
Diversity of nature's assembly lines – recent discoveries in non-ribosomal peptide synthesis
Jennifer A. E.
Payne†
 ab,
Melanie
Schoppet†
ab,
Mathias Henning
Hansen†
c and
Max J.
Cryle
*ab
ab,
Melanie
Schoppet†
ab,
Mathias Henning
Hansen†
c and
Max J.
Cryle
*ab
aEMBL Australia, Monash University, Clayton, Victoria 3800, Australia
bThe Monash Biomedicine Discovery Institute, Department of Biochemistry and Molecular Biology and ARC Centre of Excellence in Advanced Molecular Imaging, Monash University, Clayton, Victoria 3800, Australia. E-mail: max.cryle@monash.edu
cDepartment of Biology, University of Copenhagen, DK-2200 Copenhagen N, Denmark
First published on 17th November 2016
Abstract
The biosynthesis of complex natural products by non-ribosomal peptide synthetases (NRPSs) and the related polyketide synthases (PKSs) represents a major source of important bioactive compounds. These large, multi-domain machineries are able to produce a fascinating range of molecules due to the nature of their modular architectures, which allows natural products to be assembled and tailored in a modular, step-wise fashion. In recent years there has been significant progress in characterising the important domains and underlying mechanisms of non-ribosomal peptide synthesis. More significantly, several studies have uncovered important examples of novel activity in many NRPS domains. These discoveries not only greatly increase the structural diversity of the possible products of NRPS machineries but – possibly more importantly – they improve our understanding of what is a highly important, yet complex, biosynthetic apparatus. In this review, several recent examples of novel NRPS function will be introduced, which highlight the range of previously uncharacterised activities that have now been detected in the biosynthesis of important natural products by these mega-enzyme synthetases.
 Jennifer A. E. Payne | Jennifer A. E. Payne completed her BSc in Biological Sciences and PhD in Biochemistry at La Trobe University in Melbourne, Australia. Her doctoral research investigated the mechanism of antimicrobial peptides targeting the cell wall and membrane in fungi and was recognised by the award of the Nancy Millis Medal. Since the beginning of 2016 she has been undertaking postdoctoral research in the Cryle group, where her interests lie in the development of novel antibiotics to treat drug resistant bacterial infections. |
 Melanie Schoppet | Melanie Schoppet, born in 1989, studied Applied Chemistry and Bio- and Pharmaceutical Analysis at the Hochschule Fresenius in Idstein, Germany. Her research interests lie at the interface between organic chemistry and biology, which led her to commence her doctoral studies with the Cryle group in January 2015. Her research centres on the manipulation of glycopeptide antibiotics with particular focus on the process of non-ribosomal peptide synthesis. |
 Mathias Henning Hansen | Mathias Henning Hansen obtained his BSc and MSc degrees from the Faculty of Science, University of Copenhagen, Denmark, in 2014 and 2016 respectively. He is currently a research assistant working under the supervision of Assoc. Prof Peter Brodersen. His research is focused on the functional links between miRNA and methyladenosine mediated mRNA control in Arabidopsis thaliana and he has a strong interest in structural biology of complex biological systems. |
 Max J. Cryle | Max J. Cryle, born in 1979, obtained his PhD in Chemistry from the University of Queensland in 2006. He then moved to the Max Planck Institute for Medical Research in Heidelberg Germany as an HFSP Cross-Disciplinary Fellow and later as an Emmy Noether group leader funded by the DFG. Since 2016 he is an EMBL Australia group leader based within the Biomedicine Discovery Institute at Monash University, where his team focusses on understanding antibiotic biosynthesis (with particular interest in non-ribosomal peptide synthesis) as well as developing new antibiotics. |
Why should we be interested in non-ribosomal peptide synthesis ?
The rise of antibiotic resistance has created a critical need for new, effective compounds. This has driven the search for new natural compounds, as well as screening modifications to existing ones. The complexity of the structures of many natural compounds used in medicine means that we are often reliant on bacterial fermentation for their production: one important example is the glycopeptide antibiotics (GPAs), where even newer, second generation GPAs rely on the products of bacterial fermentation as the starting point for chemical modification. The reliance on natural biosynthesis pathways to produce compounds such as these places restrictions on their structural modification, and thus in order to access novel compounds modification of the biosynthesis machinery itself is required.At the heart of the great chemical diversity found in nature often lies common enzymatic machineries, which whilst individually responsible for production of different compounds share commonality in enzyme mechanism and protein architecture. In this regard, megaenzyme synthases – the non-ribosomal peptide synthetases (NRPSs, Fig. 1A) along with the related polyketide synthases (PKSs) – are responsible for the biosynthesis of a great many important natural products, particularly in realm of antibiotics (Fig. 1B), but also producing compounds with anticancer, antifungal and immunosuppressive properties.1–3 These machineries typically exploit modular architectures of repeating catalytic domains that add the individual building blocks in a step-wise fashion to the growing compound (Fig. 1A). The enzymology and domain interactions of these machineries are built around the covalent attachment of activated intermediates via a thioester bound to the phosphopantetheine arm of carrier protein (CP) domains, and although similarities do exist between these machineries, they need to be investigated as separate entities.3–5 Investigation of these assembly lines has been accelerated in recent years due to improvements in genome sequencing, producer strain manipulation6,7 as well as the ever increasing role chemical synthesis plays in gaining access to high value structures8–15 and biochemical data both in vitro16–23 and in vivo.24,25
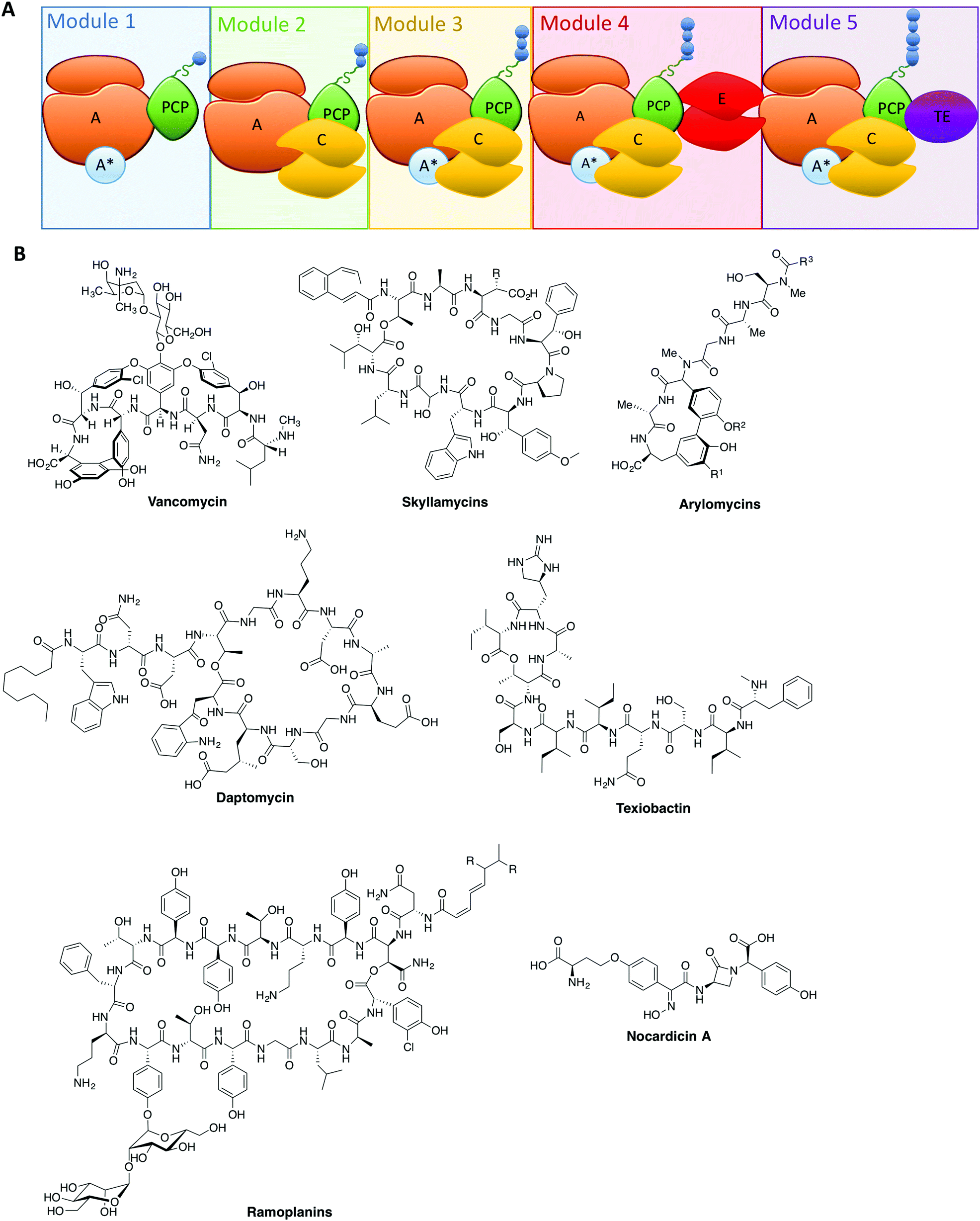 | ||
| Fig. 1 (A) Schematic representation of an NRPS assembly line showing the typical linear, modular architecture of repeating domains with the peptide (blue circles) growing along the assembly line from left to right. Adenylation (A)-domains are coloured orange, adenylation domain activating proteins (A*) are coloured blue, peptidyl carrier protein (PCP)-domains along with their phosphopantetheine linker are coloured green, condensation (C)-domains are coloured yellow, epimerisation (E)-domains are coloured red and thioesterase (TE)-domains are coloured in purple. (B) Examples of different antibiotic compounds produced by NRPS assembly lines. | ||
From these studies, it is clear that the majority of NRPS assembly lines adopt a linear, modular architecture built up of repeating groups of domains: core NRPS domains, aside from peptidyl carrier proteins (PCPs) mentioned above, include adenylation (A)-domains (responsible for monomer selection and activation) and condensation (C)-domains (which catalyse peptide bond formation from adjacent PCP-bound substrates). The final structure of an NRPS is then completed by additional domains that commonly include epimerisation (E)-domains (responsible for altering the stereochemistry of peptide residues) and thioesterase (TE)-domains (which cleave the mature peptide from the NRPS). This modular architecture allows the structure of the peptide product to be deduced from an analysis of the domain architecture and selectivity of the A-domains (Fig. 1A).3 Given our apparent understanding of how linear NRPS systems function, it is perhaps surprising that the redesign of these systems is often plagued by low product yields, although there have been some notable successes in this area that indicate the potential of successful NRPS redesign.26–30 Given that a great many products of NRPS assembly lines are still produced by bacterial fermentation, this represents a problem for the effective reengineering of such assembly lines to produce novel compounds. In order to fully harness the power of NRPS assembly lines to produce new compounds, we need to understand not only the conceptual overview of peptide biosynthesis by these machineries but also their quirks: the discovery of novel chemistry performed by “standard” NRPS systems has set the scene for future research and also dramatically increased the scope for catalytic redesign of NRPS assembly lines. In this perspective, we highlight some telling examples of such novel chemistry in NRPS systems and offer proposals for how this knowledge can be transferred into future redesign of these biosynthetic machineries.
A-domains – controlling peptide structure via engineering selectivity
Within non-ribosomal peptide synthetase assembly line, adenylation (A)-domains have received arguably the greatest amount of attention. This stems from their vital role as the agents for monomer selection and activation during peptide synthesis.31 Monomer selection in bacterial NRPS A-domains occurs within the larger, N-terminal subdomain that together with a smaller C-terminal subdomain comprises the complete A-domain. The monomer binding pocket in the larger subdomain is formed by ∼10 residues and was identified early as a means of predicting substrate specificity for monomer activation and hence the final peptide structure of the NRPS assembly line.32 The ability to activate monomers independently of the ribosome relieves the constraints placed upon the range of monomers available for selection, with more than 500 different monomers now identified as substrates for A-domain activation and hence NRPS-catalysed peptide synthesis.33 Recent examples of monomers go beyond α-amino acids and include β-amino acids,34 aminobenzoic acid units35 and even α-hydroxy acids: for α-hydroxy acids the situation is further complicated by the fact that in some cases the A-domain substrate is an α-ketoacid that is then subsequently reduced by a ketoreductase domain.36–39 Further modification domains can also be incorporated via insertions in A-domains, which are capable of additional alteration of the selected monomers.40,41A-domains often rely on small protein activators: these proteins (most commonly MbtH-like proteins,8,42–45 but there are examples of other activating proteins)46 have been demonstrated to have significant effects on A-domain activity. The role of these small protein activators remains somewhat unclear given that even within one NRPS module not all A-domains require protein activators to be folded or functional (Fig. 1A).8,42 The necessity of small protein activators also raises the question of the purpose of such proteins, with one obvious possibility being the ability to regulate the activity of NRPS assembly lines without the need to degrade and resynthesise the entire machinery. This would rely on the ability for such small protein activators to form transient complexes with A-domains: however, many examples to date appear to be involved in very tight protein–protein complexes, which would suggest the need for an external agent to be involved in such complex remodelling.42,44,45 Clearly further investigation into the role of small protein activators in A-domain – and hence NRPS – activity is needed to resolve this issue.
Within the core NRPS domains, A-domains are the only domains dependent on an external cofactor for activity, as they require ATP to activate the monomer following its selection (Fig. 2A).31 During monomer activation by A-domains, an activated aminoacyl AMP adenylate is formed in an initial step with concomitant release of pyrophosphate (Fig. 2B). The process of amino acid activation triggers a significant alteration in the relative orientations of the two subdomains within the A-domain, with the C-terminal subdomain rotating essentially through 180 degrees as a part of this process. In doing so, a tunnel into the active site of the A-domain is opened that allows access of the phosphopantetheine arm of the neighbouring peptidyl carrier protein domain (Fig. 2A).31 Interception of the activated monomer by the thiol group of the phosphopantetheine arm leads to loading of the monomer onto the carrier protein arm via a thioester bond, with release of AMP thus completing the A-domain active cycle (Fig. 2B). The connection of the small subdomain of A-domains to the downstream carrier protein domain by a short peptide linker sequence47 couples the motion of the carrier protein domain to the hydrolysis of ATP during monomer activation, which helps to explain the ability of carrier protein bound substrates to access the active sites of several catalytic NRPS domains during peptide synthesis.4,31 Recent crystal structures of complete NRPS modules in different states of peptide biosynthesis have demonstrated the effects of this motion of the carrier protein and support the importance of ATP hydrolysis in orchestrating carrier protein motion during NRPS-catalysed peptide synthesis.8–10 The limited states accessible to the carrier protein during A-domain catalysis implies that carrier protein bound substrates can also access multiple domains via domain rotation, which awaits future definitive structural characterisation.4
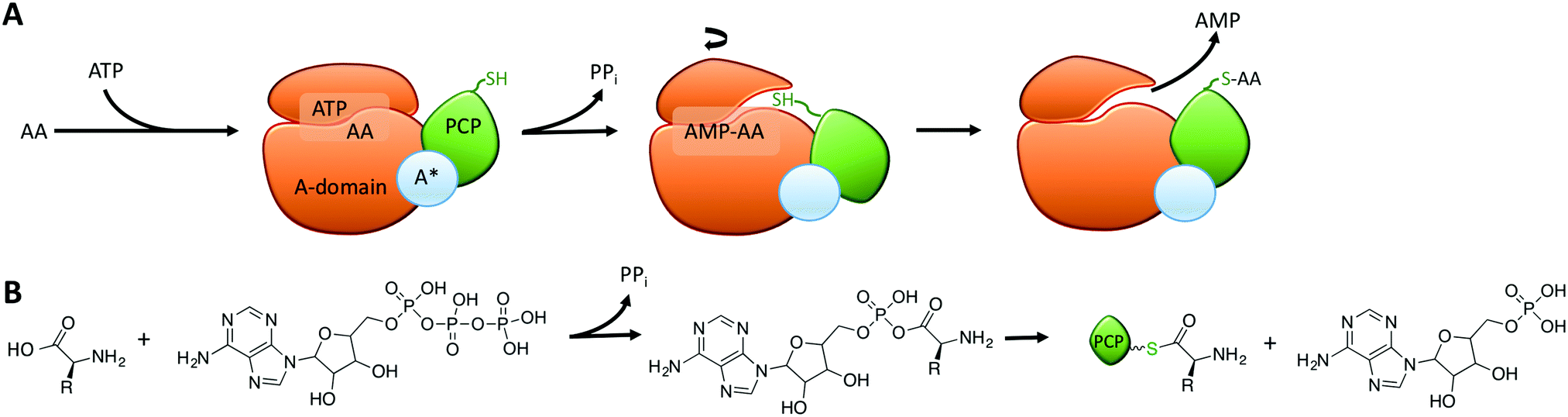 | ||
| Fig. 2 (A) Schematic representation of the catalytic cycle of adenylation domains: following the initial selection of the amino acid monomer and binding ATP (left), the A-domain (orange) catalyses the activation of the amino acid using ATP, which releases pyrophosphate and the small subdomain (top) undergoes a 180-degree rotation to allow access of the phosphopantetheine linker of the downstream PCP domain (green) to the activated amino acid; upon loading of the amino acid onto the PCP the release of AMP then completes the cycle (right). (B) Chemical structures of the substrates, intermediates and products of adenylation domains. | ||
The restriction of monomer selection to such a small pocket within A-domains clearly flags the potential to alter A-domain selectivity via mutagenesis:32 whilst many studies have been performed, the ability to maintain efficient peptide synthesis with mutated NRPS assembly lines has not always been demonstrated. Explanations for this effect have been postulated to include additional selectivity of other catalytic NRPS domains, such as the condensation domain.27,28 An alternative suggestion concentrates on the coupling of A-domain activity to carrier protein motion and hence overall peptide synthesis. This model centres on the importance of coupling the reaction rates of sequential A-domains in an NRPS to maintain efficient peptide synthesis. For example, a reduction in the activation rate of an A-domain would cause the coupled carrier protein motion to be slowed, resulting in an increased potential for hydrolysis of peptide precursors due to the interception of these intermediates by water. This makes the in vitro characterisation of A-domain activity and the coupling of activity of multiple NRPS modules of great importance for efforts to optimise NRPS reengineering. In this regard, several recent papers have made significant progress: one example is the work of Kaljunen et al. (2015) in characterising an A-domain of the Anabaenopeptin synthase from Planktothrix agaardhii strain PCC7821 (ApnA A1).48 The authors demonstrated that this A-domain is bispecific for both arginine and tyrosine and crucially could also validate how both substrates are bound and activated in the same binding pocket of the enzyme via X-ray crystallography. The A-domain pocket in ApnA A1 achieves this bispecificity by forcing the sidechain of arginine to take on a conformation that mimics the shape of the tyrosine ring, with three residues (Glu204, Ser243 and Ala307) critical for the bispecificity of the A-domain (Fig. 3A and B). The plasticity of ApnA A1 was revealed via an elegant mutational experiment, showing that specificity for arginine is achieved when Ser243 was substituted by Asp, whilst substitutions of Ala307 with Val/Leu result in improved substrate preference for tyrosine (Fig. 3C). Moreover, seven of eight Glu204 substitution variants lost activity for arginine but retained specificity for tyrosine, which emphasises the importance of Glu204 for arginine binding (Fig. 3D). Finally, by combining the larger substrate binding pockets of the substitution variants E204G and S243E, the ApnA A1 domain acquired a strong substrate specificity for tryptophan as well as 4-azidophenylalanine (Fig. 3E and F).48 The ability to select the last substrate is important as it opens the door to the generation of modified NRPS peptides bearing chemical groups that are amenable to further modification through click-type chemistry.49 Such modified A-domains have also been reported by Kries et al., and are clearly a priority for the field.50 The ability to redesign A-domains to activate a range of non-natural monomers is also an approach with great potential for enzymatic redesign, which could then allow different peptides to be produced based upon the controlled supplementation of growth media with different building blocks:29,51,52 the existence of natural A-domains that fulfil this type of function (for example in linear gramicidin biosynthesis53 and many examples from cyanobacteria54) speaks strongly in favour of the feasibility of such approaches. What is also important to note is the success of A-domain redesign by matching the size of the monomers involved to the pocket. The success of such an approach has also been observed for the production of altered calcium dependent antibiotic (CDA) peptides in vivo, which suggests this is an important consideration in such redesign.27,28 Furthermore, the effect that such modified A-domains have in the context of peptide production by the complete NRPS assembly line remains to be examined in detail. The ability to generate A-domain mutants that activate different monomers with comparable rates to that of the natural substrate certainly allow the effects of activation rate vs. NRPS domain specificity to be assessed in vitro, which is crucial information for the future effective redesign of A-domains and hence NRPS peptides.
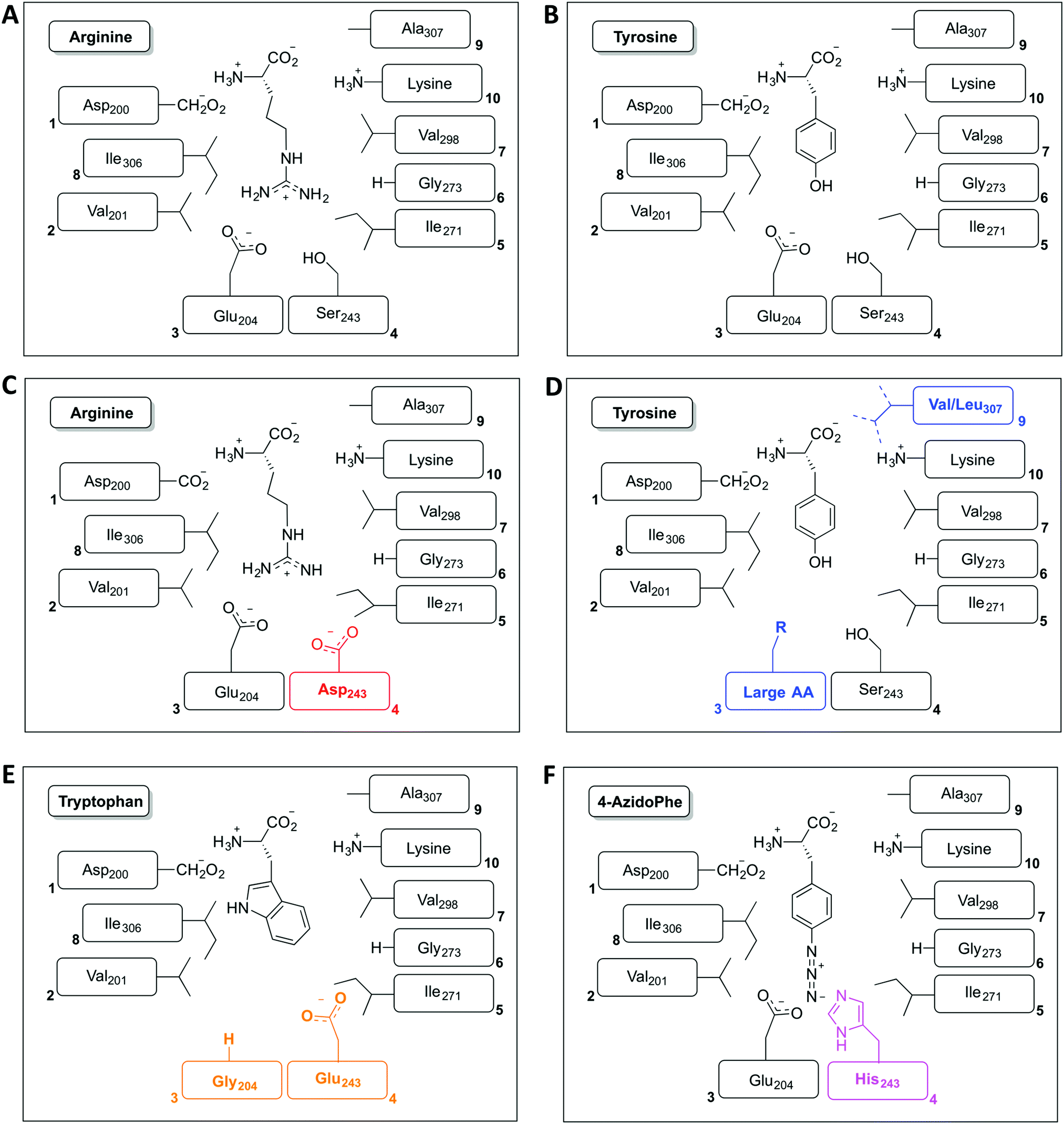 | ||
| Fig. 3 Schematic representation of the amino acid residues found in the amino acid binding pocket of the ApnA A1 domain for both wildtype substrates arginine (A) and tyrosine (B) together with altered binding pockets that alter the selectivity of this domain for monomer selection towards arginine (C), tyrosine (D), tryptophan (E) and 4-azidophenylalanine (F). | ||
C-domains – beyond peptide bond formation
Within the peptide assembly process, condensation (C)-domains play an essential role in condensing precursors to synthesise peptide bonds.55,56 C-domains and the related epimerisation (E)-domains all adopt a common structure, which is essentially a dimer of chloramphenicol acetyl transferase domains. In the centre of this structure is a large active site channel into which the donor substrate is bound, whilst at the bottom of the active site lies a loop with a conserved HHxxxDG motif. The roles of C-domain active site residues have been probed in a number of studies, which have yet to achieve a unified theory as to the mechanism of these domains.10,17,57,58 In most C-domains, the substrates are accepted in their carrier protein bound form, which presents the donor substrate as an activated thioester (Fig. 4A);59–61 there is also the potential to accept activated donor substrates free in solution, such as fattyacyl-CoAs. The acceptor substrate enters from the opposite side of the donor substrate and is positioned such that the amino group of the acceptor substrate can attack the thioester of the donor substrate (Fig. 4A). At this point, a conserved histidine residue is believed to be situated appropriately to assist in the reaction. The closest structural information of the substrate bound C-domain to support the role of the histidine residue was recently obtained through a protein modification and tagging strategy, which covalently attached an acceptor substrate mimic to an engineered cysteine residue.10 This structure was able to demonstrate the coordination of the acceptor amino group to the active site histidine residue and activity of the modified domain, although no electron density for the donor substrate could be determined, most likely due to conformational flexibility. | ||
| Fig. 4 (A) Schematic representation of the reaction performed by condensation (C)-domain (yellow), in which the amino group of the amino acid bound to the downstream PCP (the acceptor) attacks the thioester of the bound peptide attached to the upstream PCP (the donor), thus resulting in transfer of the peptide onto the acceptor PCP domain (shown in green) and elongation of the peptide chain. (B) The final C-domain in norcardicin biosynthesis not only catalyses peptide bond formation but also installs the β-lactam ring within the norcardicin pentapeptide precursor. (C) Tetracyclisation of the heptapeptide precursor to afford the glycopeptide antibiotic aglycone is performed through the sequential recruitment of four separate Cytochrome P450 (Oxy) enzymes to the PCP-bound peptide substrate (chain of blue circles) by the X-domain (shown in yellow), a conserved recruitment domain found in GPA NRPS biosynthesis. (D) Chemical structure of teicoplanin with the individual cross-links formed by the four Oxy enzymes indicated in colours that correspond to the individual Oxy enzymes. | ||
Within NRPS assembly lines, C-domains are not only responsible for amide bond formation, as examples have been demonstrated that cyclise amino acids or that generate depsipeptide (ester) bonds.62,63 Furthermore, C-domains act as bridging elements between polyketide synthase and NRPS modules in dual PKS/NRPS systems, where they are required to accept different substrates compared to those found during normal NRPS function.3 C-domains are believed to be selective for both donor and acceptor substrates in terms of the stereochemistry of the amino acids to be linked: selectivity for the acceptor amino acid domain appears to be strict for the (L)-configuration, whilst the configuration of the C-terminal donor residue can be either (L) or (D), which is typically fixed for a given C-domain.55,56,64 The ability to act as a gatekeeper for substrate stereochemistry means that there is a close relationship between C-domains and related E-domains,65–67 with the latter being responsible for the epimerisation of the acceptor substrate from the (L) to the (D) form. This is necessary due to the vast majority of A-domains selecting and activating (L)-configured monomers. The close relationship of C- and E-domains in NRPS biosynthesis is further exemplified by dual E/C domains, where the epimerisation of the C-terminal residue of the upstream peptide from the (L)- to the (D)-form and subsequent peptide bond formation occurs within a single domain.64,68 Such dual E/C domains are of great interest for NRPS reengineering, as they offer the opportunity to replace LCL domains with DCL domains without the need for an additional E-domain to be incorporated into the protein machinery. Given the importance of protein–protein interactions for NRPS-mediated catalysis, reducing changes to the overall structure of an NRPS could well prove key to successful redesign in such cases.
The roles of C-domains within NRPS assemblies have also recently been extended to performing novel chemistry on their substrates, either via direct catalysis or through recruitment of external modifying enzymes to the NRPS-bound substrate. A recent and truly remarkable example of the former has been characterised in the biosynthesis of the β-lactam antibiotic norcardicin:17 here, Gaudelli et al. could show that the origin of the β-lactam moiety is the NRPS machinery itself (Fig. 4B), rather than an independent non-heme iron oxygenase (such as isopenicillin N synthase) as is the case in the biosynthesis of many β-lactam containing antibiotics or other ATP-dependant processes (for the monobactams, carbapenems and clavulanic acid).69 The norcardicin NRPS includes two multifunctional enzymes NocA and NocB, which consist of five functional modules that assemble the β-lactam containing pentapeptide L-Hpg-L-Arg-D-Hpg-L-Ser-L-Hpg precursor; later this is further processed to generate the tripeptide pro-nocardicin G. Using module 5 of the norcardicin NRPS and PCP-bound tetrapeptide substrates, Gaudelli et al. established that the C5 domain is not only responsible for the generation of the norcardicin pentapeptide precursor, but also the formation of the β-lactam ring (that includes the amine of the C-terminal L-Hpg, Fig. 4B).17 The C5 domain has an unusual catalytic motif – HHHxxxDG compared to the representative HHxxxDG motif of most condensation domains – and this extra histidine (H790) has been demonstrated to be crucial for β-lactam formation.17 The mechanism of β-lactam formation is postulated to occur via the initial β-elimination of H2O from the L-serine of the PCP4 bound tetrapeptidyl-thioester by H790. After this has occurred, β-addition of the PCP5 bound L-Hpg occurs along with overall inversion of configuration at the resulting dehydroalanyl β-carbon.17 This process is also unique in that the initial bond formed between tethered PCP-substrates is an amine, rather than the typical amide, bond: such a mechanism has potential use for characterising trapped C-domain intermediates and will no doubt be of interest in future studies that seek to structurally characterise important C-domain intermediates. Whilst the prevalence of such C-domains in currently reported NRPS sequences is low, the ability for C-domains to perform such complex chemical rearrangements is of great interest for future reengineering efforts and stands as testimony to the synthetic potential of NRPS machineries.
Another recent example of C-domains enabling complex chemistry has been uncovered during the biosynthesis of the glycopeptide antibiotic (GPA) teicoplanin: in this case, a modified C-domain acts as a recruitment domain for external modifying enzymes that target their peptide substrates bound to the NRPS machinery (Fig. 4C).19–21,70 GPAs are highly modified heptapeptides bearing 3 or 4 crosslinks between sidechains of aromatic residues contained within the precursor peptide, with each crosslink installed by the actions of a Cytochrome P450 (Oxy) enzyme (Fig. 4C).71–76 Whilst it has been demonstrated that the substrates for these Oxy enzymes were NRPS-and hence carrier protein bound-peptides,77–79 only recently could Haslinger and Peschke et al. show that the agent responsible for the recruitment of the Oxy enzymes to their NRPS-bound peptide substrates is a modified C-domain, known as the X-domain (Fig. 4C).20 The X-domain was structurally characterised, revealing that this domain adopts the expected C-domain like fold. Furthermore, specific mutations in the typically conserved active site HHxxxDG motif (altered to HRxxxDD in the X-domain) block the active site channel to render the X-domain catalytically inactive. The X-domain does however interact with the Oxy proteins and a complex of the X-domain with the first Oxy homologue, OxyB, could be structurally characterised.20 The functional relevance of the X-domain was verified as the activity of the Oxy proteins with X-domain containing NRPS-bound peptide substrates is vastly superior to substrates without the X-domain. Oxy activity has now been extended to peptide bicyclisation20,21 and tricyclisation80 using a cascade of Oxy enzymes and X-domain containing NRPS-bound peptide substrates. How the X-domain supports the recruitment of multiple Oxy enzymes to a single substrate has recently been clarified, which indicates that there is a constant exchange of Oxy enzymes bound to the X-domain.19 The X-domain thus provides another example of how NRPS peptide chemistry can be expanded, in this case via the recruitment of multiple external modification enzymes to a single NRPS-bound peptide that enables the generation of highly complex peptide structures.
TE-domains – the possibilities offered by late stage functionalisation
All intermediates in NRPS mediated peptide synthesis are bound to carrier proteins via thioester bonds, which means that following the completion of peptide synthesis the NRPS machinery needs to liberate the final product in order to allow the assembly of subsequent peptides to proceed (Fig. 5).81 Other thioesterase enzymes, type II thioesterases, are also important for NRPS biosynthesis, where these trans acting domains regenerate active carrier proteins via cleavage of incorrect substrates.82 This is particularly important when acylated CoA is loaded onto PCP domains following synthesis of the NRPS, as this would render the synthetase inactive due to the lack of a free thiol on the PCP linker arm. NRPS TE-domains are typically found at the end of the NRPS assembly line, although in some systems the reduction of the thioester bond via a reductase domain also achieves this purpose (see e.g.ref. 46). TE-domains are far more important to NRPS synthesis than merely cleaving thioester products: they are the source of peptide cyclisation that is very common in NRPS produced peptides, which even includes unusual cyclic dipeptide products such as ingidoidine.83,84 TE-domains typically utilise an active site catalytic Ser-His-Asp triad as is found in serine proteases, where the serine residue attacks the thioester bond of the peptide product, forming an intermediate state where the peptide is loaded onto the TE domain serine residue.81,85 In this regard, TE domains are unique within NRPS assembly lines, as these domains are the only catalytic domains to covalently bind intermediates during peptide biosynthesis.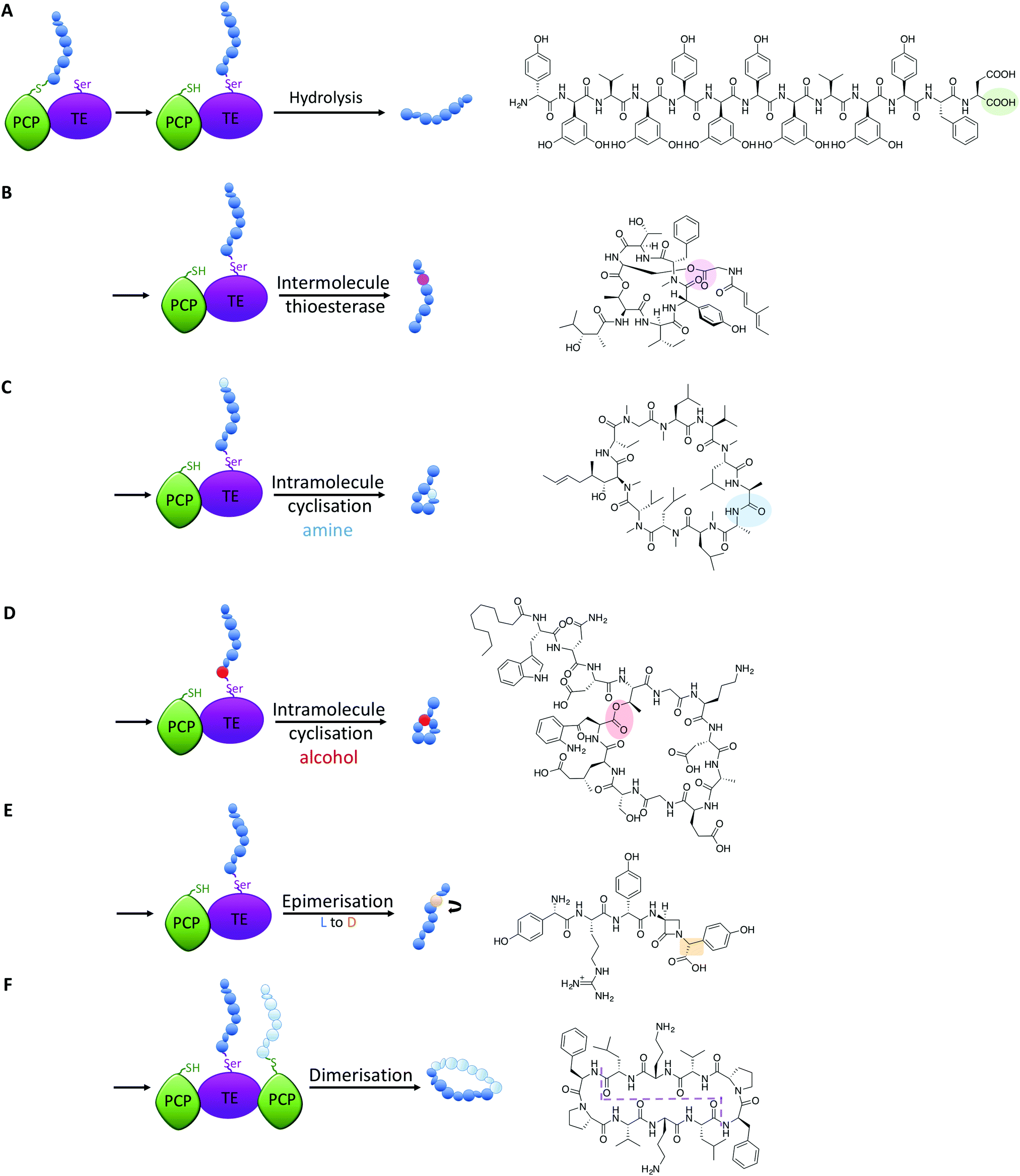 | ||
| Fig. 5 Alternate fates of peptides on different TE-domains include hydrolysis (A, feglymycin), intermolecular acylation (B, desmethylsalinamide C), intramolecular cyclisation from an amine (C, cyclosporine) or alcohol nucleophile (D, daptomycin), hydrolysis together with epimerisation of an amino acid residue (E, norcardicin G) and dimerisation/cyclisation of peptide monomers (F, gramicidin S); an example of each reaction is shown. | ||
The loaded TE intermediate has several potential fates: aside from “simple” hydrolysis of the intermediate by water to produce linear peptides (Fig. 5A), intramolecular cyclisation of the peptide is a common feature of TE-domain activity (Fig. 5). In this regard, an internal nucleophile of the TE-bound peptide attacks the ester bond of the TE-peptide intermediate to afford cyclic peptide products. The nucleophile can be an amine or an alcohol, affording amide and ester products respectively, with the source of the nucleophile the side chains of amino acid residues or the amino terminus of the peptide (Fig. 5C and D). TE-domains have even been exploited to cyclise peptides in vitro, which has fascinating potential to perform cyclisation chemistry that can otherwise be highly challenging.86 Furthermore, some NRPS TE-domains allow polymerisation of peptide monomers, which presumably occurs via initial offloading of monomers onto the TE-domain, that then undergo polymerisation with a further CP-bound monomer to produce the desired product (such as dimeric or trimeric cyclic peptides, Fig. 5F).86,87
Recent studies have demonstrated that further functions of TE-domains also exist, which extends the catalytic versatility of these domains yet further. In another example of novel NRPS chemistry from norcardicin biosynthesis, the action of the terminal TE-domain was found to not only be highly selective for β-lactam containing peptides, but it was also responsible for the epimerisation of the terminal Hpg residue (Fig. 5E).18 Epimerisation of amino acids during NRPS biosynthesis is a common process, but this is typically performed by specific epimerisation (E)-domains, which act upon carrier protein bound substrates prior to the action of C-domains.67 In the case of the norcardicin TE-domain, the epimerisation was shown to occur on the TE-bound peptide substrates and was completely selective for the epimerised product. This process is postulated to occur during the action of the catalytic triad of the TE-domain, and the increased pKa of the TE-bound oxyester is compensated for by the reduced pKa of the α-carbon of the Hpg residue due to direct substitution of the phenyl ring on the peptide α-carbon.18 These experiments conclusively showed that epimerisation of TE-bound substrates is also possible in NRPS biosynthesis, further expanding the catalytic potential of TE-domains.
A further impressive example of TE-mediated substrate modification during NRPS biosynthesis was recently identified in salinamide biosynthesis, which are a family of unusual bicyclic depsipeptide antibiotics.88 Ray et al. (2016) reported that the terminating TE domain in salinamide biosynthesis catalyses an intermolecular, transesterification reaction integral to the construction of the salinamide structure (Fig. 5B).89 In this process, and following cyclisation of the salinamide peptide precursor by TE-domain from Sln6, the TE-domain of an additional NRPS module, Sln9, catalyses the addition of a (4-methylhexa-2,4-dienoyl)-glycine unit to the serine hydroxyl moiety of the mature cyclic depsipeptide, desmethylsalinamide E to afford desmethylsalinamide C. This process is postulated to occur via intermediate loading of the (4-methylhexa-2,4-dienoyl)-glycine unit onto the TE-domain of Sln9, which is subsequently attacked by the side chain of Ser6 of the mature cyclic depsipeptide.89 The Sln9 TE is the first example of an intermolecular-acting thioesterase domain within a bacterial NRPS to mediate the transesterification of a CP-bound substrate onto a cyclic depsipeptide, and is a striking example of NRPS mediated diversification.89 These examples of non-conventional TE-domain activity extend the catalytic potential of TE-domains to the realms of C/E-domains and represent a fascinating source of future bioengineering potential in NRPS systems.
Altering NRPS interactions to generate further product diversity
The typical modular architecture adopted by linear NRPS systems is not the only one to have been identified: both iterative and non-linear systems have been reported and investigated. These systems are less prevalent and are not as commonly found to produce biomolecules of medical interest.3,62 None the less, these systems offer excellent examples of how NRPS systems can be designed to produce interesting modules: in the case of iterative systems, these generate cyclic peptides typically based on repeating dipeptide units, with different systems adopting different NRPS architectures to generate their final cyclic structures,87,90 whilst in the case of non-linear systems there is little resemblance between the NRPS architecture of the biosynthetic machinery and the final peptide product.91 Both these systems have great potential to unveil how substrate structure interacts with NRPS domains to generate specificity and will no doubt be the subject of detailed future investigations. Examples of NRPS diversity generated during linear NRPS biosynthesis have also been reported, both in terms of external interacting enzymes selecting specific carrier proteins in the NRPS for modification of their bound monomers and for entire module replacements using separately expressed proteins. In the case of the former, the biosynthesis of the cyclic depsipeptide skyllamycin has proven highly informative (Fig. 6A). In this system, three amino acid residues in the final peptide are β-hydroxylated and the origin of the hydroxylation could be traced to a single Cytochrome P450 enzyme.60,92 P450 enzymes are commonly found in both PKS and NRPS clusters,93–95 and although only a subsection of these P450s target carrier protein bound substrates71,96,97 many play important roles in structural diversification of natural products, even going beyond their monooxygenase activity to include novel functions such as nitration.98 In such cases, P450s typically are responsible for provision of β-hydroxylated amino acid precursors for eventual selection via the main NRPS machinery and these act by oxidising PCP-bound substrates on minimal A-PCP di-domain NRPS constructs.99 Uhlmann et al. could show that the P450 from skyllamycin biosynthesis does not act to produce amino acid precursors for incorporation by the main NRPS machinery, but rather targets PCP-bound amino acids within the main NRPS directly for oxidation (Fig. 6A).92 In doing so, this P450 demonstrates high selectivity for the correct PCP domains 5 (Phe), 7 (OMe-Tyr) and 11 (Leu) (Fig. 6B). Furthermore, it does not interact with PCP domain 10, which is also loaded with a leucine substrate during peptide synthesis and thus would have been a viable substrate for this P450. The interaction of the P450 was driven by recognition of the correct carrier protein domains, which could be successfully used to present alternate amino acids for oxidation by the P450.92 The use of substrate inhibitor mimics was then adopted by Haslinger et al. to then trap a complex of PCP domain 7 with the P450 from skyllamycin biosynthesis, which provided the first insight into how PCP binding to the P450 is orchestrated and hence how selectivity could be developed.11 The ability to target carrier protein substrates for selective modification during NRPS synthesis significantly increases the modifications that could be incorporated into NRPS peptides (including for example halogenation)100 without changing the A-domain selectivity of the NRPS: as such, this is certainly another area of great promise for future bioengineering of NRPS products.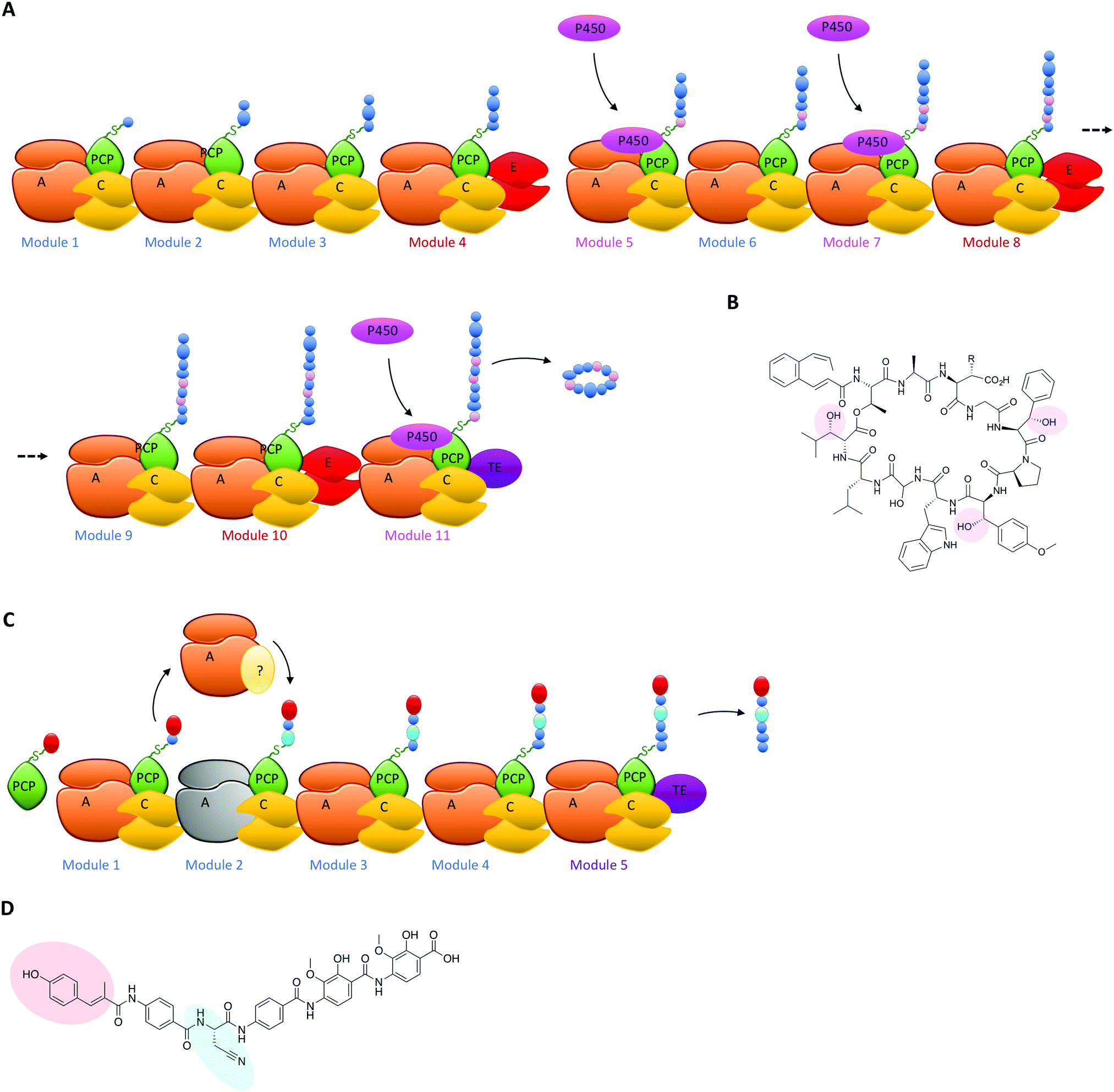 | ||
| Fig. 6 (A) NRPS assembly line for the cyclic depsipeptide skyllamycin shown together with the PCP domains that interact with the P450 contained within the cluster to produce hydroxylated amino acids at positions 5, 7 and 11 of the peptide directly during peptide formation. (B) The structure of skyllamycin with the positions of hydroxylation highlighted. (C) NRPS portion of the assembly line of the PKS/NRPS hybrid compound albicidin (with the product of the PKS portion shown in red). The inactive A-domain of NRPS module 2 is represented together with the external NRPS module Alb04 that replaces this domain and produces cyanoalanine through the oxidation of asparagine by the oxidation domain that is also present in Alb04. (D) The structure of albicidin indicating the PKS portion of the product in red and the cyanoalanine residue installed by NRPS module Alb04 highlighted in cyan. | ||
Another example of altering linear NRPS biosynthesis using an external protein is observed in albicidin biosynthesis. This hybrid PKS/NRPS system is already of great interest due to its incorporation of non-standard aminobenzoic acid precursors. In this system the second NRPS module within the main assembly line protein Alb01 was shown to possess an inactive A-domain, which was predicted to be responsible for the incorporation of the cyanoalanine residue found at this position in the final product (Fig. 6C and D).35 In the place of this inactive A-domain, an external NRPS module Alb04 activates both asparagine and cyanoalanine; tellingly, this module also contained a domain of unknown function that is most closely related to the adenosine nucleotide α-hydrolase (α-ANH-like III) superfamily. As such proteins are known to bind ATP, the role of this domain has been postulated as the dehydration of asparagine to form cyanoalanine, which is then handed off to the main NRPS assembly line via a mechanism that remains unclear with regards to the specific roles of the two carrier protein domains within Alb01/Alb04 in this process (Fig. 6C). None the less, such a trans- complementing mechanism (seen also in the loading of the PCP of module 0 in bleomycin biosynthesis by the neighbouring A-domain of module 1)101 is an important and interesting example of functional diversification within NRPS biosynthesis, as this has the potential to allow the insertion of different amino acids into a large linear NRPS via A-domain inactivation and supplementation of an standalone, interacting NRPS module used to activate alternate monomers for insertion into the growing peptide chain. A better understanding of the overall structure of NRPS assembly lines102 as well as the role of communication domains in assembling multiple proteins in the correct order will also prove vital in these efforts.103,104
Conclusions
The fascinating array of natural products produced by non-ribosomal peptide synthetase assembly lines makes understanding the enzymology behind their action of great importance. From recent reports it has become clear that linear NRPS systems are highly diverse machineries with far greater catalytic potential than has been previously demonstrated. These new insights demonstrate the vital importance of rigorous characterisation of NRPS assembly lines in order to build up a complete picture of the catalytic potential of these amazing biological machines. This knowledge is crucial given the potential to redesign NRPS-based biosynthetic pathways from important medicinal compounds and thus reinvent these compounds to tackle new and emerging challenges in human health, such as overcoming increasing antimicrobial resistance and delivering improved anticancer therapies.Acknowledgements
J. A. E. P., M. S. and M. J. C. are grateful for the support of Monash University and the EMBL Australia program. This work is dedicated to Craig Townsend in recognition of his many significant contributions to understanding the complex process of non-ribosomal peptide synthesis.References
- C. T. Walsh and T. A. Wencewicz, J. Antibiot., 2014, 67, 7–22 Search PubMed.
- E. A. Felnagle, E. E. Jackson, Y. A. Chan, A. M. Podevels, A. D. Berti, M. D. McMahon and M. G. Thomas, Mol. Pharmaceutics, 2008, 5, 191–211 Search PubMed.
- M. A. Fischbach and C. T. Walsh, Chem. Rev., 2006, 106, 3468–3496 Search PubMed.
- T. Kittilä, A. Mollo, L. K. Charkoudian and M. J. Cryle, Angew. Chem., Int. Ed., 2016, 55, 9834–9840 Search PubMed.
- F. Lipmann, in Advances in Microbial Physiology, ed. A. H. Rose and J. G. Morris, Academic Press, 1981, vol. 21, pp. 227–266 Search PubMed.
- W. Wohlleben, Y. Mast, G. Muth, M. Roettgen, E. Stegmann and T. Weber, FEBS Lett., 2012, 586, 2171–2176 Search PubMed.
- E. Stegmann, H.-J. Frasch and W. Wohlleben, Curr. Opin. Microbiol., 2010, 13, 595–602 Search PubMed.
- B. R. Miller, E. J. Drake, C. Shi, C. C. Aldrich and A. M. Gulick, J. Biol. Chem., 2016, 291, 22559–22571 Search PubMed.
- E. J. Drake, B. R. Miller, C. Shi, J. T. Tarrasch, J. A. Sundlov, C. Leigh Allen, G. Skiniotis, C. C. Aldrich and A. M. Gulick, Nature, 2016, 529, 235–238 Search PubMed.
- K. Bloudoff, Diego A. Alonzo and T. M. Schmeing, Cell Chem. Biol., 2016, 23, 331–339 Search PubMed.
- K. Haslinger, C. Brieke, S. Uhlmann, L. Sieverling, R. D. Süssmuth and M. J. Cryle, Angew. Chem., Int. Ed., 2014, 53, 8518–8522 Search PubMed.
- C. Nguyen, R. W. Haushalter, D. J. Lee, P. R. L. Markwick, J. Bruegger, G. Caldara-Festin, K. Finzel, D. R. Jackson, F. Ishikawa, B. O'Dowd, J. A. McCammon, S. J. Opella, S.-C. Tsai and M. D. Burkart, Nature, 2014, 505, 427–431 CrossRef CAS PubMed.
- A. Tanovic, S. A. Samel, L.-O. Essen and M. A. Marahiel, Science, 2008, 321, 659–663 Search PubMed.
- K. J. Weissman, Nat. Chem. Biol., 2014, 11, 660–670 CrossRef PubMed.
- C. Qiao, D. J. Wilson, E. M. Bennett and C. C. Aldrich, J. Am. Chem. Soc., 2007, 129, 6350–6351 Search PubMed.
- N. M. Gaudelli and C. A. Townsend, J. Org. Chem., 2013, 78, 6412–6426 Search PubMed.
- N. M. Gaudelli, D. H. Long and C. A. Townsend, Nature, 2015, 520, 383–387 Search PubMed.
- N. M. Gaudelli and C. A. Townsend, Nat. Chem. Biol., 2014, 10, 251–258 Search PubMed.
- M. Peschke, K. Haslinger, C. Brieke, J. Reinstein and M. Cryle, J. Am. Chem. Soc., 2016, 138, 6746–6753 Search PubMed.
- K. Haslinger, M. Peschke, C. Brieke, E. Maximowitsch and M. J. Cryle, Nature, 2015, 521, 105–109 Search PubMed.
- C. Brieke, M. Peschke, K. Haslinger and M. J. Cryle, Angew. Chem., Int. Ed., 2015, 54, 15715–15719 CrossRef CAS PubMed.
- C. Brieke and M. J. Cryle, Org. Lett., 2014, 16, 2454–2457 CrossRef CAS PubMed.
- C. Brieke, V. Kratzig, K. Haslinger, A. Winkler and M. J. Cryle, Org. Biomol. Chem., 2015, 13, 2012–2021 Search PubMed.
- I. Wilkening, S. Gazzola, E. Riva, J. S. Parascandolo, L. Song and M. Tosin, Chem. Commun., 2016, 52, 10392–10395 Search PubMed.
- E. Riva, I. Wilkening, S. Gazzola, W. M. A. Li, L. Smith, P. F. Leadlay and M. Tosin, Angew. Chem., Int. Ed., 2014, 53, 11944–11949 Search PubMed.
- M. Winn, J. K. Fyans, Y. Zhuo and J. Micklefield, Nat. Prod. Rep., 2016, 33, 317–347 Search PubMed.
- J. Thirlway, R. Lewis, L. Nunns, M. Al Nakeeb, M. Styles, A.-W. Struck, C. P. Smith and J. Micklefield, Angew. Chem., Int. Ed., 2012, 51, 7181–7184 Search PubMed.
- G. C. Uguru, C. Milne, M. Borg, F. Flett, C. P. Smith and J. Micklefield, J. Am. Chem. Soc., 2004, 126, 5032–5033 CrossRef CAS PubMed.
- Z. Hojati, C. Milne, B. Harvey, L. Gordon, M. Borg, F. Flett, B. Wilkinson, P. J. Sidebottom, B. A. M. Rudd, M. A. Hayes, C. P. Smith and J. Micklefield, Chem. Biol., 2002, 9, 1175–1187 Search PubMed.
- R. H. Baltz, ACS Synth. Biol., 2014, 3, 748–758 Search PubMed.
- A. M. Gulick, ACS Chem. Biol., 2009, 4, 811–827 Search PubMed.
- G. L. Challis, J. Ravel and C. A. Townsend, Chem. Biol., 2000, 7, 211–224 Search PubMed.
- C. T. Walsh, R. V. O'Brien and C. Khosla, Angew. Chem., Int. Ed., 2013, 52, 7098–7124 CrossRef CAS PubMed.
- A. Miyanaga, J. Cieślak, Y. Shinohara, F. Kudo and T. Eguchi, J. Biol. Chem., 2014, 289, 31448–31457 CrossRef CAS PubMed.
- S. Cociancich, A. Pesic, D. Petras, S. Uhlmann, J. Kretz, V. Schubert, L. Vieweg, S. Duplan, M. Marguerettaz, J. Noëll, I. Pieretti, M. Hügelland, S. Kemper, A. Mainz, P. Rott, M. Royer and R. D. Süssmuth, Nat. Chem. Biol., 2015, 11, 195–197 CrossRef CAS PubMed.
- A. V. Ramaswamy, C. M. Sorrels and W. H. Gerwick, J. Nat. Prod., 2007, 70, 1977–1986 Search PubMed.
- N. A. Magarvey, M. Ehling-Schulz and C. T. Walsh, J. Am. Chem. Soc., 2006, 128, 10698–10699 Search PubMed.
- C. T. Calderone, S. B. Bumpus, N. L. Kelleher, C. T. Walsh and N. A. Magarvey, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 12809–12814 Search PubMed.
- S. C. Feifel, T. Schmiederer, T. Hornbogen, H. Berg, R. D. Süssmuth and R. Zocher, ChemBioChem, 2007, 8, 1767–1770 Search PubMed.
- S. K. Shrestha and S. Garneau-Tsodikova, ChemBioChem, 2016, 17, 1328–1332 CrossRef CAS PubMed.
- K. J. Labby, S. G. Watsula and S. Garneau-Tsodikova, Nat. Prod. Rep., 2015, 32, 641–653 RSC.
- J. M. Davidsen, D. M. Bartley and C. A. Townsend, J. Am. Chem. Soc., 2013, 135, 1749–1759 CrossRef CAS PubMed.
- B. Boll, T. Taubitz and L. Heide, J. Biol. Chem., 2011, 286, 36281–36290 Search PubMed.
- E. A. Felnagle, J. J. Barkei, H. Park, A. M. Podevels, M. D. McMahon, D. W. Drott and M. G. Thomas, Biochemistry, 2010, 49, 8815–8817 Search PubMed.
- T. Kittilä, M. Schoppet and M. J. Cryle, ChemBioChem, 2016, 17, 576–584 Search PubMed.
- S. Saha and S. E. Rokita, ChemBioChem, 2016, 17, 1818–1823 Search PubMed.
- B. R. Miller, J. A. Sundlov, E. J. Drake, T. A. Makin and A. M. Gulick, Proteins: Struct., Funct., Bioinf., 2014, 82, 2691–2702 CrossRef CAS PubMed.
- H. Kaljunen, S. H. H. Schiefelbein, D. Stummer, S. Kozak, R. Meijers, G. Christiansen and A. Rentmeister, Angew. Chem., Int. Ed., 2015, 54, 8833–8836 Search PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 Search PubMed.
- H. Kries, R. Wachtel, A. Pabst, B. Wanner, D. Niquille and D. Hilvert, Angew. Chem., Int. Ed., 2014, 53, 10105–10108 Search PubMed.
- S. Weist, C. Kittel, D. Bischoff, B. Bister, V. Pfeifer, G. J. Nicholson, W. Wohlleben and R. D. Süssmuth, J. Am. Chem. Soc., 2004, 126, 5942–5943 Search PubMed.
- S. Weist, B. Bister, O. Puk, D. Bischoff, S. Pelzer, G. J. Nicholson, W. Wohlleben, G. Jung and R. D. Süssmuth, Angew. Chem., Int. Ed., 2002, 41, 3383–3385 Search PubMed.
- N. Kessler, H. Schuhmann, S. Morneweg, U. Linne and M. A. Marahiel, J. Biol. Chem., 2004, 279, 7413–7419 Search PubMed.
- M. Welker and H. Von Döhren, FEMS Microbiol. Rev., 2006, 30, 530–563 Search PubMed.
- P. J. Belshaw, C. T. Walsh and T. Stachelhaus, Science, 1999, 284, 486–489 Search PubMed.
- T. Stachelhaus, H. D. Mootz, V. Bergendahl and M. A. Marahiel, J. Biol. Chem., 1998, 273, 22773–22781 Search PubMed.
- S. A. Samel, G. Schoenafinger, T. A. Knappe, M. A. Marahiel and L.-O. Essen, Structure, 2007, 15, 781–792 Search PubMed.
- T. A. Keating, C. G. Marshall, C. T. Walsh and A. E. Keating, Nat. Struct. Mol. Biol., 2002, 9, 522–526 Search PubMed.
- L. Robbel and M. A. Marahiel, J. Biol. Chem., 2010, 285, 27501–27508 Search PubMed.
- S. Pohle, C. Appelt, M. Roux, H.-P. Fiedler and R. D. Süssmuth, J. Am. Chem. Soc., 2011, 133, 6194–6205 Search PubMed.
- K. Bloudoff, D. Rodionov and T. M. Schmeing, J. Mol. Biol., 2013, 425, 3137–3150 Search PubMed.
- G. H. Hur, C. R. Vickery and M. D. Burkart, Nat. Prod. Rep., 2012, 29, 1074–1098 Search PubMed.
- C. J. Balibar and C. T. Walsh, ChemBioChem, 2008, 9, 42–45 Search PubMed.
- C. Rausch, I. Hoof, T. Weber, W. Wohlleben and D. Huson, BMC Evol. Biol., 2007, 7, 78 Search PubMed.
- W.-H. Chen, K. Li, N. S. Guntaka and S. D. Bruner, ACS Chem. Biol., 2016, 11, 2293–2303 Search PubMed.
- S. A. Samel, P. Czodrowski and L.-O. Essen, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2014, 70, 1442–1452 Search PubMed.
- D. B. Stein, U. Linne and M. A. Marahiel, FEBS J., 2005, 272, 4506–4520 Search PubMed.
- C. J. Balibar, F. H. Vaillancourt and C. T. Walsh, Chem. Biol., 2005, 12, 1189–1200 Search PubMed.
- R. B. Hamed, J. R. Gomez-Castellanos, L. Henry, C. Ducho, M. A. McDonough and C. J. Schofield, Nat. Prod. Rep., 2013, 30, 21–107 Search PubMed.
- V. Ulrich, M. Peschke, C. Brieke and M. Cryle, Mol. BioSyst., 2016, 12, 2992–3004 Search PubMed.
- M. Peschke, M. Gonsior, R. D. Süssmuth and M. J. Cryle, Curr. Opin. Struct. Biol., 2016, 41, 46–53 Search PubMed.
- B. Hadatsch, D. Butz, T. Schmiederer, J. Steudle, W. Wohlleben, R. Süssmuth and E. Stegmann, Chem. Biol., 2007, 14, 1078–1089 Search PubMed.
- E. Stegmann, S. Pelzer, D. Bischoff, O. Puk, S. Stockert, D. Butz, K. Zerbe, J. Robinson, R. D. Süssmuth and W. Wohlleben, J. Biotechnol., 2006, 124, 640–653 Search PubMed.
- D. Bischoff, S. Pelzer, A. Holtzel, G. J. Nicholson, S. Stockert, W. Wohlleben, G. Jung and R. D. Süssmuth, Angew. Chem., Int. Ed., 2001, 40, 1693–1696 Search PubMed.
- D. Bischoff, S. Pelzer, B. Bister, G. J. Nicholson, S. Stockert, M. Schirle, W. Wohlleben, G. Jung and R. D. Süssmuth, Angew. Chem., Int. Ed., 2001, 40, 4688–4691 CrossRef CAS.
- R. D. Süssmuth, S. Pelzer, G. Nicholson, T. Walk, W. Wohlleben and G. Jung, Angew. Chem., Int. Ed., 1999, 38, 1976–1979 Search PubMed.
- K. Woithe, N. Geib, K. Zerbe, D. B. Li, M. Heck, S. Fournier-Rousset, O. Meyer, F. Vitali, N. Matoba, K. Abou-Hadeed and J. A. Robinson, J. Am. Chem. Soc., 2007, 129, 6887–6895 Search PubMed.
- K. Zerbe, K. Woithe, D. B. Li, F. Vitali, L. Bigler and J. A. Robinson, Angew. Chem., Int. Ed., 2004, 43, 6709–6713 Search PubMed.
- D. Bischoff, B. Bister, M. Bertazzo, V. Pfeifer, E. Stegmann, G. J. Nicholson, S. Keller, S. Pelzer, W. Wohlleben and R. D. Süssmuth, ChemBioChem, 2005, 6, 267–272 Search PubMed.
- M. Peschke, C. Brieke and M. J. Cryle, Sci. Rep., 2016, 6, 35584 Search PubMed.
- M. E. Horsman, T. P. A. Hari and C. N. Boddy, Nat. Prod. Rep., 2016, 33, 183–202 RSC.
- D. Schwarzer, H. D. Mootz, U. Linne and M. A. Marahiel, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14083–14088 Search PubMed.
- J. G. Owen, M. J. Calcott, K. J. Robins and D. F. Ackerly, Cell Chem. Biol., 2016 DOI:10.1016/j.chembiol.2016.09.014.
- H. Takahashi, T. Kumagai, K. Kitani, M. Mori, Y. Matoba and M. Sugiyama, J. Biol. Chem., 2007, 282, 9073–9081 Search PubMed.
- Y. Liu, T. Zheng and S. D. Bruner, Chem. Biol., 2011, 18, 1482–1488 Search PubMed.
- J. W. Trauger, R. M. Kohli, H. D. Mootz, M. A. Marahiel and C. T. Walsh, Nature, 2000, 407, 215–218 Search PubMed.
- J. Jaitzig, J. Li, R. D. Süssmuth and P. Neubauer, ACS Synth. Biol., 2014, 3, 432–438 Search PubMed.
- R. S. Al Toma, C. Brieke, M. J. Cryle and R. D. Süssmuth, Nat. Prod. Rep., 2015, 32, 1207–1235 Search PubMed.
- L. Ray, K. Yamanaka and B. S. Moore, Angew. Chem., Int. Ed., 2016, 55, 364–367 Search PubMed.
- S. Zobel, S. Boecker, D. Kulke, D. Heimbach, V. Meyer and R. D. Süssmuth, ChemBioChem, 2016, 17, 283–287 Search PubMed.
- J. H. Crosa and C. T. Walsh, Microbiol. Mol. Biol. Rev., 2002, 66, 223–249 Search PubMed.
- S. Uhlmann, R. D. Süssmuth and M. J. Cryle, ACS Chem. Biol., 2013, 8, 2586–2596 CrossRef CAS PubMed.
- M. J. Cryle, C. Brieke and K. Haslinger, Amino Acids, Pept., Proteins, 2014, 38, 1–36 Search PubMed.
- M. J. Cryle, J. E. Stok and J. J. De Voss, Aust. J. Chem., 2003, 56, 749–762 Search PubMed.
- L. M. Podust and D. H. Sherman, Nat. Prod. Rep., 2012, 29, 1251–1266 Search PubMed.
- M. J. Cryle, Metallomics, 2011, 3, 323–326 Search PubMed.
- M. J. Cryle and I. Schlichting, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 15696–15701 Search PubMed.
- S. M. Barry, J. A. Kers, E. G. Johnson, L. Song, P. R. Aston, B. Patel, S. B. Krasnoff, B. R. Crane, D. M. Gibson, R. Loria and G. L. Challis, Nat. Chem. Biol., 2012, 8, 814–816 CrossRef CAS PubMed.
- M. J. Cryle, A. Meinhart and I. Schlichting, J. Biol. Chem., 2010, 285, 24562–24574 Search PubMed.
- V. Weichold, D. Milbredt and K.-H. van Pée, Angew. Chem., Int. Ed., 2016, 55, 6374–6389 Search PubMed.
- L. Du, C. Sánchez, M. Chen, D. J. Edwards and B. Shen, Chem. Biol., 2000, 7, 623–642 Search PubMed.
- M. A. Marahiel, Nat. Prod. Rep., 2016, 33, 136–140 Search PubMed.
- M. Hahn and T. Stachelhaus, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 275–280 Search PubMed.
- E. Dehling, G. Volkmann, J. C. J. Matern, W. Dörner, J. Alfermann, J. Diecker and H. D. Mootz, J. Mol. Biol., 2016, 428, 4345–4360 Search PubMed.
Footnote |
| † Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |