello and E. Mylonakis, PLoS One, 2016, 11, e0153912 Search PubMed.
- M. Gersch, R. Kolb, F. Alte, M. Groll and S. A. Sieber, J. Am. Chem. Soc., 2014, 136, 1360–1366 CrossRef CAS PubMed.
- D. W. Carney, K. R. Schmitz, A. C. Scruse, R. T. Sauer and J. K. Sello, ChemBioChem, 2015, 16, 1875–1879 CrossRef CAS PubMed.
- J. D. Goodreid, J. Janetzko, J. P. Santa Maria, Jr., K. S. Wong, E. Leung, B. T. Eger, S. Bryson, E. F. Pai, S. D. Gray-Owen, S. Walker, W. A. Houry and R. A. Batey, J. Med. Chem., 2016, 59, 624–646 CrossRef CAS PubMed.
- B. G. Lee, E. Y. Park, K. E. Lee, H. Jeon, K. H. Sung, H. Paulsen, H. Rubsamen-Schaeff, H. Brötz-Oesterhelt and H. K. Song, Nat. Struct. Mol. Biol., 2010, 17, 471–478 CAS.
- D. H. Li, Y. S. Chung, M. Gloyd, E. Joseph, R. Ghirlando, G. D. Wright, Y. Q. Cheng, M. R. Maurizi, A. Guarne and J. Ortega, Chem. Biol., 2010, 17, 959–969 CrossRef CAS PubMed.
- K. R. Schmitz, D. W. Carney, J. K. Sello and R. T. Sauer, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E4587–4595 CrossRef CAS PubMed.
- M. Gersch, K. Famulla, M. Dahmen, C. Gobl, I. Malik, K. Richter, V. S. Korotkov, P. Sass, H. Rubsamen-Schaeff, T. Madl, H. Brötz-Oesterhelt and S. A. Sieber, Nat. Commun., 2015, 6, 6320 CrossRef CAS PubMed.
- T. Ni, F. Ye, X. Liu, J. Zhang, H. Liu, J. Li, Y. Zhang, Y. Sun, M. Wang, C. Luo, H. Jiang, L. Lan, J. Gan, A. Zhang, H. Zhou and C. G. Yang, ACS Chem. Biol., 2016, 11, 1964–1972 CrossRef CAS PubMed.
- D. W. Carney, C. L. Compton, P. V. Groomes and J. K. Sello, ACS Infect. Dis., 2015, 1, 53–58 CrossRef PubMed.
- D. A. Dougan, Chem. Biol., 2011, 18, 1072–1074 CrossRef CAS PubMed.
- N. P. Lavey, J. A. Coker, E. A. Ruben and A. S. Duerfeldt, J. Nat. Prod., 2016, 79, 1193–1197 CrossRef CAS PubMed.
- J. Pei, N. Yin, X. Ma and L. Lai, J. Am. Chem. Soc., 2014, 136, 11556–11565 CrossRef CAS PubMed.
- M. J. Evans and B. F. Cravatt, Chem. Rev., 2006, 106, 3279–3301 CrossRef CAS PubMed.
- B. F. Cravatt, A. T. Wright and J. W. Kozarich, Annu. Rev. Biochem., 2008, 77, 383–414 CrossRef CAS PubMed.
- T. Bottcher and S. A. Sieber, Angew. Chem., 2008, 47, 4600–4603 CrossRef PubMed.
- R. Manetsch, A. Krasinski, Z. Radic, J. Raushel, P. Taylor, K. B. Sharpless and H. C. Kolb, J. Am. Chem. Soc., 2004, 126, 12809–12818 CrossRef CAS PubMed.
- A. E. Speers, G. C. Adam and B. F. Cravatt, J. Am. Chem. Soc., 2003, 125, 4686–4687 CrossRef CAS PubMed.
- M. Whiting, J. Muldoon, Y. C. Lin, S. M. Silverman, W. Lindstrom, A. J. Olson, H. C. Kolb, M. G. Finn, K. B. Sharpless, J. H. Elder and V. V. Fokin, Angew. Chem., 2006, 45, 1435–1439 CrossRef CAS PubMed.
- S. Escaich, Curr. Opin. Chem. Biol., 2008, 12, 400–408 CrossRef CAS PubMed.
- D. A. Rasko and V. Sperandio, Nat. Rev. Drug Discovery, 2010, 9, 117–128 CrossRef CAS PubMed.
- E. Zeiler, V. S. Korotkov, K. Lorenz-Baath, T. Bottcher and S. A. Sieber, Bioorg. Med. Chem., 2012, 20, 583–591 CrossRef CAS PubMed.
- M. M. Dinges, P. M. Orwin and P. M. Schlievert, Clin. Microbiol. Rev., 2000, 13, 16–34 CrossRef CAS PubMed , table of contents.
- C. W. Tseng and G. C. Stewart, J. Bacteriol., 2005, 187, 5301–5309 CrossRef CAS PubMed.
- T. Bottcher and S. A. Sieber, ChemMedChem, 2009, 4, 1260–1263 CrossRef PubMed.
- M. Hamon, H. Bierne and P. Cossart, Nat. Rev. Microbiol., 2006, 4, 423–434 CrossRef CAS PubMed.
- J. A. Vazquez-Boland, M. Kuhn, P. Berche, T. Chakraborty, G. Dominguez-Bernal, W. Goebel, B. Gonzalez-Zorn, J. Wehland and J. Kreft, Clin. Microbiol. Rev., 2001, 14, 584–640 CrossRef CAS PubMed.
- S. Rathore, D. Sinha, M. Asad, T. Bottcher, F. Afrin, V. S. Chauhan, D. Gupta, S. A. Sieber and A. Mohmmed, Mol. Microbiol., 2010, 77, 873–890 CAS.
- F. Weinandy, K. Lorenz-Baath, V. S. Korotkov, T. Bottcher, S. Sethi, T. Chakraborty and S. A. Sieber, ChemMedChem, 2014, 9, 710–713 CrossRef CAS PubMed.
- A. Cole, Z. Wang, E. Coyaud, V. Voisin, M. Gronda, Y. Jitkova, R. Mattson, R. Hurren, S. Babovic, N. Maclean, I. Restall, X. Wang, D. V. Jeyaraju, M. A. Sukhai, S. Prabha, S. Bashir, A. Ramakrishnan, E. Leung, Y. H. Qia, N. Zhang, K. R. Combes, T. Ketela, F. Lin, W. A. Houry, A. Aman, R. Al-Awar, W. Zheng, E. Wienholds, C. J. Xu, J. Dick, J. C. Wang, J. Moffat, M. D. Minden, C. J. Eaves, G. D. Bader, Z. Hao, S. M. Kornblau, B. Raught and A. D. Schimmer, Cancer Cell, 2015, 27, 864–876 CrossRef CAS PubMed.
- K. Larkin and J. C. Byrd, Cancer Cell, 2015, 27, 747–749 CrossRef CAS PubMed.
- M. Gersch, F. Gut, V. S. Korotkov, J. Lehmann, T. Bottcher, M. Rusch, C. Hedberg, H. Waldmann, G. Klebe and S. A. Sieber, Angew. Chem., 2013, 52, 3009–3014 CrossRef CAS PubMed.
- M. W. Hackl, M. Lakemeyer, M. Dahmen, M. Glaser, A. Pahl, K. Lorenz-Baath, T. Menzel, S. Sievers, T. Bottcher, I. Antes, H. Waldmann and S. A. Sieber, J. Am. Chem. Soc., 2015, 137, 8475–8483 CrossRef CAS PubMed.
- W. Moreira, G. J. Ngan, J. L. Low, A. Poulsen, B. C. Chia, M. J. Ang, A. Yap, J. Fulwood, U. Lakshmanan, J. Lim, A. Y. Khoo, H. Flotow, J. Hill, R. M. Raju, E. J. Rubin and T. Dick, mBio, 2015, 6, e00253-15 CrossRef PubMed.
- A. Pahl, M. Lakemeyer, M. T. Vielberg, M. W. Hackl, J. Vomacka, V. S. Korotkov, M. L. Stein, C. Fetzer, K. Lorenz-Baath, K. Richter, H. Waldmann, M. Groll and S. A. Sieber, Angew. Chem., 2015, 54, 15892–15896 CrossRef CAS PubMed.
- T. Akopian, O. Kandror, R. M. Raju, M. Unnikrishnan, E. J. Rubin and A. L. Goldberg, EMBO J., 2012, 31, 1529–1541 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |